
Lauric Acid, a Dietary Saturated Medium-Chain Fatty Acid, Elicits Calcium-Dependent Eryptosis



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Blood Collection and RBC Purification
2.2. Chemicals and Reagents
2.3. Hemolysis
2.4. Phosphatidylserine Exposure
2.5. Erythrocyte Sedimentation Rate (ESR)
2.6. Intracellular Calcium
2.7. Cellular Morphology
2.8. Oxidative Stress
2.9. Hydrogen Peroxides (H2O2) and Superoxide Anions (SOA)
2.10. Antioxidant Enzymes
2.11. Glutathione Status
2.12. Lipid Peroxidation
2.13. Protein Carbonylation (PCC)
2.14. Complete Blood Count
2.15. Statistical Analysis
3. Results
3.1. LA stimulates Calcium-Independent Hemolysis
3.2. LA Elicits Calcium-Dependent Eryptosis
3.3. LA promotes Extracellular Calcium Influx
3.4. LA Causes Cell Shrinkage and Granularity
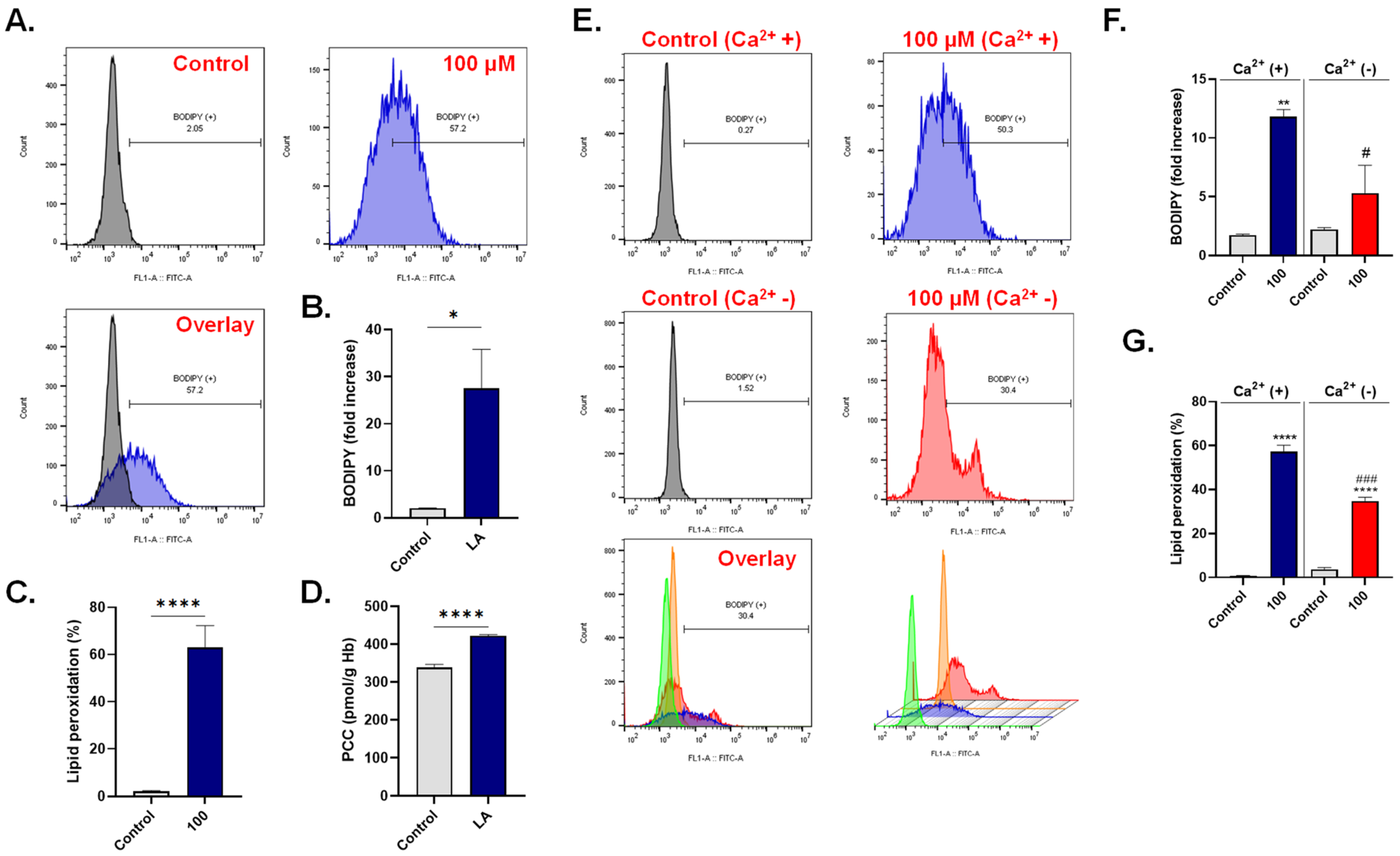
3.5. LA Triggers Oxidative Stress
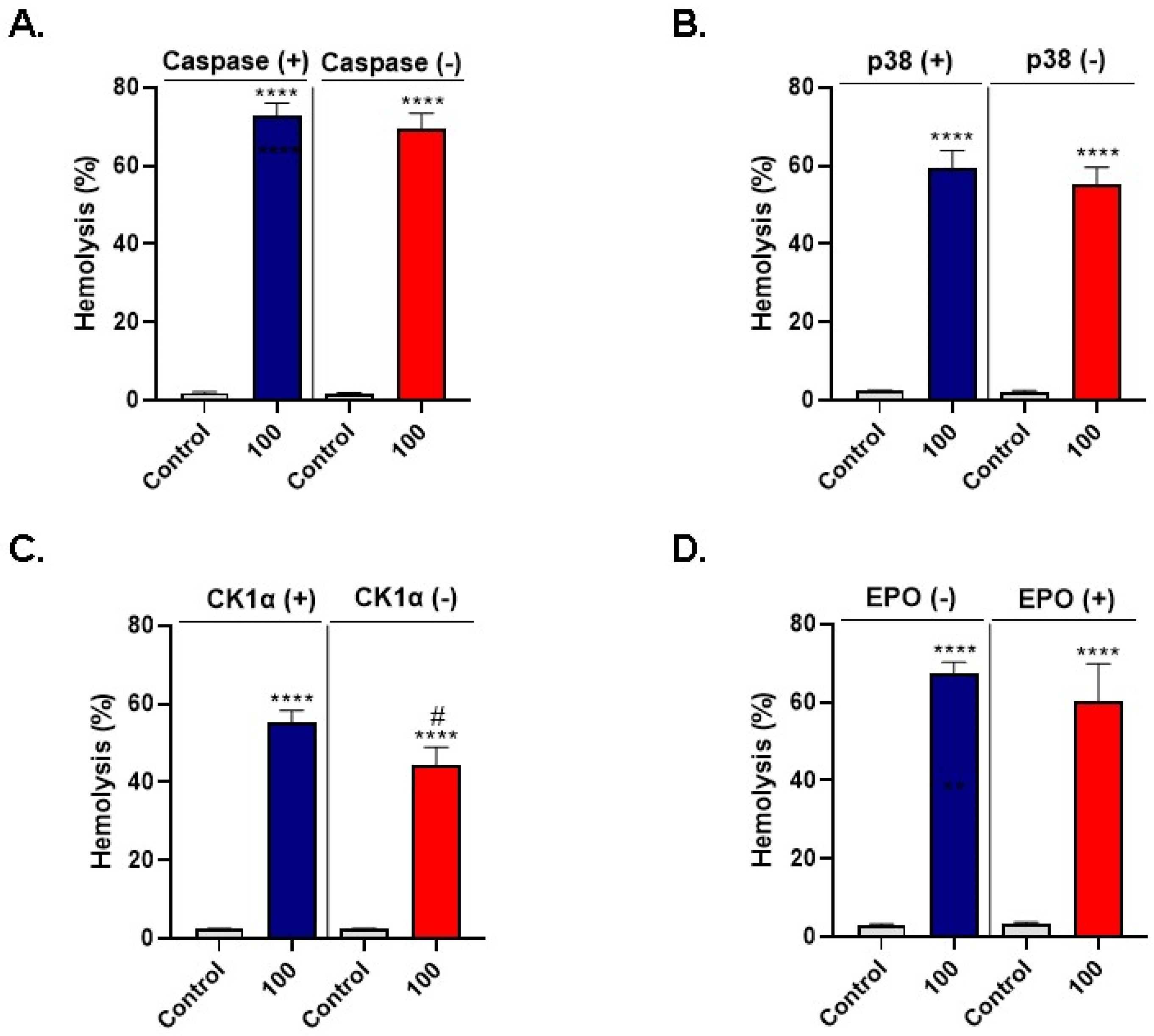
3.6. LA-Induced Hemolysis Is Mitigated by D4476
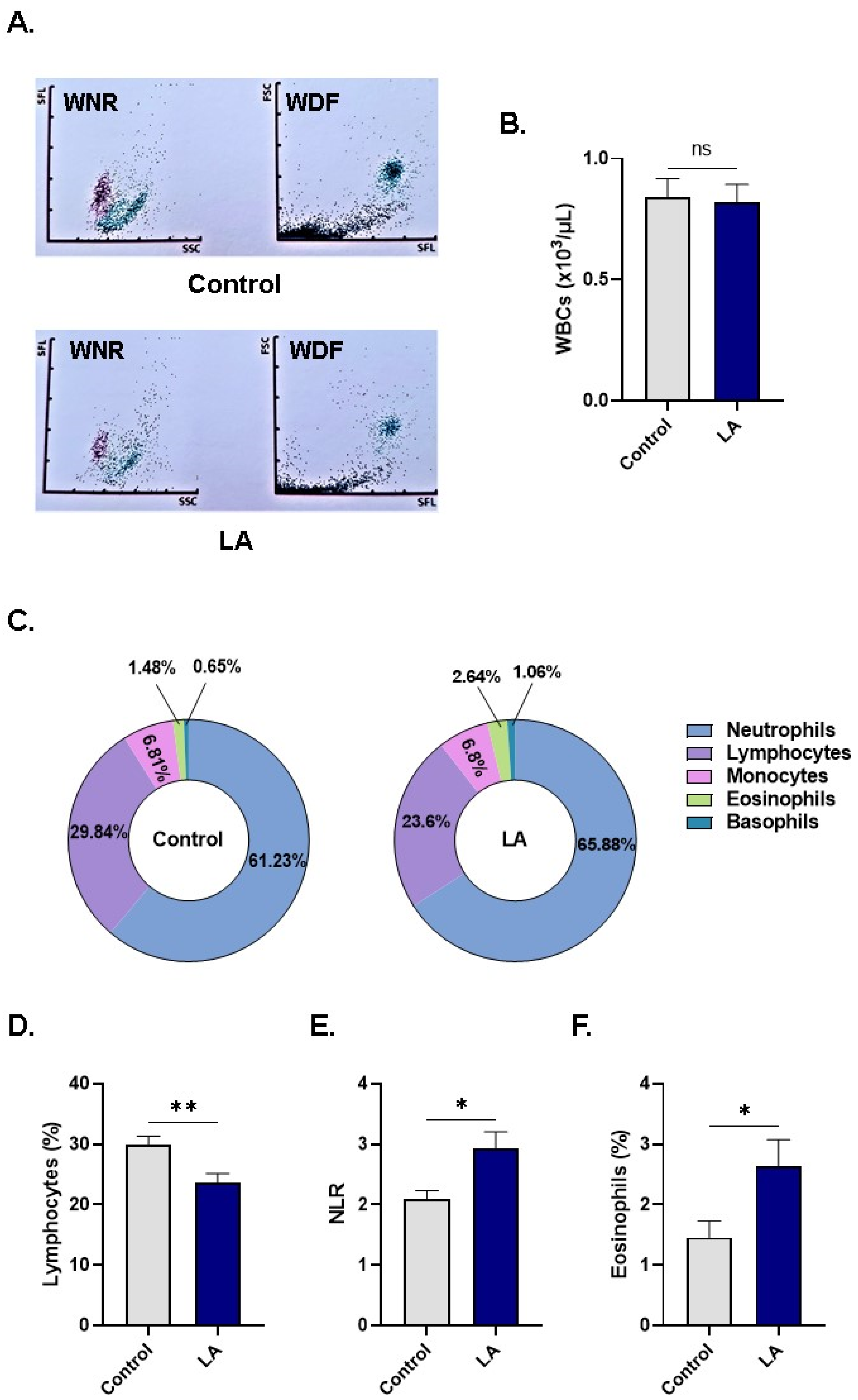
3.7. LA Elevates the Neutrophil-Lymphocyte Ratio (NLR)
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Roth, G.A.; Mensah, G.A.; Johnson, C.O.; Addolorato, G.; Ammirati, E.; Baddour, L.M.; Barengo, N.C.; Beaton, A.Z.; Benjamin, E.J.; Benziger, C.P.; et al. Global burden of cardiovascular diseases and risk factors, 1990–2019: Update from the GBD 2019 study. J. Am. Coll. Cardiol. 2020, 76, 2982–3021. [Google Scholar] [CrossRef] [PubMed]
- Van Horn, L.; McCoin, M.; Kris-Etherton, P.M.; Burke, F.; Carson, J.A.S.; Champagne, C.M.; Karmally, W.; Sikand, G. The evidence for dietary prevention and treatment of cardiovascular disease. J. Am. Diet. Assoc. 2008, 108, 287–331. [Google Scholar] [CrossRef] [PubMed]
- Riccardi, G.; Giosuè, A.; Calabrese, I.; Vaccaro, O. Dietary recommendations for prevention of atherosclerosis. Dietary recommendations for prevention of atherosclerosis. Cardiovasc. Res. 2021. [Google Scholar] [CrossRef]
- Ramírez, M.; Amate, L.; Gil, A. Absorption and distribution of dietary fatty acids from different sources. Early Hum. Dev. 2001, 65, S95–S101. [Google Scholar] [CrossRef]
- Mozaffarian, D. Free Fatty Acids, Cardiovascular Mortality, and Cardiometabolic Stress. Eur Heart J. 2007, 28, 2699–2700. [Google Scholar] [CrossRef] [Green Version]
- Otton, R.; Graziola, F.; De Souza, J.A.A.; Curi, T.C.P.; Hirata, M.H.; Curi, R. Effect of dietary fat on lymphocyte proliferation and metabolism. Cell Biochem. Funct. Cell. Biochem. Its Modul. Act. Agents Dis. 1998, 16, 253–259. [Google Scholar] [CrossRef]
- Granato, D.; Blum, S.; Rössle, C.; Le Boucher, J.; Malnoë, A.; Dutot, G. Effects of parenteral lipid emulsions with different fatty acid composition on immune cell functions in vitro. J. Parenter. Enter. Nutr. 2000, 24, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Cury-Boaventura, M.F.; Pompéia, C.; Curi, R. Comparative toxicity of oleic acid and linoleic acid on Jurkat cells. Clin. Nutr. 2004, 23, 721–732. [Google Scholar] [CrossRef]
