
Iron Metabolism as a Potential Mechanism for Inducing TRAIL-Mediated Extrinsic Apoptosis Using Methylsulfonylmethane in Embryonic Cancer Stem Cells




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Antibodies and Cell Culture Reagents
2.2. Cell Culture and Treatment
2.3. Cell Viability Assay
2.4. Western Blotting
2.5. Quantitative Real-Time Polymerase Chain Reaction (qPCR)
2.6. Fluorescence-Activated Cell Sorting (FACS) Analysis for Mitochondrial and Cellular ROS
2.7. Cell Cycle Analysis
2.8. Comet Assay
2.9. Apoptosis Analysis
2.10. Human TRAIL Enzyme-Linked Immunosorbent Assay (ELISA)
2.11. Caspase-Glo 3/7 Assay
2.12. Iron Assay Analysis
2.13. FACS Analysis for Ferrous Ion (Fe2+)
2.14. Transfections of miRNA Inhibitor
2.15. Statistical Analyses
3. Results
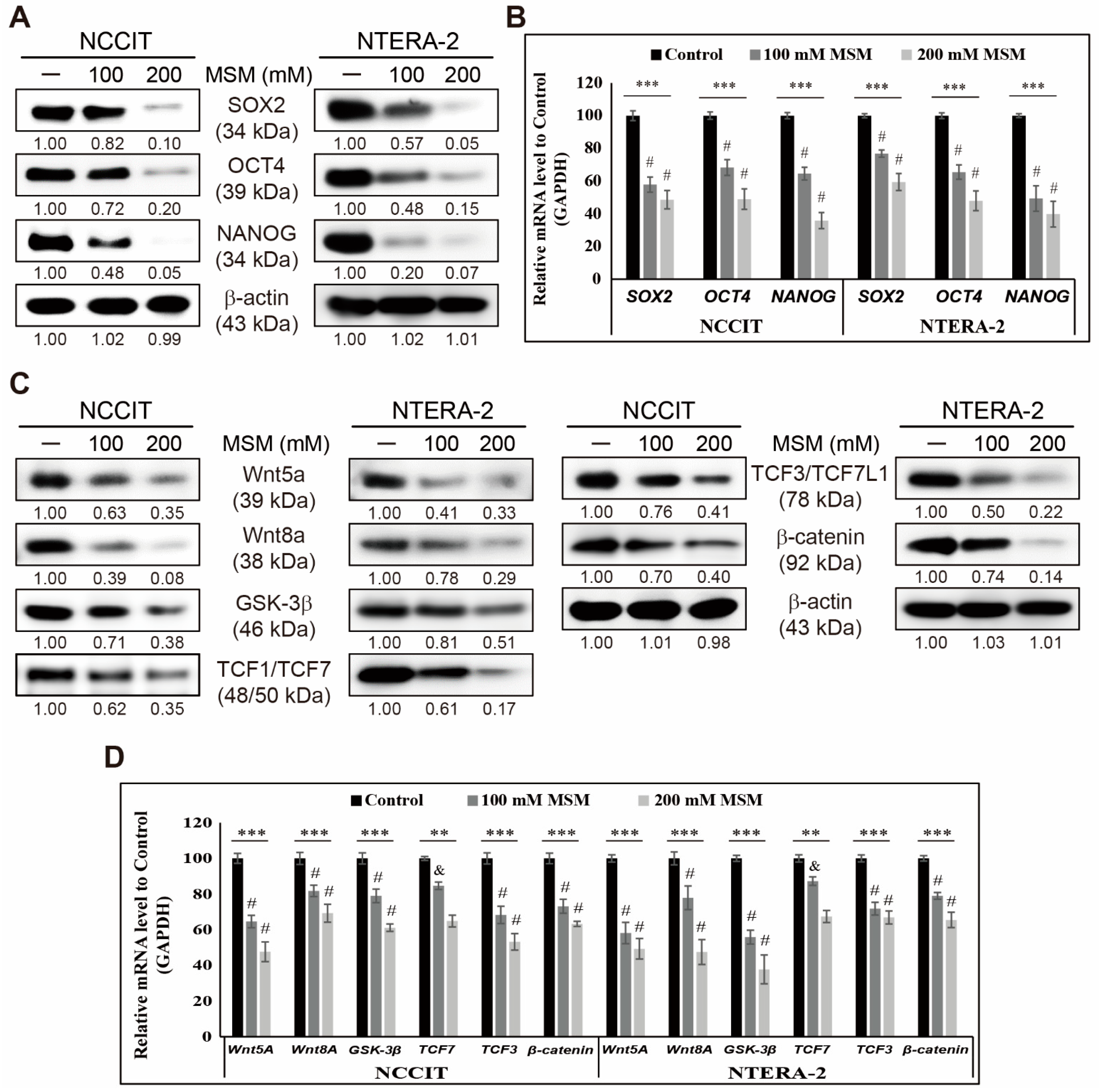
3.1. MSM Inhibited CSC Markers and Wnt/β-Catenin Signaling in Embryonic CSCs
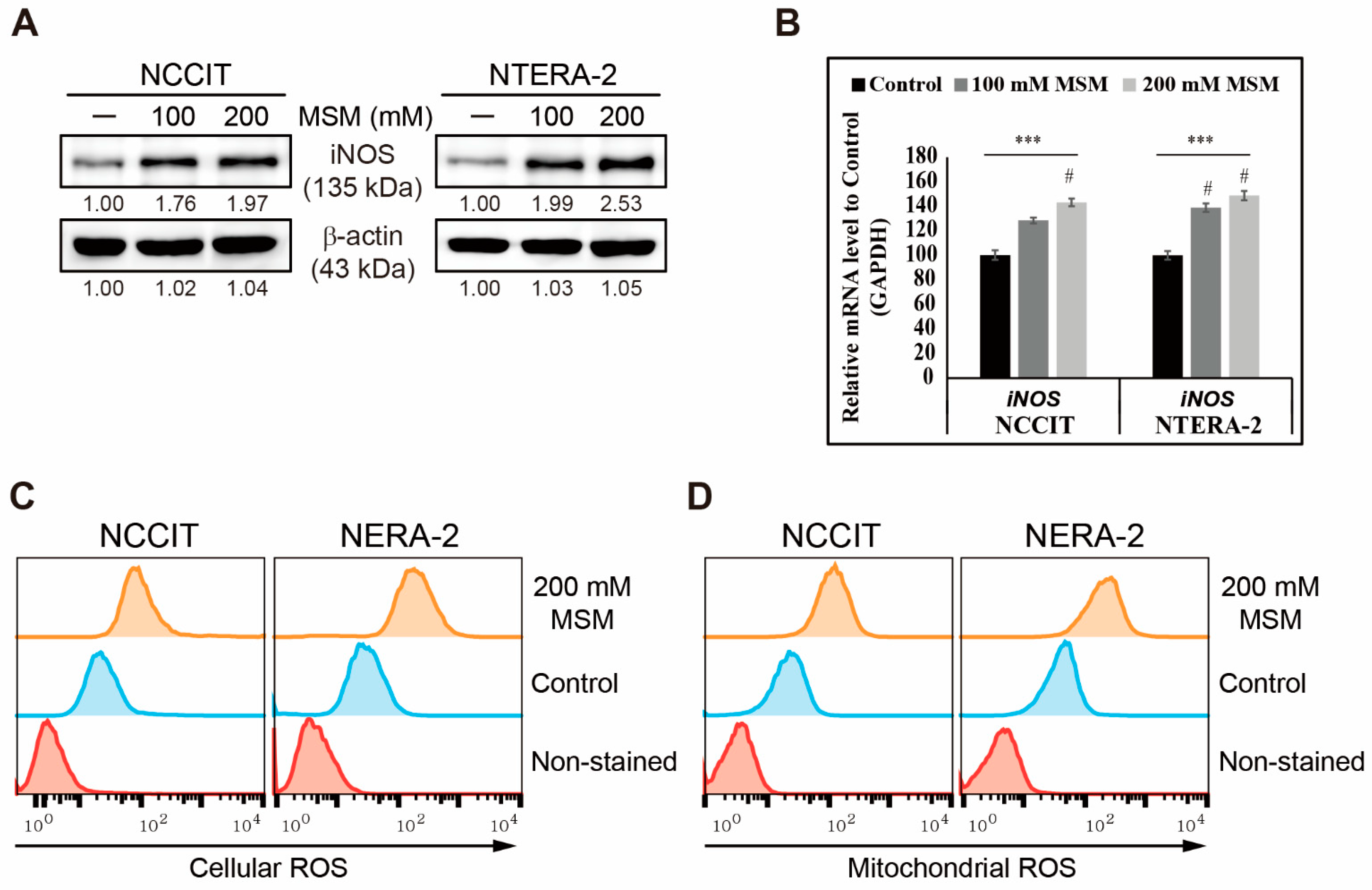
3.2. MSM Induced ROS and DNA Damage Response in Embryonic CSC
3.3. MSM Induced Cell Cycle Arrest and TRAIL-Mediated Extrinsic Apoptosis in Embryonic CSC
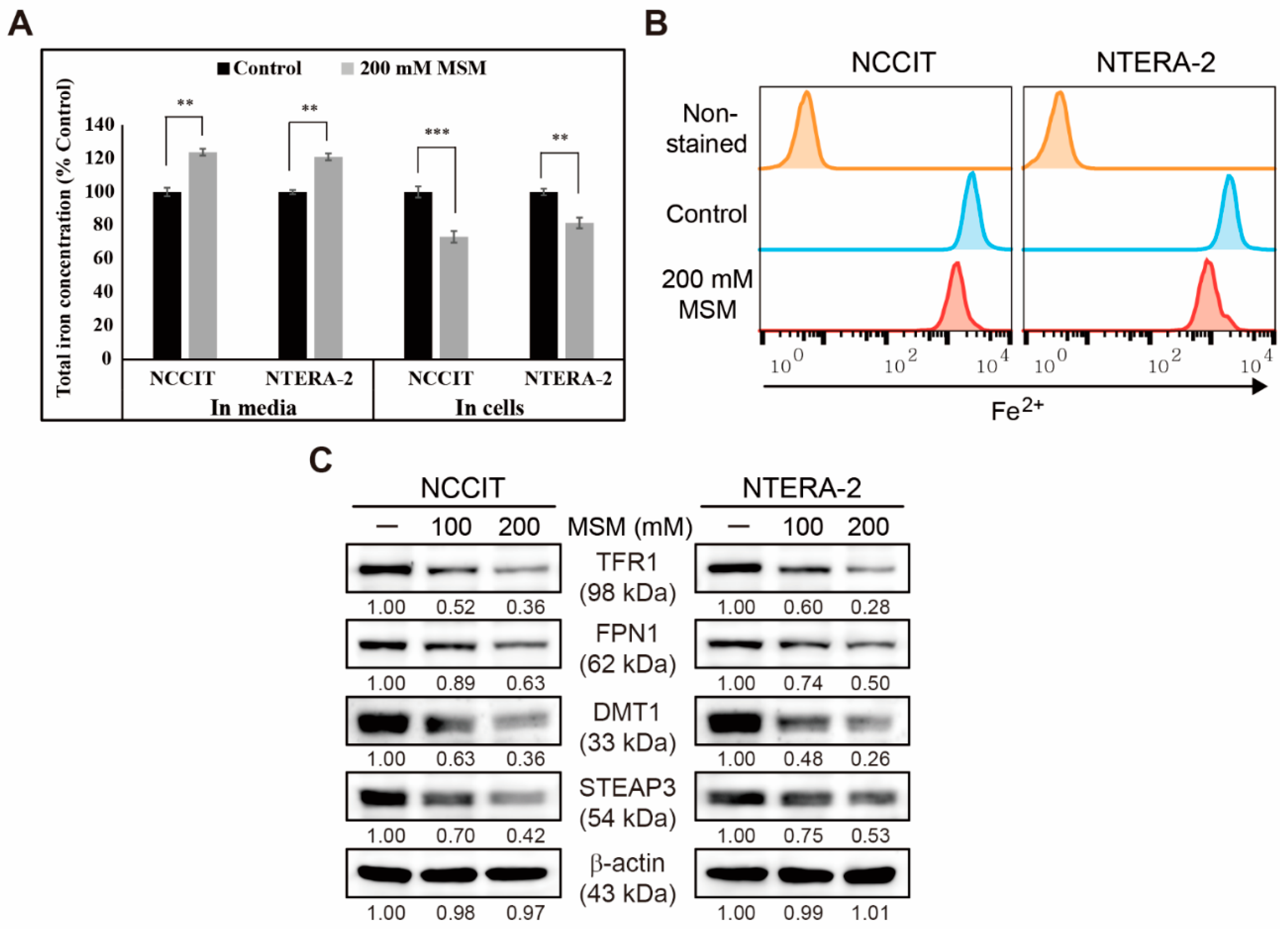
3.4. Inhibition of Iron Metabolism by MSM in Embryonic CSC
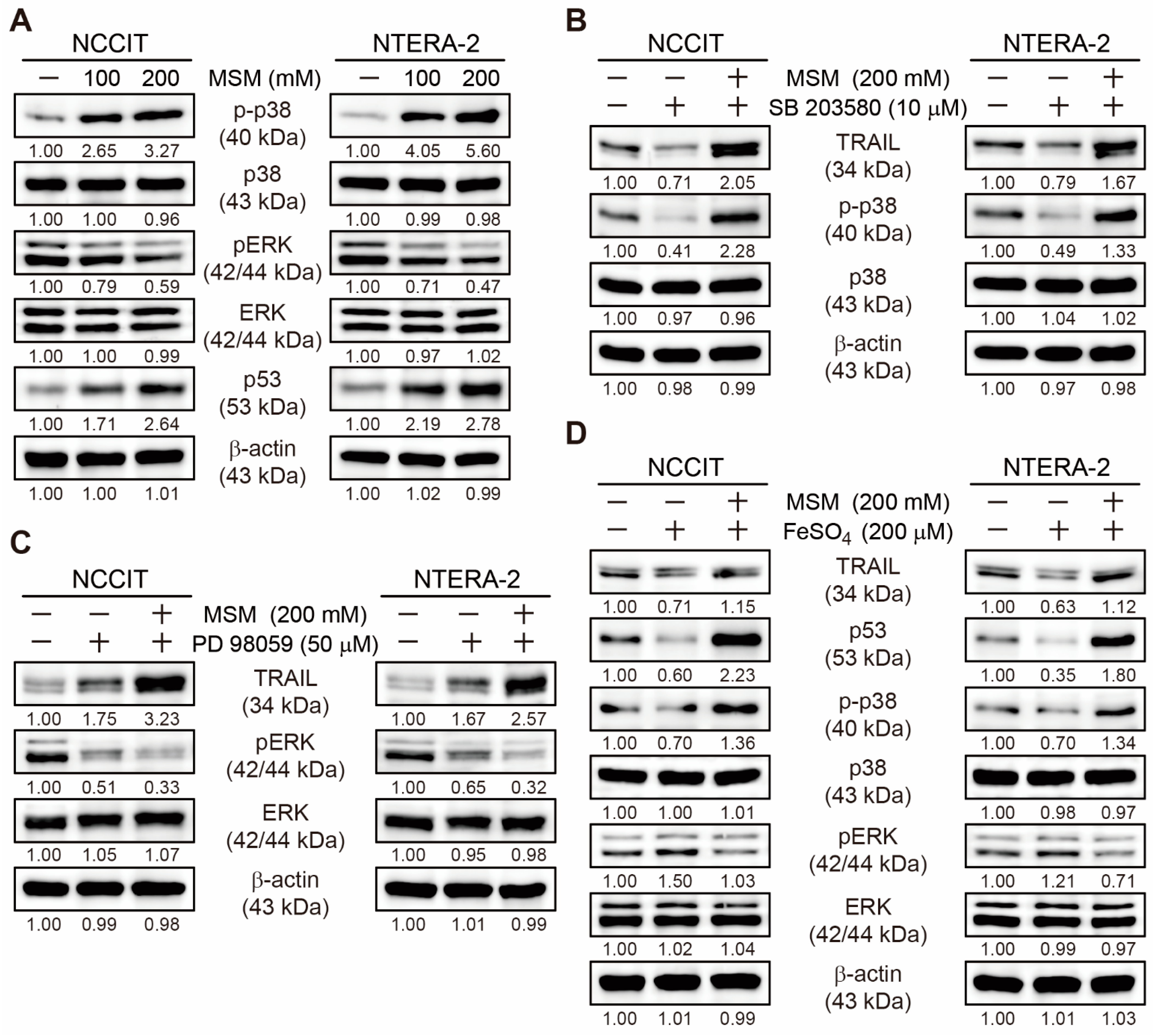
3.5. MSM Upregulated TRAIL Expression by Regulating p38/ERK/p53 Signaling
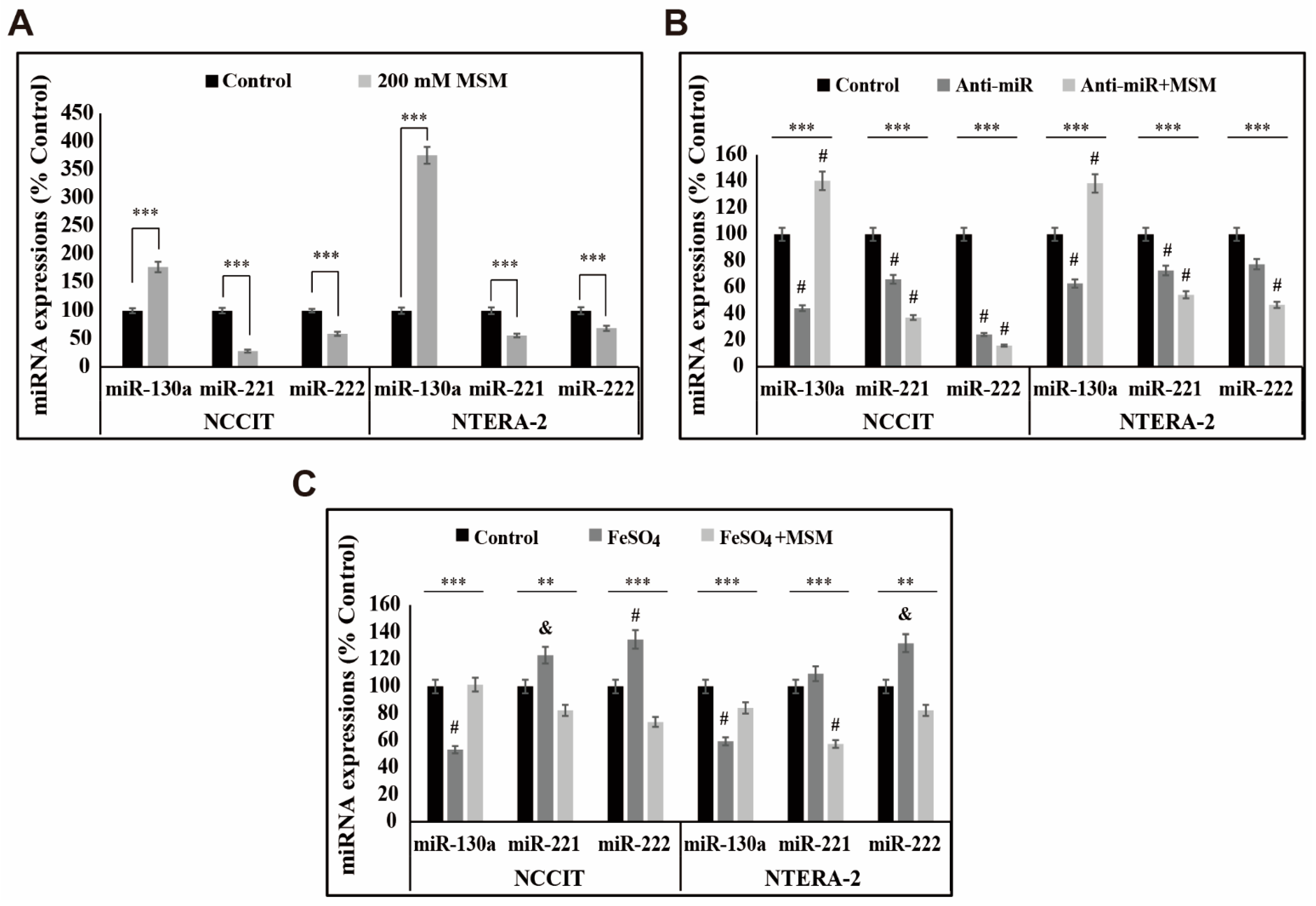
3.6. MSM Upregulated the Expression of miR-130a and Downregulated miR-221 and miR-222
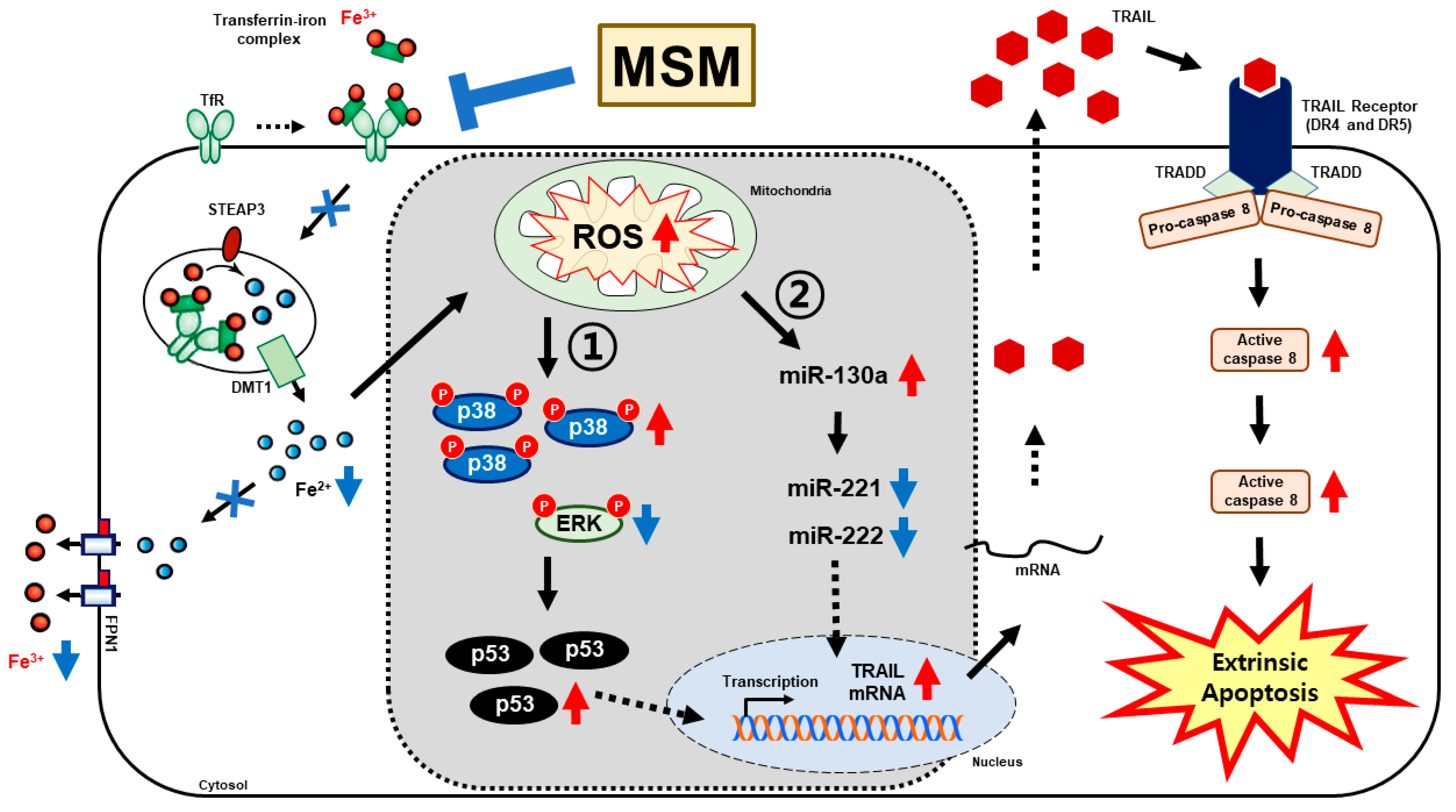
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Andrews, P.W.; Damjanov, I.; Berends, J.; Kumpf, S.; Zappavigna, V.; Mavilio, F.; Sampath, K. Inhibition of proliferation and induction of differentiation of pluripotent human embryonal carcinoma cells by osteogenic protein-1 (or bone morphogenetic protein-7). Lab. Investig. 1994, 71, 243–251. [Google Scholar] [PubMed]
