
Parkin beyond Parkinson’s Disease—A Functional Meaning of Parkin Downregulation in TDP-43 Proteinopathies



Abstract
:1. Introduction
2. Parkin and PINK1—The Key Players in Mitophagy Initiation
3. Clinical Phenotypes Associated with Mutations in PARK2 and PARK6 Genes
3.1. Biallelic PARK2 (Parkin) and PARK6 (PINK1) Mutations Cause Young/Juvenile-Onset Parkinson’s Disease
3.2. Single Heterozygous PARK2 or PARK6 Mutations Lead to Subclinical PD
3.3. Parkin Inactivation in Sporadic PD
4. Loss-of-Function (LOF) and Gain-of-Function (GOF) Mechanisms in TDP-43 Proteinopathies
5. Interwoven Relations between TDP-43 and Parkin
5.1. Consistent Effects of TDP-43 Depletion on Parkin Levels
5.2. Discrepant Effects of TDP-43 Overexpression on Parkin Levels
5.3. Parkin as an E3-Ubiquitin Ligase Affects TDP-43 Aggregation
5.4. Unanswered Question 1: Is Parkin Downregulation in TDP-43 Proteinopathies Functionally Relevant (Molecular Biology Perspective)?
5.5. Unanswered Question 2: What Is the Evidence of Parkinsonism in TDP-43 Proteinopathies (Clinical Perspective)?
6. Additional Mechanisms of TDP-43—Mediated Mitochondrial Dysfunction
6.1. How Different Genetic Backgrounds of TDP-43 Proteinopathies Might Modulate Mitophagy?
6.2. Sporadic ALS/FTLD with TDP-43 Inclusions
6.3. C9ORF72 and TARDBP Mutations
6.4. PGRN Mutations Leading to Haploinsufficiency
7. Increasing Mitophagy as a Therapeutic Approach for TDP-43 Proteinopathies
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gorno-Tempini, M.L.; Hillis, A.E.; Weintraub, S.; Kertesz, A.; Mendez, M.; Cappa, S.F.; Ogar, J.M.; Rohrer, J.D.; Black, S.; Boeve, B.F.; et al. Classification of primary progressive aphasia and its variants. Neurology 2011, 76, 1006–1014. [Google Scholar] [CrossRef] [Green Version]
- Pottier, C.; Ravenscroft, T.A.; Sanchez-Contreras, M.; Rademakers, R. Genetics of FTLD: Overview and what else we can expect from genetic studies. J. Neurochem. 2016, 138, 32–53. [Google Scholar] [CrossRef] [PubMed]
- Mann, D.M.A.; Snowden, J.S. Frontotemporal lobar degeneration: Pathogenesis, pathology and pathways to phenotype. Brain Pathol. 2017, 27, 723–736. [Google Scholar] [CrossRef]
- Nguyen, H.P.; Van Broeckhoven, C.; van der Zee, J. ALS Genes in the Genomic Era and their Implications for FTD. Trends Genet. 2018, 34, 404–423. [Google Scholar] [CrossRef] [Green Version]
- Taylor, J.P.; Brown, R.H., Jr.; Cleveland, D.W. Decoding ALS: From genes to mechanism. Nature 2016, 539, 197–206. [Google Scholar] [CrossRef] [Green Version]
- Cook, C.; Petrucelli, L. Genetic Convergence Brings Clarity to the Enigmatic Red Line in ALS. Neuron 2019, 101, 1057–1069. [Google Scholar] [CrossRef] [Green Version]
- Ferrari, R.; Manzoni, C.; Hardy, J. Genetics and molecular mechanisms of frontotemporal lobar degeneration: An update and future avenues. Neurobiol. Aging 2019, 78, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.B.; Almeida, S.; Lopez-Gonzalez, R. Dysregulated molecular pathways in amyotrophic lateral sclerosis-frontotemporal dementia spectrum disorder. EMBO J. 2017, 36, 2931–2950. [Google Scholar] [CrossRef] [PubMed]
- Strong, M.J.; Abrahams, S.; Goldstein, L.H.; Woolley, S.; McLaughlin, P.; Snowden, J.; Mioshi, E.; Roberts-South, A.; Benatar, M.; HortobaGyi, T.; et al. Amyotrophic lateral sclerosis—Frontotemporal spectrum disorder (ALS-FTSD): Revised diagnostic criteria. Amyotroph. Lateral Scler. Front. Degener. 2017, 18, 153–174. [Google Scholar] [CrossRef]
- Borroni, B.; Alberici, A.; Buratti, E. Review: Molecular pathology of frontotemporal lobar degenerations. Neuropathol. Appl. Neurobiol. 2019, 45, 41–57. [Google Scholar] [CrossRef] [Green Version]
- Neumann, M.; Sampathu, D.M.; Kwong, L.K.; Truax, A.C.; Micsenyi, M.C.; Chou, T.T.; Bruce, J.; Schuck, T.; Grossman, M.; Clark, C.M.; et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 2006, 314, 130–133. [Google Scholar] [CrossRef] [Green Version]
- Ling, S.C.; Polymenidou, M.; Cleveland, D.W. Converging mechanisms in ALS and FTD: Disrupted RNA and protein homeostasis. Neuron 2013, 79, 416–438. [Google Scholar] [CrossRef] [Green Version]
- De Boer, E.M.J.; Orie, V.K.; Williams, T.; Baker, M.R.; De Oliveira, H.M.; Polvikoski, T.; Silsby, M.; Menon, P.; van den Bos, M.; Halliday, G.M.; et al. TDP-43 proteinopathies: A new wave of neurodegenerative diseases. J. Neurol. Neurosurg. Psychiatry 2020, 92, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Visanji, N.P.; Lang, A.E.; Kovacs, G.G. Beyond the synucleinopathies: Alpha synuclein as a driving force in neurodegenerative comorbidities. Transl. Neurodegener. 2019, 8, 28. [Google Scholar] [CrossRef] [Green Version]
