
Facing CAR T Cell Challenges on the Deadliest Paediatric Brain Tumours

 , , ,
, , , 
Abstract
:1. Introduction
2. CAR T Cell Immunotherapy
3. Overview of High-Grade CNS Paediatric Tumours
3.1. Medulloblastoma
3.2. Paediatric High-Grade Gliomas
3.3. Ependymomas
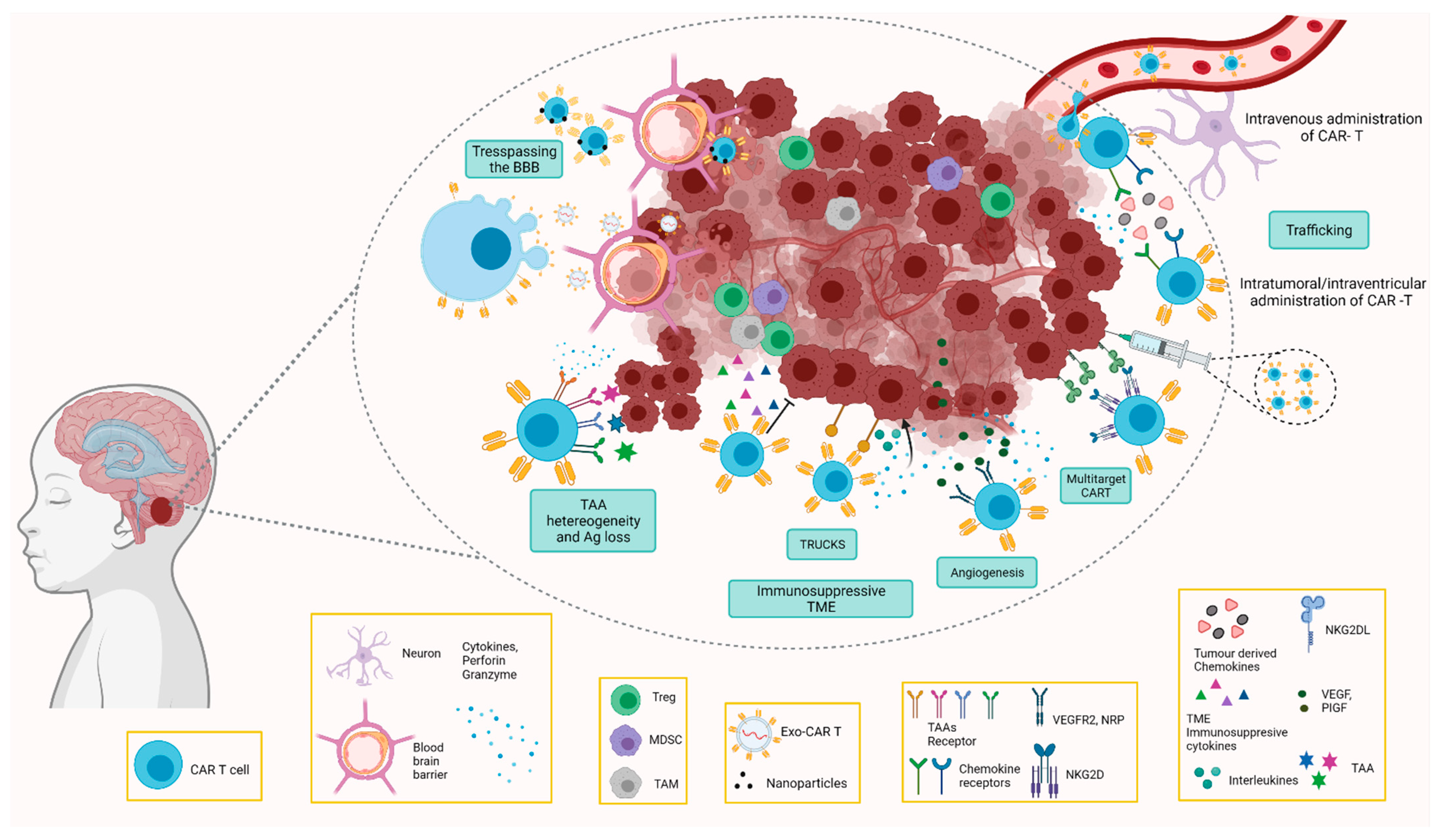
4. Challenges and Opportunities of Car T Cell Therapy in Paediatric CNS Tumours
4.1. Challenges for Paediatric Brain Tumours
4.1.1. Location
4.1.2. Blood–Brain Barrier
4.1.3. Tumour Microenvironment
4.1.4. Lack of Specific Tumour Antigens
4.1.5. Toxicity
4.2. Overcoming the Challenges by CAR T Cells
4.2.1. CAR T Cells Directed to the Immunosuppressive TME
4.2.2. Trespassing the BBB
CAR T Cell Delivery
CAR T Cell-Derived Exosomes
Routes of Administration of CAR T Cell Therapy
5. Antigen Escape
6. Toxicity
7. Other Considerations
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Johnson, K.J.; Cullen, J.; Barnholtz-Sloan, J.S.; Ostrom, Q.T.; Langer, C.E.; Turner, M.C.; Mckean-Cowdin, R.; Fisher, J.L.; Lupo, P.J.; Partap, S.; et al. Childhood Brain Tumor Epidemiology: A Brain Tumor Epidemiology Consortium Review. Cancer Epidemiol. Biomark. Prev. 2014, 23, 2716–2736. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.S.; Bandopadhayay, P.; Jenkins, M.R. Towards Immunotherapy for Pediatric Brain Tumors. Trends Immunol. 2019, 40, 748–761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pérez-Martínez, A.; Lassaletta, A.; Gonzaíez-Vicent, M.; Sevilla, J.J.; Dıáz, M.A.; Madero, L. High-dose chemotherapy with autologous stem cell rescue for children with high risk and recurrent medulloblastoma and supratentorial primitive neuroectodermal tumors. J. Neurooncol. 2005, 71, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Pearson, A.D.J.; Rossig, C.; Lesa, G.; Diede, S.J.; Weiner, S.; Anderson, J.; Gray, J.; Geoerger, B.; Minard-Colin, V.; Marshall, L.V.; et al. ACCELERATE and European Medicines Agency Paediatric Strategy Forum for medicinal product development of checkpoint inhibitors for use in combination therapy in paediatric patients. Eur. J. Cancer 2020, 127, 52–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abedalthagafi, M.; Mobark, N.; Al-Rashed, M.; AlHarbi, M. Epigenomics and immunotherapeutic advances in pediatric brain tumors. NPJ Precis. Oncol. 2021, 5, 34. [Google Scholar] [CrossRef]
- Hong, C.S.; Ho, W.; Piazza, M.G.; Ray-Chaudhury, A.; Zhuang, Z.; Heiss, J.D. Characterization of the blood brain barrier in pe-diatric central nervous system neoplasms. J. Interdiscip. Histopathol. 2016, 4, 29–33. [Google Scholar] [CrossRef] [Green Version]
- Plant, A.S.; Koyama, S.; Sinai, C.; Solomon, I.H.; Griffin, G.K.; Ligon, K.L.; Bandopadhayay, P.; Betensky, R.; Emerson, R.; Dranoff, G.; et al. Immunophenotyping of pediatric brain tumors: Correlating immune infiltrate with histology, mutational load, and survival and assessing clonal T cell response. J. Neurooncol. 2018, 137, 269–278. [Google Scholar] [CrossRef]
- Mueller, T.; Stucklin, A.S.G.; Postlmayr, A.; Metzger, S.; Gerber, N.; Kline, C.; Grotzer, M.; Nazarian, J.; Mueller, S. Advances in Targeted Therapies for Pediatric Brain Tumors. Curr. Treat. Options Neurol. 2020, 22, 43. [Google Scholar] [CrossRef]
- Ransohoff, R.M.; Kivisäkk, P.; Kidd, G. Three or more routes for leukocyte migration into the central nervous system. Nat. Rev. Immunol. 2003, 3, 569–581. [Google Scholar] [CrossRef]
- Foster, J.B.; Madsen, P.J.; Hegde, M.; Ahmed, N.; Cole, K.A.; Maris, J.M.; Resnick, A.C.; Storm, P.B.; Waanders, A.J. Immunotherapy for pediatric brain tumors: Past and present. Neuro-Oncol. 2019, 21, 1226–1238. [Google Scholar] [CrossRef]
- Biswas, A.; Kashyap, L.; Kakkar, A.; Sarkar, C.; Kumar Julka, P. Cancer Management and Research Dovepress Atypical teratoid/rhabdoid tumors: Challenges and search for solutions. Cancer Manag. Res. 2016, 8, 115–125. [Google Scholar] [CrossRef] [Green Version]
- Munhoz, R.R.; Postow, M.A. Recent advances in understanding antitumor immunity. F1000Research 2016, 5, 2545. [Google Scholar] [CrossRef] [Green Version]
- Choi, B.D.; Maus, M.V.; June, C.H.; Sampson, J.H. Immunotherapy for Glioblastoma: Adoptive T-cell Strategies. Clin. Cancer Res. 2019, 25, 2042–2048. [Google Scholar] [CrossRef]
- Verdegaal, E.M.E. Adoptive cell therapy: A highly successful individualized therapy for melanoma with great potential for other malignancies. Curr. Opin. Immunol. 2016, 39, 90–95. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Yang, J.C.; Sherry, R.M.; Kammula, U.S.; Marybeth, S.; Phan, G.Q.; Citrin, D.E.; Restifo, N.P.; Robbins, P.F.; John, R.; et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunothera-py. Clin. Cancer Res. 2012, 17, 4550–4557. [Google Scholar] [CrossRef] [Green Version]
- Larson, R.C.; Maus, M.V. Recent advances and discoveries in the mechanisms and functions of CAR T cells. Nat. Rev. Cancer 2021, 21, 145–161. [Google Scholar] [CrossRef]
- Sadelain, M.; Brentjens, R.; Rivière, I. The basic principles of chimeric antigen receptor (CAR) design. Cancer Discov. 2008, 141, 520–529. [Google Scholar]
- Shirasu, N.; Kuroki, M. Functional design of chimeric T-cell antigen receptors for adoptive immunotherapy of cancer: Architecture and outcomes. Anticancer Res. 2012, 32, 2377–2383. [Google Scholar]
- Shum, T.; Kruse, R.L.; Rooney, C.M. Strategies for enhancing adoptive T-cell immunotherapy against solid tumors using engineered cytokine signaling and other modalities. Expert Opin. Biol. Ther. 2018, 18, 653–664. [Google Scholar] [CrossRef] [PubMed]
- Yeku, O.O.; Brentjens, R.J.; Soc, B.; Author, T. Armored CAR T-cells: Utilizing cytokines and pro-inflammatory ligands to enhance CAR T-cell anti-tumour efficacy HHS Public Access Author manuscript. Biochem. Soc. Trans. 2016, 44, 412–418. [Google Scholar] [CrossRef]
- Wang, Z.; Wu, Z.; Liu, Y.; Han, W. New development in CAR-T cell therapy. J. Hematol. Oncol. 2017, 10, 53. [Google Scholar] [CrossRef] [Green Version]
- Akhavan, D.; Alizadeh, D.; Wang, D.; Weist, M.R.; Shepphird, J.K.; Brown, C.E. CAR T cells for brain tumors: Lessons learned and road ahead. Immunol. Rev. 2019, 290, 60–84. [Google Scholar] [CrossRef] [Green Version]
- Marofi, F.; Motavalli, R.; Safonov, V.A.; Thangavelu, L.; Yumashev, A.V.; Alexander, M.; Shomali, N.; Chartrand, M.S.; Pathak, Y.; Jarahian, M.; et al. CAR T cells in solid tumors: Challenges and opportunities. Stem Cell Res. Ther. 2021, 12, 81. [Google Scholar] [CrossRef] [PubMed]
- Sterner, R.C.; Sterner, R.M. CAR-T cell therapy: Current limitations and potential strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef]
- Yong, C.S.M.; Dardalhon, V.; Devaud, C.; Taylor, N.; Darcy, P.K.; Kershaw, M.H. CAR T-cell therapy of solid tumors. Immunol. Cell Biol. 2017, 95, 356–363. [Google Scholar] [CrossRef]
- Brown, C.E.; Alizadeh, D.; Starr, R.; Weng, L.; Wagner, J.R.; Naranjo, A.; Ostberg, J.R.; Blanchard, M.S.; Kilpatrick, J.; Simpson, J.; et al. Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. N. Engl. J. Med. 2016, 375, 2561–2569. [Google Scholar] [CrossRef]
- Ahmed, N.; Brawley, V.; Hegde, M.; Bielamowicz, K.; Kalra, M.; Landi, D.; Robertson, C.; Gray, T.L.; Diouf, O.; Wakefield, A.; et al. HER2-Specific Chimeric Antigen Receptor-Modified Virus-Specific T Cells for Progressive Glioblastoma A Phase 1 Dose-Escalation Trial. JAMA Oncol. 2017, 3, 1094–1101. [Google Scholar] [CrossRef]
- Louis, C.U.; Savoldo, B.; Dotti, G.; Pule, M.; Yvon, E.; Myers, G.D.; Rossig, C.; Russell, H.V.; Diouf, O.; Liu, E.; et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood 2011, 118, 6050–6056. [Google Scholar] [CrossRef]
