Abstract
Following efficient tumor therapy, some cancer cells may survive through a dormancy process, contributing to tumor recurrence and worse outcomes. Dormancy is considered a process where most cancer cells in a tumor cell population are quiescent with no, or only slow, proliferation. Recent advances indicate that redox mechanisms control the dormant cancer cell life cycle, including dormancy entrance, long-term dormancy, and metastatic relapse. This regulatory network is orchestrated mainly through redox modification on key regulators or global change of reactive oxygen species (ROS) levels in dormant cancer cells. Encouragingly, several strategies targeting redox signaling, including sleeping, awaking, or killing dormant cancer cells are currently under early clinical evaluation. However, the molecular mechanisms underlying redox control of the dormant cancer cell cycle are poorly understood and need further exploration. In this review, we discuss the underlying molecular basis of redox signaling in the cell life cycle of dormant cancer and the potential redox-based targeting strategies for eliminating dormant cancer cells.
1. Introduction
The past three decades have witnessed rapid developments in cancer therapy, including advances in surgery, chemotherapy, radiotherapy, targeted therapy, and immunotherapy [1,2,3,4]. However, tumor metastasis and relapse may happen years or even decades after surgical eradication due to cancer cell dormancy, a process in which cells remain viable but cease to display or only display slow proliferation [5,6]. For cancer cells, this process is also called quiescence; when most cells in a tumor go through quiescence, it is called tumor dormancy. Cancer treatment may eradicate most cancer cells at the primary site, but some cancer cells may enter a quiescent state to survive and stop proliferation, which is considered one of the reasons for minimal residual disease [7]. At the same time, cancer cell dissemination from the primary tumor may occur at an early stage to form a remote dormant population, contributing to the recurrence of refractory tumors [8]. The beginning of disseminated cancer cell dormancy is considered a niche-dependent process when disseminated cancer cells engage with supportive niches, including bone marrow and metastatic and perivascular niches, where disseminated cancer cells go through a reprogramming process to enter dormancy [9,10]. After a cellular reprogramming process, the cancer cells enter into long-term dormancy [11]. When dormant cancer cells gather sufficient nutrients or are exposed to specific stimuli (for example, hormones, oxidative stress), the dormant cancer cells may reactivate and cause tumor relapse [12,13]. During secondary treatment on the relapsed tumor, the disseminated cancer cells may re-enter dormancy, which contributes to poor survival and a refractory state in late-stage patients [14]. Intriguingly, dormant cancer cells show similar characteristics to drug-tolerant cells, persister cells, and cancer stem cells, indicating that the regulation of cancer dormancy may rely on mutation-independent mechanisms, including epigenetic regulation or adaptive stress response [15,16,17,18]. For decades, researchers have widely recognized that the principle of dormant cancer cell life cycles is related to both intracellular signaling and extracellular stimuli [19], indicating that the control of the dormant cancer cell life cycle may contain a stress-dependent mechanism.
Oxidative stress is usually caused by the accumulation of reactive oxygen species (ROS). The intracellular sources of ROS are mainly generated from nicotinamide adenine dinucleotide phosphate (NADPH) oxidases, the mitochondrial electron transport chain (ETC), and endoplasmic reticulum (ER) stress. Extrinsic factors, including radiation, chemicals, or damage also play essential roles in ROS generation [20]. Increased ROS level may inactivate Kelch-like ECH-associated protein 1 (KEAP1), an essential component of E3 ubiquitin ligase complex targeting (NRF2), thus promoting the translation of antioxidant proteins, including superoxide dismutase (SOD), catalase, glutathione peroxidase (GPX), peroxiredoxins (PRX), thioredoxins (TRX), and glutathione reductase (GR), which maintains redox homeostasis [21,22]. Early studies of redox biology encouraged the notion that ROS are the toxic byproduct in cell metabolism, while recent advances provide provocative insights into their role as intracellular signaling molecules at a low physiological level [23,24]. Redox modifications on thiol of cysteine residues, including S-sulfenic acids (–SOH), S-sulfinic acids (–SO2H), and intramolecular or intermolecular disulfides (–SS–), can modulate protein structure, enzyme activity, and protein–protein interactions, which modulates key regulators in multiple signaling pathways [25,26]. ROS and antioxidants are in balance (redox homeostasis) in normal cells, while increased ROS levels are considered a hallmark of cancer cells, leading to genomic instability, facilitated cell proliferation, increased motility, and activated oncogenic signaling [24,27]. Excess ROS produced by chemotherapy may induce irreversible cell damage and cause tumor cell death [28,29]. However, some cancer cells may stay in quiescence by overexpressing antioxidant enzymes to maintain low levels of ROS, even lower than that of normal cells, to evade chemotherapy-induced cell death [30,31]. Such quiescent cancer cells are highly relevant to tumor recurrence and have several characteristics similar to dormant cancer cells, indicating potential relationships between redox and cancer dormancy [32]. Emerging evidence suggests that redox signaling is strongly associated with the dormant cancer cell life cycle (Figure 1). In this review, we focus on the mechanisms that control redox mechanisms in the dormant cancer cell life cycle, as well as strategies for targeting dormant cancer cells from a redox perspective.
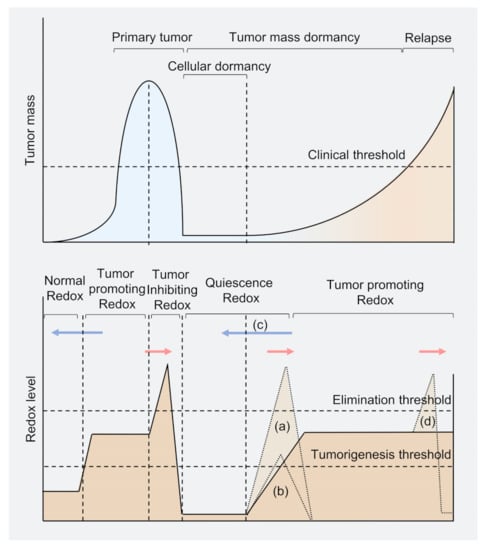
Figure 1.
Model of tumor burden and redox level in cancer cell dormancy. Primary tumors rely on mild oxidative stress that increases ROS levels beyond the tumorigenesis threshold to proliferation. While under therapeutic conditions, the ROS level may be beyond the elimination threshold to rapidly decrease tumor mass. Several cancer cells (including residual cells and disseminated cells) may reprogram and survive in a redox level lower than normal cells and enter dormancy. When some dormant cancer cells awaken in the tumor population, the redox level may again exceed the tumorigenesis threshold. The tumor proliferates slowly under the clinical threshold, called tumor mass dormancy. Once the tumor mass exceeds the clinical threshold, the tumor has relapsed. Blue arrows represent potential strategies to conduct antioxidant therapy to treat tumors; red arrows represent potential oxidant-dependent therapy. (a) Strategy to kill dormant cancer cells through excessive ROS; (b) Strategy for reawaking dormant cancer cells to sensitize cancer cells to anti-proliferation drugs; (c) Strategy using antioxidant to keep dormant cancer cells from awaking; (d) Chemotherapy on recurrent tumors, but may enter into another life cycle of dormant cancer cells.
2. Dormancy Entrance in Redox Perspective
Disseminated cancer cells from the primary tumor site may transfer to a supportive niche for dormancy through intravascular processes [33,34]. Traditionally, researchers considered dissemination as a late stage of the metastatic cascade, which happened when cancer cells had acquired several key mutations to complete the colonization process [35]. However, multiple lines of evidence indicate that about 80% of metastasis is partly caused by cancer cell dissemination at an early stage, meaning that the dormant transform process may not be solely driven by genetic change [8,36,37]. Recent advances suggest that the regulation of dormancy entrance is closely related to redox signaling (Figure 2).
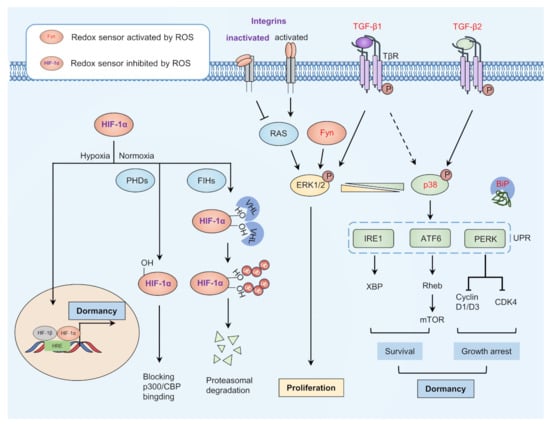
Figure 2.
Model of dormancy entrance. The balance between proliferation and dormancy may be partly dependent on the ratio of p-p38 and p-ERK1/2. Redox can activate TGF-β2 and inactivate integrins to activate p38 and inhibit activation of ERK1/2, as well as to modify ER chaperone BiP for release from key regulators of ER-stress signaling, thus activating ER-stress signaling and promoting cell survival and growth arrest. Redox-activated TGF-β1 and Fyn can activate ERK1/2 to promote cell proliferation, while integrins activate when it is not redox-modified. HIF-1α is inhibited under normoxia through redox modification mediated by PHDs and FIHs. Under hypoxia conditions, HIF-1α may translocate to the nucleus and promote transcription of dormant-related genes. ER, endoplasmic reticulum; ERK1/2, extracellular signal-regulated kinase 1/2; FIHs, factor inhibiting HIF-1; HIF-1α, hypoxia-inducible transcription factors 1α; TGF-β2, transforming growth factor β2.
2.1. Balance between Dormancy and Proliferation
The ratio between the active form of two mitogen-activated protein kinases (MAPKs), extracellular signal-regulated kinase 1/2 (ERK1/2), and p38 are considered to be an indicator of cancer cell dormancy [38]. Specifically, the cancer cells favor proliferation when p-ERK1/2 play a dominant role (high p-ERK/p-p38 ratio); otherwise, the cancer cells prefer to enter dormancy (low p-ERK/p-p38 ratio). Recent studies indicate that a redox sensor tyrosine-protein kinase Fyn regulates both p-ERK1/2 and p-p38 through Ras/Raf/MEK/ERK pathway and MEK3/6, respectively. Intriguingly, the activation of p38 is considered redox-dependent rather than ERK1/2, since mutating the redox modified site Cys488 on Fyn turns the p38-relevant phenotype into ERK1/2-dominate phenotype [39]. Consistently, a study on global profiling of cysteine modification in C. elegans revealed that the enrichment score of p38 MAPK signaling was most significant [40], indicating that p38 MAPK signaling may take priority under redox regulation. Cys173 has been identified as a conserved redox-sensitive site for p38 activation regulation in C. elegans [40]. Furthermore, Cys119, and Cys162 in p38α also act as redox sensors, managing the formation of a heterodimer with mitogen-activated protein kinase kinase 3 (MKK3) [41]. However, the biological function and regulatory mechanisms of these redox-sensitive sites need further elucidation.