- Blauwendraat, C.; Nalls, M.A.; Singleton, A.B. The genetic architecture of Parkinson’s disease. Lancet Neurol. 2020, 19, 170–178. [Google Scholar] [CrossRef]
- Deng, H.; Wang, P.; Jankovic, J. The genetics of Parkinson disease. Ageing Res. Rev. 2018, 42, 72–85. [Google Scholar] [CrossRef] [PubMed]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [CrossRef]
- Narendra, D.; Tanaka, A.; Suen, D.F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef] [Green Version]
- Arai, T.; Hasegawa, M.; Akiyama, H.; Ikeda, K.; Nonaka, T.; Mori, H.; Mann, D.; Tsuchiya, K.; Yoshida, M.; Hashizume, Y.; et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 2006, 351, 602–611. [Google Scholar] [CrossRef]
- Polymenidou, M.; Lagier-Tourenne, C.; Hutt, K.R.; Huelga, S.C.; Moran, J.; Liang, T.Y.; Ling, S.C.; Sun, E.; Wancewicz, E.; Mazur, C.; et al. Long pre-mRNA depletion and RNA missplicing contribute to neuronal vulnerability from loss of TDP-43. Nat. Neurosci. 2011, 14, 459–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lagier-Tourenne, C.; Polymenidou, M.; Hutt, K.R.; Vu, A.Q.; Baughn, M.; Huelga, S.C.; Clutario, K.M.; Ling, S.C.; Liang, T.Y.; Mazur, C.; et al. Divergent roles of ALS-linked proteins FUS/TLS and TDP-43 intersect in processing long pre-mRNAs. Nat. Neurosci. 2012, 15, 1488–1497. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; Narendra, D.P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell Biol. 2011, 12, 9–14. [Google Scholar] [CrossRef]
- Pickles, S.; Vigie, P.; Youle, R.J. Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr. Biol. 2018, 28, R170–R185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitworth, A.J.; Pallanck, L.J. PINK1/Parkin mitophagy and neurodegeneration-what do we really know in vivo? Curr. Opin. Genet. Dev. 2017, 44, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.W.; Hang, L.; Yao, T.P.; Lim, K.L. Parkin Regulation and Neurodegenerative Disorders. Front. Aging Neurosci. 2015, 7, 248. [Google Scholar] [CrossRef] [Green Version]
- Ge, P.; Dawson, V.L.; Dawson, T.M. PINK1 and Parkin mitochondrial quality control: A source of regional vulnerability in Parkinson’s disease. Mol. Neurodegener. 2020, 15, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meissner, C.; Lorenz, H.; Weihofen, A.; Selkoe, D.J.; Lemberg, M.K. The mitochondrial intramembrane protease PARL cleaves human Pink1 to regulate Pink1 trafficking. J. Neurochem. 2011, 117, 856–867. [Google Scholar] [CrossRef]
- Liu, J.; Liu, W.; Li, R.; Yang, H. Mitophagy in Parkinson’s Disease: From Pathogenesis to Treatment. Cells 2019, 8, 712. [Google Scholar] [CrossRef] [Green Version]
- Deas, E.; Plun-Favreau, H.; Gandhi, S.; Desmond, H.; Kjaer, S.; Loh, S.H.; Renton, A.E.; Harvey, R.J.; Whitworth, A.J.; Martins, L.M.; et al. PINK1 cleavage at position A103 by the mitochondrial protease PARL. Hum. Mol. Genet. 2011, 20, 867–879. [Google Scholar] [CrossRef] [PubMed]
- Yamano, K.; Youle, R.J. PINK1 is degraded through the N-end rule pathway. Autophagy 2013, 9, 1758–1769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaweda-Walerych, K.; Zekanowski, C. Integrated pathways of parkin control over mitochondrial maintenance—Relevance to Parkinson’s disease pathogenesis. Acta Neurobiol. Exp. 2013, 73, 199–224. [Google Scholar]
- Eiyama, A.; Okamoto, K. PINK1/Parkin-mediated mitophagy in mammalian cells. Curr. Opin. Cell Biol. 2015, 33, 95–101. [Google Scholar] [CrossRef]
- Roca-Portoles, A.; Tait, S.W.G. Mitochondrial quality control: From molecule to organelle. Cell. Mol. Life Sci. 2021, 78, 3853–3866. [Google Scholar] [CrossRef]
- Pallanck, L.J. Culling sick mitochondria from the herd. J. Cell Biol. 2010, 191, 1225–1227. [Google Scholar] [CrossRef] [Green Version]
- Miller, S.; Muqit, M.M.K. Therapeutic approaches to enhance PINK1/Parkin mediated mitophagy for the treatment of Parkinson’s disease. Neurosci. Lett. 2019, 705, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.; Harvey, K.; Gispert, S.; Ali, Z.; Del Turco, D.; Bentivoglio, A.R.; Healy, D.G.; et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 2004, 304, 1158–1160. [Google Scholar] [CrossRef] [Green Version]