- Donovan, P.J.; Gearhart, J. The end of the beginning for pluripotent stem cells. Nature 2001, 414, 92–97. [Google Scholar] [CrossRef] [PubMed]
- Mansouri, A.; Fukumitsu, H.; Schindehuette, J.; Krieglstein, K. Differentiation of embryonic stem cells. Curr. Protoc. Neurosci. 2009, 3.6. [Google Scholar] [CrossRef] [PubMed]
- Sp, N.; Kang, D.Y.; Jo, E.S.; Rugamba, A.; Kim, W.S.; Park, Y.M.; Hwang, D.Y.; Yoo, J.S.; Liu, Q.; Jang, K.J.; et al. Tannic acid promotes TRAIL-induced extrinsic apoptosis by regulating mitochondrial ROS in human embryonic carcinoma cells. Cells 2020, 9, 282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Sousa, E.M.F.; Vermeulen, L. Wnt signaling in cancer stem cell biology. Cancers 2016, 8, 60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mavila, N.; Thundimadathil, J. The emerging roles of cancer stem cells and Wnt/beta-catenin signaling in hepatoblastoma. Cancers 2019, 11, 1406. [Google Scholar] [CrossRef] [Green Version]
- MacDonald, B.T.; Tamai, K.; He, X. Wnt/beta-catenin signaling: Components, mechanisms, and diseases. Dev. Cell 2009, 17, 9–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aberle, H.; Bauer, A.; Stappert, J.; Kispert, A.; Kemler, R. Beta-catenin is a target for the ubiquitin-proteasome pathway. EMBO J. 1997, 16, 3797–3804. [Google Scholar] [CrossRef] [Green Version]
- Sp, N.; Kang, D.Y.; Kim, D.H.; Park, J.H.; Lee, H.G.; Kim, H.J.; Darvin, P.; Park, Y.M.; Yang, Y.M. Nobiletin inhibits CD36-dependent tumor angiogenesis, migration, invasion, and sphere formation through the CD36/Stat3/NF-kappaB signaling axis. Nutrients 2018, 10, 772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eini, R.; Stoop, H.; Gillis, A.J.; Biermann, K.; Dorssers, L.C.; Looijenga, L.H. Role of SOX2 in the etiology of embryonal carcinoma, based on analysis of the NCCIT and NT2 cell lines. PLoS ONE 2014, 9, e83585. [Google Scholar] [CrossRef] [Green Version]