- Choe, J.H.; Watchmaker, P.B.; Simic, M.S.; Gilbert, R.D.; Li, A.W.; Krasnow, N.A.; Downey, K.M.; Yu, W.; Carrera, D.A.; Celli, A.; et al. SynNotch-CAR T cells overcome challenges of specificity, heterogeneity, and persistence in treating glioblastoma. Sci. Transl. Med. 2021, 13, eabe7378. [Google Scholar] [CrossRef]
- Abramson, J.S.; McGree, B.; Noyes, S.; Plummer, S.; Wong, C.; Chen, Y.B.; Palmer, E.; Albertson, T.; Ferry, J.A.; Arrillaga-Romany, I.C. Anti-CD19 CAR T Cells in CNS Diffuse Large-B-Cell Lym-phoma. N. Engl. J. Med. 2017, 377, 783–784. [Google Scholar] [CrossRef]
- Engelhardt, B.; Vajkoczy, P.; Weller, R.O. The movers and shapers in immune privilege of the CNS. Nat. Immunol. 2017, 18, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Slaney, C.Y.; Kershaw, M.H.; Darcy, P.K. Trafficking of T cells into tumors. Cancer Res. 2014, 74, 7168–7174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, Y.; Pan, J.; Deng, B.; Ling, Z.; Song, W.; Xu, J.; Duan, J.; Wang, Z.; Yu, X.; Chang, A.H.; et al. Toxicity and effectiveness of CD19 CAR T therapy in children with high-burden central nervous system refractory B-ALL. Cancer Immunol. Immunother. 2021, 70, 1979–1993. [Google Scholar] [CrossRef] [PubMed]
- Byer, L.; Kline, C.; Mueller, S. Clinical trials in pediatric neuro-oncology: What is missing and how we can improve. CNS Oncol. 2016, 5, 5. [Google Scholar] [CrossRef] [Green Version]
- Cacciotti, C.; Fleming, A.; Ramaswamy, V. Advances in the molecular classification of pediatric brain tumors: A guide to the galaxy. J. Pathol. 2020, 251, 249–261. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.W.; Pajtler, K.W.; Worst, B.C.; Pfister, S.M.; Wechsler-Reya, R.J. Molecular mechanisms and therapeutic targets in pediatric brain tumors. Sci. Signal. 2017, 10, eaaf759. [Google Scholar] [CrossRef] [Green Version]
- Pollack, I.F.; Agnihotri, S.; Broniscer, A. Childhood brain tumors: Current management, biological insights, and future directions. J. Neurosurg. Pediatr. 2019, 23, 261–273. [Google Scholar] [CrossRef] [Green Version]
- Martin, A.M.; Raabe, E.; Eberhart, C.; Cohen, K.J. Management of Pediatric and Adult Patients with Medulloblastoma. Curr. Treat. Options Oncol. 2014, 15, 581–594. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro-Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
- Thomas, A.; Noël, G. Medulloblastoma: Optimizing care with a multidisciplinary approach. J. Multidiscip. Healthc. 2019, 12, 335–347. [Google Scholar] [CrossRef] [Green Version]
- Braunstein, S.; Raleigh, D.; Bindra, R.; Mueller, S.; Haas-Kogan, D. Pediatric high-grade glioma: Current molecular landscape and therapeutic approaches. J. Neurooncol. 2017, 134, 541–549. [Google Scholar] [CrossRef]
- Fangusaro, J.; Warren, K.; Hipp, S.J.; Jallo, G.; Gururangan, S. Pediatric high grade glioma: A review and update on tumor clinical characteristics and biology. Front. Oncol. 2012, 2, 105. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Guo, X.; Gao, L.; Wang, Y.; Guo, Y.; Xing, B.; Ma, W. Classification of pediatric gliomas based on immunological profiling: Implications for immunotherapy strategies. Mol. Ther.-Oncol. 2021, 20, 34–47. [Google Scholar] [CrossRef]
- McCrea, H.J.; Bander, E.D.; Venn, R.A.; Reiner, A.S.; Iorgulescu, J.B.; Puchi, L.A.; Schaefer, P.M.; Cederquist, G.; Greenfield, J.P. Sex, age, anatomic location, and extent of resection influence outcomes in children with high-grade glioma. Neurosurgery 2015, 77, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.; Karajannis, M.A.; Jones, D.T.W.; Kieran, M.W.; Monje, M.; Baker, S.J.; Becher, O.J.; Cho, Y.J.; Gupta, N.; Hawkins, C.; et al. Pediatric high-grade glioma: Biologically and clinically in need of new thinking. Neuro-Oncol. 2017, 19, 153–161. [Google Scholar] [CrossRef] [Green Version]
- Mackay, A.; Burford, A.; Carvalho, D.; Izquierdo, E.; Fazal-Salom, J.; Taylor, K.R.; Bjerke, L.; Clarke, M.; Vinci, M.; Nandhabalan, M.; et al. Integrated Molecular Meta-Analysis of 1,000 Pediatric High-Grade and Diffuse Intrinsic Pontine Glioma. Cancer Cell 2017, 32, 520–537.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarke, M.; Mackay, A.; Ismer, B.; Pickles, J.C.; Tatevossian, R.G.; Newman, S.; Bale, T.A.; Stoler, I.; Izquierdo, E.; Temelso, S.; et al. Infant high-grade gliomas comprise multiple subgroups characterized by novel targetable gene fusions and favorable outcomes. Cancer Discov. 2020, 10, 942–963. [Google Scholar] [CrossRef] [Green Version]
- Varlet, P.; Le Teuff, G.; Le Deley, M.C.; Giangaspero, F.; Haberler, C.; Jacques, T.S.; Figarella-Branger, D.; Pietsch, T.; Andreiuolo, F.; Deroulers, C.; et al. WHO grade has no prognostic value in the pediatric high-grade glioma included in the HERBY trial. Neuro-Oncol. 2020, 22, 116–127. [Google Scholar] [CrossRef]
- Rizzo, D.; Ruggiero, A.; Martini, M.; Rizzo, V.; Maurizi, P.; Riccardi, R. Molecular Biology in Pediatric High-Grade Glioma: Impact on Prognosis and Treatment. Biomed Res. Int. 2015, 2015, 215135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [Green Version]
- Srikanthan, D.; Taccone, M.S.; Van Ommeren, R.; Ishida, J.; Krumholtz, S.L.; Rutka, J.T. Diffuse intrinsic pontine glioma: Current insights and future directions. Chin. Neurosurg. J. 2021, 7, 6. [Google Scholar] [CrossRef]
- Himes, B.T.; Zhang, L.; Daniels, D.J. Treatment strategies in diffuse midline gliomas with the H3K27M mutation: The role of convection-enhanced delivery in overcoming anatomic challenges. Front. Oncol. 2019, 9, 31. [Google Scholar] [CrossRef] [Green Version]
- Mack, S.C.; Pajtler, K.W.; Chavez, L.; Okonechnikov, K.; Bertrand, K.C.; Wang, X.; Erkek, S.; Federation, A.; Song, A.; Lee, C.; et al. Therapeutic targeting of ependymoma as informed by oncogenic enhancer profiling. Nature 2018, 553, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Jia, Y.; Ye, Y.; Kang, E.; Chen, H.; Wang, J.; He, X. Identification of key methylation differentially expressed genes in posterior fossa ependymoma based on epigenomic and transcriptome analysis. J. Transl. Med. 2021, 19, 174. [Google Scholar] [CrossRef]
- Plant-Fox, A.S.; O’Halloran, K.; Goldman, S. Pediatric brain tumors: The era of molecular diagnostics, targeted and immune-based therapeutics, and a focus on long term neurologic sequelae. Curr. Probl. Cancer 2021, 45, 100777. [Google Scholar] [CrossRef] [PubMed]
- Klonou, A.; Piperi, C.; Gargalionis, A.N.; Papavassiliou, A.G. Molecular Basis of Pediatric Brain Tumors. NeuroMol. Med. 2017, 19, 256–270. [Google Scholar] [CrossRef]
- Muzumdar, D.; Ventureyra, E.C.G. Treatment of posterior fossa tumors in children. Expert Rev. Neurother. 2010, 10, 525–546. [Google Scholar] [CrossRef]
- Moussalem, C.; Ftouni, L.; Mrad, Z.A.; Amine, A.; Hamideh, D.; Baassiri, W.; Bali, B.; Najjar, M. Pediatric posterior fossa tumors outcomes: Experience in a tertiary care center in the Middle East. Clin. Neurol. Neurosurg. 2020, 197, 106170. [Google Scholar] [CrossRef]
- Lawther, B.K.; Kumar, S.; Krovvidi, H. Blood-brain barrier. Contin. Educ. Anaesth. Crit. Care Pain 2011, 11, 128–132. [Google Scholar] [CrossRef]
- Sampson, J.H.; Gunn, M.D.; Fecci, P.E.; Ashley, D.M. Brain immunology and immunotherapy in brain tumours. Nat. Rev. Cancer 2020, 20, 12–25. [Google Scholar] [CrossRef] [PubMed]
- Grabovska, Y.; Mackay, A.; O’Hare, P.; Crosier, S.; Finetti, M.; Schwalbe, E.C.; Pickles, J.C.; Fairchild, A.R.; Avery, A.; Cockle, J.; et al. Pediatric pan-central nervous system tumor analysis of immune-cell infiltration identifies correlates of antitumor immunity. Nat. Commun. 2020, 11, 4324. [Google Scholar] [CrossRef]
- Kipnis, J. Multifaceted interactions between adaptive immunity and the central nervous system. Science 2016, 353, 766–771. [Google Scholar] [CrossRef] [Green Version]