- Lucking, C.B.; Durr, A.; Bonifati, V.; Vaughan, J.; De Michele, G.; Gasser, T.; Harhangi, B.S.; Meco, G.; Denefle, P.; Wood, N.W.; et al. Association between early-onset Parkinson’s disease and mutations in the parkin gene. N. Engl. J. Med. 2000, 342, 1560–1567. [Google Scholar] [CrossRef]
- Schrag, A.; Ben-Shlomo, Y.; Brown, R.; Marsden, C.D.; Quinn, N. Young-onset Parkinson’s disease revisited—Clinical features, natural history, and mortality. Mov. Disord. 1998, 13, 885–894. [Google Scholar] [CrossRef]
- Khan, N.L.; Graham, E.; Critchley, P.; Schrag, A.E.; Wood, N.W.; Lees, A.J.; Bhatia, K.P.; Quinn, N. Parkin disease: A phenotypic study of a large case series. Brain 2003, 126, 1279–1292. [Google Scholar] [CrossRef] [PubMed]
- Tijero, B.; Gabilondo, I.; Lezcano, E.; Teran-Villagra, N.; Llorens, V.; Ruiz-Martinez, J.; Marti-Masso, J.F.; Carmona, M.; Luquin, M.R.; Berganzo, K.; et al. Autonomic involvement in Parkinsonian carriers of PARK2 gene mutations. Parkinsonism Relat. Disord. 2015, 21, 717–722. [Google Scholar] [CrossRef] [PubMed]
- Albanese, A.; Valente, E.M.; Romito, L.M.; Bellacchio, E.; Elia, A.E.; Dallapiccola, B. The PINK1 phenotype can be indistinguishable from idiopathic Parkinson disease. Neurology 2005, 64, 1958–1960. [Google Scholar] [CrossRef] [PubMed]
- Houlden, H.; Singleton, A.B. The genetics and neuropathology of Parkinson’s disease. Acta Neuropathol. 2012, 124, 325–338. [Google Scholar] [CrossRef] [Green Version]
- Schneider, S.A.; Alcalay, R.N. Neuropathology of genetic synucleinopathies with parkinsonism: Review of the literature. Mov. Disord. 2017, 32, 1504–1523. [Google Scholar] [CrossRef]
- Zimmermann, M.; Wilke, C.; Schulte, C.; Hoffmann, J.; Klopfer, J.; Reimold, M.; Brockmann, K.; Synofzik, M. Biallelic Parkin (PARK2) mutations can cause a bvFTD phenotype without clinically relevant parkinsonism. Parkinsonism Relat. Disord. 2018, 55, 145–147. [Google Scholar] [CrossRef] [PubMed]
- Klein, C.; Lohmann-Hedrich, K.; Rogaeva, E.; Schlossmacher, M.G.; Lang, A.E. Deciphering the role of heterozygous mutations in genes associated with parkinsonism. Lancet Neurol. 2007, 6, 652–662. [Google Scholar] [CrossRef]
- Klein, C.; Westenberger, A. Genetics of Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a008888. [Google Scholar] [CrossRef] [Green Version]
- Bruggemann, N.; Mitterer, M.; Lanthaler, A.J.; Djarmati, A.; Hagenah, J.; Wiegers, K.; Winkler, S.; Pawlack, H.; Lohnau, T.; Pramstaller, P.P.; et al. Frequency of heterozygous Parkin mutations in healthy subjects: Need for careful prospective follow-up examination of mutation carriers. Parkinsonism Relat. Disord. 2009, 15, 425–429. [Google Scholar] [CrossRef]
- Krohn, L.; Grenn, F.P.; Makarious, M.B.; Kim, J.J.; Bandres-Ciga, S.; Roosen, D.A.; Gan-Or, Z.; Nalls, M.A.; Singleton, A.B.; Blauwendraat, C. Comprehensive assessment of PINK1 variants in Parkinson’s disease. Neurobiol. Aging 2020, 91, 168.e1–168.e5. [Google Scholar] [CrossRef]
- Hilker, R.; Klein, C.; Ghaemi, M.; Kis, B.; Strotmann, T.; Ozelius, L.J.; Lenz, O.; Vieregge, P.; Herholz, K.; Heiss, W.D.; et al. Positron emission tomographic analysis of the nigrostriatal dopaminergic system in familial parkinsonism associated with mutations in the parkin gene. Ann. Neurol. 2001, 49, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Khan, N.L.; Scherfler, C.; Graham, E.; Bhatia, K.P.; Quinn, N.; Lees, A.J.; Brooks, D.J.; Wood, N.W.; Piccini, P. Dopaminergic dysfunction in unrelated, asymptomatic carriers of a single parkin mutation. Neurology 2005, 64, 134–136. [Google Scholar] [CrossRef]
- Anders, S.; Sack, B.; Pohl, A.; Munte, T.; Pramstaller, P.; Klein, C.; Binkofski, F. Compensatory premotor activity during affective face processing in subclinical carriers of a single mutant Parkin allele. Brain 2012, 135, 1128–1140. [Google Scholar] [CrossRef]
- Van Nuenen, B.F.; Weiss, M.M.; Bloem, B.R.; Reetz, K.; van Eimeren, T.; Lohmann, K.; Hagenah, J.; Pramstaller, P.P.; Binkofski, F.; Klein, C.; et al. Heterozygous carriers of a Parkin or PINK1 mutation share a common functional endophenotype. Neurology 2009, 72, 1041–1047. [Google Scholar] [CrossRef] [Green Version]