After activation of p38 under oxidative stress, the downstream ER-stress signaling may contribute to the quiescence of disseminated cancer cells [42,43]. Binding immunoglobulin protein (BiP) is an ER chaperone that binds under normal conditions with three downstream transducers of ER-stress signaling, including inositol-requiring enzyme 1α (IRE1α), activating transcription factor 6α (ATF6α) and proline-rich receptor-like protein kinase (PERK) [44]. When unfolded protein reaction (UPR) is induced, BiP then binds with misfolded peptides with release from ER transducers, thus activating ER-stress signaling [45]. Redox activated glutathione peroxidase 7 (GPx7) can oxidize BiP on Cys41 and Cys420 to form an intramolecular disulfide bond, resulting in an enhanced ability to bind to misfolded peptides and thus ER-stress signaling activation [46]. Redox-mediated activation of IRE1α and ATF6α may support dormant cancer cell survival, while activating PERK phosphorylates eukaryotic initiation factor 2α (eIF2α) and inhibiting its translation of cyclin-dependent kinases (CDK4), Cyclin D1/D3, leading to quiescence of dormant cancer cells [47]. These findings indicate that redox mechanisms may trigger dormant processes through redox modification on BiP, while redox modifications on other key regulators in ER-stress signaling need further exploration [48]. Moreover, other p38 downstream factors, including nuclear receptor subfamily 2 group F member 1 (NR2F1) and p53, may also participate in the entrance of cancer cell quiescence [49,50].
2.2. Reforming the Related Microenvironment
The tumor microenvironment contributes to tumor progression, immune evasion, metastasis, drug resistance, and dormancy, where redox reactions play a pivotal role [51,52]. Recent advances indicate redox regulates the entrance of cancer dormancy through regulating several key factors in the microenvironment, including hypoxia-inducible transcription factors (HIFs), integrins, and transforming growth factor β (TGF-β) signaling.
Due to the rapid growth of tumor cells and the aberrant formation of blood vessels, the tumor microenvironment exhibits hypoxia regions, resulting in cancer cell dormancy [53,54,55]. Redox controls hypoxic signaling by regulating the stability of HIFs. Under normal oxidative conditions, prolyl-4-hydroxylases (PHDs) hydroxylate HIF-1α on proline residues so that the von Hippel–Lindau protein (pVHL) polyubiquitinates HIF-1α and leads to 26S proteasome system-dependent degradation [56]. In addition, factor inhibiting HIF-1 (FIH-1) can hydroxylate HIF-1α on asparagine residues, thus preventing its binding to the p300/CBP complex and subsequent transcription [57]. While under hypoxia, HIF-1α can translocate into the nucleus and conduct transcription of dormant-related genes (for example, Cyclin G2). In addition, recent studies indicate constitutively photomorphogenic 9 signalosome subunit 8 (CSN8) can stabilize HIF-1α protein through deubiquitylation and upregulates HIF-1α transcription in a nuclear factor-κB (NF-κB)-dependent manner in the hypoxia tumor microenvironment, leading to epithelial–mesenchymal transition (EMT) and dormancy of colorectal cancer cells [58].
Apart from HIF-1α, redox in the tumor microenvironment can affect intracellular signaling by regulating several membrane receptors, including integrins and TGF-βreceptors, leading to cancer dormancy [59,60]. Redox can mediate intramolecular disulfide bond formation in integrins, promoting an inactivated bent conformation, resulting in cancer cell dormancy [61,62]. However, when the disulfide bond is reduced, integrins switch to the activated upright conformation, thus maintaining cell cycle progression through RAS-ERK/MAPK pathways or through cytoskeleton reorganization [63,64]. Multiple lines of evidence indicate that the TGF-β ligand secreted by tumor cells and niche cells in the tumor microenvironment can control cancer cell dormancy [59]. Of note, a recent advance shows that redox and TGF-β/ bone morphogenetic protein (BMP) signaling are sufficient to fully induce prostate cancer cell dormancy, indicating the pivotal role of redox in promoting cancer cell dormancy [65]. ROS can turn latent TGF-β into an active form and facilitate the phosphorylation of the TGF-β receptor I (ALK5), thus activate the TGF-β pathway [66,67]. Furthermore, ROS can inactivate phosphatase and tensin homolog (PTEN)/PPM1A-dephosphorylation of SMAD2/3, thus promoting the transcription of dormancy-inducing genes [68]. Intriguingly, activated TGF-β1 promotes T4-2 breast cancer cells to escape from dormancy in a 3D culture model while inhibiting T4-2 breast cancer cell proliferation in the 2D model, according to recent studies, which may be caused by the different redox states of these culture models [59,69]. Unlike TGF-β1, TGF-β2 to date has only been shown to exhibit its pro-dormant function in various cancer cells [70,71]. Other than intracellular signaling, redox effects can control TGF-β signaling in the local niches, thus affecting the dormancy of disseminated tumor cells. For example, bone marrow-derived TGF-β2 induces dormancy in both head and neck squamous cell carcinoma and a prostate cancer model in a tumor-specific and ligand-specific manner [70,71]. Moreover, redox controls the disulfide bond formation between homotrimers of thrombospondin-1 (TSP-1) in the perivascular niche, facilitating the activation of downstream TGF-β signaling and leading to quiescence of breast cancer cells [72]. Importantly, redox has the ability to disseminate cancer cells into dormancy by inactivating integrins or activating TGF-β2. Intriguingly, TGF-β1 exhibits a context-dependent function on cancer cell dormancy; how redox is involved in this process should be further evaluated.
3. Redox-Mediated Long-Term Dormancy
When disseminated cancer cells enter the dormancy process, they have to keep themselves in quiescence and away from harmful stimuli. Redox mechanisms can sustain cancer cell dormancy in both an intrinsic (for example, sustaining stemness, inducing autophagy) and extrinsic way (immune cells in microenvironment) (Figure 3).
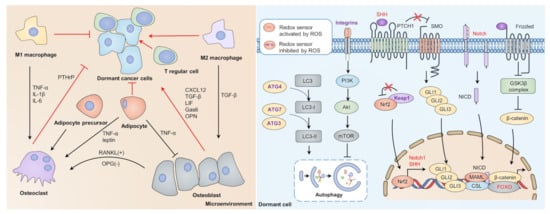
Figure 3.
Dormant-dependent microenvironment and intrinsic supportive signaling. Interactions between immune cells and cytokines balance the entrance and escape of dormancy. Osteoblasts, M2 macrophages, and T regular cells are considered protectors of cancer dormancy, while osteoclasts, M1 macrophage, and adipocytes may break cancer dormancy. Redox control of dormant sustaining mechanisms. Redox may activate FOXO to bind with β-catenin to promote transcription of dormancy-related genes competitively. Upon oxidative stress, KEAP1 can be inactivated and release NRF2 to translocate into the nucleus, thus promoting transcription of Notch1 and SHH. Wnt, Notch, and Hedgehog signaling may sustain cancer dormancy through transcription of stemness-related genes. Oxidative stress may activate autophagy by impairing the integrin/PI3K/Akt/mTOR axis, which impairs autophagy by inactivating ATG4, ATG7, and ATG3. FOXO, forkhead box O; KEAP1, Kelch-like ECH-associated protein 1; mTOR, mammalian target of rapamycin; NRF2, nuclear factor erythroid 2-related factor 2; PI3K, phosphoinositide 3-kinase; SHH, sonic hedgehog.
3.1. Redox Sustaining Cancer Cell Quiescence
Dormant cancer cells exhibit slow cycling, reversibility, and drug tolerance, thus sharing many similarities with cancer stem cells (CSCs) [17,73]. Recent advances indicate that redox-dependent intrinsic signaling (for example, Wnt signaling, Notch signaling, and Hedgehog signaling) plays an essential role in sustaining cancer stemness as well as cancer dormancy [74]. ROS mediates the nuclear translocation and affiliation between β-catenin and forkhead box O (FOXO) proteins by activating c-Jun N-terminal kinase (JNK) signaling, leading to apoptosis or quiescence [75,76]. When FOXO proteins are inactivated, β-catenin can promote proliferation by binding with T cell factor (TCF) transcription factors [77], suggesting the regulatory role of redox in Wnt signaling-dependent cancer dormancy. When cancer cells enter dormancy, the ROS levels may remain relatively low due to the high expression of antioxidant enzymes (for example, NRF2, SOD2) [32,78]. Intriguingly, NRF2 was found to coactivate with β-catenin in hepatocellular cancer (HCC), which may contribute to the refractory nature of HCC [79]. In addition, the expression of NRF2 may lead to downstream transcription of Notch1 and sonic hedgehog, which may activate Notch and Hedgehog signaling [80,81,82]. Multiple lines of evidence indicate the relationship between stemness-sustaining signaling and dormancy. For example, treating prostate cancer cells with Wnt5A promotes their dormancy in bone through the Wnt5A/ROR2/SIAH2 axis [83]. Itraconazole disrupts colorectal cancer dormancy by Wnt inhibition, indicating the essential function of Wnt signaling in dormancy maintenance [84]. In addition, IL-23 treatment induces cancer cell dormancy via Wnt/Notch signaling in esophageal squamous carcinoma cells (ESCCs) [85]. Moreover, Notch1 and Notch2 showed a regulatory role in breast cancer dormancy and recurrence [86,87]. The Hedgehog pathway also participates in cancer dormancy, inhibition of GLI2 abrogating leukemia stem cell (LSC) dormancy and chemoresistance [88]. These data suggest the pro-dormant role of stemness-sustaining signaling; however, the crosstalk and the detailed molecular mechanism involved in redox and quiescence still need further exploration.
Oxidative stress also modulates cancer cell dormancy through autophagy [89]. Autophagy is considered to be a lysosome-dependent degradation process to cope with stress [90]. Upon ER stress, autophagy can recycle misfolded proteins and damaged organelles, thus promoting survival signaling and maintaining dormancy [91,92]. In addition, redox can activate autophagy through the integrins/PI3K/Akt/mTOR axis and transcriptional control, thereby managing dormancy induction [63,93,94]. Intriguingly, oxidative stress also inhibits autophagy by multiple mechanisms. For example, the LC3 processer ATG4B showed attenuated activity after redox modification at Cys292 and Cys361, causing decreased autophagic flux [89,95]. Furthermore, oxidation of Atg3 and Atg7 abrogates phosphatidylethanolamine (PE) conjugation to microtubule-associated proteins 1A/1B light chain 3 (LC3), required for autophagy elongation in a mouse model [96]. Consistently, a hypoxic condition inhibits mTOR to activate autophagy [97]. The context-dependent role of ROS in regulating autophagy suggests that ROS may contribute to both entrance and abrogation of dormancy.