- Carrithers, M.D.; Visintin, I.; Kang, S.J.; Janeway, C.A. Differential adhesion molecule requirements for immune surveillance and inflammatory recruitment. Brain 2000, 123, 1092–1101. [Google Scholar] [CrossRef] [Green Version]
- Greenwood, J.; Heasman, S.J.; Alvarez, J.I.; Prat, A.; Lyck, R.; Engelhardt, B. Review: Leucocyte-endothelial cell crosstalk at the blood-brain barrier: A prerequisite for successful immune cell entry to the brain. Neuropathol. Appl. Neurobiol. 2011, 37, 24–39. [Google Scholar] [CrossRef]
- Negi, N.; Das, B.K. CNS: Not an immunoprivilaged site anymore but a virtual secondary lymphoid organ. Int. Rev. Immunol. 2018, 37, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Prinz, M.; Priller, J. The role of peripheral immune cells in the CNS in steady state and disease. Nat. Neurosci. 2017, 20, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Roesch, S.; Rapp, C.; Dettling, S.; Herold-Mende, C. Molecular Sciences When Immune Cells Turn Bad-Tumor-Associated Microglia/Macrophages in Glioma. Int. J. Mol. Sci. 2018, 19, 436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quail, D.F.; Joyce, J.A. The Microenvironmental Landscape of Brain Tumors. Cancer Cell 2017, 31, 326–341. [Google Scholar] [CrossRef] [Green Version]
- Phoenix, T.N.; Patmore, D.M.; Boop, S.; Boulos, N.; Jacus, M.O.; Patel, Y.T.; Roussel, M.F.; Finkelstein, D.; Goumnerova, L.; Perreault, S.; et al. Medulloblastoma Genotype Dictates Blood Brain Barrier Phenotype. Cancer Cell 2016, 29, 508–522. [Google Scholar] [CrossRef] [Green Version]
- Batista, A.; Riedemann, L.; Vardam, T.; Jain, R.K. Targeting the Tumor Microenvironment to Enhance Pediatric Brain Cancer Treatment. Cancer J. (USA) 2015, 21, 307–313. [Google Scholar] [CrossRef]
- Terry, R.L.; Meyran, D.; Ziegler, D.S.; Haber, M.; Ekert, P.G.; Trapani, J.A.; Neeson, P.J. Immune profiling of pediatric solid tumors. J. Clin. Investig. 2020, 130, 3391–3402. [Google Scholar] [CrossRef]
- Wu, S.Y.; Watabe, K. The roles of microglia/macrophages in tumor progression of brain cancer and metastatic disease. Front. Biosci.-Landmark 2017, 22, 1805–1829. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Geng, X.; Hou, J.; Wu, G. New insights into M1/M2 macrophages: Key modulators in cancer progression. Cancer Cell Int. 2021, 21, 389. [Google Scholar] [CrossRef]
- Upreti, D.; Bakhshinyan, D.; Bloemberg, D.; Vora, P.; Venugopal, C.; Singh, S.K. Strategies to Enhance the Efficacy of T-Cell Therapy for Central Nervous System Tumors. Front. Immunol. 2020, 11, 599253. [Google Scholar] [CrossRef] [PubMed]
- Diao, S.; Gu, C.; Zhang, H.; Yu, C. Immune cell infiltration and cytokine secretion analysis reveal a non-inflammatory microenvironment of medulloblastoma. Oncol. Lett. 2020, 20, 397. [Google Scholar] [CrossRef] [PubMed]
- Maximov, V.; Chen, Z.; Wei, Y.; Robinson, M.H.; Herting, C.J.; Shanmugam, N.S.; Rudneva, V.A.; Goldsmith, K.C.; MacDonald, T.J.; Northcott, P.A.; et al. Tumour-associated macrophages exhibit anti-tumoural properties in Sonic Hedgehog medulloblastoma. Nat. Commun. 2019, 10, 2410. [Google Scholar] [CrossRef]
- Pyonteck, S.M.; Akkari, L.; Schuhmacher, A.J.; Bowman, R.L.; Sevenich, L.; Quail, D.F.; Olson, O.C.; Quick, M.L.; Huse, J.T.; Teijeiro, V.; et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med. 2013, 19, 1264–1272. [Google Scholar]
- Lampson, L.A. Immunotherapy of Malignant Tumors in the Brain: How Different from Other Sites? Front. Oncol. 2016, 8. [Google Scholar] [CrossRef]
- Lieberman, N.A.P.; Degolier, K.; Kovar, H.M.; Davis, A.; Hoglund, V.; Stevens, J.; Winter, C.; Deutsch, G.; Furlan, S.N.; Vitanza, N.A.; et al. Characterization of the immune microenvironment of diffuse intrinsic pontine glioma: Implications for development of immunotherapy. Neuro-Oncol. 2019, 21, 83–94. [Google Scholar] [CrossRef] [Green Version]
- Chalmers, Z.R.; Connelly, C.F.; Fabrizio, D.; Gay, L.; Ali, S.M.; Ennis, R.; Schrock, A.; Campbell, B.; Shlien, A.; Chmielecki, J.; et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017, 9, 34. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.T.; Jin, W.L. B7-H3/CD276: An Emerging Cancer Immunotherapy. Front. Immunol. 2021, 12, 701006. [Google Scholar] [CrossRef] [PubMed]
- Majzner, R.G.; Theruvath, J.L.; Nellan, A.; Heitzeneder, S.; Cui, Y.; Mount, C.W.; Rietberg, S.P.; Linde, M.H.; Xu, P.; Rota, C.; et al. CAR T Cells Targeting B7-H3, a Pan-Cancer Antigen, Demonstrate Potent Preclinical Activity Against Pediatric Solid Tumors and Brain Tumors. Clin. Cancer Res. 2019, 25, 2560–2574. [Google Scholar] [CrossRef]
- Maachani, U.B.; Tosi, U.; Pisapia, D.J.; Mukherjee, S.; Marnell, C.S.; Voronina, J.; Martinez, D.; Santi, M.; Dahmane, N.; Zhou, Z.; et al. B7-H3 as a Prognostic Biomarker and Therapeutic Target in Pediatric central nervous system Tumors. Transl. Oncol. 2020, 13, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Patterson, J.D.; Henson, J.C.; Breese, R.O.; Bielamowicz, K.J.; Rodriguez, A. CAR T Cell Therapy for Pediatric Brain Tumors. Front. Oncol. 2020, 10, 1582. [Google Scholar] [CrossRef] [PubMed]
- Donovan, L.K.; Delaidelli, A.; Joseph, S.K.; Bielamowicz, K.; Fousek, K.; Holgado, B.L.; Manno, A.; Srikanthan, D.; Gad, A.Z.; Van Ommeren, R.; et al. Locoregional delivery of CAR T cells to the cerebrospinal fluid for treatment of metastatic medulloblastoma and ependymoma. Nat. Med. 2020, 26, 720–731. [Google Scholar] [CrossRef]
- Okada, H.; Low, K.L.; Kohanbash, G.; McDonald, H.A.; Hamilton, R.L.; Pollack, I.F. Expression of glioma-associated antigens in pediatric brain stem and non-brain stem gliomas. J. Neurooncol. 2008, 88, 245–250. [Google Scholar] [CrossRef] [Green Version]
- Kawakami, M.; Kawakami, K.; Takahashi, S.; Abe, M.; Puri, R.K. Analysis of interleukin-13 receptor α2 expression in human pediatric brain tumors. Cancer 2004, 101, 1036–1042. [Google Scholar] [CrossRef]
- Nausch, N.; Cerwenka, A. NKG2D ligands in tumor immunity. Oncogene 2008, 27, 5944–5958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández, L.; Portugal, R.; Valentín, J.; Martín, R.; Maxwell, H.; González-Vicent, M.; Díaz, M.Á.; de Prada, I.; Pérez-Martínez, A. In vitro natural killer cell immunotherapy for medulloblastoma. Front. Oncol. 2013, 3, 94. [Google Scholar] [CrossRef] [Green Version]
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.D.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A.; et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 2017, 9, 57–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mount, C.W.; Majzner, R.G.; Sundaresh, S.; Arnold, E.P.; Kadapakkam, M.; Haile, S.; Labanieh, L.; Hulleman, E.; Woo, P.J.; Rietberg, S.P.; et al. Potent antitumor efficacy of anti-GD2 CAR T cells in H3-K27M+ diffuse midline gliomas letter. Nat. Med. 2018, 24, 572–579. [Google Scholar] [CrossRef]
- Nazha, B.; Inal, C.; Owonikoko, T.K. Disialoganglioside GD2 Expression in Solid Tumors and Role as a Target for Cancer Therapy. Front. Oncol. 2020, 10, 1000. [Google Scholar] [CrossRef]
- Basciani, S.; Mariani, S.; Arizzi, M.; Ulisse, S.; Rucci, N.; Jannini, E.A.; Della Rocca, C.; Manicone, A.; Carani, C.; Spera, G.; et al. Expression of Platelet-Derived Growth Factor-A (PDGF-A), PDGF-B, and PDGF Receptor-and-during Human Testicular Development and Disease. J. Clin. Endocrinol. Metab. 2002, 87, 2310–2319. [Google Scholar] [PubMed] [Green Version]
- Paugh, B.S.; Qu, C.; Jones, C.; Liu, Z.; Adamowicz-Brice, M.; Zhang, J.; Bax, D.A.; Coyle, B.; Barrow, J.; Hargrave, D.; et al. Integrated molecular genetic profiling of pediatric high-grade gliomas reveals key differences with the adult disease. J. Clin. Oncol. 2010, 28, 3061–3068. [Google Scholar] [CrossRef] [PubMed]