- Buhmann, C.; Binkofski, F.; Klein, C.; Buchel, C.; van Eimeren, T.; Erdmann, C.; Hedrich, K.; Kasten, M.; Hagenah, J.; Deuschl, G.; et al. Motor reorganization in asymptomatic carriers of a single mutant Parkin allele: A human model for presymptomatic parkinsonism. Brain 2005, 128, 2281–2290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrish, P.K.; Sawle, G.V.; Brooks, D.J. Regional changes in [18F]dopa metabolism in the striatum in Parkinson’s disease. Brain 1996, 119 Pt 6, 2097–2103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hilker, R.; Klein, C.; Hedrich, K.; Ozelius, L.J.; Vieregge, P.; Herholz, K.; Pramstaller, P.P.; Heiss, W.D. The striatal dopaminergic deficit is dependent on the number of mutant alleles in a family with mutations in the parkin gene: Evidence for enzymatic parkin function in humans. Neurosci. Lett. 2002, 323, 50–54. [Google Scholar] [CrossRef]
- Pavese, N.; Khan, N.L.; Scherfler, C.; Cohen, L.; Brooks, D.J.; Wood, N.W.; Bhatia, K.P.; Quinn, N.P.; Lees, A.J.; Piccini, P. Nigrostriatal dysfunction in homozygous and heterozygous parkin gene carriers: An 18F-dopa PET progression study. Mov. Disord. 2009, 24, 2260–2266. [Google Scholar] [CrossRef] [PubMed]
- Puschmann, A.; Fiesel, F.C.; Caulfield, T.R.; Hudec, R.; Ando, M.; Truban, D.; Hou, X.; Ogaki, K.; Heckman, M.G.; James, E.D.; et al. Heterozygous PINK1 p.G411S increases risk of Parkinson’s disease via a dominant-negative mechanism. Brain 2017, 140, 98–117. [Google Scholar] [CrossRef]
- Reed, X.; Bandres-Ciga, S.; Blauwendraat, C.; Cookson, M.R. The role of monogenic genes in idiopathic Parkinson’s disease. Neurobiol. Dis. 2019, 124, 230–239. [Google Scholar] [CrossRef]
- Borsche, M.; Konig, I.R.; Delcambre, S.; Petrucci, S.; Balck, A.; Bruggemann, N.; Zimprich, A.; Wasner, K.; Pereira, S.L.; Avenali, M.; et al. Mitochondrial damage-associated inflammation highlights biomarkers in PRKN/PINK1 parkinsonism. Brain 2020, 143, 3041–3051. [Google Scholar] [CrossRef] [PubMed]
- Dawson, T.M.; Dawson, V.L. Parkin plays a role in sporadic Parkinson’s disease. Neurodegener. Dis. 2014, 13, 69–71. [Google Scholar] [CrossRef] [Green Version]
- Ou, S.H.; Wu, F.; Harrich, D.; Garcia-Martinez, L.F.; Gaynor, R.B. Cloning and characterization of a novel cellular protein, TDP-43, that binds to human immunodeficiency virus type 1 TAR DNA sequence motifs. J. Virol. 1995, 69, 3584–3596. [Google Scholar] [CrossRef] [Green Version]
- Buratti, E. TDP-43 post-translational modifications in health and disease. Expert Opin. Ther. Targets 2018, 22, 279–293. [Google Scholar] [CrossRef]
- Afroz, T.; Perez-Berlanga, M.; Polymenidou, M. Structural Transition, Function and Dysfunction of TDP-43 in Neurodegenerative Diseases. Chimia Int. J. Chem. 2019, 73, 380–390. [Google Scholar] [CrossRef]
- Buratti, E. Trends in Understanding the Pathological Roles of TDP-43 and FUS Proteins. Adv. Exp. Med. Biol. 2021, 1281, 243–267. [Google Scholar] [PubMed]
- Francois-Moutal, L.; Perez-Miller, S.; Scott, D.D.; Miranda, V.G.; Mollasalehi, N.; Khanna, M. Structural Insights Into TDP-43 and Effects of Post-translational Modifications. Front. Mol. Neurosci. 2019, 12, 301. [Google Scholar] [CrossRef]
- Mompean, M.; Romano, V.; Pantoja-Uceda, D.; Stuani, C.; Baralle, F.E.; Buratti, E.; Laurents, D.V. The TDP-43 N-terminal domain structure at high resolution. FEBS J. 2016, 283, 1242–1260. [Google Scholar] [CrossRef] [PubMed]
- Qin, H.; Lim, L.Z.; Wei, Y.; Song, J. TDP-43 N terminus encodes a novel ubiquitin-like fold and its unfolded form in equilibrium that can be shifted by binding to ssDNA. Proc. Natl. Acad. Sci. USA 2014, 111, 18619–18624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afroz, T.; Hock, E.M.; Ernst, P.; Foglieni, C.; Jambeau, M.; Gilhespy, L.A.B.; Laferriere, F.; Maniecka, Z.; Pluckthun, A.; Mittl, P.; et al. Functional and dynamic polymerization of the ALS-linked protein TDP-43 antagonizes its pathologic aggregation. Nat. Commun. 2017, 8, 45. [Google Scholar] [CrossRef] [Green Version]
- Buratti, E.; Baralle, F.E. Characterization and functional implications of the RNA binding properties of nuclear factor TDP-43, a novel splicing regulator of CFTR exon 9. J. Biol. Chem. 2001, 276, 36337–36343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lukavsky, P.J.; Daujotyte, D.; Tollervey, J.R.; Ule, J.; Stuani, C.; Buratti, E.; Baralle, F.E.; Damberger, F.F.; Allain, F.H. Molecular basis of UG-rich RNA recognition by the human splicing factor TDP-43. Nat. Struct. Mol. Biol. 2013, 20, 1443–1449. [Google Scholar] [CrossRef] [PubMed]
- Babinchak, W.M.; Haider, R.; Dumm, B.K.; Sarkar, P.; Surewicz, K.; Choi, J.K.; Surewicz, W.K. The role of liquid-liquid phase separation in aggregation of the TDP-43 low-complexity domain. J. Biol. Chem. 2019, 294, 6306–6317. [Google Scholar] [CrossRef] [Green Version]