- Lima, T.; Kanunfre, C.; Pompéia, C.; Verlengia, R.; Curi, R. Ranking the toxicity of fatty acids on Jurkat and Raji cells by flow cytometric analysis. Toxicol. In Vitro 2002, 16, 741–747. [Google Scholar] [CrossRef]
- Martins de Lima, T.; Cury-Boaventura, M.F.; Giannocco, G.; Nunes, M.T.; Curi, R. Comparative toxicity of fatty acids on a macrophage cell line (J774). Clin. Sci. 2006, 111, 307–317. [Google Scholar] [CrossRef] [Green Version]
- Pilz, S.; Scharnagl, H.; Tiran, B.; Wellnitz, B.; Seelhorst, U.; Boehm, B.O.; März, W. Elevated plasma free fatty acids predict sudden cardiac death: A 6.85-year follow-up of 3315 patients after coronary angiography. Eur. Heart J. 2007, 28, 2763–2769. [Google Scholar] [CrossRef] [Green Version]
- Djoussé, L.; Benkeser, D.; Arnold, A.; Kizer, J.R.; Zieman, S.J.; Lemaitre, R.N.; Tracy, R.P.; Gottdiener, J.S.; Mozaffarian, D.; Siscovick, D.S.; et al. Plasma Free Fatty Acids and Risk of Heart Failure. Circ. Heart Fail. 2013, 6, 964–969. [Google Scholar] [CrossRef] [Green Version]
- Sobczak, I.S.; Blindauer, A.; Stewart, J. Changes in plasma free fatty acids associated with type-2 diabetes. Nutrients 2019, 11, 2022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Astrup, A.; Bertram, H.C.; Bonjour, J.P.; de Groot, L.C.; de Oliveira Otto, M.C.; Feeney, E.L.; Garg, M.L.; Givens, L.; Kok, F.J.; Krauss, R.M.; et al. WHO draft guidelines on dietary saturated and trans fatty acids: Time for a new approach? BMJ 2019, 366, l4137. [Google Scholar] [CrossRef] [Green Version]
- Forouhi, N.G.; Krauss, R.M.; Taubes, G.; Willett, W. Dietary fat and cardiometabolic health: Evidence, controversies, and consensus for guidance. BMJ 2018, 361, k2139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dayrit, F.M. The properties of lauric acid and their significance in coconut oil. J. Am. Oil Chem. Soc. 2015, 92, 1–15. [Google Scholar] [CrossRef]
- Mensink, R.P.; Zock, P.; Kester, A.D.M.; Katan, M.B. Effects of dietary fatty acids and carbohydrates on the ratio of serum total to HDL cholesterol and on serum lipids and apolipoproteins: A meta-analysis of 60 controlled trials. Am. J. Clin. Nutr. 2003, 77, 1146–1155. [Google Scholar] [CrossRef]
- Stampfer, M.J.; Sacks, F.M.; Salvini, S.; Willett, W.C.; Hennekens, C.H. A prospective study of cholesterol, apolipoproteins, and the risk of myocardial infarction. N. Engl. J. Med. 1991, 325, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Stein, O.; Stein, Y. Atheroprotective mechanisms of HDL. Atherosclerosis 1999, 144, 285–301. [Google Scholar] [CrossRef]
- Saraswathi, V.; Kumar, N.; Gopal, T.; Bhatt, S.; Ai, W.; Ma, C.; Talmon, G.; Desouza, C. Lauric Acid versus Palmitic Acid: Effects on Adipose Tissue Inflammation, Insulin Resistance, and Non-Alcoholic Fatty Liver Disease in Obesity. Biology 2020, 9, 346. [Google Scholar] [CrossRef]
- Denke, M.A.; Grundy, S.M. Comparison of effects of lauric acid and palmitic acid on plasma lipids and lipoproteins. Am. J. Clin. Nutr. 1992, 56, 895–898. [Google Scholar] [CrossRef] [Green Version]
