
Corosolic Acid Attenuates the Invasiveness of Glioblastoma Cells by Promoting CHIP-Mediated AXL Degradation and Inhibiting GAS6/AXL/JAK Axis


 ,
,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents and Antibodies
2.2. CA Treatment
2.3. Cell Culture
2.4. Cell Viability Assay
2.5. Colony Formation Assay
2.6. Migration and Invasion Ability by Boyden Chamber Assay
2.7. Immunofluorescence Staining
2.8. Western Blot
2.9. Quantitative Real-Time Polymerase Chain Reaction (qPCR)
2.10. Knockdown of CHIP by Small Inhibitory RNAs
2.11. Molecular Docking Approach
2.12. Statistical Analysis
3. Results
3.1. Effects of CA on the Cell Viability and Colony Formation Potential of Normal Astrocyte and GBM Cells
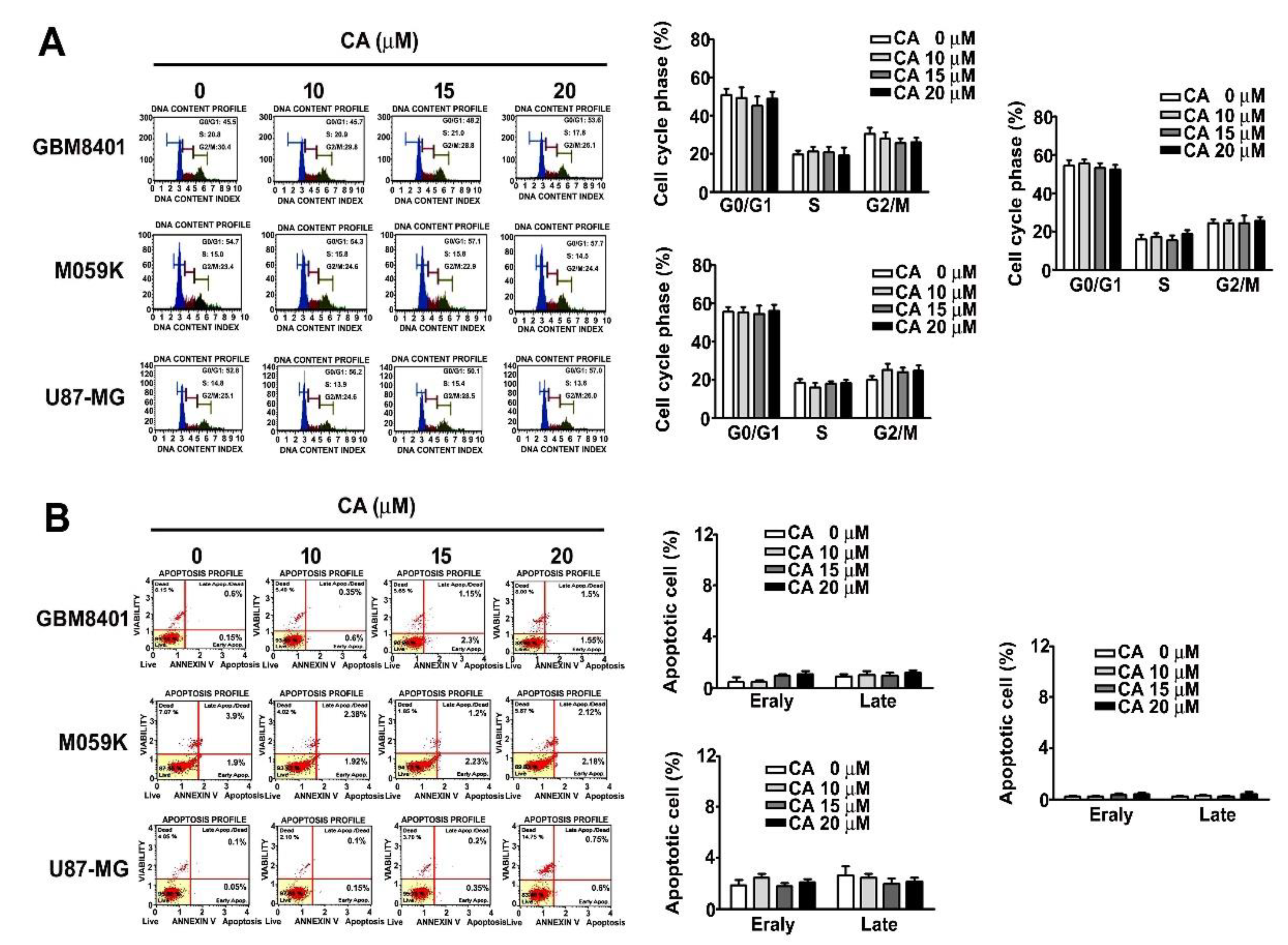
3.2. Effects of CA on the Cell Cycle and Cell Death of Three GBM Cells
3.3. CA Attenuates the Invasiveness of Human GBM Cells and Reduces F-Actin Expression
3.4. CA Reduces the Protein Level of AXL by Promoting Ubiquitin-Mediated Proteasome Degradation
3.5. Involvement of CHIP in CA-Reduced AXL and F-Actin and CA-Attenuated Invasiveness of GBM8401 Cell
3.6. Involvement of GAS6 in CA-Attenuated Invasiveness of GBM Cells
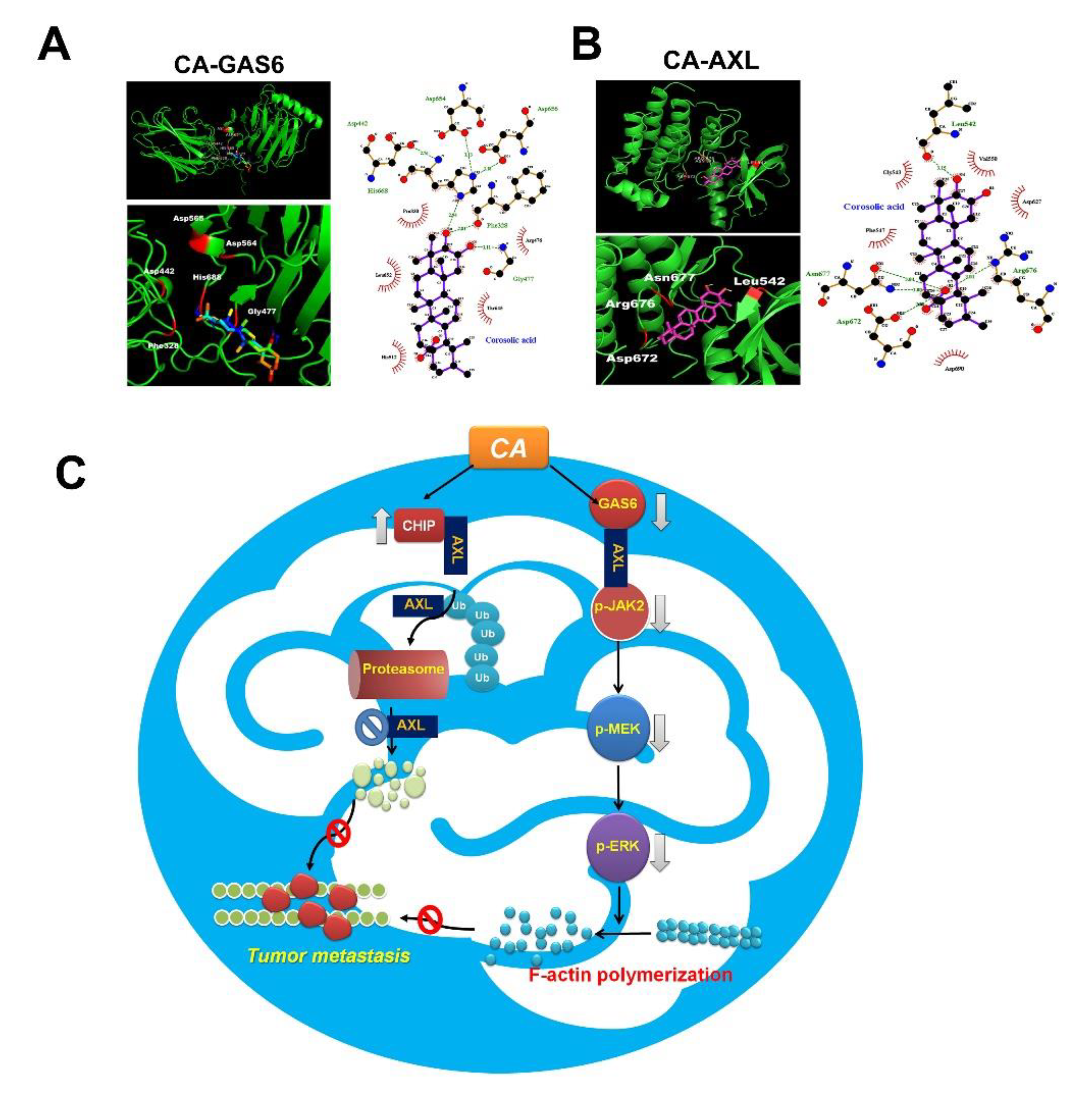
3.7. Docking Study of CA with AXL and GAS6
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K.; Burger, P.C.; Jouvet, A.; Scheithauer, B.W.; Kleihues, P. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007, 114, 97–109. [Google Scholar] [CrossRef] [Green Version]
- McLendon, R.E.; Halperin, E.C. Is the long-term survival of patients with intracranial glioblastoma multiforme overstated? Cancer 2003, 98, 1745–1748. [Google Scholar] [CrossRef]
- Stewart, L.A. Chemotherapy in adult high-grade glioma: A systematic review and meta-analysis of individual patient data from 12 randomised trials. Lancet 2002, 359, 1011–1018. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Phillips, R.E.; Soshnev, A.A.; Allis, C.D. Epigenomic Reprogramming as a Driver of Malignant Glioma. Cancer Cell 2020, 38, 647–660. [Google Scholar] [CrossRef] [PubMed]