- Conicella, A.E.; Zerze, G.H.; Mittal, J.; Fawzi, N.L. ALS Mutations Disrupt Phase Separation Mediated by alpha-Helical Structure in the TDP-43 Low-Complexity C-Terminal Domain. Structure 2016, 24, 1537–1549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mompean, M.; Chakrabartty, A.; Buratti, E.; Laurents, D.V. Electrostatic Repulsion Governs TDP-43 C-terminal Domain Aggregation. PLoS Biol. 2016, 14, e1002447. [Google Scholar] [CrossRef]
- Pantoja-Uceda, D.; Stuani, C.; Laurents, D.V.; McDermott, A.E.; Buratti, E.; Mompean, M. Phe-Gly motifs drive fibrillization of TDP-43’s prion-like domain condensates. PLoS Biol. 2021, 19, e3001198. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.S.; Tsai, K.J.; Chang, Y.J.; Kao, P.; Woods, R.; Kuo, P.H.; Wu, C.C.; Liao, J.Y.; Chou, S.C.; Lin, V.; et al. Full-length TDP-43 forms toxic amyloid oligomers that are present in frontotemporal lobar dementia-TDP patients. Nat. Commun. 2014, 5, 4824. [Google Scholar] [CrossRef]
- Capitini, C.; Conti, S.; Perni, M.; Guidi, F.; Cascella, R.; De Poli, A.; Penco, A.; Relini, A.; Cecchi, C.; Chiti, F. TDP-43 inclusion bodies formed in bacteria are structurally amorphous, non-amyloid and inherently toxic to neuroblastoma cells. PLoS ONE 2014, 9, e86720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, B.S.; Snead, D.; Lee, J.J.; McCaffery, J.M.; Shorter, J.; Gitler, A.D. TDP-43 is intrinsically aggregation-prone, and amyotrophic lateral sclerosis-linked mutations accelerate aggregation and increase toxicity. J. Biol. Chem. 2009, 284, 20329–20339. [Google Scholar] [CrossRef] [Green Version]
- Woerner, A.C.; Frottin, F.; Hornburg, D.; Feng, L.R.; Meissner, F.; Patra, M.; Tatzelt, J.; Mann, M.; Winklhofer, K.F.; Hartl, F.U.; et al. Cytoplasmic protein aggregates interfere with nucleocytoplasmic transport of protein and RNA. Science 2016, 351, 173–176. [Google Scholar] [CrossRef] [PubMed]
- Igaz, L.M.; Kwong, L.K.; Chen-Plotkin, A.; Winton, M.J.; Unger, T.L.; Xu, Y.; Neumann, M.; Trojanowski, J.Q.; Lee, V.M. Expression of TDP-43 C-terminal Fragments in Vitro Recapitulates Pathological Features of TDP-43 Proteinopathies. J. Biol. Chem. 2009, 284, 8516–8524. [Google Scholar] [CrossRef] [Green Version]
- Berning, B.A.; Walker, A.K. The Pathobiology of TDP-43 C-Terminal Fragments in ALS and FTLD. Front. Neurosci. 2019, 13, 335. [Google Scholar] [CrossRef] [Green Version]
- Dammer, E.B.; Fallini, C.; Gozal, Y.M.; Duong, D.M.; Rossoll, W.; Xu, P.; Lah, J.J.; Levey, A.I.; Peng, J.; Bassell, G.J.; et al. Coaggregation of RNA-binding proteins in a model of TDP-43 proteinopathy with selective RGG motif methylation and a role for RRM1 ubiquitination. PLoS ONE 2012, 7, e38658. [Google Scholar] [CrossRef] [Green Version]
- Collins, M.; Riascos, D.; Kovalik, T.; An, J.; Krupa, K.; Hood, B.L.; Conrads, T.P.; Renton, A.E.; Traynor, B.J.; Bowser, R. The RNA-binding motif 45 (RBM45) protein accumulates in inclusion bodies in amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration with TDP-43 inclusions (FTLD-TDP) patients. Acta Neuropathol. 2012, 124, 717–732. [Google Scholar] [CrossRef] [Green Version]
- Mackenzie, I.R.; Neumann, M. FET proteins in frontotemporal dementia and amyotrophic lateral sclerosis. Brain Res. 2012, 1462, 40–43. [Google Scholar] [CrossRef] [Green Version]
- Shelkovnikova, T.A.; Robinson, H.K.; Troakes, C.; Ninkina, N.; Buchman, V.L. Compromised paraspeckle formation as a pathogenic factor in FUSopathies. Hum. Mol. Genet. 2014, 23, 2298–2312. [Google Scholar] [CrossRef]
- Bolognesi, B.; Faure, A.J.; Seuma, M.; Schmiedel, J.M.; Tartaglia, G.G.; Lehner, B. The mutational landscape of a prion-like domain. Nat. Commun. 2019, 10, 4162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanden Broeck, L.; Callaerts, P.; Dermaut, B. TDP-43-mediated neurodegeneration: Towards a loss-of-function hypothesis? Trends Mol. Med. 2014, 20, 66–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klim, J.R.; Williams, L.A.; Limone, F.; Guerra San Juan, I.; Davis-Dusenbery, B.N.; Mordes, D.A.; Burberry, A.; Steinbaugh, M.J.; Gamage, K.K.; Kirchner, R.; et al. ALS-implicated protein TDP-43 sustains levels of STMN2, a mediator of motor neuron growth and repair. Nat. Neurosci. 2019, 22, 167–179. [Google Scholar] [CrossRef]
- Hardy, J.; Rogaeva, E. Motor neuron disease and frontotemporal dementia: Sometimes related, sometimes not. Exp. Neurol. 2014, 262 Pt B, 75–83. [Google Scholar] [CrossRef]