- Tholstrup, T.; Marckmann, P.; Jespersen, J.; Sandström, B. Fat high in stearic acid favorably affects blood lipids and factor VII coagulant activity in comparison with fats high in palmitic acid or high in myristic and lauric acids. Am. J. Clin. Nutr. 1994, 59, 371–377. [Google Scholar] [CrossRef]
- Schwab, U.S.; Niskanen, L.K.; Maliranta, H.M.; Savolainen, M.J.; Kesäniemi, Y.A.; Uusitupa, M.I. Lauric and palmitic acid-enriched diets have minimal impact on serum lipid and lipoprotein concentrations and glucose metabolism in healthy young women. J. Nutr. 1995, 125, 466–473. [Google Scholar]
- De Roos, N.M.; Schouten, E.G.; Katan, M.B. Consumption of a solid fat rich in lauric acid results in a more favorable serum lipid profile in healthy men and women than consumption of a solid fat rich in trans-fatty acids. J. Nutr. 2001, 131, 242–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Temme, E.; Mensink, R.P.; Hornstra, G. Comparison of the effects of diets enriched in lauric, palmitic, or oleic acids on serum lipids and lipoproteins in healthy women and men. Am. J. Clin. Nutr. 1996, 63, 897–903. [Google Scholar] [CrossRef] [Green Version]
- Shramko, V.S.; Polonskaya, Y.V.; Kashtanova, E.V.; Stakhneva, E.M.; Ragino, Y.I. The Short Overview on the Relevance of Fatty Acids for Human Cardiovascular Disorders. Biomolecules 2020, 10, 1127. [Google Scholar] [CrossRef]
- Michalski, M.-C.; Genotc, C.; Gayetd, C.; Lopeze, C.; Finef, F.; Joffreg, F.; Vendeuvreh, J.L.; Bouvieri, J.; Chardignyjk, J.M.; Raynal-Ljutovaci, K.; et al. Multiscale structures of lipids in foods as parameters affecting fatty acid bioavailability and lipid metabolism. Prog. Lipid Res. 2013, 52, 354–373. [Google Scholar] [CrossRef] [Green Version]
- Watson, H. Biological membranes. Essays Biochem. 2015, 59, 43–69. [Google Scholar] [CrossRef]
- Kay, J.G.; Grinstein, S. Sensing phosphatidylserine in cellular membranes. Sensors 2011, 11, 744–1755. [Google Scholar] [CrossRef] [Green Version]
- Seeman, P.; Kwant, W.; Sauks, T. Membrane expansion of erythrocyte ghosts by tranquilizers and anesthetics. Biochim. Biophys. Acta (Bba)-Biomembr. 1969, 183, 499–511. [Google Scholar] [CrossRef]
- Fortes, P.; Ellory, J. Asymmetric membrane expansion and modification of active and passive cation permeability of human red cells by the fluorescent probe 1-anilino-8-napththalene sulfonate. Biochim. Biophys. Acta 1975, 413, 65–78. [Google Scholar] [CrossRef]
- Hägerstrand, H.; Isomaa, B. Amphiphile-induced antihaemolysis is not causally related to shape changes and vesiculation. Chem. Interact. 1991, 79, 335–347. [Google Scholar] [CrossRef]
- Løvstad, R.A. Fatty acid induced hemolysis. Protective action of ceruloplasmin, albumins, thiols and vitamin C. Int. J. Biochem. 1986, 18, 771–775. [Google Scholar] [CrossRef]
- Pignatelli, P.; Menichelli, D.; Pastori, D.; Violi, F. Oxidative stress and cardiovascular disease: New insights. Kardiol. Pol. (Pol. Heart J.) 2018, 76, 713–722. [Google Scholar] [CrossRef] [Green Version]