3.2. Redox Managing Dormant Microenvironment
Apart from intrinsic factors, the redox level in the microenvironment of the dormant cancer niche is also essential for the maintenance of cancer dormancy [98]. The microenvironment in dormant cancer niches is considered a complex system, where different immune cells and cytokines orchestrate the balance of the inflammatory or anti-inflammatory response [99]. For instance, osteoblasts may secrete CXC chemokine ligand 12 (CXCL12) [100,101], TGF-β [71], growth arrest-specific protein 6 (GAS6) [102], osteoprotegerin (OPG) [103], osteopontin (OPN) [104], and leukemia inhibitory factor (LIF) [105], to promote colonization and dormancy of disseminating cancer cells in bone marrow. Osteoclasts may play a dual role by secreting TGF-β to promote dormancy [106], while reverse dormancy occurs through parathyroid hormone-related protein (PTHrP) [107]. In addition, adipocyte precursors produce receptor activators of NF-κB ligand (RANKL) to stimulate osteoclast differentiation, thus awakening dormant cancer cells [108]. Similarly, adipocytes may promote osteoclastogenesis by secreting tumor necrosis factor α (TNF-α) [109] and may induce proliferation with leptin [110], which impairs the dormant state of quiescent cancer cells. Moreover, different subtypes of macrophages play different roles in modulating cancer dormancy through different secreted cytokines [111]. Furthermore, recent advances indicate that elevated CD73 in CD150high CD4+ T cells and CD150high T regulatory cells may protect dormant HSCs in the bone marrow niche from oxidative stress and immune elimination [112,113]. Managing redox level in the dormant microenvironment may become a potential strategy to control cancer dormancy. However, the crosstalk among these immune cells and inflammatory-dependent factors requires further study.
4. Redox Mechanisms Control the Metastatic Relapse of Dormant Cancer Cells
The entrance into and maintenance of cancer cell dormancy contribute to the survival of residual cancer cells after treatment, while the reactivation of dormant cancer cells is closely related to the metastatic relapse of various cancer types [6]. However, the mechanism of dormant cancer cell reactivation still needs further exploration. One possible explanation is that it is caused by both intrinsic and extrinsic factors. When disseminated cancer cells enter dormancy, they begin accumulating nutrients to support the reinitiation of tumor growth. Once they have gathered sufficient nutrients, the dormant cancer cells may trigger the reactivation program and maintain proliferation [19]. From the extrinsic aspect, adaptive stress responses, including hypoxia, oxidative stress, and UPR, have been shown to contribute to the reactivation of cancer cell dormancy [13].
4.1. Redox Regulates Metabolism-Related Reactivation of Dormant Cancer Cells
Although there are multiple studies on the proliferation/dormancy balance through the p-ERK/p-p38 ratio, direct studies on dormant cancer cell reactivation are still limited. Recent studies reveal that lipid metabolism participates in reactivation from dormancy [114]. For instance, the fatty acid translocase CD36 is upregulated in oral and prostate cancer cells, thus increasing the synthesis and uptake of β-oxidized lipid and driving metastatic outgrowth [115,116,117]. Intriguingly, redox modifications on Cys333 and Cys272 support the activity of CD36 and accelerate gastric cancer metastasis [118]. In addition, epoxyeicosatrienoic acids produced by P450 epoxygenases may be upregulated under oxidative stress, contributing to tumor dormancy escape [119]. Consistently, recent studies confirmed that oxidized lipids derived from stress-activated neutrophils reactivate dormant cancer cells during early recurrence in cancer [120]. Of note, NRF2 promotes reactivation of dormant breast cancer cells by sustaining redox homeostasis and nucleotide metabolism, indicating the importance of the antioxidant system in studying dormancy reactivation [32].
4.2. Redox Related Cell-Extrinsic Environmental Changes
Remodeling of the extrinsic environment is another pivotal cause of redox-mediated cancer dormancy escape [121,122]. Recent studies indicate that the induction of a microenvironment by nasal instillation of lipopolysaccharide (LPS) or tobacco smoke exposure causes the formation of neutrophil extracellular traps (NETs), leading to the proteolysis of the extracellular matrix (ECM) protein laminin and thrombospondin-1 (TSP-1) degradation, which mediates dormant cancer cells reactivation through integrin-dependent pathways [6,123]. For hormone-dependent breast cancer, estrogen can reactivate dormant breast cancer cells and promote cancer recurrence, where redox signaling may also play a pivotal role [124,125]. This mechanism may contribute to breast cancer relapse and the poor prognosis for recurrent breast cancer [12]. Moreover, remodeling of the endosteal niche by osteoclasts activates dormant myeloma cells [126]. Of note, sprouting neovasculature in the perivascular niche promotes dormancy escape through TGF-β1 and periostin derived from endothelial tip cells [72].
5. Targeting Dormant Cancer Cell Life Cycle by the Redox-Mediated Mechanism
Due to the limitation of any systematic studies on the redox state in dormant cancer cells, it is not yet possible to provide a precise strategy for targeting dormant cancer cells in different situations. However, there are typically three strategies to choose from for redox management, that is, targeting redox to sleep dormant cancer cells, awaking dormant cancer cells to increase the efficiency of anti-proliferation drugs, and eliminating dormant cancer cells through excessive oxidative stress (Figure 4).
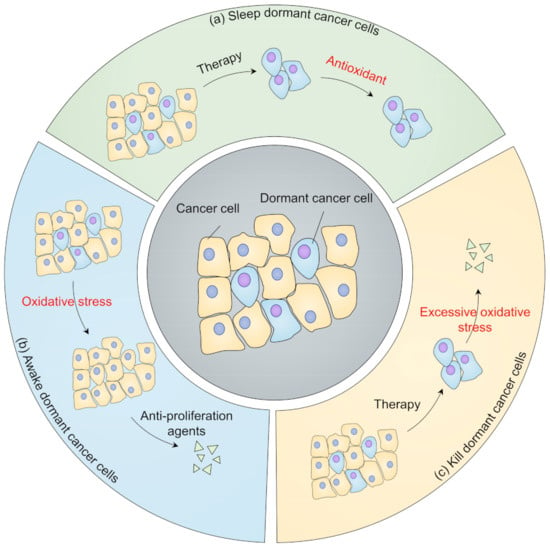
Figure 4.
Strategies targeting dormancy from a redox perspective. (a) Keep dormant cancer cells asleep. After cancer therapy, dormant cancer cells may remain viable and remain at low redox levels. Antioxidant treatment may prevent dormant cancer cells from reactivating; (b) Awaken dormant cancer cells. Oxidants are used to reawaken dormant cancer cells, thus sensitizing them to anti-proliferation agents; (c) Kill dormant cancer cells. Oxidative phosphorylation inhibitors, autophagy inhibitors, and ferroptosis inducers can be used to eliminate dormant cancer cells.
5.1. Sleep or Reactivation in Dormant Cancer Cells
Preventing dormant cancer cells from reactivation by treatment with antioxidant agents may prolong the survival of patients who have a high probability of recurrence. Recent studies indicate that retinoic acid can reduce ROS under stress, sustaining hematopoietic stem cell (HSC) dormancy [127]. Of note, retinoic acid combined with 5-azacytidine as a dormancy-maintaining treatment has been evaluated in phase II clinical trial for head and neck squamous cell carcinoma (HNSCC) therapy (NCT03572387). For hormone-dependent prostate cancer and breast cancer, tumor recurrence is redox-dependent, contributing to poor patient survival [128,129]. A combination of the antioxidant muscadine grape extract with androgen deprivation therapy to treat recurrent prostate cancer has entered a phase II clinical trial (NCT03496805). Similarly, antioxidant green tea catechin extracts treating hormone receptor-negative stage I–III breast cancer are being evaluated in phase I clinical trial (NCT00516243). In addition, strategies to maintain cancer cell dormancy, including estrogen antagonists, inhibition of CDK4/6, ERK, and integrin signaling, may prevent reactivation of dormant cancer cells and tumor relapse, as shown by an acceptable clinical response in breast cancer [60,130,131,132]. However, targeting redox modification of key regulators in dormant signaling may lack specificity, which needs further exploration. Sustaining dormancy may require lifetime treatment, and some dormant cancer cells may not respond to therapeutic agents, causing low proliferation of tumors.
Another strategy is to reactivate dormant cancer cells so that anti-proliferation agents can be used to inhibit these dormant cancer cells. For example, recent studies indicate that enhanced lipid metabolism sensitizes dormant cancer cells to 5-aminolevulinic acid-based photodynamic therapy [133]. Intriguingly, dormant HSCs undergoing acute interferon α (IFN-α) treatment are sensitive to anti-proliferation agents. Consistently, the combination of pegylated IFNs with nilotinib provides a better molecular response than nilotinib alone in chronic leukemia (CML) [134], and is under clinical evaluation for CML patients who have stable detectable molecular residual disease after imatinib treatment (NCT01866553). By contrast, chronic IFN-α treatment may cause HSCs to re-enter into the dormant state during chemotherapy, due to the different cellular ROS levels related to the two treatments [135,136,137]. Unlike strategies to keep dormant cancer cells asleep, reactivating dormant cancer cells may be more challenging, since activating dormant cancer cells is dose-dependent and could even promote tumor progression, limiting the clinical use of reactivation strategies. For example, a study on estrogen receptor alpha-positive dormant breast cancer treatment found that AMPK activated by metformin or high fatty acid intake may induce persistency of breast cancer cells under endocrine therapy; however, fatty acid oxidation inhibitors in combination with endocrine therapy may eliminate dormant cancer cells [138].
5.2. Killing Dormant Cancer Cells in a Redox Way
Recent advances indicate that dormant pancreatic ductal adenocarcinoma cells (PDACs) rely on oxidative phosphorylation, which is highly sensitive to the oxidative phosphorylation inhibitor oligomycin [139], for survival and recurrence. Similarly, researchers found that quiescent human leukemia stem cells (LSCs) are highly reliant on B-cell lymphoma 2 (BCL-2)-dependent oxidative phosphorylation; using BCL-2 inhibitors can selectively eradicate quiescent LSCs [140]. In addition, the novel oxidative phosphorylation inhibitor IACS010759 has been found to benefit patients with minimal residual acute myeloid leukemia (AML) cells after chemotherapy [141,142]. IACS010759 is entering phase I clinical trials for relapsed or refractory AML and other advanced cancers (NCT02882321, NCT03291938).