- Balendra, R.; Isaacs, A.M. C9orf72-mediated ALS and FTD: Multiple pathways to disease. Nat. Rev. Neurol. 2018, 14, 544–558. [Google Scholar] [CrossRef] [PubMed]
- Solomon, D.A.; Mitchell, J.C.; Salcher-Konrad, M.T.; Vance, C.A.; Mizielinska, S. Review: Modelling the pathology and behaviour of frontotemporal dementia. Neuropathol. Appl. Neurobiol. 2019, 45, 58–80. [Google Scholar] [CrossRef] [Green Version]
- Davis, S.A.; Itaman, S.; Khalid-Janney, C.M.; Sherard, J.A.; Dowell, J.A.; Cairns, N.J.; Gitcho, M.A. TDP-43 interacts with mitochondrial proteins critical for mitophagy and mitochondrial dynamics. Neurosci. Lett. 2018, 678, 8–15. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Duan, Y.; Qin, C.; Li, J.C.; Duan, G.; Deng, X.; Ni, J.; Cao, X.; Xiang, K.; Tian, K.; et al. Distinct multilevel misregulations of Parkin and PINK1 revealed in cell and animal models of TDP-43 proteinopathy. Cell Death Dis. 2018, 9, 953. [Google Scholar] [CrossRef]
- Hebron, M.L.; Lonskaya, I.; Sharpe, K.; Weerasinghe, P.P.; Algarzae, N.K.; Shekoyan, A.R.; Moussa, C.E. Parkin ubiquitinates Tar-DNA binding protein-43 (TDP-43) and promotes its cytosolic accumulation via interaction with histone deacetylase 6 (HDAC6). J. Biol. Chem. 2013, 288, 4103–4115. [Google Scholar] [CrossRef] [Green Version]
- Gaweda-Walerych, K.; Walerych, D.; Berdynski, M.; Buratti, E.; Zekanowski, C. Parkin Levels Decrease in Fibroblasts with Progranulin (PGRN) Pathogenic Variants and in a Cellular Model of PGRN Deficiency. Front. Mol. Neurosci. 2021, 14, 676478. [Google Scholar] [CrossRef] [PubMed]
- Rabin, S.J.; Kim, J.M.; Baughn, M.; Libby, R.T.; Kim, Y.J.; Fan, Y.; La Spada, A.; Stone, B.; Ravits, J. Sporadic ALS has compartment-specific aberrant exon splicing and altered cell-matrix adhesion biology. Hum. Mol. Genet. 2010, 19, 313–328. [Google Scholar] [CrossRef] [PubMed]
- Stribl, C.; Samara, A.; Trumbach, D.; Peis, R.; Neumann, M.; Fuchs, H.; Gailus-Durner, V.; Hrabe de Angelis, M.; Rathkolb, B.; Wolf, E.; et al. Mitochondrial dysfunction and decrease in body weight of a transgenic knock-in mouse model for TDP-43. J. Biol. Chem. 2014, 289, 10769–10784. [Google Scholar] [CrossRef] [Green Version]
- Wenqiang, C.; Lonskaya, I.; Hebron, M.L.; Ibrahim, Z.; Olszewski, R.T.; Neale, J.H.; Moussa, C.E. Parkin-mediated reduction of nuclear and soluble TDP-43 reverses behavioral decline in symptomatic mice. Hum. Mol. Genet. 2014, 23, 4960–4969. [Google Scholar] [CrossRef] [Green Version]
- Zilocchi, M.; Colugnat, I.; Lualdi, M.; Meduri, M.; Marini, F.; Corasolla Carregari, V.; Moutaoufik, M.T.; Phanse, S.; Pieroni, L.; Babu, M.; et al. Exploring the Impact of PARK2 Mutations on the Total and Mitochondrial Proteome of Human Skin Fibroblasts. Front. Cell Dev. Biol. 2020, 8, 423. [Google Scholar] [CrossRef] [PubMed]
- Pacelli, C.; De Rasmo, D.; Signorile, A.; Grattagliano, I.; di Tullio, G.; D’Orazio, A.; Nico, B.; Comi, G.P.; Ronchi, D.; Ferranini, E.; et al. Mitochondrial defect and PGC-1alpha dysfunction in parkin-associated familial Parkinson’s disease. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2011, 1812, 1041–1053. [Google Scholar] [CrossRef] [Green Version]
- Mortiboys, H.; Thomas, K.J.; Koopman, W.J.; Klaffke, S.; Abou-Sleiman, P.; Olpin, S.; Wood, N.W.; Willems, P.H.; Smeitink, J.A.; Cookson, M.R.; et al. Mitochondrial function and morphology are impaired in parkin-mutant fibroblasts. Ann. Neurol. 2008, 64, 555–565. [Google Scholar] [CrossRef]
- Grunewald, A.; Voges, L.; Rakovic, A.; Kasten, M.; Vandebona, H.; Hemmelmann, C.; Lohmann, K.; Orolicki, S.; Ramirez, A.; Schapira, A.H.; et al. Mutant Parkin impairs mitochondrial function and morphology in human fibroblasts. PLoS ONE 2010, 5, e12962. [Google Scholar] [CrossRef]
- Zucca, S.; Gagliardi, S.; Pandini, C.; Diamanti, L.; Bordoni, M.; Sproviero, D.; Arigoni, M.; Olivero, M.; Pansarasa, O.; Ceroni, M.; et al. RNA-Seq profiling in peripheral blood mononuclear cells of amyotrophic lateral sclerosis patients and controls. Sci. Data 2019, 6, 190006. [Google Scholar] [CrossRef]
- Briese, M.; Saal-Bauernschubert, L.; Luningschror, P.; Moradi, M.; Dombert, B.; Surrey, V.; Appenzeller, S.; Deng, C.; Jablonka, S.; Sendtner, M. Loss of Tdp-43 disrupts the axonal transcriptome of motoneurons accompanied by impaired axonal translation and mitochondria function. Acta Neuropathol. Commun. 2020, 8, 116. [Google Scholar] [CrossRef]