- Violi, F.; Loffredo, L.; Carnevale, R.; Pignatelli, P.; Pastori, D. Atherothrombosis and Oxidative Stress: Mechanisms and Management in Elderly. Antioxid. Redox Signal. 2017, 27, 1083–1124. [Google Scholar] [CrossRef]
- Harrison, D.; Griendling, K.; Landmesser, U.; Hornig, B.; Drexler, H. Role of oxidative stress in atherosclerosis. Am. J. Cardiol. 2003, 91, 7–11. [Google Scholar] [CrossRef]
- Alfhili, M.A.; Lee, M.H. Flow Cytofluorometric Analysis of Molecular Mechanisms of Premature Red Blood Cell Death. Methods Mol. Biol. 2021, 2326, 155–165. [Google Scholar]
- Alfhili, M.A.; Alsughayyir, J.; Basudan, A.B. Epidemic dropsy toxin, sanguinarine chloride, stimulates sucrose-sensitive hemolysis and breakdown of membrane phospholipid asymmetry in human erythrocytes. Toxicon 2021, 199, 41–48. [Google Scholar] [CrossRef]
- Alfhili, M.A.; Alsalmi, E.; Aljedai, A.; Alsughayyir, J.; Abudawood, M.; Basudan, A.M. Calcium-oxidative stress signaling axis and casein kinase 1α mediate eryptosis and hemolysis elicited by novel p53 agonist inauhzin. J. Chemother. 2021, 1–11. [Google Scholar] [CrossRef]
- Narang, V.; Grover, S.; Kang, A.K.; Garg, A.; Sood, N. Comparative Analysis of Erythrocyte Sedimentation Rate Measured by Automated and Manual Methods in Anaemic Patients. J. Lab. Physicians 2020, 12, 239–243. [Google Scholar] [CrossRef]
- Alfhili, M.A.; Alamri, H.S.; Alsughayyir, J.; Basudan, A.M. Induction of hemolysis and eryptosis by occupational pollutant nickel chloride is mediated through calcium influx and p38 MAP kinase signaling. Int. J. Occup. Med. Environ. Health 2021, 35. [Google Scholar] [CrossRef]
- Alfhili, M.A.; Alsughayyir, J.; Basudan, A.M. Reprogramming of erythrocyte lifespan by NFκB-TNFα naphthoquinone antagonist beta-lapachone is regulated by calcium overload and CK1α. J. Food Biochem. 2021, 45, e13710. [Google Scholar] [CrossRef]
- Ghneim, H.K.; Alshebly, M.M. Biochemical Markers of Oxidative Stress in Saudi Women with Recurrent Miscarriage. J. Korean Med. Sci. 2016, 31, 98–105. [Google Scholar] [CrossRef]
- Ghneim, H.K.; Al-Sheikh, Y.A.; Alshebly, M.M.; Aboul-Soud, M.A.M. Superoxide dismutase activity and gene expression levels in Saudi women with recurrent miscarriage. Mol. Med. Rep. 2016, 13, 2606–2612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akiel, M.; Alsughayyir, J.; Basudan, A.M.; Alamri, H.S.; Dera, A.; Barhoumi, T.; Al Subayyil, A.M.; Basmaeil, Y.S.; Aldakheel, F.M.; Alakeel, R.; et al. Physcion Induces Hemolysis and Premature Phosphatidylserine Externalization in Human Erythrocytes. Biol. Pharm. Bull. 2021, 44, 372–378. [Google Scholar] [CrossRef]
- Kumar, P.; Maurya, P.K. L-Cysteine Efflux in Erythrocytes As A Function of Human Age: Correlation with Reduced Glutathione and Total Anti-Oxidant Potential. Rejuvenation Res. 2013, 16, 179–184. [Google Scholar] [CrossRef]