Impaired autophagy induced by hydroxychloroquine (HCQ), 3-methyladenine, or bafilomycin decreases the viability of dormant breast cancer cells through redox-dependent apoptosis, while for relapsed tumors, autophagy inhibition may increase the risk of metastasis, which indicates the context-dependent role of redox in these two cases [91,143]. A combination of autophagy inhibitor HCQ, CDK4/6 inhibitor Palbociclib, and the immune checkpoint programmed cell death-1 (PD-1)/Programmed death-ligand 1 (PD-L1) inhibitor avelumab to eradicate dormant breast cancer has entered a phase II clinical trial (NCT04841148). For patients who have detectable disseminated breast cancer cells in bone marrow, the combination therapy of HCQ and everolimus (NCT03400254) or the combination therapy of HCQ and gedatolisib (NCT03400254) are currently under clinical evaluation. Furthermore, the inhibition of ER-stress sensor PERK selectively eliminates dormant cancer cells under hypoxia conditions, suggesting targeting ER stress may also be a potential therapeutic strategy [144].
Ferroptosis is a redox-dependent process caused by redundant accumulated oxidized lipids, which has attracted much attention for tumor therapy [145,146]. Cancer cells that survive from targeted therapy or chemotherapy are efficiently eliminated by inhibiting glutathione peroxidase 4 (GPX4), the major antioxidant enzyme for reducing oxidated lipids [147,148]. Erlotinib, vemurafenib, and lapatinib eradicate PC9, A375, and BT474 cell lines through GPX4 inhibition, respectively, indicating the clinical potential of GPX4 inhibitors for dormant cancer therapy [148]. It is encouraging that GPX4 inhibitors [145], including altretamine and ashwagandha, are currently under clinical evaluation (NCT00002936, NCT04092647). In addition, aldehyde dehydrogenase (ALDH) can also sustain cancer survival under redundant lipid peroxidation by eliminating the downstream toxic aldehyde products [149]. The ALDH inhibitor crizotinib can abrogate the transition from toxic aldehydes to a harmless acid form, resulting in increased oxidative stress and the elimination of dormant cancer cells [150]. Other than GPX4 or ALDH inhibitors, there exist numerous strategies for inducing ferroptosis, including glutathione (GSH) inhibitors, iron activators, SLC7A11 inhibitors. The therapeutic effects of these strategies for killing dormant cancer cells need further evaluation.
6. Conclusions
In this review, we have summarized the recent advances in redox signaling in dormant cancer cells. Redox displays context-dependent roles in the control of the dormant cancer cell life cycle, indicating the complexity of the molecular mechanisms involved. Cancer cells rely on oxidative stress to promote proliferation and progression; however, under excess oxidative conditions (for instance, chemotherapy, radiotherapy), ROS may inactivate integrins and activate TGF-β2 on the membrane of some tumor cells, leading to the activation of p38 and downstream signaling (for example, ER-stress signaling), which ultimately causes cancer cell dormancy. Dormant cancer cells may highly express antioxidant enzymes, including NRF2, to maintain low ROS levels and upregulate stemness-sustaining signaling (for example, Wnt, Notch, Hedgehog signaling). During the dormant state, autophagy may cope with stress conditions to maintain cell quiescence. Apart from intracellular factors, the immune cells and cytokines in the dormant cancer microenvironment also participate in preserving cancer dormancy. When dormant cancer cells accumulate sufficient nutrients or suffer from oxidative stress from the extracellular microenvironment, cancer cells may escape from dormancy through activation of TGF-β1 or proliferation signaling. These highly reversible processes contribute to refractory or recurrent tumors, which currently lack efficient therapeutic strategies. From a redox perspective, there are three possible strategies: A combination of antioxidants (for example, retinoic acid, muscadine grape extract, green tea catechin extract) with targeted therapy to prevent reactivation of dormant cancer cells; Killing dormant cancer cells through excessive oxidative stress caused by inhibition of pro-survival signaling (oxidative phosphorylation and autophagy) and promoting ferroptosis; Combination of dormancy reactivating agents (for example, IFN-α) with an anti-proliferation agent. However, the latter has been limited in clinical use due to the difficulty of managing context-dependent redox levels and the risk of cancer progression. Encouragingly, several strategies have been shown to be effective and have entered clinical trials, which may favor the clinical treatment of cancer dormancy (Table 1).

Table 1.
Summary of clinical trials targeting dormant cancer cells from redox perspectives.
Despite the rapid development of novel strategies to overcome dormant tumors, our molecular understanding of redox-dependent dormancy is still in its infancy. The main obstacle in studying redox-dependent dormancy is determining the redox threshold indicators in the dormant cancer cell life cycle (for example, dormancy entrance, maintaining, reactivation, and redox-dependent death). In addition, current strategies targeting redox are rather broad-based regulating methods, while targeted therapy and precision medicine are the current trends in cancer therapy. Exploring the biological function of redox modification on key regulators and the crosstalk between redox and dormancy-related signaling may assist the development of strategies for targeting allosteric disulfides to eliminate dormant tumors. Furthermore, since redox control of dormancy is context-dependent, intensive studies on redox-regulated networks in different organs and individuals should be considered. With increased understanding of the molecular mechanisms involved in clinical treatment, targeting dormant cancer cells through a redox perspective may shed new light on cancer therapy.
Author Contributions
Conceptualization, C.H. and R.X.; Writing—original draft preparation, B.L., Y.H. and H.M.; Writing—review and editing, E.C.N.; visualization, B.L. and H.M.; supervision, C.H. and R.X. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Guangdong Basic and Applied Basic Research Foundation, grant number 2019B030302012; National Key Research and Development Project of China, grant number 2020YFA0509400; the Chinese NSFC, grant number 81821002, 81790251, and 82130082; Sichuan Applied Basic Research Project, grant number 2020YJ0107; Ningbo Health Young Talents Fund, grant number 2020SWSQNGG; and The Affiliated Hospital of Medical School of Ningbo University Qingmiao Talent Cultivation Fund, grant number FYQM-KY-202003.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Tian, H.; Zhang, J.; Zhang, H.; Jiang, Y.; Song, A.; Luan, Y. Low side-effect and heat-shock protein-inhibited chemo-phototherapy nanoplatform via co-assembling strategy of biotin-tailored IR780 and quercetin. Chem. Eng. J. 2020, 382, 123043. [Google Scholar] [CrossRef]
- Tian, H.; Zhao, S.; Nice, E.C.; Huang, C.; He, W.; Zou, B.; Lin, J. A cascaded copper-based nanocatalyst by modulating glutathione and cyclooxygenase-2 for hepatocellular carcinoma therapy. J. Colloid. Interf. Sci. 2021, 607, 1516–1526. [Google Scholar] [CrossRef]
- Marx, V. How to pull the blanket off dormant cancer cells. Nat. Methods 2018, 15, 249–252. [Google Scholar] [CrossRef]
- Aguirre-Ghiso, J.A. How dormant cancer persists and reawakens. Science 2018, 361, 1314–1315. [Google Scholar] [CrossRef] [PubMed]
- Nicolini, A.; Rossi, G.; Ferrari, P.; Carpi, A. Minimal residual disease in advanced or metastatic solid cancers: The G0-G1 state and immunotherapy are key to unwinding cancer complexity. Semin. Cancer Biol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Hosseini, H.; Obradović, M.M.S.; Hoffmann, M.; Harper, K.L.; Sosa, M.S.; Werner-Klein, M.; Nanduri, L.K.; Werno, C.; Ehrl, C.; Maneck, M.; et al. Early dissemination seeds metastasis in breast cancer. Nature 2016, 540, 552–558. [Google Scholar] [CrossRef]
- Goddard, E.T.; Bozic, I.; Riddell, S.R.; Ghajar, C.M. Dormant tumour cells, their niches and the influence of immunity. Nat. Cell Biol. 2018, 20, 1240–1249. [Google Scholar] [CrossRef]
- Hu, Z.; Ding, J.; Ma, Z.; Sun, R.; Seoane, J.A.; Scott Shaffer, J.; Suarez, C.J.; Berghoff, A.S.; Cremolini, C.; Falcone, A.; et al. Quantitative evidence for early metastatic seeding in colorectal cancer. Nat. Genet. 2019, 51, 1113–1122. [Google Scholar] [CrossRef] [PubMed]