- Jang, G.B.; Kim, J.Y.; Cho, S.D.; Park, K.S.; Jung, J.Y.; Lee, H.Y.; Hong, I.S.; Nam, J.S. Blockade of Wnt/beta-catenin signaling suppresses breast cancer metastasis by inhibiting CSC-like phenotype. Sci. Rep. 2015, 5, 12465. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Shi, L.; Zhang, L.; Li, R.; Liang, J.; Yu, W.; Sun, L.; Yang, X.; Wang, Y.; Zhang, Y.; et al. The molecular mechanism governing the oncogenic potential of SOX2 in breast cancer. J. Biol. Chem. 2008, 283, 17969–17978. [Google Scholar] [CrossRef] [Green Version]
- Thorburn, A. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) pathway signaling. J. Thorac. Oncol. 2007, 2, 461–465. [Google Scholar] [CrossRef] [Green Version]
- LeBlanc, H.N.; Ashkenazi, A. Apo2L/TRAIL and its death and decoy receptors. Cell Death Differ. 2003, 10, 66–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnstone, R.W.; Frew, A.J.; Smyth, M.J. The TRAIL apoptotic pathway in cancer onset, progression and therapy. Nat. Rev. Cancer 2008, 8, 782–798. [Google Scholar] [CrossRef]
- Beaudouin, J.; Liesche, C.; Aschenbrenner, S.; Hörner, M.; Eils, R. Caspase-8 cleaves its substrates from the plasma membrane upon CD95-induced apoptosis. Cell Death Differ. 2013, 20, 599–610. [Google Scholar] [CrossRef] [Green Version]
- Rahman, M.; Davis, S.R.; Pumphrey, J.G.; Bao, J.; Nau, M.M.; Meltzer, P.S.; Lipkowitz, S. TRAIL induces apoptosis in triple-negative breast cancer cells with a mesenchymal phenotype. Breast Cancer Res. Treat. 2009, 113, 217–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muckenthaler, M.U.; Rivella, S.; Hentze, M.W.; Galy, B. A red carpet for iron metabolism. Cell 2017, 168, 344–361. [Google Scholar] [CrossRef] [Green Version]