- Koschmann, C.; Zamler, D.; Mackay, A.; Robinson, D.; Wu, Y.-M.; Doherty, R.; Marini, B.; Tran, D.; Garton, H.; Muraszko, K.; et al. Characterizing and targeting PDGFRA alterations in pediatric high-grade glioma. Oncotarget 2016, 7, 65696–65706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paugh, B.S.; Zhu, X.; Qu, C.; Endersby, R.; Diaz, A.K.; Zhang, J.; Bax, D.A.; Carvalho, D.; Reis, R.M.; Onar-Thomas, A.; et al. Molecular and Cellular Pathobiology Novel Oncogenic PDGFRA Mutations in Pediatric High-Grade Gliomas. Cancer Res. 2013, 73, 6219–6229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puget, S.; Philippe, C.; Bax, D.A.; Varlet, J.B. Mesenchymal Transition and PDGFRA Amplification/Mutation Are Key Distinct Oncogenic Events in Pediatric Diffuse Intrinsic Pontine Gliomas. PLoS ONE 2012, 7, 30313. [Google Scholar] [CrossRef] [Green Version]
- Snuderl, M.; Batista, A.; Kirkpatrick, N.D.; De Almodovar, C.R.; Riedemann, L.; Walsh, E.C.; Anolik, R.; Huang, Y.; Martin, J.D.; Kamoun, W.; et al. Targeting Placental Growth Factor/Neuropilin 1 Pathway Inhibits Growth and Spread of Medulloblastoma. Cell 2013, 152, 1065–1076. [Google Scholar] [CrossRef] [Green Version]
- De Falco, S. The discovery of placenta growth factor and its biological activity. Exp. Mol. Med. 2012, 44, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Krenciute, G.; Krebs, S.; Torres, D.; Wu, M.F.; Liu, H.; Dotti, G.; Li, X.N.; Lesniak, M.S.; Balyasnikova, I.V.; Gottschalk, S. Characterization and functional analysis of scFv-based chimeric antigen receptors to redirect T Cells to IL13Rα2-positive glioma. Mol. Ther. 2016, 24, 354–363. [Google Scholar] [CrossRef] [Green Version]
- Chow, K.K.; Naik, S.; Kakarla, S.; Brawley, V.S.; Shaffer, D.R.; Yi, Z.; Rainusso, N.; Wu, M.F.; Liu, H.; Kew, Y.; et al. T cells redirected to EphA2 for the immunotherapy of glioblastoma. Mol. Ther. 2013, 21, 629–637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haydar, D.; Houke, H.; Chiang, J.; Yi, Z.; Odé, Z.; Caldwell, K.; Zhu, X.; Mercer, K.S.; Stripay, J.L.; Shaw, T.I.; et al. Cell-surface antigen profiling of pediatric brain tumors: B7-H3 is consistently expressed and can be targeted via local or systemic CAR T-cell delivery. Neuro-Oncology 2021, 23, 999–1011. [Google Scholar] [CrossRef]
- Jafarzadeh, L.; Masoumi, E.; Fallah-Mehrjardi, K.; Mirzaei, H.R.; Hadjati, J. Prolonged Persistence of Chimeric Antigen Receptor (CAR) T Cell in Adoptive Cancer Immunotherapy: Challenges and Ways Forward. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Chen, W. Mechanisms of failure of chimeric antigen receptor T-cell therapy. Curr. Opin. Hematol. 2019, 26, 427–433. [Google Scholar] [CrossRef]
- Hsieh, E.M.; Scherer, L.D.; Rouce, R.H. Replacing CAR-T cell resistance with persistence by changing a single residue. J. Clin. Investig. 2020, 130, 2806–2808. [Google Scholar] [CrossRef]
- Cheng, J.; Zhao, L.; Zhang, Y.; Qin, Y.; Guan, Y.; Zhang, T.; Liu, C.; Zhou, J. Understanding the Mechanisms of Resistance to CAR T-Cell Therapy in Malignancies. Front. Oncol. 2019, 9, 1237. [Google Scholar] [CrossRef] [Green Version]
- Goff, S.L.; Morgan, R.A.; Yang, J.C.; Sherry, R.M.; Robbins, P.F.; Restifo, N.P.; Feldman, S.A.; Lu, Y.-C.; Lu, L.; Zheng, Z.; et al. Pilot trial of adoptive transfer of chimeric antigen receptor transduced T cells targeting EGFRvIII in patients with glioblastoma HHS Public Access. J. Immunother. 2019, 42, 126–135. [Google Scholar] [CrossRef]
- Suter, R.K.; Rodriguez-Blanco, J.; Ayad, N.G. Epigenetic pathways and plasticity in brain tumors. Neurobiol. Dis. 2020, 145, 105060. [Google Scholar] [CrossRef]
- Morris, E.C.; Neelapu, S.S.; Giavridis, T.; Sadelain, M. Cytokine release syndrome and associated neurotoxicity in cancer immunotherapy. Nat. Rev. Immunol. 2021, 0123456789. [Google Scholar] [CrossRef]
- Neelapu, S.S.; Tummala, S.; Kebriaei, P.; Wierda, W.; Gutierrez, C.; Locke, F.L.; Komanduri, K.V.; Lin, Y.; Jain, N.; Daver, N.; et al. Chimeric antigen receptor T-cell therapy—Assessment and management of toxicities. Nat. Rev. Clin. Oncol. 2018, 15, 47–62. [Google Scholar] [CrossRef] [PubMed]
- June, C.H.; Sadelain, M. Chimeric Antigen Receptor Therapy | Enhanced Reader. N. Engl. J. Med. Rev. 2018, 379, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Siegler, E.L.; Kenderian, S.S. Neurotoxicity and Cytokine Release Syndrome After Chimeric Antigen Receptor T Cell Therapy: Insights into Mechanisms and Novel Therapies. Front. Immunol. 2020, 11, 1973. [Google Scholar] [CrossRef]
- Sharma, A.; De Leon, G.; Porter, A.; Grill, M.F.; Rosenthal, A.; Brown, C.E.; Swanson, K.; Mrugala, M.M. CAR-T cell therapy in neuro-oncology: Applications and toxicity. Neuroimmunol. Neuroinflamm. 2018, 5, 43. [Google Scholar] [CrossRef] [Green Version]
- Wu, C.Y.; Roybal, K.T.; Puchner, E.M.; Onuffer, J.; Lim, W.A. Remote control of therapeutic T cells through a small molecule-gated chimeric receptor. Science 2015, 350, aab4077. [Google Scholar] [CrossRef] [Green Version]
- Santomasso, B.D.; Park, J.H.; Salloum, D.; Riviere, I.; Mead, E.; Halton, E.; Wang, X.; Senechal, B.; Cross, J.R.; Liu, H.; et al. Clinical and Biological Correlates of Neurotoxicity Associated with CAR T-cell Therapy in Patients with B-cell Acute Lymphoblastic Leukemia. Cancer Discov. 2019, 8, 958–971. [Google Scholar] [CrossRef] [Green Version]
- Gust, J.; Ponce, R.; Liles, W.C.; Garden, G.A.; Turtle, C.J. Cytokines in CAR T Cell–Associated Neurotoxicity. Front. Immunol. 2020, 11, 577027. [Google Scholar] [CrossRef]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef]
- Schuster, S.J.; Bishop, M.R.; Tam, C.S.; Waller, E.K.; Borchmann, P.; McGuirk, J.P.; Jäger, U.; Jaglowski, S.; Andreadis, C.; Westin, J.R.; et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2019, 380, 45–56. [Google Scholar] [CrossRef]
- Teachey, D.T.; Lacey, S.F.; Shaw, P.A.; Melenhorst, J.J.; Maude, S.L.; Frey, N.; Pequignot, E.; Gonzalez, V.E.; Chen, F.; Finklestein, J.; et al. Identification of Predictive Biomarkers for Cytokine Release Syndrome after Chimeric Antigen Receptor T-cell Therapy for Acute Lymphoblastic Leukemia. Cancer Discov. 2016, 6, 664–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norelli, M.; Camisa, B.; Barbiera, G.; Falcone, L.; Purevdorj, A.; Genua, M.; Sanvito, F.; Ponzoni, M.; Doglioni, C.; Cristofori, P.; et al. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat. Med. 2018, 24, 739–748. [Google Scholar] [CrossRef] [PubMed]
- Giavridis, T.; van der Stegen, S.J.; Eyquem, J.; Hamieh, M.; Piersigilli, A.; Sadelain, M. CAR T cell-induced cytokine release syndrome is mediated by macrophages. Physiol. Behav. 2019, 176, 139–148. [Google Scholar]
- Lee, D.W.; Santomasso, B.D.; Locke, F.L.; Ghobadi, A.; Turtle, C.J.; Brudno, J.N.; Maus, M.V.; Park, J.H.; Mead, E.; Pavletic, S.; et al. ASTCT Consensus Grading for Cytokine Release Syndrome and Neurologic Toxicity Associated with Immune Effector Cells. Biol. Blood Marrow Transplant. 2019, 25, 625–638. [Google Scholar] [CrossRef] [Green Version]
- Rafiq, S.; Hackett, C.S.; Brentjens, R.J. Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat. Rev. Clin. Oncol. 2020, 17, 147–167. [Google Scholar] [CrossRef]
- Gust, J.; Hay, K.A.; Hanafi, L.-A.; Li, D.; Myerson, D.; Gonzalez-Cuyar, L.F.; Yeung, C.; Liles, W.C.; Wurfel, M.; Lopez, J.A.; et al. Endothelial Activation and Blood-Brain Barrier Disruption in Neurotoxicity after Adoptive Immunotherapy with CD19 CAR-T Cells. Cancer Discov. 2021, 7, 1404–1419. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.W.; Kochenderfer, J.N.; Stetler-Stevenson, M.; Cui, Y.K.; Delbrook, C.; Feldman, S.A.; Fry, T.J.; Orentas, R.; Sabatino, M.; Shah, N.N.; et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: A phase 1 dose-escalation trial. Lancet 2015, 385, 517–528. [Google Scholar] [CrossRef]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D.; et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Hombach, A.A.; Abken, H. Shared target antigens on cancer cells and tissue stem cells: Go or no-go for CAR T cells? Expert Rev. Clin. Immunol. 2017, 13, 151–155. [Google Scholar] [CrossRef]