- Linger, R.M.; Keating, A.K.; Earp, H.S.; Graham, D.K. TAM receptor tyrosine kinases: Biologic functions, signaling, and potential therapeutic targeting in human cancer. Adv. Cancer Res. 2008, 100, 35–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graham, D.K.; DeRyckere, D.; Davies, K.D.; Earp, H.S. The TAM family: Phosphatidylserine sensing receptor tyrosine kinases gone awry in cancer. Nat. Rev. Cancer 2014, 14, 769–785. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Siemann, D.W. Gas6/Axl Signaling Pathway in the Tumor Immune Microenvironment. Cancers 2020, 12, 1850. [Google Scholar] [CrossRef]
- Wium, M.; Paccez, J.D.; Zerbini, L.F. The Dual Role of TAM Receptors in Autoimmune Diseases and Cancer: An Overview. Cells 2018, 7, 166. [Google Scholar] [CrossRef] [Green Version]
- Nakada, M.; Kita, D.; Teng, L.; Pyko, I.V.; Watanabe, T.; Hayashi, Y.; Hamada, J.I. Receptor Tyrosine Kinases: Principles and Functions in Glioma Invasion. Adv. Exp. Med. Biol. 2020, 1202, 151–178. [Google Scholar] [CrossRef]
- Hutterer, M.; Knyazev, P.; Abate, A.; Reschke, M.; Maier, H.; Stefanova, N.; Knyazeva, T.; Barbieri, V.; Reindl, M.; Muigg, A.; et al. Axl and growth arrest-specific gene 6 are frequently overexpressed in human gliomas and predict poor prognosis in patients with glioblastoma multiforme. Clin. Cancer Res. 2008, 14, 130–138. [Google Scholar] [CrossRef] [Green Version]
- Linger, R.M.; Keating, A.K.; Earp, H.S.; Graham, D.K. Taking aim at Mer and Axl receptor tyrosine kinases as novel therapeutic targets in solid tumors. Expert Opin. Ther. Targets 2010, 14, 1073–1090. [Google Scholar] [CrossRef] [PubMed]
- Cheng, P.; Phillips, E.; Kim, S.H.; Taylor, D.; Hielscher, T.; Puccio, L.; Hjelmeland, A.B.; Lichter, P.; Nakano, I.; Goidts, V. Kinome-wide shRNA screen identifies the receptor tyrosine kinase AXL as a key regulator for mesenchymal glioblastoma stem-like cells. Stem Cell Rep. 2015, 4, 899–913. [Google Scholar] [CrossRef]
- Dong, M.; Xiao, Q.; Hu, J.; Cheng, F.; Zhang, P.; Zong, W.; Tang, Q.; Li, X.; Mao, F.; He, Y.; et al. Targeting LRIG2 overcomes resistance to EGFR inhibitor in glioblastoma by modulating GAS6/AXL/SRC signaling. Cancer Gene Ther. 2020, 27, 878–897. [Google Scholar] [CrossRef]