- Pretorius, E.; Du Plooy, J.N.; Bester, J. A Comprehensive Review on Eryptosis. Cell. Physiol. Biochem. 2016, 39, 1977–2000. [Google Scholar] [CrossRef]
- Jemaà, M.; Fezai, M.; Bissinger, R.; Lang, F. Methods Employed in Cytofluorometric Assessment of Eryptosis, the Suicidal Erythrocyte Death. Cell. Physiol. Biochem. 2017, 43, 431–444. [Google Scholar] [CrossRef] [Green Version]
- Colombo, G.; Clerici, M.; Garavaglia, M.E.; Giustarini, D.; Rossi, R.; Milzani, A.D.G.; Dalle-Donne, I. A step-by-step protocol for assaying protein carbonylation in biological samples. J. Chromatogr. B 2015, 1019, 178–190. [Google Scholar] [CrossRef]
- van Wijk, R.; van Solinge, W.W. The energy-less red blood cell is lost: Erythrocyte enzyme abnormalities of glycolysis. Blood 2005, 106, 4034–4042. [Google Scholar] [CrossRef]
- Alfhili, M.A.; Basudan, A.M.; Alsughayyir, J. Antiproliferative Wnt inhibitor wogonin prevents eryptosis following ionophoric challenge, hyperosmotic shock, oxidative stress, and metabolic deprivation. J. Food Biochem. 2021, 45, e13977. [Google Scholar] [CrossRef]
- Mahmud, H.; Ruifrok, W.P.T.; Westenbrink, B.D.; Cannon, M.V.; Vreeswijk-Baudoin, I.; van Gilst, W.H.; Silljé, H.H.W.; de Boer, R.A. Suicidal erythrocyte death, eryptosis, as a novel mechanism in heart failure-associated anaemia. Cardiovasc. Res. 2013, 98, 37–46. [Google Scholar] [CrossRef] [Green Version]
- Virani, S.S.; Alonso, A.; Aparicio, H.J.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Cheng, S.; Delling, F.N.; et al. Heart Disease and Stroke Statistics–2021 Update: A Report From the American Heart Association. Circulation 2021, 143, e254–e743. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, G.F.; Meier-Augenstein, W.; Stöckler, S.; Surtees, R.; Rating, D.; Nyhan, W.L. Physiology and pathophysiology of organic acids in cerebrospinal fluid. J. Inherit. Metab. Dis. 1993, 16, 648–669. [Google Scholar] [CrossRef]
- Sheela, D.L.; Narayanankutty, A.; Nazeem, P.A.; Raghavamenon, A.C.; Muthangaparambil, S.R. Lauric acid induce cell death in colon cancer cells mediated by the epidermal growth factor receptor downregulation: An in silico and in vitro study. Hum. Exp. Toxicol. 2019, 38, 753–761. [Google Scholar] [CrossRef] [PubMed]
- Lappano, R.; Sebastiani, A.; Cirillo, F.; Rigiracciolo, D.C.; Galli, G.R.; Curcio, R.; Malaguarnera, R.; Belfiore, A.; Cappello, A.R.; Maggiolini, M. The lauric acid-activated signaling prompts apoptosis in cancer cells. Cell Death Discov. 2017, 3, 17063. [Google Scholar] [CrossRef]
- Fauser, J.K.; Matthews, G.M.; Cummins, A.G.; Howarth, G.S. Induction of apoptosis by the medium-chain length fatty acid lauric acid in colon cancer cells due to induction of oxidative stress. Chemotherapy 2013, 59, 214–224. [Google Scholar] [CrossRef]
- Dhaliwal, G.; Cornett, P.A.; Tierney, L.M., Jr. Hemolytic anemia. Am. Fam. Physician 2004, 69, 2599–2606. [Google Scholar]