- Majzner, R.G.; Ramakrishna, S.; Mochizuki, A.; Patel, S.; Chinnasamy, H.; Yeom, K.; Schultz, L.; Richards, R.; Campen, C.; Reschke, A.; et al. Abstract CT031: GD2 CAR T cells mediate clinical activity and manageable toxicity in children and young adults with DIPG and H3K27M-mutated diffuse midline gliomas. Clin. Trials 2021, 81, CT031. [Google Scholar]
- Schuelke, M.R.; Wongthida, P.; Thompson, J.; Kottke, T.; Driscoll, C.B.; Huff, A.L.; Shim, K.G.; Coffey, M.; Pulido, J.; Evgin, L.; et al. Diverse immunotherapies can effectively treat syngeneic brainstem tumors in the absence of overt toxicity. J. Immunother. Cancer 2019, 7, 188. [Google Scholar] [CrossRef] [PubMed]
- Chmielewski, M.; Abken, H. CAR T Cells Releasing IL-18 Convert to T-Bethigh FoxO1low Effectors that Exhibit Augmented Activity against Advanced Solid Tumors. Cell Rep. 2017, 21, 3205–3219. [Google Scholar] [CrossRef] [Green Version]
- Zimmermann, K.; Kuehle, J.; Dragon, A.C.; Galla, M.; Kloth, C.; Rudek, L.S.; Sandalcioglu, I.E.; Neyazi, B.; Moritz, T.; Meyer, J.; et al. Design and characterization of an “all-in-one” lentiviral vector system combining constitutive anti-gd2 car expression and inducible cytokines. Cancers 2020, 12, 375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chmielewski, M.; Abken, H. TRUCKS, the fourth-generation CAR T cells: Current developments and clinical translation. Adv. Cell Gene Ther. 2020, 3, e84. [Google Scholar] [CrossRef]
- Hawkins, E.R.; D’souza, R.R.; Klampatsa, A. Armored CAR T-cells: The next chapter in T-cell cancer immunotherapy. Biol. Targets Ther. 2021, 15, 95–105. [Google Scholar] [CrossRef]
- Agliardi, G.; Liuzzi, A.R.; Hotblack, A.; De Feo, D.; Núñez, N.; Stowe, C.L.; Friebel, E.; Nannini, F.; Rindlisbacher, L.; Roberts, T.A.; et al. Intratumoral IL-12 delivery empowers CAR-T cell immunotherapy in a pre-clinical model of glioblastoma. Nat. Commun. 2021, 12, 444. [Google Scholar] [CrossRef]
- Koneru, M.; O’Cearbhaill, R.; Pendharkar, S.; Spriggs, D.R.; Brentjens, R.J. A phase I clinical trial of adoptive T cell therapy using IL-12 secreting MUC-16ecto directed chimeric antigen receptors for recurrent ovarian cancer. J. Transl. Med. 2015, 13, 102. [Google Scholar] [CrossRef] [Green Version]
- Nair, S.; Wang, J.-B.; Tsao, S.-T.; Liu, Y.; Zhu, W.; Slayton, W.B.; Moreb, J.S.; Dong, L.; Chang, L.-J. Functional Improvement of Chimeric Antigen Receptor Through Intrinsic Interleukin-15Rα Signaling. Curr. Gene Ther. 2018, 19, 40–53. [Google Scholar] [CrossRef]
- Jin, L.; Tao, H.; Karachi, A.; Long, Y.; Hou, A.Y.; Na, M.; Dyson, K.A.; Grippin, A.J.; Deleyrolle, L.P.; Zhang, W.; et al. CXCR1- or CXCR2-modified CAR T cells co-opt IL-8 for maximal antitumor efficacy in solid tumors. Nat. Commun. 2019, 10, 4016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Wu, H.; Chen, G.; Wei, X.; Wang, C.; Zhou, S.; Huang, A.; Zhang, Z.; Zhan, C.; Wu, Y.; et al. Arming Anti-EGFRvIII CAR-T with TGFβ Trap Improves Antitumor Efficacy in Glioma Mouse Models. Front. Oncol. 2020, 10, 1117. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Yan, J.; Liu, B. Targeting VEGF/VEGFR to modulate antitumor immunity. Front. Immunol. 2018, 9, 978. [Google Scholar] [CrossRef] [Green Version]
- Gururangan, S.; Chi, S.N.; Poussaint, T.Y.; Onar-Thomas, A.; Gilbertson, R.J.; Vajapeyam, S.; Friedman, H.S.; Packer, R.J.; Rood, B.N.; Boyett, J.M.; et al. Lack of efficacy of bevacizumab plus irinotecan in children with recurrent malignant glioma and diffuse brainstem glioma: A pediatric brain tumor consortium study. J. Clin. Oncol. 2010, 28, 3069–3075. [Google Scholar] [CrossRef]
- Englisch, A.; Altvater, B.; Kailayangiri, S.; Hartmann, W.; Rossig, C. VEGFR2 as a target for CAR T cell therapy of Ewing sarcoma. Pediatr. Blood Cancer 2020, 67, e28313. [Google Scholar] [CrossRef]
- Han, X.; Wang, Y.; Han, W.-D. Chimeric antigen receptor modified T-cells for cancer treatment. Chronic Dis. Transl. Med. 2018, 4, 225–243. [Google Scholar] [CrossRef]
- Liu, H.; Sun, Q.; Zhang, M.; Zhang, Z.; Fan, X.; Yuan, H.; Li, C.; Guo, Y.; Ning, W.; Sun, Y.; et al. Differential expression of folate receptor 1 in medulloblastoma and the correlation with clinicopathological characters and target therapeutic potential. Oncotarget 2017, 8, 23048–23060. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.J.; Chu, H.; Wheeler, L.W.; Nelson, M.; Westrick, E.; Matthaei, J.F.; Cardle, I.I.; Johnson, A.; Gustafson, J.; Parker, N.; et al. Preclinical evaluation of bispecific adaptor molecule controlled folate receptor CAR-T cell therapy with special focus on pediatric malignancies. Front. Oncol. 2019, 9, 151. [Google Scholar] [CrossRef]
- Xue, D.; Han, J.; Liu, Y.; Tuo, H.; Peng, Y. Current perspectives on exosomes in the diagnosis and treatment of hepatocellular carcinoma (review). Cancer Biol. Ther. 2021, 22, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.J.; Sun, X.Y.; Huang, K.M.; Zhang, L.; Yang, Z.S.; Zou, D.D.; Wang, B.; Warnock, G.L.; Dai, L.J.; Luo, J. Therapeutic potential of CAR-T cell-derived exosomes: A cell-free modality for targeted cancer therapy. Oncotarget 2015, 6, 44179–44190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, P.; Cao, X.; Cai, H.; Feng, P.; Chen, X.; Zhu, Y.; Yang, Y.; An, W.; Yang, Y.; Jie, J. The exosomes derived from CAR-T cell efficiently target mesothelin and reduce triple-negative breast cancer growth. Cell. Immunol. 2021, 360, 104262. [Google Scholar] [CrossRef]
- Fu, W.; Lei, C.; Liu, S.; Cui, Y.; Wang, C.; Qian, K.; Li, T.; Shen, Y.; Fan, X.; Lin, F.; et al. CAR exosomes derived from effector CAR-T cells have potent antitumour effects and low toxicity. Nat. Commun. 2019, 10, 4355. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Erviti, L.; Seow, Y.; Yin, H.; Betts, C.; Lakhal, S.; Wood, M.J.A. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat. Biotechnol. 2011, 29, 341–345. [Google Scholar] [CrossRef]
- Theruvath, J.; Sotillo, E.; Mount, C.W.; Graef, C.M.; Delaidelli, A.; Heitzeneder, S.; Labanieh, L.; Dhingra, S.; Leruste, A.; Majzner, R.G.; et al. Locoregionally administered B7-H3-targeted CAR T cells for treatment of atypical teratoid/rhabdoid tumors. Nat. Med. 2020, 26, 712–719. [Google Scholar] [CrossRef]
- Mulazzani, M.; Fräßle, S.P.; Von Mücke-Heim, I.; Langer, S.; Zhou, X.; Ishikawa-Ankerhold, H.; Leube, J.; Zhang, W.; Dötsch, S.; Svec, M.; et al. Long-term in vivo microscopy of CAR T cell dynamics during eradication of CNS lymphoma in mice. Proc. Natl. Acad. Sci. USA 2019, 116, 24275–24284. [Google Scholar] [CrossRef] [Green Version]
- Nellan, A.; Rota, C.; Majzner, R.; Lester-McCully, C.M.; Griesinger, A.M.; Mulcahy Levy, J.M.; Foreman, N.K.; Warren, K.E.; Lee, D.W. Durable regression of Medulloblastoma after regional and intravenous delivery of anti-HER2 chimeric antigen receptor T cells. J. Immunother. Cancer 2018, 6, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Priceman, S.J.; Tilakawardane, D.; Jeang, B.; Aguilar, B.; Murad, J.P.; Park, A.K.; Chang, W.-C.; Ostberg, J.R.; Neman, J.; Jandial, R.; et al. Regional Delivery of Chimeric Antigen Receptor-Engineered T Cells Effectively Targets HER2+ Breast Cancer Metastasis to the Brain. Clin. Cancer Res. 2018, 24, 95–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zottel, A.; Videtic Paska, A.; Jovčevska, I. Nanotechnology Meets Oncology: Nanomaterials in brain cancer research, diagnosis and therapy. Materials 2019, 12, 1588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balakrishnan, P.B.; Sweeney, E.E. Nanoparticles for Enhanced Adoptive T Cell Therapies and Future Perspectives for CNS Tumors. Front. Immunol. 2021, 12, 600659. [Google Scholar] [CrossRef]