- Cornelissen, A.; Guo, L.; Sakamoto, A.; Virmani, R.; Finn, A.V. New insights into the role of iron in inflammation and atherosclerosis. EBiomedicine 2019, 47, 598–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Yu, L.; Ding, J.; Chen, Y. Iron metabolism in cancer. Int. J. Mol. Sci 2018, 20, 95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Fan, Z.; Yang, Y.; Gu, C. Iron metabolism and its contribution to cancer (Review). Int. J. Oncol. 2019, 54, 1143–1154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Harrington, L.; Trebicka, E.; Shi, H.N.; Kagan, J.C.; Hong, C.C.; Lin, H.Y.; Babitt, J.L.; Cherayil, B.J. Selective modulation of TLR4-activated inflammatory responses by altered iron homeostasis in mice. J. Clin. Investig. 2009, 119, 3322–3328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sp, N.; Kang, D.Y.; Joung, Y.H.; Park, J.H.; Kim, W.S.; Lee, H.K.; Song, K.D.; Park, Y.M.; Yang, Y.M. Nobiletin inhibits angiogenesis by regulating Src/FAK/STAT3-mediated signaling through PXN in ER(+) breast cancer cells. Int. J. Mol. Sci 2017, 18, 935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sp, N.; Kang, D.Y.; Lee, J.M.; Bae, S.W.; Jang, K.J. Potential antitumor effects of 6-gingerol in p53-dependent mitochondrial apoptosis and inhibition of tumor sphere formation in breast cancer cells. Int. J. Mol. Sci. 2021, 22, 660. [Google Scholar] [CrossRef] [PubMed]
- Silva Ferreira, A.C.; Rodrigues, P.; Hogg, T.; Guedes De Pinho, P. Influence of some technological parameters on the formation of dimethyl sulfide, 2-mercaptoethanol, methionol, and dimethyl sulfone in port wines. J. Agric. Food Chem. 2003, 51, 727–732. [Google Scholar] [CrossRef]
- Kim, Y.H.; Kim, D.H.; Lim, H.; Baek, D.Y.; Shin, H.K.; Kim, J.K. The anti-inflammatory effects of methylsulfonylmethane on lipopolysaccharide-induced inflammatory responses in murine macrophages. Biol. Pharm. Bull. 2009, 32, 651–656. [Google Scholar] [CrossRef] [Green Version]
- Jo, E.S.; Sp, N.; Kang, D.Y.; Rugamba, A.; Kim, I.H.; Bae, S.W.; Liu, Q.; Jang, K.J.; Yang, Y.M. Sulfur compounds inhibit high glucose-induced inflammation by regulating NF-kappaB signaling in human monocytes. Molecules 2020, 25, 342. [Google Scholar] [CrossRef]