- Balla, G.; Jacob, H.S.; Eaton, J.W.; Belcher, J.D.; Vercellotti, G.M. Hemin: A possible physiological mediator of low density lipoprotein oxidation and endothelial injury. Arter. Thromb. A J. Vasc. Biol. 1991, 11, 1700–1711. [Google Scholar] [CrossRef] [Green Version]
- Anderson, J.; Glynn, L.G.; Newell, J.; Iglesias, A.A.; Reddan, D.; Murphy, A.W. The impact of renal insufficiency and anaemia on survival in patients with cardiovascular disease: A cohort study. Bmc Cardiovasc. Disord. 2009, 9, 51. [Google Scholar] [CrossRef] [Green Version]
- Bindra, K.; Berry, C.; Rogers, J.; Stewart, N.; Watts, M.; Christie, J.; Cobbe, S.; Eteiba, H. Abnormal haemoglobin levels in acute coronary syndromes. J. Assoc. Physicians 2006, 99, 851–862. [Google Scholar] [CrossRef] [PubMed]
- Zeidman, A.; Fradin, Z.; Blecher, A.; Oster, H.S.; Avrahami, Y.; Mittelman, M. Anemia as a risk factor for ischemic heart disease. Imaj-Ramat Gan 2004, 6, 16–18. [Google Scholar]
- Metivier, F.; Marchais, S.J.; Guerin, A.P.; Pannier, B.; London, G.M. Pathophysiology of anaemia: Focus on the heart and blood vessels. Nephrol. Dial. Transplant. 2000, 15, 14–18. [Google Scholar] [CrossRef] [Green Version]
- Lang, F.; Lang, K.; Lang, P.A.; Huber, S.M.; Wieder, T. Mechanisms and Significance of Eryptosis. Antioxid. Redox Signal. 2006, 8, 1183–1192. [Google Scholar] [CrossRef] [PubMed]
- Repsold, L.; Joubert, A.M. Eryptosis: An Erythrocyte’s Suicidal Type of Cell Death. Biomed. Res. Int. 2018, 2018, 9405617. [Google Scholar] [CrossRef] [PubMed]
- Attanasio, P.; Bissinger, R.; Haverkamp, W.; Pieske, B.; Wutzler, A.; Lang, F. Enhanced suicidal erythrocyte death in acute cardiac failure. Eur. J. Clin. Investig. 2015, 45, 1316–1324. [Google Scholar] [CrossRef]
- Danesh, J.; Collins, R.; Peto, R.; Lowe, G. Haematocrit, viscosity, erythrocyte sedimentation rate: Meta-analyses of prospective studies of coronary heart disease. Eur. Heart J. 2000, 21, 515–520. [Google Scholar] [CrossRef]
- IIngelsson, E.; Ärnlöv, J.; Sundstrom, J.; Lind, L. Inflammation, as Measured by the Erythrocyte Sedimentation Rate, Is an Independent Predictor for the Development of Heart Failure. J. Am. Coll. Cardiol. 2005, 45, 1802–1806. [Google Scholar] [CrossRef] [Green Version]
- Andresdottir, M.B.; Sigfusson, N.; Sigvaldason, H.; Gudnason, V. Erythrocyte Sedimentation Rate, an Independent Predictor of Coronary Heart Disease in Men and Women: The Reykjavik Study. Am. J. Epidemiol. 2003, 158, 844–851. [Google Scholar] [CrossRef]
- Erikssen, G.; Liestøl, K.; Bjørnholt, J.; Stormorken, H.; Thaulow, E. Erythrocyte sedimentation rate: A possible marker of atherosclerosis and a strong predictor of coronary heart disease mortality. Eur. Heart J. 2000, 21, 1614–1620. [Google Scholar] [CrossRef]
- Alfhili, M.A.; Weidner, D.A.; Lee, M.-H. Disruption of erythrocyte membrane asymmetry by triclosan is preceded by calcium dysregulation and p38 MAPK and RIP1 stimulation. Chemosphere 2019, 229, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Alfhili, M.A.; Nkany, M.B.; Weidner, D.A.; Lee, M.-H. Stimulation of eryptosis by broad-spectrum insect repellent N,N-Diethyl-3-methylbenzamide (DEET). Toxicol. Appl. Pharmacol. 2019, 370, 36–43. [Google Scholar] [CrossRef]