- Reinhard, K.; Rengstl, B.; Oehm, P.; Türeci, Ö.; Sahin, U. Abstract LB-383: An RNA vaccine drives expansion and efficacy of claudin-CAR-T cells against solid tumors. Science 2020, 367, 446–453. [Google Scholar] [CrossRef]
- Sanz-Ortega, L.; Rojas, J.M.; Barber, D.F. Improving tumor retention of effector cells in adoptive cell transfer therapies by magnetic targeting. Pharmaceutics 2020, 12, 812. [Google Scholar] [CrossRef]
- Majzner, R.G.; Mackall, C.L. Tumor antigen escape from car t-cell therapy. Cancer Discov. 2018, 8, 1219–1226. [Google Scholar] [CrossRef] [Green Version]
- Drent, E.; Themeli, M.; Poels, R.; de Jong-Korlaar, R.; Yuan, H.; de Bruijn, J.; Martens, A.C.M.; Zweegman, S.; van de Donk, N.W.C.J.; Groen, R.W.J.; et al. A Rational Strategy for Reducing On-Target Off-Tumor Effects of CD38-Chimeric Antigen Receptors by Affinity Optimization. Mol. Ther. 2017, 25, 1946–1958. [Google Scholar] [CrossRef]
- Turatti, F.; Figini, M.; Balladore, E.; Alberti, P.; Casalini, P.; Marks, J.D.; Canevari, S.; Mezzanzanica, D. Redirected activity of human antitumor chimeric immune receptors is governed by antigen and receptor expression levels and affinity of interaction. J. Immunother. 2007, 30, 684–693. [Google Scholar] [CrossRef]
- Walker, A.J.; Majzner, R.G.; Zhang, L.; Wanhainen, K.; Long, A.H.; Nguyen, S.M.; Lopomo, P.; Vigny, M.; Fry, T.J.; Orentas, R.J.; et al. Tumor Antigen and Receptor Densities Regulate Efficacy of a Chimeric Antigen Receptor Targeting Anaplastic Lymphoma Kinase. Mol. Ther. 2017, 25, 2189–2201. [Google Scholar] [CrossRef] [Green Version]
- Barber, A.; Rynda, A.; Sentman, C.L. Chimeric NKG2D Expressing T Cells Eliminate Immunosuppression and Activate Immunity within the Ovarian Tumor Microenvironment. J. Immunol. 2009, 183, 6939–6947. [Google Scholar] [CrossRef] [Green Version]
- Fernández, L.; Metais, J.Y.; Escudero, A.; Vela, M.; Valentín, J.; Vallcorba, I.; Leivas, A.; Torres, J.; Valeri, A.; Patiño-García, A.; et al. Memory T cells expressing an NKG2D-CAR efficiently target osteosarcoma cells. Clin. Cancer Res. 2017, 23, 5824–5835. [Google Scholar] [CrossRef]
- Zhu, X.; Prasad, S.; Gaedicke, S.; Hettich, M.; Firat, E.; Niedermann, G. Patient-derived glioblastoma stem cells are killed by CD133-specific CAR T cells but induce the T cell aging marker CD57. Oncotarget 2015, 6, 171–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masoumi, J.; Jafarzadeh, A.; Abdolalizadeh, J.; Khan, H.; Philippe, J.; Mirzaei, H.; Mirzaei, H.R. Cancer stem cell-targeted chimeric antigen receptor (CAR)-T cell therapy: Challenges and prospects. Acta Pharm. Sin. B 2021, 11, 1721–1739. [Google Scholar] [CrossRef]
- Rossig, C.; Kailayangiri, S.; Jamitzky, S.; Altvater, B. Carbohydrate targets for CAR T cells in solid childhood cancers. Front. Oncol. 2018, 8, 513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arcangeli, S.; Falcone, L.; Camisa, B.; De Girardi, F.; Biondi, M.; Giglio, F.; Ciceri, F.; Bonini, C.; Bondanza, A.; Casucci, M. Next-Generation Manufacturing Protocols Enriching TSCM CAR T Cells Can Overcome Disease-Specific T Cell Defects in Cancer Patients. Front. Immunol. 2020, 11, 1217. [Google Scholar] [CrossRef]
- Fernández, L.; Fernández, A.; Mirones, I.; Escudero, A.; Cardoso, L.; Vela, M.; Lanzarot, D.; de Paz, R.; Leivas, A.; Gallardo, M.; et al. GMP-Compliant Manufacturing of NKG2D CAR Memory T Cells Using CliniMACS Prodigy. Front. Immunol. 2019, 10, 2361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tantalo, D.G.M.; Oliver, A.J.; Von Scheidt, B.; Harrison, A.J.; Mueller, S.N.; Kershaw, M.H.; Slaney, C.Y. Understanding T cell phenotype for the design of effective chimeric antigen receptor T cell therapies. J. Immunother. Cancer 2021, 9, e002555. [Google Scholar] [CrossRef]
- Alvarez-Fernández, C.; Escribà-Garcia, L.; Caballero, A.C.; Escudero-López, E.; Ujaldón-Miró, C.; Montserrat-Torres, R.; Pujol-Fernández, P.; Sierra, J.; Briones, J. Memory stem T cells modified with a redesigned CD30-chimeric antigen receptor show an enhanced antitumor effect in Hodgkin lymphoma. Clin. Transl. Immunol. 2021, 10, e1268. [Google Scholar] [CrossRef]
- Watanabe, K.; Kuramitsu, S.; Posey, A.D.; June, C.H. Expanding the therapeutic window for CAR T cell therapy in solid tumors: The knowns and unknowns of CAR T cell biology. Front. Immunol. 2018, 9, 2486. [Google Scholar] [CrossRef] [Green Version]
- Andrea, A.E.; Chiron, A.; Bessoles, S.; Hacein-Bey-Abina, S. Molecular Sciences Engineering Next-Generation CAR-T Cells for Better Toxicity Management. J. Mol. Sci. 2020, 21, 8620. [Google Scholar] [CrossRef]
- Brandt, L.J.B.; Barnkob, M.B.; Michaels, Y.S.; Heiselberg, J.; Barington, T. Emerging Approaches for Regulation and Control of CAR T Cells: A Mini Review. Front. Immunol. 2020, 11, 326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, B.S.; Lamb, L.S.; Goldman, F.; Di Stasi, A. Improving the safety of cell therapy products by suicide gene transfer. Front. Pharmacol. 2014, 5, 1–8. [Google Scholar] [CrossRef]
- Marin, V.; Cribioli, E.; Philip, B.; Tettamanti, S.; Pizzitola, I.; Biondi, A.; Biagi, E.; Pule, M. Comparison of different suicide-gene strategies for the safety improvement of genetically manipulated T cells. Hum. Gene Ther. Methods 2012, 23, 376–386. [Google Scholar] [CrossRef]
- Fedorov, V.D.; Themeli, M.; Sadelain, M. PD-1- and CTLA-4-based inhibitory chimeric antigen receptors (iCARs) divert off-target immunotherapy responses. Sci. Transl. Med. 2014, 11, 215ra172. [Google Scholar] [CrossRef] [Green Version]
- Moghanloo, E.; Mollanoori, H.; Talebi, M.; Pashangzadeh, S.; Faraji, F.; Hadjilooei, F.; Mahmoodzadeh, H. Remote controlling of CAR-T cells and toxicity management: Molecular switches and next generation CARs. Transl. Oncol. 2021, 14, 101070. [Google Scholar] [CrossRef]
- Locke, F.L.; Altrock, P.M.; Kimmel, G.J. The roles of T cell competition and stochastic extinction events in chimeric antigen receptor T cell therapy. Proc. Biol. Sci. 2021, 288, 20210229. [Google Scholar]
- León-Triana, O.; Pérez-Martínez, A.; Ramírez-Orellana, M.; Pérez-García, V.M. Dual-Target CAR-Ts with On- and Off-Tumour Activity May Override Immune Suppression in Solid Cancers: A Mathematical Proof of Concept. Cancers (Basel) 2021, 13, 703. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, P.; Yang, X.; Abler, D.; Maestrini, D.; Adhikarla, V.; Frankhouser, D.; Cho, H.; Machuca, V.; Wang, D.; Barish, M.; et al. Mathematical deconvolution of CAR T-cell proliferation and exhaustion from real-time killing assay data. J. R. Soc. Interface 2020, 17, 20190734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez-Garcia, A.; Lynn, R.C.; Poussin, M.; Eiva, M.A.; Shaw, L.C.; O’Connor, R.S.; Minutolo, N.G.; Casado-Medrano, V.; Lopez, G.; Matsuyama, T.; et al. CAR-T cell-mediated depletion of immunosuppressive tumor-associated macrophages promotes endogenous antitumor immunity and augments adoptive immunotherapy. Nat. Commun. 2021, 12, 877. [Google Scholar] [CrossRef]
- Alizadeh, D.; Wong, R.A.; Gholamin, S.; Maker, M.; Aftabizadeh, M.; Yang, X.; Pecoraro, J.R.; Jeppson, J.D.; Wang, D.; Aguilar, B.; et al. IFNγ Is Critical for CAR T Cell–Mediated Myeloid Activation and Induction of Endogenous Immunity. Cancer Discov. 2021, 11, 2248–2265. [Google Scholar] [CrossRef] [PubMed]