- Dobson, S.M.; García-Prat, L.; Vanner, R.J.; Wintersinger, J.; Waanders, E.; Gu, Z.; McLeod, J.; Gan, O.I.; Grandal, I.; Payne-Turner, D.; et al. Relapse-Fated Latent Diagnosis Subclones in Acute B Lineage Leukemia Are Drug Tolerant and Possess Distinct Metabolic Programs. Cancer Discov. 2020, 10, 568–587. [Google Scholar] [CrossRef]
- Pan, H.; Gray, R.; Braybrooke, J.; Davies, C.; Taylor, C.; McGale, P.; Peto, R.; Pritchard, K.I.; Bergh, J.; Dowsett, M.; et al. 20-Year Risks of Breast-Cancer Recurrence after Stopping Endocrine Therapy at 5 Years. N. Engl. J. Med. 2017, 377, 1836–1846. [Google Scholar] [CrossRef]
- Senft, D.; Ronai, Z.E. Adaptive Stress Responses During Tumor Metastasis and Dormancy. Trends Cancer 2016, 2, 429–442. [Google Scholar] [CrossRef] [PubMed]
- Mahvi, D.A.; Liu, R.; Grinstaff, M.W.; Colson, Y.L.; Raut, C.P. Local Cancer Recurrence: The Realities, Challenges, and Opportunities for New Therapies. CA Cancer J. Clin. 2018, 68, 488–505. [Google Scholar] [CrossRef] [PubMed]
- Vallette, F.M.; Olivier, C.; Lézot, F.; Oliver, L.; Cochonneau, D.; Lalier, L.; Cartron, P.F.; Heymann, D. Dormant, quiescent, tolerant and persister cells: Four synonyms for the same target in cancer. Biochem. Pharmacol. 2019, 162, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Jiang, J.; Assaraf, Y.G.; Xiao, H.; Chen, Z.S.; Huang, C. Surmounting cancer drug resistance: New insights from the perspective of N(6)-methyladenosine RNA modification. Drug Resist. Updates 2020, 53, 100720. [Google Scholar] [CrossRef] [PubMed]
- Hen, O.; Barkan, D. Dormant disseminated tumor cells and cancer stem/progenitor-like cells: Similarities and opportunities. Semin. Cancer Biol. 2020, 60, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, A.I.; Trinidad, J.R.; Sandiford, O.; Etchegaray, J.P.; Rameshwar, P. Epigenetic dynamics in cancer stem cell dormancy. Cancer Metastasis Rev. 2020, 39, 721–738. [Google Scholar] [CrossRef]
- Phan, T.G.; Croucher, P.I. The dormant cancer cell life cycle. Nat. Rev. Cancer 2020, 20, 398–411. [Google Scholar] [CrossRef]
- Hayes, J.D.; Dinkova-Kostova, A.T.; Tew, K.D. Oxidative Stress in Cancer. Cancer Cell 2020, 38, 167–197. [Google Scholar] [CrossRef]
- Harris, I.S.; DeNicola, G.M. The Complex Interplay between Antioxidants and ROS in Cancer. Trends Cell Biol. 2020, 30, 440–451. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Kensler, T.W.; Motohashi, H. The KEAP1-NRF2 System: A Thiol-Based Sensor-Effector Apparatus for Maintaining Redox Homeostasis. Physiol. Rev. 2018, 98, 1169–1203. [Google Scholar] [CrossRef] [PubMed]
- Srinivas, U.S.; Tan, B.W.Q.; Vellayappan, B.A.; Jeyasekharan, A.D. ROS and the DNA damage response in cancer. Redox Biol. 2019, 25, 101084. [Google Scholar] [CrossRef] [PubMed]
- Moloney, J.N.; Cotter, T.G. ROS signalling in the biology of cancer. Semin. Cell Dev. Biol. 2018, 80, 50–64. [Google Scholar] [CrossRef] [PubMed]
- Paulsen, C.E.; Carroll, K.S. Cysteine-mediated redox signaling: Chemistry, biology, and tools for discovery. Chem. Rev. 2013, 113, 4633–4679. [Google Scholar] [CrossRef] [PubMed]
- Alcock, L.J.; Perkins, M.V.; Chalker, J.M. Chemical methods for mapping cysteine oxidation. Chem. Soc. Rev. 2018, 47, 231–268. [Google Scholar] [CrossRef]
- Forman, H.J.; Zhang, H. Targeting oxidative stress in disease: Promise and limitations of antioxidant therapy. Nat. Rev. Drug Discov. 2021, 20, 689–709. [Google Scholar] [CrossRef]
- Hegedűs, C.; Kovács, K.; Polgár, Z.; Regdon, Z.; Szabó, É.; Robaszkiewicz, A.; Forman, H.J.; Martner, A.; Virág, L. Redox control of cancer cell destruction. Redox Biol. 2018, 16, 59–74. [Google Scholar] [CrossRef]
- Vaccaro, A.; Kaplan Dor, Y.; Nambara, K.; Pollina, E.A.; Lin, C.; Greenberg, M.E.; Rogulja, D. Sleep Loss Can Cause Death through Accumulation of Reactive Oxygen Species in the Gut. Cell 2020, 181, 1307–1328.e1315. [Google Scholar] [CrossRef]
- Huang, H.; Zhang, S.; Li, Y.; Liu, Z.; Mi, L.; Cai, Y.; Wang, X.; Chen, L.; Ran, H.; Xiao, D.; et al. Suppression of mitochondrial ROS by prohibitin drives glioblastoma progression and therapeutic resistance. Nat. Commun. 2021, 12, 3720. [Google Scholar] [CrossRef]
- de la Vega, M.R.; Chapman, E.; Zhang, D.D. NRF2 and the Hallmarks of Cancer. Cancer Cell 2018, 34, 21–43. [Google Scholar] [CrossRef]
- Fox, D.B.; Garcia, N.M.G.; McKinney, B.J.; Lupo, R.; Noteware, L.C.; Newcomb, R.; Liu, J.; Locasale, J.W.; Hirschey, M.D.; Alvarez, J.V. NRF2 activation promotes the recurrence of dormant tumour cells through regulation of redox and nucleotide metabolism. Nat. Metab. 2020, 2, 318–334. [Google Scholar] [CrossRef] [PubMed]
- Ghajar, C.M. Metastasis prevention by targeting the dormant niche. Nat. Rev. Cancer 2015, 15, 238–247. [Google Scholar] [CrossRef]
- Price, T.T.; Burness, M.L.; Sivan, A.; Warner, M.J.; Cheng, R.; Lee, C.H.; Olivere, L.; Comatas, K.; Magnani, J.; Kim Lyerly, H.; et al. Dormant breast cancer micrometastases reside in specific bone marrow niches that regulate their transit to and from bone. Sci. Transl. Med. 2016, 8, 340ra373. [Google Scholar] [CrossRef]
- Yates, L.R.; Knappskog, S.; Wedge, D.; Farmery, J.H.R.; Gonzalez, S.; Martincorena, I.; Alexandrov, L.B.; Van Loo, P.; Haugland, H.K.; Lilleng, P.K.; et al. Genomic Evolution of Breast Cancer Metastasis and Relapse. Cancer Cell 2017, 32, 169–184.e167. [Google Scholar] [CrossRef]
- Ming, H.; Li, B.; Zhou, L.; Goel, A.; Huang, C. Long non-coding RNAs and cancer metastasis: Molecular basis and therapeutic implications. Biochim. Biophys. Acta Rev. Cancer 2021, 1875, 188519. [Google Scholar] [CrossRef]
- Lu, Z.; Zou, J.; Li, S.; Topper, M.J.; Tao, Y.; Zhang, H.; Jiao, X.; Xie, W.; Kong, X.; Vaz, M.; et al. Epigenetic therapy inhibits metastases by disrupting premetastatic niches. Nature 2020, 579, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Sosa, M.S.; Avivar-Valderas, A.; Bragado, P.; Wen, H.C.; Aguirre-Ghiso, J.A. ERK1/2 and p38α/β signaling in tumor cell quiescence: Opportunities to control dormant residual disease. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2011, 17, 5850–5857. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.E.; Roh, E.; Lee, M.H.; Yu, D.H.; Kim, D.J.; Lim, T.G.; Jung, S.K.; Peng, C.; Cho, Y.Y.; Dickinson, S.; et al. Fyn is a redox sensor involved in solar ultraviolet light-induced signal transduction in skin carcinogenesis. Oncogene 2016, 35, 4091–4101. [Google Scholar] [CrossRef]
- Meng, J.; Fu, L.; Liu, K.; Tian, C.; Wu, Z.; Jung, Y.; Ferreira, R.B.; Carroll, K.S.; Blackwell, T.K.; Yang, J. Global profiling of distinct cysteine redox forms reveals wide-ranging redox regulation in C. elegans. Nat. Commun. 2021, 12, 1415. [Google Scholar] [CrossRef]
- Bassi, R.; Burgoyne, J.R.; DeNicola, G.F.; Rudyk, O.; DeSantis, V.; Charles, R.L.; Eaton, P.; Marber, M.S. Redox-dependent dimerization of p38α mitogen-activated protein kinase with mitogen-activated protein kinase kinase 3. J. Biol. Chem. 2017, 292, 16161–16173. [Google Scholar] [CrossRef]
- Lee, J.; Song, C.H. Effect of Reactive Oxygen Species on the Endoplasmic Reticulum and Mitochondria during Intracellular Pathogen Infection of Mammalian Cells. Antioxidants 2021, 10, 872. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Kim, Y.; Lim, M.J.; Park, Y.G.; Park, S.I.; Sohn, J. The p38-activated ER stress-ATF6α axis mediates cellular senescence. FASEB J. 2019, 33, 2422–2434. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Zhang, K.; Kaufman, R.J. Mechanisms, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell Biol. 2020, 21, 421–438. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Cubillos-Ruiz, J.R. Endoplasmic reticulum stress signals in the tumour and its microenvironment. Nat. Rev. Cancer 2021, 21, 71–88. [Google Scholar] [CrossRef] [PubMed]
- Wei, P.C.; Hsieh, Y.H.; Su, M.I.; Jiang, X.; Hsu, P.H.; Lo, W.T.; Weng, J.Y.; Jeng, Y.M.; Wang, J.M.; Chen, P.L.; et al. Loss of the oxidative stress sensor NPGPx compromises GRP78 chaperone activity and induces systemic disease. Mol. Cell 2012, 48, 747–759. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Hokinson, D.; Park, S.; Elvira, R.; Kusuma, F.; Lee, J.M.; Yun, M.; Lee, S.G.; Han, J. ER Stress Induces Cell Cycle Arrest at the G2/M Phase Through eIF2α Phosphorylation and GADD45α. Int. J. Mol. Sci. 2019, 20, 6309. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhang, L.; Zhou, L.; Lei, Y.; Zhang, Y.; Huang, C. Redox signaling and unfolded protein response coordinate cell fate decisions under ER stress. Redox Biol. 2019, 25, 101047. [Google Scholar] [CrossRef]