- Wong, P. A basis of the acanthocytosis in inherited and acquired disorders. Med. Hypotheses 2004, 62, 966–969. [Google Scholar] [CrossRef]
- Cloos, A.-S.; Daenen, L.G.M.; Maja, M.; Stommen, A.; Vanderroost, J.; Van Der Smissen, P.; Rab, M.; Westerink, J.; Mignolet, E.; Larondelle, Y.; et al. Impaired Cytoskeletal and Membrane Biophysical Properties of Acanthocytes in Hypobetalipoproteinemia –A Case Study. Front. Physiol. 2021, 12, 638027. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Kang, H.J.; Kim, S.Z.; Kwon, T.O.; Jeong, S.-I.; Jang, S.I. Antioxidant effect of astragalin isolated from the leaves of Morus alba L. against free radical-induced oxidative hemolysis of human red blood cells. Arch. Pharmacal Res. 2013, 36, 912–917. [Google Scholar] [CrossRef]
- Ray, P.D.; Huang, B.-W.; Tsuji, Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell. Signal. 2012, 24, 981–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senoner, T.; Dichtl, W. Oxidative stress in cardiovascular diseases: Still a therapeutic target? Nutrients 2019, 11, 2090. [Google Scholar] [CrossRef] [Green Version]
- Lü, J.-M.; Lin, P.H.; Yao, Q.; Chen, C. Chemical and molecular mechanisms of antioxidants: Experimental approaches and model systems. J. Cell. Mol. Med. 2009, 14, 840–860. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B. Antioxidants in Human Health and Disease. Annu. Rev. Nutr. 1996, 16, 33–50. [Google Scholar] [CrossRef]
- Halliwell, B.; Aeschbach, R.; Löliger, J.; Aruoma, O.I. The characterization of antioxidants. Food Chem. Toxicol. 1995, 33, 601–617. [Google Scholar] [CrossRef]
- Sies, H. Glutathione and its role in cellular functions. Free. Radic. Biol. Med. 1999, 27, 916–921. [Google Scholar] [CrossRef]
- D’Oria, R.; Schipani, R.; Leonardini, A.; Natalicchio, A.; Perrini, S.; Cignarelli, A.; Laviola, L.; Giorgino, F. The Role of Oxidative Stress in Cardiac Disease: From Physiological Response to Injury Factor. Oxidative Med. Cell. Longev. 2020, 2020, 5732956. [Google Scholar] [CrossRef] [PubMed]
- Nakata, Y.; Maeda, N. Vulnerable atherosclerotic plaque morphology in apolipoprotein E-deficient mice unable to make ascorbic Acid. Circulation 2002, 105, 1485–1490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meagher, E.; Rader, D.J. Antioxidant therapy and atherosclerosis: Animal and human studies. Trends Cardiovasc. Med. 2001, 11, 162–165. [Google Scholar] [CrossRef]
- Aune, D.; Keum, N.; Giovannucci, E.; Fadnes, L.T.; Boffetta, P.; Greenwood, D.C.; Tonstad, S.; Vatten, L.J.; Riboli, E.; Norat, T. Dietary intake and blood concentrations of antioxidants and the risk of cardiovascular disease, total cancer, and all-cause mortality: A systematic review and dose-response meta-analysis of prospective studies. Am. J. Clin. Nutr. 2018, 108, 1069–1091. [Google Scholar] [CrossRef]