- Duluc, D.; Corvaisier, M.; Blanchard, S.; Catala, L.; Descamps, P.; Gamelin, E.; Ponsoda, S.; Delneste, Y.; Hebbar, M.; Jeannin, P. Interferon-γ reverses the immunosuppressive and protumoral properties and prevents the generation of human tumor-associated macrophages. Int. J. Cancer 2009, 125, 367–373. [Google Scholar] [CrossRef] [PubMed]
- Chiocca, E.A.; Yu, J.S.; Lukas, R.V.; Solomon, I.H.; Ligon, K.L.; Nakashima, H.; Triggs, D.A.; Reardon, D.A.; Wen, P.; Stopa, B.M.; et al. Regulatable interleukin-12 gene therapy in patients with recurrent high-grade glioma: Results of a phase 1 trial. Sci. Transl. Med. 2019, 11, eaaw5680. [Google Scholar] [CrossRef]
- Hu, B.; Ren, J.; Luo, Y.; Keith, B.; Young, R.M.; Scholler, J.; Zhao, Y.; June, C.H. Augmentation of Antitumor Immunity by Human and Mouse CAR T Cells Secreting IL-18. Cell Rep. 2017, 20, 3025–3033. [Google Scholar] [CrossRef] [Green Version]
- Avanzi, M.P.; Yeku, O.; Li, X.; Wijewarnasuriya, D.P.; van Leeuwen, D.G.; Cheung, K.; Park, H.; Purdon, T.J.; Daniyan, A.F.; Spitzer, M.H.; et al. Engineered Tumor-Targeted T Cells Mediate Enhanced Anti-Tumor Efficacy Both Directly and through Activation of the Endogenous Immune System. Cell Rep. 2018, 23, 2130–2141. [Google Scholar] [CrossRef]
- Alizadeh, D.; Wong, R.A.; Yang, X.; Wang, D.; Pecoraro, J.R.; Kuo, C.F.; Aguilar, B.; Qi, Y.; Ann, D.K.; Starr, R.; et al. IL15 enhances CAR-T cell antitumor activity by reducing mTORC1 activity and preserving their stem cell memory phenotype. Cancer Immunol. Res. 2019, 7, 759–772. [Google Scholar] [CrossRef]
- Chen, Y.; Sun, C.; Landoni, E.; Metelitsa, L.; Dotti, G.; Savoldo, B. Eradication of neuroblastoma by T cells redirected with an optimized GD2-specific chimeric antigen receptor and interleukin-15. Clin. Cancer Res. 2019, 25, 2915–2924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golumba-Nagy, V.; Kuehle, J.; Hombach, A.A.; Abken, H. CD28-ζ CAR T Cells Resist TGF-β Repression through IL-2 Signaling, Which Can Be Mimicked by an Engineered IL-7 Autocrine Loop. Mol. Ther. 2018, 26, 2218–2230. [Google Scholar] [CrossRef] [Green Version]
- Ivey, M.J.; Kuwabara, J.T.; Riggsbee, K.L.; Tallquist, M.D. Platelet-derived growth factor receptor-is essential for cardiac fibroblast survival. Am. J. Physiol.-Heart Circ. Physiol. 2019, 317, 330–344. [Google Scholar] [CrossRef]
- Xiao, W.; Wang, J.; Wen, X.; Xu, B.; Que, Y.; Yu, K.; Xu, L.; Zhao, J.; Pan, Q.; Zhou, P.; et al. Chimeric antigen receptor-modified T-cell therapy for platelet-derived growth factor receptor α-positive rhabdomyosarcoma. Cancer 2020, 126, 2093–2100. [Google Scholar] [CrossRef] [Green Version]
- Akkari, L.; Decker, W.K.; Torrado, E.; Sevenich, L.; Schulz, M.; Salamero-Boix, A.; Niesel, K.; Alekseeva, T. Microenvironmental Regulation of Tumor Progression and Therapeutic Response in Brain Metastasis. Front. Immunol. 2019, 1, 1713. [Google Scholar]
- Choi, B.D.; Yu, X.; Castano, A.P.; Darr, H.; Henderson, D.B.; Bouffard, A.A.; Larson, R.C.; Scarfò, I.; Bailey, S.R.; Gerhard, G.M.; et al. CRISPR-Cas9 disruption of PD-1 enhances activity of universal EGFRvIII CAR T cells in a preclinical model of human glioblastoma. J. Immunother. Cancer 2019, 7, 304. [Google Scholar] [CrossRef]
- Ollauri-Ibáñez, C.; Astigarraga, I. Use of antiangiogenic therapies in pediatric solid tumors. Cancers 2021, 13, 253. [Google Scholar] [CrossRef]
- Sie, M.; den Dunnen, W.F.A.; Hoving, E.W.; de Bont, E.S.J.M. Anti-angiogenic therapy in pediatric brain tumors: An effective strategy? Crit. Rev. Oncol. Hematol. 2014, 89, 418–432. [Google Scholar] [CrossRef] [PubMed]
- Bao, S.; Wu, Q.; Sathornsumetee, S.; Hao, Y.; Li, Z.; Hjelmeland, A.B.; Shi, Q.; McLendon, R.E.; Bigner, D.D.; Rich, J.N. Stem cell-like glioma cells promote tumor angiogenesis through vascular endothelial growth factor. Cancer Res. 2006, 66, 7843–7848. [Google Scholar] [CrossRef] [Green Version]
- Beccaria, K.; Canney, M.; Bouchoux, G.; Puget, S.; Grill, J.; Carpentier, A. Blood-brain barrier disruption with low-intensity pulsed ultrasound for the treatment of pediatric brain tumors: A review and perspectives. Neurosurg. Focus 2020, 48, E10. [Google Scholar] [CrossRef] [Green Version]
- Hersh, D.S.; Wadajkar, A.S.; Roberts, N.; Perez, J.G.; Connolly, N.P.; Frenkel, V.; Winkles, J.A.; Woodworth, G.F.; Kim, A.J. Evolving Drug Delivery Strategies to Overcome the Blood Brain Barrier. Curr. Pharm. Des. 2016, 22, 1177–1193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabbagh, A.; Beccaria, K.; Ling, X.; Marisetty, A.; Ott, M.; Caruso, H.; Barton, E.; Kong, L.-Y.; Fang, D.; Latha, K.; et al. Opening of the Blood–Brain Barrier Using Low-Intensity Pulsed Ultrasound Enhances Responses to Immunotherapy in Preclinical Glioma Models. Clin. Cancer Res. 2021, 27, 4325–4337. [Google Scholar] [CrossRef]
- Sanz-Ortega, L.; Portilla, Y.; Pérez-Yagüe, S.; Barber, D.F. Magnetic targeting of adoptively transferred tumour-specific nanoparticle-loaded CD8+ T cells does not improve their tumour infiltration in a mouse model of cancer but promotes the retention of these cells in tumour-draining lymph nodes. J. Nanobiotechnol. 2019, 17, 87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maier-Hauff, K.; Ulrich, F.; Nestler, D.; Niehoff, H.; Wust, P.; Thiesen, B.; Orawa, H.; Budach, V.; Jordan, A. Efficacy and safety of intratumoral thermotherapy using magnetic iron-oxide nanoparticles combined with external beam radiotherapy on patients with recurrent glioblastoma multiforme. J. Neurooncol. 2011, 103, 317–324. [Google Scholar] [CrossRef] [Green Version]
- Kolosnjaj-Tabi, J.; Wilhelm, C. Magnetic nanoparticles in cancer therapy: How can thermal approaches help? Nanomedicine 2017, 12, 573–575. [Google Scholar] [CrossRef]
- Tran, T.H.; Mattheolabakis, G.; Aldawsari, H.; Amiji, M. Exosomes as nanocarriers for immunotherapy of cancer and inflammatory diseases. Clin. Immunol. 2015, 160, 46–58. [Google Scholar] [CrossRef]
- Yang, T.; Martin, P.; Fogarty, B.; Brown, A.; Schurman, K.; Phipps, R.; Yin, V.P.; Lockman, P.; Bai, S. Exosome delivered anticancer drugs across the blood-brain barrier for brain cancer therapy in Danio rerio. Pharm Res. 2015, 32, 2003–2014. [Google Scholar] [CrossRef]
- Brown, C.E.; Aguilar, B.; Starr, R.; Yang, X.; Chang, W.C.; Weng, L.; Chang, B.; Sarkissian, A.; Brito, A.; Sanchez, J.F.; et al. Optimization of IL13Rα2-Targeted Chimeric Antigen Receptor T Cells for Improved Anti-tumor Efficacy against Glioblastoma. Mol. Ther. 2018, 26, 31–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaur, G.; Sharma, P.; Dogra, N.; Singh, S. Eradicating Cancer Stem Cells: Concepts, Issues, and Challenges. Options Oncol. 2018, 19, 20. [Google Scholar] [CrossRef] [PubMed]
- Colak, S.; Medema, J.P. Cancer stem cells—Important players in tumor therapy resistance. FEBS J. 2014, 281, 4779–4791. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Lee, T.K.; Zheng, B.-J.; Chan, K.W.; Guan, X.-Y. CD133 þ HCC cancer stem cells confer chemoresistance by preferential expression of the Akt/PKB survival pathway. Oncogene 2008, 27, 1749–1758. [Google Scholar] [CrossRef] [Green Version]
- Batlle, E.; Clevers, H. Cancer stem cells revisited. Nat. Med. 2017, 23, 1124–1134. [Google Scholar] [CrossRef]
- Davila, M.L.; Riviere, I.; Wang, X.; Bartido, S.; Park, J.; Chung, S.S.; Stefanski, J.; Borquez-ojeda, O.; Qu, J.; Wasielewska, T.; et al. Efficacy and Toxicity Management of 19-28z CAR T Cell Therapy. Sci. Transl. Med. 2014, 6, 224–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weijtens, M.E.M.; Hart, E.H.; Bolhuis, R.L.H. Functional balance between T cell chimeric receptor density and tumor associated antigen density: CTL mediated cytolysis and lymphokine production. Gene Ther. 2000, 7, 35–42. [Google Scholar] [CrossRef] [Green Version]