- Sosa, M.S.; Parikh, F.; Maia, A.G.; Estrada, Y.; Bosch, A.; Bragado, P.; Ekpin, E.; George, A.; Zheng, Y.; Lam, H.M.; et al. NR2F1 controls tumour cell dormancy via SOX9- and RARβ-driven quiescence programmes. Nat. Commun. 2015, 6, 6170. [Google Scholar] [CrossRef] [PubMed]
- Engeland, K. Cell cycle arrest through indirect transcriptional repression by p53: I have a DREAM. Cell Death Differ. 2018, 25, 114–132. [Google Scholar] [CrossRef]
- Laplane, L.; Duluc, D.; Larmonier, N.; Pradeu, T.; Bikfalvi, A. The Multiple Layers of the Tumor Environment. Trends Cancer 2018, 4, 802–809. [Google Scholar] [CrossRef]
- Wu, P.; Gao, W.; Su, M.; Nice, E.C.; Zhang, W.; Lin, J.; Xie, N. Adaptive Mechanisms of Tumor Therapy Resistance Driven by Tumor Microenvironment. Front. Cell Dev. Biol. 2021, 9, 357. [Google Scholar] [CrossRef]
- Butturini, E.; Carcereri de Prati, A.; Boriero, D.; Mariotto, S. Tumor Dormancy and Interplay with Hypoxic Tumor Microenvironment. Int. J. Mol. Sci. 2019, 20, 4305. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, A.; Roser, C.T.; El-Far, M.H.; Savanur, V.H.; Eljarrah, A.; Gergues, M.; Kra, J.A.; Etchegaray, J.P.; Rameshwar, P. Hypoxia-mediated changes in bone marrow microenvironment in breast cancer dormancy. Cancer Lett. 2020, 488, 9–17. [Google Scholar] [CrossRef]
- Lee, H.R.; Leslie, F.; Azarin, S.M. A facile in vitro platform to study cancer cell dormancy under hypoxic microenvironments using CoCl(2). J. Biol. Eng. 2018, 12, 12. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Zhang, Y.; Wang, Z.; Dong, H.; Fu, N.; Han, X. The roles and signaling pathways of prolyl-4-hydroxylase 2 in the tumor microenvironment. Chem. Biol. Interact. 2019, 303, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Ren, Z.; Ye, L.C.; Zhou, P.H.; Xu, J.M.; Shi, Q.; Yao, L.Q.; Zhong, Y.S. Factor inhibiting HIF1α (FIH-1) functions as a tumor suppressor in human colorectal cancer by repressing HIF1α pathway. Cancer Biol. Ther. 2015, 16, 244–252. [Google Scholar] [CrossRef]
- Ju, S.; Wang, F.; Wang, Y.; Ju, S. CSN8 is a key regulator in hypoxia-induced epithelial-mesenchymal transition and dormancy of colorectal cancer cells. Mol. Cancer 2020, 19, 168. [Google Scholar] [CrossRef]
- Prunier, C.; Baker, D.; Ten Dijke, P.; Ritsma, L. TGF-β Family Signaling Pathways in Cellular Dormancy. Trends Cancer 2019, 5, 66–78. [Google Scholar] [CrossRef]
- Barkan, D.; Chambers, A.F. β1-integrin: A potential therapeutic target in the battle against cancer recurrence. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2011, 17, 7219–7223. [Google Scholar] [CrossRef]
- Eble, J.A.; de Rezende, F.F. Redox-relevant aspects of the extracellular matrix and its cellular contacts via integrins. Antioxid Redox Signal. 2014, 20, 1977–1993. [Google Scholar] [CrossRef]
- Rosenberg, N.; Mor-Cohen, R.; Sheptovitsky, V.H.; Romanenco, O.; Hess, O.; Lahav, J. Integrin-mediated cell adhesion requires extracellular disulfide exchange regulated by protein disulfide isomerase. Exp. Cell Res. 2019, 381, 77–85. [Google Scholar] [CrossRef]
- Yeh, A.C.; Ramaswamy, S. Mechanisms of Cancer Cell Dormancy--Another Hallmark of Cancer? Cancer Res. 2015, 75, 5014–5022. [Google Scholar] [CrossRef]
- Kechagia, J.; Ivaska, J.; Roca-Cusachs, P. Integrins as biomechanical sensors of the microenvironment. Nat. Rev. Mol. Cell Biol. 2019, 20, 457–473. [Google Scholar] [CrossRef] [PubMed]
- Bui, A.T.; Laurent, F.; Havard, M.; Dautry, F.; Tchénio, T. SMAD signaling and redox imbalance cooperate to induce prostate cancer cell dormancy. Cell Cycle 2015, 14, 1218–1231. [Google Scholar] [CrossRef][Green Version]
- Zhang, D.; Jin, W.; Wu, R.; Li, J.; Park, S.A.; Tu, E.; Zanvit, P.; Xu, J.; Liu, O.; Cain, A.; et al. High Glucose Intake Exacerbates Autoimmunity through Reactive-Oxygen-Species-Mediated TGF-β Cytokine Activation. Immunity 2019, 51, 671–681.e675. [Google Scholar] [CrossRef]
- van Caam, A.; Madej, W.; de Vinuesa, A.G.; Goumans, M.J.; Dijke, P.T.; Davidson, E.B.; van der Kraan, P. TGFβ1-induced SMAD2/3 and SMAD1/5 phosphorylation are both ALK5-kinase-dependent in primary chondrocytes and mediated by TAK1 kinase activity. Arthritis Res. Ther. 2017, 19, 112. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Liu, G.S.; Dusting, G.J.; Chan, E.C. NADPH oxidase-dependent redox signaling in TGF-β-mediated fibrotic responses. Redox Biol. 2014, 2, 267–272. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, N.; Cho, P.; Selfors, L.M.; Kuiken, H.J.; Kaul, R.; Fujiwara, T.; Harris, I.S.; Zhang, T.; Gygi, S.P.; Brugge, J.S. 3D Culture Models with CRISPR Screens Reveal Hyperactive NRF2 as a Prerequisite for Spheroid Formation via Regulation of Proliferation and Ferroptosis. Mol. Cell 2020, 80, 828–844.e826. [Google Scholar] [CrossRef]
- Bragado, P.; Estrada, Y.; Parikh, F.; Krause, S.; Capobianco, C.; Farina, H.G.; Schewe, D.M.; Aguirre-Ghiso, J.A. TGF-β2 dictates disseminated tumour cell fate in target organs through TGF-β-RIII and p38α/β signalling. Nat. Cell Biol. 2013, 15, 1351–1361. [Google Scholar] [CrossRef]
- Yu-Lee, L.Y.; Yu, G.; Lee, Y.C.; Lin, S.C.; Pan, J.; Pan, T.; Yu, K.J.; Liu, B.; Creighton, C.J.; Rodriguez-Canales, J.; et al. Osteoblast-Secreted Factors Mediate Dormancy of Metastatic Prostate Cancer in the Bone via Activation of the TGFβRIII-p38MAPK-pS249/T252RB Pathway. Cancer Res. 2018, 78, 2911–2924. [Google Scholar] [CrossRef]
- Ghajar, C.M.; Peinado, H.; Mori, H.; Matei, I.R.; Evason, K.J.; Brazier, H.; Almeida, D.; Koller, A.; Hajjar, K.A.; Stainier, D.Y.; et al. The perivascular niche regulates breast tumour dormancy. Nat. Cell Biol. 2013, 15, 807–817. [Google Scholar] [CrossRef]
- van Velthoven, C.T.J.; Rando, T.A. Stem Cell Quiescence: Dynamism, Restraint, and Cellular Idling. Cell Stem Cell 2019, 24, 213–225. [Google Scholar] [CrossRef] [PubMed]
- Martin, V.; Turos-Cabal, M.; Sanchez-Sanchez, A.M.; Rodríguez, C. Metabolism-Redox Interplay in Tumor Stem Cell Signaling. In Handbook of Oxidative Stress in Cancer: Mechanistic Aspects; Springer: Berlin/Heidelberg, Germany, 2020; pp. 1–22. [Google Scholar]
- Ma, X.; Su, P.; Yin, C.; Lin, X.; Wang, X.; Gao, Y.; Patil, S.; War, A.R.; Qadir, A.; Tian, Y.; et al. The Roles of FoxO Transcription Factors in Regulation of Bone Cells Function. Int. J. Mol. Sci. 2020, 21, 692. [Google Scholar] [CrossRef] [PubMed]
- Eijkelenboom, A.; Burgering, B.M. FOXOs: Signalling integrators for homeostasis maintenance. Nat. Rev. Mol. Cell Biol. 2013, 14, 83–97. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Zhu, X.; Qin, F.; Zhang, Y.; Lin, J.; Ding, Y.; Yang, Z.; Shang, Y.; Wang, L.; Zhang, Q.; et al. Linc00210 drives Wnt/β-catenin signaling activation and liver tumor progression through CTNNBIP1-dependent manner. Mol. Cancer 2018, 17, 73. [Google Scholar] [CrossRef]
- Collins, S.J.; Tumpach, C.; Groveman, B.R.; Drew, S.C.; Haigh, C.L. Prion protein cleavage fragments regulate adult neural stem cell quiescence through redox modulation of mitochondrial fission and SOD2 expression. Cell. Mol. Life Sci. 2018, 75, 3231–3249. [Google Scholar] [CrossRef]
- Tao, J.; Krutsenko, Y.; Moghe, A.; Singh, S.; Poddar, M.; Bell, A.; Oertel, M.; Singhi, A.D.; Geller, D.; Chen, X.; et al. Nuclear factor erythroid 2-related factor 2 and β-Catenin Coactivation in Hepatocellular Cancer: Biological and Therapeutic Implications. Hepatology 2021, 74, 741–759. [Google Scholar] [CrossRef]
- Wakabayashi, N.; Chartoumpekis, D.V.; Kensler, T.W. Crosstalk between Nrf2 and Notch signaling. Free. Radic. Biol. Med. 2015, 88, 158–167. [Google Scholar] [CrossRef]
- Kim, J.H.; Thimmulappa, R.K.; Kumar, V.; Cui, W.; Kumar, S.; Kombairaju, P.; Zhang, H.; Margolick, J.; Matsui, W.; Macvittie, T.; et al. NRF2-mediated Notch pathway activation enhances hematopoietic reconstitution following myelosuppressive radiation. J. Clin. Investig. 2014, 124, 730–741. [Google Scholar] [CrossRef]
- Leung, H.W.; Lau, E.Y.T.; Leung, C.O.N.; Lei, M.M.L.; Mok, E.H.K.; Ma, V.W.S.; Cho, W.C.S.; Ng, I.O.L.; Yun, J.P.; Cai, S.H.; et al. NRF2/SHH signaling cascade promotes tumor-initiating cell lineage and drug resistance in hepatocellular carcinoma. Cancer Lett. 2020, 476, 48–56. [Google Scholar] [CrossRef]
- Ren, D.; Dai, Y.; Yang, Q.; Zhang, X.; Guo, W.; Ye, L.; Huang, S.; Chen, X.; Lai, Y.; Du, H.; et al. Wnt5a induces and maintains prostate cancer cells dormancy in bone. J. Exp. Med. 2019, 216, 428–449. [Google Scholar] [CrossRef] [PubMed]
- Popova, S.A.; Buczacki, S.J.A. Itraconazole perturbs colorectal cancer dormancy through SUFU-mediated WNT inhibition. Mol. Cell. Oncol. 2018, 5, e1494950. [Google Scholar] [CrossRef]