- Wang, Q.; Sun, Q.; Ma, X.; Rao, Z.; Li, H. Probing the binding interaction of human serum albumin with three bioactive constituents of Eriobotrta japonica leaves: Spectroscopic and molecular modeling approaches. J. Photochem. Photobiol. B 2015, 148, 268–276. [Google Scholar] [CrossRef] [PubMed]
- Ahn, K.S.; Hahm, M.S.; Park, E.J.; Lee, H.K.; Kim, I.H. Corosolic acid isolated from the fruit of Crataegus pinnatifida var. psilosa is a protein kinase C inhibitor as well as a cytotoxic agent. Planta Med. 1998, 64, 468–470. [Google Scholar] [CrossRef]
- Ku, C.Y.; Wang, Y.R.; Lin, H.Y.; Lu, S.C.; Lin, J.Y. Corosolic Acid Inhibits Hepatocellular Carcinoma Cell Migration by Targeting the VEGFR2/Src/FAK Pathway. PLoS ONE 2015, 10, e0126725. [Google Scholar] [CrossRef] [Green Version]
- Fujiwara, Y.; Komohara, Y.; Ikeda, T.; Takeya, M. Corosolic acid inhibits glioblastoma cell proliferation by suppressing the activation of signal transducer and activator of transcription-3 and nuclear factor-kappa B in tumor cells and tumor-associated macrophages. Cancer Sci. 2011, 102, 206–211. [Google Scholar] [CrossRef]
- Nho, K.J.; Chun, J.M.; Kim, H.K. Corosolic acid induces apoptotic cell death in human lung adenocarcinoma A549 cells in vitro. Food Chem. Toxicol. 2013, 56, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Ge, R.; Du, J.; Xin, H.; Yi, T.; Sheng, J.; Wang, Y.; Ling, C. Corosolic acid induces apoptosis through mitochondrial pathway and caspase activation in human cervix adenocarcinoma HeLa cells. Cancer Lett. 2009, 284, 229–237. [Google Scholar] [CrossRef]
- Lin, C.S.; Lin, C.L.; Ying, T.H.; Chiou, H.L.; Hung, C.H.; Liao, W.S.; Hsieh, Y.H.; Kao, S.H. Beta-Mangostin inhibits the metastatic power of cervical cancer cells attributing to suppression of JNK2/AP-1/Snail cascade. J. Cell. Physiol. 2020, 235, 8446–8460. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.C.; Yang, S.F.; Chiou, H.L.; Hsieh, S.C.; Wen, S.H.; Lu, K.H.; Hsieh, Y.H. Licochalcone A-Induced Apoptosis Through the Activation of p38MAPK Pathway Mediated Mitochondrial Pathways of Apoptosis in Human Osteosarcoma Cells In Vitro and In Vivo. Cells 2019, 8, 1441. [Google Scholar] [CrossRef] [Green Version]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamaguchi, H.; Condeelis, J. Regulation of the actin cytoskeleton in cancer cell migration and invasion. Biochim. Biophys. Acta 2007, 1773, 642–652. [Google Scholar] [CrossRef] [Green Version]