- Straathof, K.; Flutter, B.; Wallace, R.; Jain, N.; Loka, T.; Depani, S.; Wright, G.; Thomas, S.; Cheung, G.W.K.; Gileadi, T.; et al. Antitumor activity without on-target off-tumor toxicity of GD2-chimeric antigen receptor T cells in patients with neuroblastoma. Sci. Transl. Med. 2020, 12, eabd6169. [Google Scholar] [CrossRef]
- Ying, Z.; Huang, X.F.; Xiang, X.; Liu, Y.; Kang, X.; Song, Y.; Guo, X.; Liu, H.; Ding, N.; Zhang, T.; et al. A safe and potent anti-CD19 CAR T cell therapy. Nat. Med. 2019, 25, 947–953. [Google Scholar] [CrossRef]
- Barros, L.R.C.; Paixão, E.A.; Valli, A.M.P.; Naozuka, G.T.; Fassoni, A.C.; Almeida, R.C. Cartmath—A mathematical model of car-t immunotherapy in preclinical studies of hematological cancers. Cancers 2021, 13, 2914. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, N.; Fernández, V.; Pereira, R.C.; Rancati, S.; Pelizzoli, R.; De Pietri Tonelli, D. A Xenotransplant Model of Human Brain Tumors in Wild-Type Mice. iScience 2020, 23, 100831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacob, F.; Salinas, R.D.; Zhang, D.Y.; Nguyen, P.T.T.; Schnoll, J.G.; Wong, S.Z.H.; Thokala, R.; Sheikh, S.; Saxena, D.; Prokop, S.; et al. A Patient-Derived Glioblastoma Organoid Model and Biobank Recapitulates Inter- and Intra-tumoral Heterogeneity. Cell 2020, 180, 188–204.e22. [Google Scholar] [CrossRef]
- Mariappan, A.; Goranci-Buzhala, G.; Ricci-Vitiani, L.; Pallini, R.; Gopalakrishnan, J. Trends and challenges in modeling glioma using 3D human brain organoids. Cell Death Differ. 2021, 28, 15–23. [Google Scholar] [CrossRef]
- Ballabio, C.; Anderle, M.; Gianesello, M.; Lago, C.; Miele, E.; Cardano, M.; Aiello, G.; Piazza, S.; Caron, D.; Gianno, F.; et al. Modeling medulloblastoma in vivo and with human cerebellar organoids. Nat. Commun. 2020, 11, 583. [Google Scholar] [CrossRef]
- Wu, Y.; Yu, X.Z. Modelling CAR-T therapy in humanized mice. EBioMedicine 2019, 40, 25–26. [Google Scholar] [CrossRef] [Green Version]
- Siegler, E.L.; Wang, P. Preclinical Models in Chimeric Antigen Receptor-Engineered T-Cell Therapy. Hum. Gene Ther. 2018, 29, 534–546. [Google Scholar] [CrossRef] [PubMed]
- Sanmamed, M.F.; Chester, C.; Melero, I.; Kohrt, H. Defining the optimal murine models to investigate immune checkpoint blockers and their combination with other immunotherapies. Ann. Oncol. 2016, 27, 1190–1198. [Google Scholar] [CrossRef] [PubMed]
- Textor, A.; Listopad, J.J.; Le Wührmann, L.; Perez, C.; Kruschinski, A.; Chmielewski, M.; Abken, H.; Blankenstein, T.; Charo, J. Efficacy of CAR T-cell therapy in large tumors relies upon stromal targeting by IFNγ. Cancer Res. 2014, 74, 6796–6805. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
| Age | NCT | Phase | Tumour Type | Target Antigen | Administration | Sponsor |
|---|---|---|---|---|---|---|
| 2–30 years old | NCT04196413 | 1 | DIPG and DMG | GD2 | Intravenously | Lucile Packard Children’s Hospital |
| 1–26 years old | NCT04185038 | 1 | DIPG/DMG, and recurrent or refractory Paediatric CNS tumours | B7-H3 | Catheter into the tumour or ventricular system | Seattle Children’s Hospital |
| 1–18 years old | NCT04099797 | 1 | DIPG, Embryonal Tumour, HGG, medulloblastoma | GD2 | Intravenously | Texas Children’s Hospital |
| 1–26 years old | NCT03638167 | 1 | Recurrent or refractory Paediatric CNS tumours | EGFR | Tumour resection cavity or the ventricular system | Seattle Children’s Hospital |
| 1–26 years old | NCT03500991 | 1 | Recurrent or refractory Paediatric CNS tumours | HER-2 | Tumour resection cavity or into their ventricular system | Seattle Children’s Hospital |
| 4–70 years old | NCT03638206 | 1 and 2 | Several solid tumours. Gliomas from the CNS | EGFR V III | Intravenously | Shenzhen BinDeBio Ltd. |
| 12–75 years old | NCT02208362 | 1 | Recurrent or refractory malignant glioma | IL13Rα2 | Intracavitary, intratumoral or intraventricular | City of Hope Comprehensive Cancer Center |
| 4–25 years old | NCT04510051 | 1 | Recurrent or refractory brain tumours in children | IL13Rα2 | Intraventricular | City of Hope Medical Center National Cancer Institute (NCI) |
| 1–22 years old | NCT04903080 * | 1 | Ependymoma | HER-2 | Intravenously | Pediatric Brain Tumor Consortium Texas Children’s Cancer Center Baylor College of Medicine |
| 3 years old and older | NCT02442297 | 1 | CNS tumours | HER-2 | Tumour, tumour resection cavity, and/or cerebrospinal fluid (CSF) space | Baylor College of Medicine |
| Target | CNS Paediatric Tumour Expression | Expression in Healthy Tissues | Bibliography |
|---|---|---|---|
| B7-H3 | Moderate-to-high levels in MB, ependymoma, and gliomas | Very low or undetected | [81,82,83] |
| HER2 | 40% of MB | Not expressed in healthy brain tissue | [84] |
| IL-13Ralpha2 | Highly expressed (60–100%) in gliomas, MB, and ependymoma | Not expressed in healthy brain tissue or other organs except testis | [85,86,87] |
| EphA2, | Gliomas | Not expressed in healthy brain tissue | [85,86] |
| NKG2DL | Variable expression in MB | Rarely detectable in healthy tissue | [88,89] |
| EGFRvIII | Variable expression in pHGG | Not expressed in healthy tissue | [90] |
| GD2 | High levels in pHGG | GD2 in normal tissues is limited essentially at low levels on neurons and peripheral nerve fibres, dermal melanocytes, lymphocytes, and mesenchymal stem cell | [91,92] |
| PDGFRA | Variable expression in pHGG | In healthy tissues expressed on development | [93,94,95,96,97] |
| PIGF | Highly expressed in MB | Expressed in placenta and at low levels in several other organs, including the heart, lung, thyroid, skeletal muscle, and adipose tissue under normal physiological conditions | [98,99] |
| Challenges | Potential Strategies |
|---|---|
| Tumour microenvironment | Modulation of the TME using TRUCKs [130,131,132,133,134,135,136] Chemokines [137] Blocking immunosuppressive molecules: TGFβ-resistant [138] VEGFR2 CAR T [139,140,141] PDGFRA CAR T [94,95,96,97,142] CAR T cells targeting TAMs [68,76,77,143,144] PIGF CAR T [98] |
| Trespassing the BBB | CAR T derived exosomes [145,146,147,148] Local delivery [149,150,151,152,153] Nanoparticles [154,155,156,157] |
| Antigen escape | Targeting multiple antigens [88,89,158,159,160,161,162,163] synNotch CAR T [29] Targeting the CSC [164,165] New neoantigens [166] |
| CAR T associated toxicities | T cell subpopulation [167,168,169,170,171] iCas9 CAR T [172,173,174,175,176,177] Prediction with mathematical modelling [178,179,180] Manufacturing process [102,168] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferreras, C.; Fernández, L.; Clares-Villa, L.; Ibáñez-Navarro, M.; Martín-Cortázar, C.; Esteban-Rodríguez, I.; Saceda, J.; Pérez-Martínez, A. Facing CAR T Cell Challenges on the Deadliest Paediatric Brain Tumours. Cells 2021, 10, 2940. https://doi.org/10.3390/cells10112940
Ferreras C, Fernández L, Clares-Villa L, Ibáñez-Navarro M, Martín-Cortázar C, Esteban-Rodríguez I, Saceda J, Pérez-Martínez A. Facing CAR T Cell Challenges on the Deadliest Paediatric Brain Tumours. Cells. 2021; 10(11):2940. https://doi.org/10.3390/cells10112940
Chicago/Turabian StyleFerreras, Cristina, Lucía Fernández, Laura Clares-Villa, Marta Ibáñez-Navarro, Carla Martín-Cortázar, Isabel Esteban-Rodríguez, Javier Saceda, and Antonio Pérez-Martínez. 2021. "Facing CAR T Cell Challenges on the Deadliest Paediatric Brain Tumours" Cells 10, no. 11: 2940. https://doi.org/10.3390/cells10112940
APA StyleFerreras, C., Fernández, L., Clares-Villa, L., Ibáñez-Navarro, M., Martín-Cortázar, C., Esteban-Rodríguez, I., Saceda, J., & Pérez-Martínez, A. (2021). Facing CAR T Cell Challenges on the Deadliest Paediatric Brain Tumours. Cells, 10(11), 2940. https://doi.org/10.3390/cells10112940