- Zhou, Y.; Su, Y.; Zhu, H.; Wang, X.; Li, X.; Dai, C.; Xu, C.; Zheng, T.; Mao, C.; Chen, D. Interleukin-23 receptor signaling mediates cancer dormancy and radioresistance in human esophageal squamous carcinoma cells via the Wnt/Notch pathway. J. Mol. Med. 2019, 97, 177–188. [Google Scholar] [CrossRef] [PubMed]
- Abravanel, D.L.; Belka, G.K.; Pan, T.C.; Pant, D.K.; Collins, M.A.; Sterner, C.J.; Chodosh, L.A. Notch promotes recurrence of dormant tumor cells following HER2/neu-targeted therapy. J. Clin. Investig. 2015, 125, 2484–2496. [Google Scholar] [CrossRef]
- Capulli, M.; Hristova, D.; Valbret, Z.; Carys, K.; Arjan, R.; Maurizi, A.; Masedu, F.; Cappariello, A.; Rucci, N.; Teti, A. Notch2 pathway mediates breast cancer cellular dormancy and mobilisation in bone and contributes to haematopoietic stem cell mimicry. Br. J. Cancer 2019, 121, 157–171. [Google Scholar] [CrossRef]
- Sadarangani, A.; Pineda, G.; Lennon, K.M.; Chun, H.J.; Shih, A.; Schairer, A.E.; Court, A.C.; Goff, D.J.; Prashad, S.L.; Geron, I.; et al. GLI2 inhibition abrogates human leukemia stem cell dormancy. J. Transl. Med. 2015, 13, 98. [Google Scholar] [CrossRef] [PubMed]
- Ligeon, L.A.; Pena-Francesch, M.; Vanoaica, L.D.; Núñez, N.G.; Talwar, D.; Dick, T.P.; Münz, C. Oxidation inhibits autophagy protein deconjugation from phagosomes to sustain MHC class II restricted antigen presentation. Nat. Commun. 2021, 12, 1508. [Google Scholar] [CrossRef]
- Li, X.; He, S.; Ma, B. Autophagy and autophagy-related proteins in cancer. Mol. Cancer 2020, 19, 12. [Google Scholar] [CrossRef]
- Vera-Ramirez, L.; Vodnala, S.K.; Nini, R.; Hunter, K.W.; Green, J.E. Autophagy promotes the survival of dormant breast cancer cells and metastatic tumour recurrence. Nat. Commun. 2018, 9, 1944. [Google Scholar] [CrossRef]
- Zhao, D.; Zou, C.X.; Liu, X.M.; Jiang, Z.D.; Yu, Z.Q.; Suo, F.; Du, T.Y.; Dong, M.Q.; He, W.; Du, L.L. A UPR-Induced Soluble ER-Phagy Receptor Acts with VAPs to Confer ER Stress Resistance. Mol. Cell 2020, 79, 963–977.e963. [Google Scholar] [CrossRef]
- Qiu, Y.; Qiu, S.; Deng, L.; Nie, L.; Gong, L.; Liao, X.; Zheng, X.; Jin, K.; Li, J.; Tu, X.; et al. Biomaterial 3D collagen I gel culture model: A novel approach to investigate tumorigenesis and dormancy of bladder cancer cells induced by tumor microenvironment. Biomaterials 2020, 256, 120217. [Google Scholar] [CrossRef]
- Wang, H.; Wang, N.; Xu, D.; Ma, Q.; Chen, Y.; Xu, S.; Xia, Q.; Zhang, Y.; Prehn, J.H.M.; Wang, G.; et al. Oxidation of multiple MiT/TFE transcription factors links oxidative stress to transcriptional control of autophagy and lysosome biogenesis. Autophagy 2020, 16, 1683–1696. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Yang, Z.; Gu, Q.; Xia, F.; Fu, Y.; Liu, P.; Yin, X.M.; Li, M. The protease activity of human ATG4B is regulated by reversible oxidative modification. Autophagy 2020, 16, 1838–1850. [Google Scholar] [CrossRef] [PubMed]
- Frudd, K.; Burgoyne, T.; Burgoyne, J.R. Oxidation of Atg3 and Atg7 mediates inhibition of autophagy. Nat. Commun. 2018, 9, 95. [Google Scholar] [CrossRef] [PubMed]
- Cosin-Roger, J.; Simmen, S.; Melhem, H.; Atrott, K.; Frey-Wagner, I.; Hausmann, M.; de Vallière, C.; Spalinger, M.R.; Spielmann, P.; Wenger, R.H.; et al. Hypoxia ameliorates intestinal inflammation through NLRP3/mTOR downregulation and autophagy activation. Nat. Commun. 2017, 8, 98. [Google Scholar] [CrossRef]
- Tian, H.; Zhang, M.; Jin, G.; Jiang, Y.; Luan, Y. Cu-MOF chemodynamic nanoplatform via modulating glutathione and H2O2 in tumor microenvironment for amplified cancer therapy. J. Colloid Interface Sci. 2021, 587, 358–366. [Google Scholar] [CrossRef]
- Hu, W.; Zhang, L.; Dong, Y.; Tian, Z.; Chen, Y.; Dong, S. Tumour dormancy in inflammatory microenvironment: A promising therapeutic strategy for cancer-related bone metastasis. Cell Mol. Life Sci. 2020, 77, 5149–5169. [Google Scholar] [CrossRef]
- Gao, X.L.; Zheng, M.; Wang, H.F.; Dai, L.L.; Yu, X.H.; Yang, X.; Pang, X.; Li, L.; Zhang, M.; Wang, S.S.; et al. NR2F1 contributes to cancer cell dormancy, invasion and metastasis of salivary adenoid cystic carcinoma by activating CXCL12/CXCR4 pathway. BMC Cancer 2019, 19, 743. [Google Scholar] [CrossRef]
- Adamski, V.; Hattermann, K.; Kubelt, C.; Cohrs, G.; Lucius, R.; Synowitz, M.; Sebens, S.; Held-Feindt, J. Entry and exit of chemotherapeutically-promoted cellular dormancy in glioblastoma cells is differentially affected by the chemokines CXCL12, CXCL16, and CX3CL1. Oncogene 2020, 39, 4421–4435. [Google Scholar] [CrossRef]
- Decker, A.M.; Decker, J.T.; Jung, Y.; Cackowski, F.C.; Daignault-Newton, S.; Morgan, T.M.; Shea, L.D.; Taichman, R.S. Adrenergic Blockade Promotes Maintenance of Dormancy in Prostate Cancer Through Upregulation of GAS6. Transl. Oncol. 2020, 13, 100781. [Google Scholar] [CrossRef] [PubMed]
- Ottewell, P.D.; Wang, N.; Brown, H.K.; Fowles, C.A.; Croucher, P.I.; Eaton, C.L.; Holen, I. OPG-Fc inhibits ovariectomy-induced growth of disseminated breast cancer cells in bone. Int. J. Cancer 2015, 137, 968–977. [Google Scholar] [CrossRef]
- Boyerinas, B.; Zafrir, M.; Yesilkanal, A.E.; Price, T.T.; Hyjek, E.M.; Sipkins, D.A. Adhesion to osteopontin in the bone marrow niche regulates lymphoblastic leukemia cell dormancy. Blood 2013, 121, 4821–4831. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.W.; Finger, E.C.; Olcina, M.M.; Vilalta, M.; Aguilera, T.; Miao, Y.; Merkel, A.R.; Johnson, J.R.; Sterling, J.A.; Wu, J.Y.; et al. Induction of LIFR confers a dormancy phenotype in breast cancer cells disseminated to the bone marrow. Nat. Cell Biol. 2016, 18, 1078–1089. [Google Scholar] [CrossRef] [PubMed]
- Levoye, A.; Balabanian, K.; Baleux, F.; Bachelerie, F.; Lagane, B. CXCR7 heterodimerizes with CXCR4 and regulates CXCL12-mediated G protein signaling. Blood 2009, 113, 6085–6093. [Google Scholar] [CrossRef] [PubMed]
- Martin, T.J.; Johnson, R.W. Multiple actions of parathyroid hormone-related protein in breast cancer bone metastasis. Br. J. Pharmacol. 2021, 178, 1923–1935. [Google Scholar] [CrossRef]
- Beekman, K.M.; Zwaagstra, M.; Veldhuis-Vlug, A.G.; van Essen, H.W.; den Heijer, M.; Maas, M.; Kerckhofs, G.; Parac-Vogt, T.N.; Bisschop, P.H.; Bravenboer, N. Ovariectomy increases RANKL protein expression in bone marrow adipocytes of C3H/HeJ mice. Am. J. Physiol. Endocrinol. Metab 2019, 317, e1050–e1054. [Google Scholar] [CrossRef]
- Abuna, R.P.; De Oliveira, F.S.; Santos Tde, S.; Guerra, T.R.; Rosa, A.L.; Beloti, M.M. Participation of TNF-α in Inhibitory Effects of Adipocytes on Osteoblast Differentiation. J. Cell. Physiol. 2016, 231, 204–214. [Google Scholar] [CrossRef]
- Delort, L.; Rossary, A.; Farges, M.C.; Vasson, M.P.; Caldefie-Chézet, F. Leptin, adipocytes and breast cancer: Focus on inflammation and anti-tumor immunity. Life Sci. 2015, 140, 37–48. [Google Scholar] [CrossRef]
- Dou, C.; Ding, N.; Zhao, C.; Hou, T.; Kang, F.; Cao, Z.; Liu, C.; Bai, Y.; Dai, Q.; Ma, Q.; et al. Estrogen Deficiency-Mediated M2 Macrophage Osteoclastogenesis Contributes to M1/M2 Ratio Alteration in Ovariectomized Osteoporotic Mice. J. Bone Miner. Res. 2018, 33, 899–908. [Google Scholar] [CrossRef]
- Hirata, Y.; Kakiuchi, M.; Robson, S.C.; Fujisaki, J. CD150(high) CD4 T cells and CD150(high) regulatory T cells regulate hematopoietic stem cell quiescence via CD73. Haematologica 2019, 104, 1136–1142. [Google Scholar] [CrossRef]
- Hirata, Y.; Furuhashi, K.; Ishii, H.; Li, H.W.; Pinho, S.; Ding, L.; Robson, S.C.; Frenette, P.S.; Fujisaki, J. CD150(high) Bone Marrow Tregs Maintain Hematopoietic Stem Cell Quiescence and Immune Privilege via Adenosine. Cell Stem Cell 2018, 22, 445–453.e445. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Cheng, C.; Tan, Z.; Li, N.; Tang, M.; Yang, L.; Cao, Y. Emerging roles of lipid metabolism in cancer metastasis. Mol. Cancer 2017, 16, 76. [Google Scholar] [CrossRef] [PubMed]
- Pascual, G.; Avgustinova, A.; Mejetta, S.; Martín, M.; Castellanos, A.; Attolini, C.S.; Berenguer, A.; Prats, N.; Toll, A.; Hueto, J.A.; et al. Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature 2017, 541, 41–45. [Google Scholar] [CrossRef]
- Watt, M.J.; Clark, A.K.; Selth, L.A.; Haynes, V.R.; Lister, N.; Rebello, R.; Porter, L.H.; Niranjan, B.; Whitby, S.T.; Lo, J.; et al. Suppressing fatty acid uptake has therapeutic effects in preclinical models of prostate cancer. Sci. Transl. Med. 2019, 11, eaau5758. [Google Scholar] [CrossRef]