- Andres-Benito, P.; Gelpi, E.; Povedano, M.; Santpere, G.; Ferrer, I. Gene Expression Profile in Frontal Cortex in Sporadic Frontotemporal Lobar Degeneration-TDP. J. Neuropathol. Exp. Neurol. 2018, 77, 608–627. [Google Scholar] [CrossRef]
- Iridoy, M.O.; Zubiri, I.; Zelaya, M.V.; Martinez, L.; Ausin, K.; Lachen-Montes, M.; Santamaria, E.; Fernandez-Irigoyen, J.; Jerico, I. Neuroanatomical Quantitative Proteomics Reveals Common Pathogenic Biological Routes between Amyotrophic Lateral Sclerosis (ALS) and Frontotemporal Dementia (FTD). Int. J. Mol. Sci. 2018, 20, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mol, M.O.; Miedema, S.S.M.; van Swieten, J.C.; van Rooij, J.G.J.; Dopper, E.G.P. Molecular Pathways Involved in Frontotemporal Lobar Degeneration with TDP-43 Proteinopathy: What Can We Learn from Proteomics? Int. J. Mol. Sci. 2021, 22, 10298. [Google Scholar] [CrossRef]
- Mackenzie, I.R.; Neumann, M. Reappraisal of TDP-43 pathology in FTLD-U subtypes. Acta Neuropathol. 2017, 134, 79–96. [Google Scholar] [CrossRef] [PubMed]
- Tan, R.H.; Kril, J.J.; Fatima, M.; McGeachie, A.; McCann, H.; Shepherd, C.; Forrest, S.L.; Affleck, A.; Kwok, J.B.; Hodges, J.R.; et al. TDP-43 proteinopathies: Pathological identification of brain regions differentiating clinical phenotypes. Brain 2015, 138, 3110–3122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Espay, A.J.; Litvan, I. Parkinsonism and frontotemporal dementia: The clinical overlap. J. Mol. Neurosci. 2011, 45, 343–349. [Google Scholar] [CrossRef] [Green Version]
- Pickrell, A.M.; Youle, R.J. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 2015, 85, 257–273. [Google Scholar] [CrossRef] [Green Version]
- Rowe, J.B. Parkinsonism in frontotemporal dementias. Int. Rev. Neurobiol. 2019, 149, 249–275. [Google Scholar]
- Park, H.K.; Chung, S.J. New perspective on parkinsonism in frontotemporal lobar degeneration. J. Mov. Disord. 2013, 6, 1–8. [Google Scholar] [CrossRef]
- Rayaprolu, S.; Fujioka, S.; Traynor, S.; Soto-Ortolaza, A.I.; Petrucelli, L.; Dickson, D.W.; Rademakers, R.; Boylan, K.B.; Graff-Radford, N.R.; Uitti, R.J.; et al. TARDBP mutations in Parkinson’s disease. Parkinsonism Relat. Disord. 2013, 19, 312–315. [Google Scholar] [CrossRef] [Green Version]
- Foster, N.L.; Wilhelmsen, K.; Sima, A.A.; Jones, M.Z.; D’Amato, C.J.; Gilman, S. Frontotemporal dementia and parkinsonism linked to chromosome 17: A consensus conference. Conference Participants. Ann. Neurol. 1997, 41, 706–715. [Google Scholar] [CrossRef]
- Baker, M.; Mackenzie, I.R.; Pickering-Brown, S.M.; Gass, J.; Rademakers, R.; Lindholm, C.; Snowden, J.; Adamson, J.; Sadovnick, A.D.; Rollinson, S.; et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature 2006, 442, 916–919. [Google Scholar] [CrossRef] [PubMed]
- Boeve, B.F.; Baker, M.; Dickson, D.W.; Parisi, J.E.; Giannini, C.; Josephs, K.A.; Hutton, M.; Pickering-Brown, S.M.; Rademakers, R.; Tang-Wai, D.; et al. Frontotemporal dementia and parkinsonism associated with the IVS1 + 1G->A mutation in progranulin: A clinicopathologic study. Brain 2006, 129, 3103–3114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boeve, B.F.; Hutton, M. Refining frontotemporal dementia with parkinsonism linked to chromosome 17: Introducing FTDP-17 (MAPT) and FTDP-17 (PGRN). Arch. Neurol. 2008, 65, 460–464. [Google Scholar] [CrossRef] [PubMed]
- Forrest, S.L.; Kril, J.J.; Stevens, C.H.; Kwok, J.B.; Hallupp, M.; Kim, W.S.; Huang, Y.; McGinley, C.V.; Werka, H.; Kiernan, M.C.; et al. Retiring the term FTDP-17 as MAPT mutations are genetic forms of sporadic frontotemporal tauopathies. Brain 2018, 141, 521–534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gasca-Salas, C.; Masellis, M.; Khoo, E.; Shah, B.B.; Fisman, D.; Lang, A.E.; Kleiner-Fisman, G. Characterization of Movement Disorder Phenomenology in Genetically Proven, Familial Frontotemporal Lobar Degeneration: A Systematic Review and Meta-Analysis. PLoS ONE 2016, 11, e0153852. [Google Scholar]
- Xu, X.; Su, Y.; Zou, Z.; Zhou, Y.; Yan, J. Correlation between C9ORF72 mutation and neurodegenerative diseases: A comprehensive review of the literature. Int. J. Med. Sci. 2021, 18, 378–386. [Google Scholar] [CrossRef]
- Estevez-Fraga, C.; Magrinelli, F.; Hensman Moss, D.; Mulroy, E.; Di Lazzaro, G.; Latorre, A.; Mackenzie, M.; Houlden, H.; Tabrizi, S.J.; Bhatia, K.P. Expanding the Spectrum of Movement Disorders Associated With C9orf72 Hexanucleotide Expansions. Neurol. Genet. 2021, 7, e575. [Google Scholar] [CrossRef] [PubMed]