- Jenkins, D.J.A.; Kitts, D.; Giovannucci, E.L.; Sahye-Pudaruth, S.; Paquette, M.; Mejia, S.B.; Patel, D.; Kavanagh, M.; Tsirakis, T.; Kendall, C.W.C.; et al. Selenium, antioxidants, cardiovascular disease, and all-cause mortality: A systematic review and meta-analysis of randomized controlled trials. Am. J. Clin. Nutr. 2020, 112, 1642–1652. [Google Scholar] [CrossRef] [PubMed]
- Tribble, D.L. AHA Science Advisory. Antioxidant consumption and risk of coronary heart disease: Emphasison vitamin C, vitamin E, and beta-carotene: A statement for healthcare professionals from the American Heart Association. Circulation 1999, 99, 591–595. [Google Scholar] [CrossRef] [Green Version]
- Sekhar, R.V.; Patel, S.G.; Guthikonda, A.P.; Reid, M.; Balasubramanyam, A.; Taffet, G.E.; Jahoor, F. Deficient synthesis of glutathione underlies oxidative stress in aging and can be corrected by dietary cysteine and glycine supplementation. Am. J. Clin. Nutr. 2011, 94, 847–853. [Google Scholar] [CrossRef] [Green Version]
- Sekhar, R.V.; McKay, S.V.; Patel, S.G.; Guthikonda, A.P.; Reddy, V.T.; Balasubramanyam, A.; Jahoor, F. Glutathione Synthesis Is Diminished in Patients With Uncontrolled Diabetes and Restored by Dietary Supplementation With Cysteine and Glycine. Diabetes Care 2010, 34, 162–167. [Google Scholar] [CrossRef] [Green Version]
- Moris, D.; Spartalis, M.; Spartalis, E.; Karachaliou, G.-S.; Karaolanis, G.I.; Tsourouflis, G.; Tsilimigras, D.I.; Tzatzaki, E.; Theocharis, S. The role of reactive oxygen species in the pathophysiology of cardiovascular diseases and the clinical significance of myocardial redox. Ann. Transl. Med. 2017, 5, 326. [Google Scholar] [CrossRef] [Green Version]
- Biswas, S.K.; Newby, D.E.; Rahman, I.; Megson, I.L. Depressed glutathione synthesis precedes oxidative stress and atherogenesis in Apo-E−/−mice. Biochem. Biophys. Res. Commun. 2005, 338, 1368–1373. [Google Scholar] [CrossRef] [PubMed]
- Bajic, V.P.; Van Neste, C.; Obradovic, M.; Zafirovic, S.; Radak, D.; Bajic, V.B.; Essack, M.; Isenovic, E.R. Glutathione “Redox Homeostasis” and Its Relation to Cardiovascular Disease. Oxidative Med. Cell. Longev. 2019, 2019, 5028181. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alfhili, M.A.; Aljuraiban, G.S. Lauric Acid, a Dietary Saturated Medium-Chain Fatty Acid, Elicits Calcium-Dependent Eryptosis. Cells 2021, 10, 3388. https://doi.org/10.3390/cells10123388
Alfhili MA, Aljuraiban GS. Lauric Acid, a Dietary Saturated Medium-Chain Fatty Acid, Elicits Calcium-Dependent Eryptosis. Cells. 2021; 10(12):3388. https://doi.org/10.3390/cells10123388
Chicago/Turabian StyleAlfhili, Mohammad A., and Ghadeer S. Aljuraiban. 2021. "Lauric Acid, a Dietary Saturated Medium-Chain Fatty Acid, Elicits Calcium-Dependent Eryptosis" Cells 10, no. 12: 3388. https://doi.org/10.3390/cells10123388
APA StyleAlfhili, M. A., & Aljuraiban, G. S. (2021). Lauric Acid, a Dietary Saturated Medium-Chain Fatty Acid, Elicits Calcium-Dependent Eryptosis. Cells, 10(12), 3388. https://doi.org/10.3390/cells10123388