- Lim, E.J.; Hong, D.Y.; Park, J.H.; Joung, Y.H.; Darvin, P.; Kim, S.Y.; Na, Y.M.; Hwang, T.S.; Ye, S.K.; Moon, E.S.; et al. Methylsulfonylmethane suppresses breast cancer growth by down-regulating STAT3 and STAT5b pathways. PLoS ONE 2012, 7, e33361. [Google Scholar] [CrossRef]
- Kim, D.H.; Sp, N.; Kang, D.Y.; Jo, E.S.; Rugamba, A.; Jang, K.J.; Yang, Y.M. Effect of methylsulfonylmethane on proliferation and apoptosis of A549 lung cancer cells through G2/M cell-cycle arrest and intrinsic cell death pathway. Anticancer Res. 2020, 40, 1905–1913. [Google Scholar] [CrossRef]
- Kim, D.H.; Kang, D.Y.; Sp, N.; Jo, E.S.; Rugamba, A.; Jang, K.J.; Yang, Y.M. Methylsulfonylmethane induces cell cycle arrest and apoptosis, and suppresses the stemness potential of HT-29 cells. Anticancer Res. 2020, 40, 5191–5200. [Google Scholar] [CrossRef]
- Nipin, S.P.; Kang, D.Y.; Kim, B.J.; Joung, Y.H.; Darvin, P.; Byun, H.J.; Kim, J.G.; Park, J.U.; Yang, Y.M. Methylsulfonylmethane Induces G1 Arrest and Mitochondrial Apoptosis in YD-38 Gingival Cancer Cells. Anticancer Res. 2017, 37, 1637–1646. [Google Scholar] [CrossRef] [Green Version]
- Nipin, S.P.; Darvin, P.; Yoo, Y.B.; Joung, Y.H.; Kang, D.Y.; Kim, D.N.; Hwang, T.S.; Kim, S.Y.; Kim, W.S.; Lee, H.K.; et al. The combination of methylsulfonylmethane and tamoxifen inhibits the Jak2/STAT5b pathway and synergistically inhibits tumor growth and metastasis in ER-positive breast cancer xenografts. BMC Cancer 2015, 15, 474. [Google Scholar] [CrossRef] [Green Version]
- Joung, Y.H.; Na, Y.M.; Yoo, Y.B.; Darvin, P.; Sp, N.; Kang, D.Y.; Kim, S.Y.; Kim, H.S.; Choi, Y.H.; Lee, H.K.; et al. Combination of AG490, a Jak2 inhibitor, and methylsulfonylmethane synergistically suppresses bladder tumor growth via the Jak2/STAT3 pathway. Int. J. Oncol. 2014, 44, 883–895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butawan, M.; Benjamin, R.L.; Bloomer, R.J. Methylsulfonylmethane: Applications and safety of a novel dietary supplement. Nutrients 2017, 9, 290. [Google Scholar] [CrossRef] [PubMed]