- Izdebska, M.; Zielinska, W.; Grzanka, D.; Gagat, M. The Role of Actin Dynamics and Actin-Binding Proteins Expression in Epithelial-to-Mesenchymal Transition and Its Association with Cancer Progression and Evaluation of Possible Therapeutic Targets. BioMed Res. Int. 2018, 2018, 4578373. [Google Scholar] [CrossRef] [Green Version]
- Wium, M.; Ajayi-Smith, A.F.; Paccez, J.D.; Zerbini, L.F. The Role of the Receptor Tyrosine Kinase Axl in Carcinogenesis and Development of Therapeutic Resistance: An Overview of Molecular Mechanisms and Future Applications. Cancers 2021, 13, 1521. [Google Scholar] [CrossRef] [PubMed]
- Krishnamoorthy, G.P.; Guida, T.; Alfano, L.; Avilla, E.; Santoro, M.; Carlomagno, F.; Melillo, R.M. Molecular mechanism of 17-allylamino-17-demethoxygeldanamycin (17-AAG)-induced AXL receptor tyrosine kinase degradation. J. Biol. Chem. 2013, 288, 17481–17494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holland, S.J.; Pan, A.; Franci, C.; Hu, Y.; Chang, B.; Li, W.; Duan, M.; Torneros, A.; Yu, J.; Heckrodt, T.J.; et al. R428, a selective small molecule inhibitor of Axl kinase, blocks tumor spread and prolongs survival in models of metastatic breast cancer. Cancer Res. 2010, 70, 1544–1554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, W.; Cooke, L.S.; Stejskal, A.; Riley, C.; Croce, K.D.; Saldanha, J.W.; Bearss, D.; Mahadevan, D. MP470, a novel receptor tyrosine kinase inhibitor, in combination with Erlotinib inhibits the HER family/PI3K/Akt pathway and tumor growth in prostate cancer. BMC Cancer 2009, 9, 142. [Google Scholar] [CrossRef] [Green Version]
- Che, Y.; Wang, Z.; Yuan, Y.; Zhou, H.; Wu, H.; Wang, S.; Tang, Q. By restoring autophagic flux and improving mitochondrial function, corosolic acid protects against Dox-induced cardiotoxicity. Cell Biol. Toxicol. 2021. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Yamada, K.; Yoshikawa, N.; Nakamura, K.; Haginaka, J.; Kunitomo, M. Corosolic acid prevents oxidative stress, inflammation and hypertension in SHR/NDmcr-cp rats, a model of metabolic syndrome. Life Sci. 2006, 79, 2474–2479. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Cui, Z.; Gao, X.; Liu, H.; Wang, L.; Gong, J.; Wang, A.; Zhang, J.; Ma, Q.; Huang, Y.; et al. Corosolic acid ameliorates non-alcoholic steatohepatitis induced by high-fat diet and carbon tetrachloride by regulating TGF-beta1/Smad2, NF-kappaB, and AMPK signaling pathways. Phytother. Res. 2021, 35, 5214–5226. [Google Scholar] [CrossRef] [PubMed]
- Sivakumar, G.; Vail, D.R.; Nair, V.; Medina-Bolivar, F.; Lay, J.O., Jr. Plant-based corosolic acid: Future anti-diabetic drug? Biotechnol. J. 2009, 4, 1704–1711. [Google Scholar] [CrossRef] [PubMed]
- Zdzalik-Bielecka, D.; Poswiata, A.; Kozik, K.; Jastrzebski, K.; Schink, K.O.; Brewinska-Olchowik, M.; Piwocka, K.; Stenmark, H.; Miaczynska, M. The GAS6-AXL signaling pathway triggers actin remodeling that drives membrane ruffling, macropinocytosis, and cancer-cell invasion. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef] [PubMed]
- Abu-Thuraia, A.; Goyette, M.A.; Boulais, J.; Delliaux, C.; Apcher, C.; Schott, C.; Chidiac, R.; Bagci, H.; Thibault, M.P.; Davidson, D.; et al. AXL confers cell migration and invasion by hijacking a PEAK1-regulated focal adhesion protein network. Nat. Commun. 2020, 11, 3586. [Google Scholar] [CrossRef] [PubMed]
- Baxter, E.J.; Scott, L.M.; Campbell, P.J.; East, C.; Fourouclas, N.; Swanton, S.; Vassiliou, G.S.; Bench, A.J.; Boyd, E.M.; Curtin, N.; et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 2005, 365, 1054–1061. [Google Scholar] [CrossRef]
- Kralovics, R.; Passamonti, F.; Buser, A.S.; Teo, S.S.; Tiedt, R.; Passweg, J.R.; Tichelli, A.; Cazzola, M.; Skoda, R.C. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N. Engl. J. Med. 2005, 352, 1779–1790. [Google Scholar] [CrossRef] [Green Version]
- Ben-Batalla, I.; Erdmann, R.; Jorgensen, H.; Mitchell, R.; Ernst, T.; von Amsberg, G.; Schafhausen, P.; Velthaus, J.L.; Rankin, S.; Clark, R.E.; et al. Axl Blockade by BGB324 Inhibits BCR-ABL Tyrosine Kinase Inhibitor-Sensitive and -Resistant Chronic Myeloid Leukemia. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 2289–2300. [Google Scholar] [CrossRef] [Green Version]