- Jay, A.G.; Chen, A.N.; Paz, M.A.; Hung, J.P.; Hamilton, J.A. CD36 binds oxidized low density lipoprotein (LDL) in a mechanism dependent upon fatty acid binding. J. Biol. Chem. 2015, 290, 4590–4603. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Tao, B.; Fan, Q.; Wang, S.; Chen, L.; Zhang, J.; Hao, Y.; Dong, S.; Wang, Z.; Wang, W.; et al. Fatty-acid receptor CD36 functions as a hydrogen sulfide-targeted receptor with its Cys333-Cys272 disulfide bond serving as a specific molecular switch to accelerate gastric cancer metastasis. EBioMedicine 2019, 45, 108–123. [Google Scholar] [CrossRef]
- Panigrahy, D.; Edin, M.L.; Lee, C.R.; Huang, S.; Bielenberg, D.R.; Butterfield, C.E.; Barnés, C.M.; Mammoto, A.; Mammoto, T.; Luria, A.; et al. Epoxyeicosanoids stimulate multiorgan metastasis and tumor dormancy escape in mice. J. Clin. Investig. 2012, 122, 178–191. [Google Scholar] [CrossRef]
- Perego, M.; Tyurin, V.A.; Tyurina, Y.Y.; Yellets, J.; Nacarelli, T.; Lin, C.; Nefedova, Y.; Kossenkov, A.; Liu, Q.; Sreedhar, S.; et al. Reactivation of dormant tumor cells by modified lipids derived from stress-activated neutrophils. Sci. Transl. Med. 2020, 12, eabb5817. [Google Scholar] [CrossRef]
- McGarry, T.; Biniecka, M.; Veale, D.J.; Fearon, U. Hypoxia, oxidative stress and inflammation. Free. Radic. Biol. Med. 2018, 125, 15–24. [Google Scholar] [CrossRef]
- Liu, Y.; Li, Z.; Li, J.; Yang, S.; Zhang, Y.; Yao, B.; Song, W.; Fu, X.; Huang, S. Stiffness-mediated mesenchymal stem cell fate decision in 3D-bioprinted hydrogels. Burns. Trauma 2020, 8, tkaa029. [Google Scholar] [CrossRef] [PubMed]
- Albrengues, J.; Shields, M.A.; Ng, D.; Park, C.G.; Ambrico, A.; Poindexter, M.E.; Upadhyay, P.; Uyeminami, D.L.; Pommier, A.; Küttner, V.; et al. Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science 2018, 361, eaao4227. [Google Scholar] [CrossRef]
- El-Shennawy, L.; Dubrovskyi, O.; Kastrati, I.; Danes, J.M.; Zhang, Y.; Whiteley, H.E.; Creighton, C.J.; Frasor, J. Coactivation of Estrogen Receptor and IKKβ Induces a Dormant Metastatic Phenotype in ER-Positive Breast Cancer. Cancer Res. 2018, 78, 974–984. [Google Scholar] [CrossRef] [PubMed]
- Maiti, S.; Nazmeen, A. Impaired redox regulation of estrogen metabolizing proteins is important determinant of human breast cancers. Cancer Cell Int. 2019, 19, 111. [Google Scholar] [CrossRef] [PubMed]
- Lawson, M.A.; McDonald, M.M.; Kovacic, N.; Hua Khoo, W.; Terry, R.L.; Down, J.; Kaplan, W.; Paton-Hough, J.; Fellows, C.; Pettitt, J.A.; et al. Osteoclasts control reactivation of dormant myeloma cells by remodelling the endosteal niche. Nat. Commun. 2015, 6, 8983. [Google Scholar] [CrossRef]
- Cabezas-Wallscheid, N.; Buettner, F.; Sommerkamp, P.; Klimmeck, D.; Ladel, L.; Thalheimer, F.B.; Pastor-Flores, D.; Roma, L.P.; Renders, S.; Zeisberger, P.; et al. Vitamin A-Retinoic Acid Signaling Regulates Hematopoietic Stem Cell Dormancy. Cell 2017, 169, 807–823.e819. [Google Scholar] [CrossRef] [PubMed]
- Price, D.K. Efficacy of androgen deprivation therapy and the role of oxidative stress. Ann. Oncol. 2017, 28, 451–453. [Google Scholar] [CrossRef]
- Havas, K.M.; Milchevskaya, V.; Radic, K.; Alladin, A.; Kafkia, E.; Garcia, M.; Stolte, J.; Klaus, B.; Rotmensz, N.; Gibson, T.J.; et al. Metabolic shifts in residual breast cancer drive tumor recurrence. J. Clin. Investig. 2017, 127, 2091–2105. [Google Scholar] [CrossRef]
- El Touny, L.H.; Vieira, A.; Mendoza, A.; Khanna, C.; Hoenerhoff, M.J.; Green, J.E. Combined SFK/MEK inhibition prevents metastatic outgrowth of dormant tumor cells. J. Clin. Investig. 2014, 124, 156–168. [Google Scholar] [CrossRef]
- Abderrahman, B.; Jordan, V.C. Rethinking Extended Adjuvant Antiestrogen Therapy to Increase Survivorship in Breast Cancer. JAMA Oncol. 2018, 4, 15–16. [Google Scholar] [CrossRef]
- O’Leary, B.; Finn, R.S.; Turner, N.C. Treating cancer with selective CDK4/6 inhibitors. Nat. Rev. Clin. Oncol. 2016, 13, 417–430. [Google Scholar] [CrossRef]
- Nakayama, T.; Sano, T.; Oshimo, Y.; Kawada, C.; Kasai, M.; Yamamoto, S.; Fukuhara, H.; Inoue, K.; Ogura, S.I. Enhanced lipid metabolism induces the sensitivity of dormant cancer cells to 5-aminolevulinic acid-based photodynamic therapy. Sci. Rep. 2021, 11, 7290. [Google Scholar] [CrossRef] [PubMed]
- Nicolini, F.E.; Etienne, G.; Huguet, F.; Guerci-Bresler, A.; Charbonnier, A.; Escoffre-Barbe, M.; Dubruille, V.; Johnson-Ansah, H.; Legros, L.; Coiteux, V. The Combination of Nilotinib+ Pegylated IFN Alpha 2a Provides Somewhat Higher Cumulative Incidence Rates of MR4. 5 at M36 Versus Nilotinib Alone in Newly Diagnosed CP CML Patients. In Updated Results of the Petals Phase III National Study; American Society of Hematology Washington: Washington, DC, USA, 2019. [Google Scholar]
- Pietras, E.M.; Lakshminarasimhan, R.; Techner, J.M.; Fong, S.; Flach, J.; Binnewies, M.; Passegué, E. Re-entry into quiescence protects hematopoietic stem cells from the killing effect of chronic exposure to type I interferons. J. Exp. Med. 2014, 211, 245–262. [Google Scholar] [CrossRef]
- Tasdogan, A.; Kumar, S.; Allies, G.; Bausinger, J.; Beckel, F.; Hofemeister, H.; Mulaw, M.; Madan, V.; Scharffetter-Kochanek, K.; Feuring-Buske, M.; et al. DNA Damage-Induced HSPC Malfunction Depends on ROS Accumulation Downstream of IFN-1 Signaling and Bid Mobilization. Cell Stem Cell 2016, 19, 752–767. [Google Scholar] [CrossRef]
- Singh, S.; Jakubison, B.; Keller, J.R. Protection of hematopoietic stem cells from stress-induced exhaustion and aging. Curr. Opin. Hematol. 2020, 27, 225–231. [Google Scholar] [CrossRef]
- Hampsch, R.A.; Wells, J.D.; Traphagen, N.A.; McCleery, C.F.; Fields, J.L.; Shee, K.; Dillon, L.M.; Pooler, D.B.; Lewis, L.D.; Demidenko, E.; et al. AMPK Activation by Metformin Promotes Survival of Dormant ER(+) Breast Cancer Cells. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2020, 26, 3707–3719. [Google Scholar] [CrossRef] [PubMed]
- Viale, A.; Pettazzoni, P.; Lyssiotis, C.A.; Ying, H.; Sánchez, N.; Marchesini, M.; Carugo, A.; Green, T.; Seth, S.; Giuliani, V.; et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature 2014, 514, 628–632. [Google Scholar] [CrossRef] [PubMed]
- Lagadinou, E.D.; Sach, A.; Callahan, K.; Rossi, R.M.; Neering, S.J.; Minhajuddin, M.; Ashton, J.M.; Pei, S.; Grose, V.; O’Dwyer, K.M.; et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell 2013, 12, 329–341. [Google Scholar] [CrossRef]
- Han, L.; Cavazos, A.; Baran, N.; Zhang, Q.; Kuruvilla, V.M.; Gay, J.P.; Feng, N.; Battula, V.L.; Kantarjian, H.M.; Daver, N.G. Mitochondrial OxPhos as Survival Mechanism of Minimal Residual AML Cells after Induction Chemotherapy: Survival Benefit by Complex. I Inhibition with IACS-010759; American Society of Hematology Washington: Washington, DC, USA, 2019. [Google Scholar]
- Han, L.; Cavazos, A.; Baran, N.; Zhang, Q.; Kuruvilla, V.M.; Gay, J.P.; Feng, N.; Battula, V.L.; Andreeff, M.; Kantarjian, H.M. Targeting residual chemotherapy-resistant acute myeloid leukemia cells by a novel OXPHOS inhibitor IACS010759. Blood 2017, 130, 2623. [Google Scholar]
- Dower, C.M.; Wills, C.A.; Frisch, S.M.; Wang, H.G. Mechanisms and context underlying the role of autophagy in cancer metastasis. Autophagy 2018, 14, 1110–1128. [Google Scholar] [CrossRef]
- Sandoval, M.V.; Fluegen, G.; Staschke, K.A.; Calvo-Vidal, V.; Aguirre-Ghiso, J.A. Abstract A45: PERK-Inhibition as a possible therapy for hypoxia-induced solitary dormant tumor cells. AACR 2016, 76, A45. [Google Scholar]
- Chen, X.; Kang, R.; Kroemer, G.; Tang, D. Broadening horizons: The role of ferroptosis in cancer. Nat. Rev. Clin. Oncol. 2021, 18, 280–296. [Google Scholar] [CrossRef]
- Jiang, X.; Stockwell, B.R.; Conrad, M. Ferroptosis: Mechanisms, biology and role in disease. Nat. Rev. Mol. Cell Biol. 2021, 22, 266–282. [Google Scholar] [CrossRef]
- Recasens, A.; Munoz, L. Targeting Cancer Cell Dormancy. Trends Pharmacol. Sci. 2019, 40, 128–141. [Google Scholar] [CrossRef]
- Hangauer, M.J.; Viswanathan, V.S.; Ryan, M.J.; Bole, D.; Eaton, J.K.; Matov, A.; Galeas, J.; Dhruv, H.D.; Berens, M.E.; Schreiber, S.L.; et al. Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature 2017, 551, 247–250. [Google Scholar] [CrossRef] [PubMed]
- Barrera, G.; Pizzimenti, S.; Daga, M.; Dianzani, C.; Arcaro, A.; Cetrangolo, G.P.; Giordano, G.; Cucci, M.A.; Graf, M.; Gentile, F. Lipid Peroxidation-Derived Aldehydes, 4-Hydroxynonenal and Malondialdehyde in Aging-Related Disorders. Antioxidants 2018, 7, 102. [Google Scholar] [CrossRef] [PubMed]
- Raha, D.; Wilson, T.R.; Peng, J.; Peterson, D.; Yue, P.; Evangelista, M.; Wilson, C.; Merchant, M.; Settleman, J. The cancer stem cell marker aldehyde dehydrogenase is required to maintain a drug-tolerant tumor cell subpopulation. Cancer Res. 2014, 74, 3579–3590. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).