- Dawood, S.; Austin, L.; Cristofanilli, M. Cancer stem cells: Implications for cancer therapy. Oncology 2014, 28, 1101–1110. [Google Scholar] [PubMed]
- Caron, J.M.; Monteagudo, L.; Sanders, M.; Bannon, M.; Deckers, P.J. Methyl sulfone manifests anticancer activity in a metastatic murine breast cancer cell line and in human breast cancer tissue--part 2: Human breast cancer tissue. Chemotherapy 2013, 59, 24–34. [Google Scholar] [CrossRef] [PubMed]
- Morton, J.I.; Siegel, B.V. Effects of oral dimethyl sulfoxide and dimethyl sulfone on murine autoimmune lymphoproliferative disease. Proc. Soc. Exp. Biol. Med. 1986, 183, 227–230. [Google Scholar] [CrossRef] [PubMed]
- Joung, Y.H.; Darvin, P.; Kang, D.Y.; Sp, N.; Byun, H.J.; Lee, C.H.; Lee, H.K.; Yang, Y.M. Methylsulfonylmethane inhibits RANKL-induced osteoclastogenesis in BMMs by suppressing NF-kappaB and STAT3 activities. PLoS ONE 2016, 11, e0159891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rugamba, A.; Kang, D.Y.; Sp, N.; Jo, E.S.; Lee, J.M.; Bae, S.W.; Jang, K.J. Silibinin regulates tumor progression and Tumorsphere Formation by suppressing PD-L1 expression in non-small cell lung cancer (NSCLC) cells. Cells 2021, 10, 632. [Google Scholar] [CrossRef] [PubMed]
- Maréchal, A.; Zou, L. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb. Perspect. Biol. 2013, 5, 716. [Google Scholar] [CrossRef] [PubMed]
- Shiloh, Y. ATM and related protein kinases: Safeguarding genome integrity. Nat. Rev. Cancer 2003, 3, 155–168. [Google Scholar] [CrossRef] [PubMed]
- Lavin, M.F. Ataxia-telangiectasia: From a rare disorder to a paradigm for cell signalling and cancer. Nat. Rev. Mol. Cell Biol. 2008, 9, 759–769. [Google Scholar] [CrossRef]