- Pearson, S.; Blance, R.; Somervaille, T.C.P.; Whetton, A.D.; Pierce, A. AXL Inhibition Extinguishes Primitive JAK2 Mutated Myeloproliferative Neoplasm Progenitor Cells. HemaSphere 2019, 3, e233. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Hawkins, C.; Clarke, I.D.; Squire, J.A.; Bayani, J.; Hide, T.; Henkelman, R.M.; Cusimano, M.D.; Dirks, P.B. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar] [CrossRef]
- Mao, P.; Joshi, K.; Li, J.; Kim, S.H.; Li, P.; Santana-Santos, L.; Luthra, S.; Chandran, U.R.; Benos, P.V.; Smith, L.; et al. Mesenchymal glioma stem cells are maintained by activated glycolytic metabolism involving aldehyde dehydrogenase 1A3. Proc. Natl. Acad. Sci. USA 2013, 110, 8644–8649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadahiro, H.; Kang, K.D.; Gibson, J.T.; Minata, M.; Yu, H.; Shi, J.; Chhipa, R.; Chen, Z.; Lu, S.; Simoni, Y.; et al. Activation of the Receptor Tyrosine Kinase AXL Regulates the Immune Microenvironment in Glioblastoma. Cancer Res. 2018, 78, 3002–3013. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, L.-W.; Kao, S.-H.; Yang, S.-F.; Jhang, S.-W.; Lin, Y.-C.; Chen, C.-M.; Hsieh, Y.-H. Corosolic Acid Attenuates the Invasiveness of Glioblastoma Cells by Promoting CHIP-Mediated AXL Degradation and Inhibiting GAS6/AXL/JAK Axis. Cells 2021, 10, 2919. https://doi.org/10.3390/cells10112919
Sun L-W, Kao S-H, Yang S-F, Jhang S-W, Lin Y-C, Chen C-M, Hsieh Y-H. Corosolic Acid Attenuates the Invasiveness of Glioblastoma Cells by Promoting CHIP-Mediated AXL Degradation and Inhibiting GAS6/AXL/JAK Axis. Cells. 2021; 10(11):2919. https://doi.org/10.3390/cells10112919
Chicago/Turabian StyleSun, Li-Wei, Shao-Hsuan Kao, Shun-Fa Yang, Shang-Wun Jhang, Yi-Chen Lin, Chien-Min Chen, and Yi-Hsien Hsieh. 2021. "Corosolic Acid Attenuates the Invasiveness of Glioblastoma Cells by Promoting CHIP-Mediated AXL Degradation and Inhibiting GAS6/AXL/JAK Axis" Cells 10, no. 11: 2919. https://doi.org/10.3390/cells10112919
APA StyleSun, L.-W., Kao, S.-H., Yang, S.-F., Jhang, S.-W., Lin, Y.-C., Chen, C.-M., & Hsieh, Y.-H. (2021). Corosolic Acid Attenuates the Invasiveness of Glioblastoma Cells by Promoting CHIP-Mediated AXL Degradation and Inhibiting GAS6/AXL/JAK Axis. Cells, 10(11), 2919. https://doi.org/10.3390/cells10112919