- Pellegata, N.S.; Antoniono, R.J.; Redpath, J.L.; Stanbridge, E.J. DNA damage and p53-mediated cell cycle arrest: A reevaluation. Proc. Natl. Acad. Sci. USA 1996, 93, 15209–15214. [Google Scholar] [CrossRef] [Green Version]
- Pawlowski, J.; Kraft, A.S. Bax-induced apoptotic cell death. Proc. Natl. Acad. Sci. USA 2000, 97, 529–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sp, N.; Kang, D.Y.; Kim, D.H.; Yoo, J.S.; Jo, E.S.; Rugamba, A.; Jang, K.J.; Yang, Y.M. Tannic acid inhibits non-small cell lung cancer (NSCLC) stemness by inducing G0/G1 cell cycle arrest and intrinsic apoptosis. Anticancer Res. 2020, 40, 3209–3220. [Google Scholar] [CrossRef] [PubMed]
- Suzuki-Karasaki, Y.; Fujiwara, K.; Saito, K.; Suzuki-Karasaki, M.; Ochiai, T.; Soma, M. Distinct effects of TRAIL on the mitochondrial network in human cancer cells and normal cells: Role of plasma membrane depolarization. Oncotarget 2015, 6, 21572–21588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Wang, Y.; Lai, H.; Li, X.; Chen, T. Iron(II)-polypyridyl complexes inhibit the growth of glioblastoma tumor and enhance TRAIL-induced cell apoptosis. Chem. Asian J. 2018, 13, 2730–2738. [Google Scholar] [CrossRef]
- Forciniti, S.; Greco, L.; Grizzi, F.; Malesci, A.; Laghi, L. Iron metabolism in cancer progression. Int. J. Mol. Sci. 2020, 21, 2257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.B.; Wang, D.G.; Guo, F.F.; Xuan, C. Mitochondrial membrane potential and reactive oxygen species in cancer stem cells. Fam. Cancer 2015, 14, 19–23. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Lu, Y.; Shen, H.M. Targeting p53 as a therapeutic strategy in sensitizing TRAIL-induced apoptosis in cancer cells. Cancer Lett. 2012, 314, 8–23. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.L.; Tsai, Y.M.; Lien, C.T.; Kuo, P.L.; Hung, A.J. The roles of microRNA in lung cancer. Int. J. Mol. Sci 2019, 20, 1611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, T.; Shao, N.; Ji, C. Targeting microRNAs to modulate TRAIL-induced apoptosis of cancer cells. Cancer Gene Ther. 2013, 20, 33–37. [Google Scholar] [CrossRef] [PubMed]
- Acunzo, M.; Visone, R.; Romano, G.; Veronese, A.; Lovat, F.; Palmieri, D.; Bottoni, A.; Garofalo, M.; Gasparini, P.; Condorelli, G.; et al. miR-130a targets MET and induces TRAIL-sensitivity in NSCLC by downregulating miR-221 and 222. Oncogene 2012, 31, 634–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garofalo, M.; Di Leva, G.; Romano, G.; Nuovo, G.; Suh, S.S.; Ngankeu, A.; Taccioli, C.; Pichiorri, F.; Alder, H.; Secchiero, P.; et al. miR-221&222 regulate TRAIL resistance and enhance tumorigenicity through PTEN and TIMP3 downregulation. Cancer Cell 2009, 16, 498–509. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sp, N.; Kang, D.Y.; Jo, E.S.; Lee, J.-M.; Jang, K.-J. Iron Metabolism as a Potential Mechanism for Inducing TRAIL-Mediated Extrinsic Apoptosis Using Methylsulfonylmethane in Embryonic Cancer Stem Cells. Cells 2021, 10, 2847. https://doi.org/10.3390/cells10112847
Sp N, Kang DY, Jo ES, Lee J-M, Jang K-J. Iron Metabolism as a Potential Mechanism for Inducing TRAIL-Mediated Extrinsic Apoptosis Using Methylsulfonylmethane in Embryonic Cancer Stem Cells. Cells. 2021; 10(11):2847. https://doi.org/10.3390/cells10112847
Chicago/Turabian StyleSp, Nipin, Dong Young Kang, Eun Seong Jo, Jin-Moo Lee, and Kyoung-Jin Jang. 2021. "Iron Metabolism as a Potential Mechanism for Inducing TRAIL-Mediated Extrinsic Apoptosis Using Methylsulfonylmethane in Embryonic Cancer Stem Cells" Cells 10, no. 11: 2847. https://doi.org/10.3390/cells10112847
APA StyleSp, N., Kang, D. Y., Jo, E. S., Lee, J.-M., & Jang, K.-J. (2021). Iron Metabolism as a Potential Mechanism for Inducing TRAIL-Mediated Extrinsic Apoptosis Using Methylsulfonylmethane in Embryonic Cancer Stem Cells. Cells, 10(11), 2847. https://doi.org/10.3390/cells10112847