- Benussi, A.; Padovani, A.; Borroni, B. Phenotypic Heterogeneity of Monogenic Frontotemporal Dementia. Front. Aging Neurosci. 2015, 7, 171. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, I.R. The neuropathology and clinical phenotype of FTD with progranulin mutations. Acta Neuropathol. 2007, 114, 49–54. [Google Scholar] [CrossRef]
- Kelley, B.J.; Haidar, W.; Boeve, B.F.; Baker, M.; Graff-Radford, N.R.; Krefft, T.; Frank, A.R.; Jack, C.R., Jr.; Shiung, M.; Knopman, D.S.; et al. Prominent phenotypic variability associated with mutations in Progranulin. Neurobiol. Aging 2009, 30, 739–751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carecchio, M.; Galimberti, D.; Fenoglio, C.; Serpente, M.; Scarpini, E.; Comi, C.; Terazzi, E.; Cantello, R. Evidence of pre-synaptic dopaminergic deficit in a patient with a novel progranulin mutation presenting with atypical parkinsonism. J. Alzheimer’s Dis. 2014, 38, 747–752. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, R.L.; Zhang, W.; Che, C.H.; Feng, S.Y.; Huang, H.P.; Liu, C.Y.; Zou, Z.Y. Novel TARDBP missense mutation caused familial amyotrophic lateral sclerosis with frontotemporal dementia and parkinsonism. Neurobiol. Aging 2021, 107, 168–173. [Google Scholar] [CrossRef]
- Baizabal-Carvallo, J.F.; Jankovic, J. Parkinsonism, movement disorders and genetics in frontotemporal dementia. Nat. Rev. Neurol. 2016, 12, 175–185. [Google Scholar] [CrossRef]
- Siuda, J.; Fujioka, S.; Wszolek, Z.K. Parkinsonian syndrome in familial frontotemporal dementia. Parkinsonism Relat. Disord. 2014, 20, 957–964. [Google Scholar] [CrossRef] [Green Version]
- Arienti, F.; Lazzeri, G.; Vizziello, M.; Monfrini, E.; Bresolin, N.; Saetti, M.C.; Picillo, M.; Franco, G.; Di Fonzo, A. Unravelling Genetic Factors Underlying Corticobasal Syndrome: A Systematic Review. Cells 2021, 10, 171. [Google Scholar] [CrossRef]
- Kertesz, A.; McMonagle, P.; Jesso, S. Extrapyramidal syndromes in frontotemporal degeneration. J. Mol. Neurosci. 2011, 45, 336–342. [Google Scholar] [CrossRef] [PubMed]
- Dulski, J.; Cerquera-Cleves, C.; Milanowski, L.; Kidd, A.; Sitek, E.J.; Strongosky, A.; Vanegas Monroy, A.M.; Dickson, D.W.; Ross, O.A.; Pentela-Nowicka, J.; et al. Clinical, pathological and genetic characteristics of Perry disease-new cases and literature review. Eur. J. Neurol. 2021, 28, 4010–4021. [Google Scholar] [CrossRef] [PubMed]
- Milanowski, L.; Sitek, E.J.; Dulski, J.; Cerquera-Cleves, C.; Gomez, J.D.; Brockhuis, B.; Schinwelski, M.; Kluj-Kozlowska, K.; Ross, O.A.; Slawek, J.; et al. Cognitive and behavioral profile of Perry syndrome in two families. Parkinsonism Relat. Disord. 2020, 77, 114–120. [Google Scholar] [CrossRef]
- Mimuro, M.; Yoshida, M.; Kuzuhara, S.; Kokubo, Y. Amyotrophic lateral sclerosis and parkinsonism-dementia complex of the Hohara focus of the Kii Peninsula: A multiple proteinopathy? Neuropathology 2018, 38, 98–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucini, C.B.; Braun, R.J. Mitochondrion-Dependent Cell Death in TDP-43 Proteinopathies. Biomedicines 2021, 9, 376. [Google Scholar] [CrossRef]
- Dafinca, R.; Barbagallo, P.; Talbot, K. The Role of Mitochondrial Dysfunction and ER Stress in TDP-43 and C9ORF72 ALS. Front. Cell. Neurosci. 2021, 15, 653688. [Google Scholar] [CrossRef]
- Prasad, A.; Bharathi, V.; Sivalingam, V.; Girdhar, A.; Patel, B.K. Molecular Mechanisms of TDP-43 Misfolding and Pathology in Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2019, 12, 25. [Google Scholar] [CrossRef]
- Floare, M.L.; Allen, S.P. Why TDP-43? Why Not? Mechanisms of Metabolic Dysfunction in Amyotrophic Lateral Sclerosis. Neurosci. Insights 2020, 15, 2633105520957302. [Google Scholar] [CrossRef]
- Gao, J.; Wang, L.; Yan, T.; Perry, G.; Wang, X. TDP-43 proteinopathy and mitochondrial abnormalities in neurodegeneration. Mol. Cell. Neurosci. 2019, 100, 103396. [Google Scholar] [CrossRef]
- Tsai, Y.L.; Manley, J.L. Multiple ways to a dead end: Diverse mechanisms by which ALS mutant genes induce cell death. Cell Cycle 2021, 20, 631–646. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Yan, S.; Zhang, Z. Maintaining the balance of TDP-43, mitochondria, and autophagy: A promising therapeutic strategy for neurodegenerative diseases. Transl. Neurodegener. 2020, 9, 40. [Google Scholar] [CrossRef]
- Hong, K.; Li, Y.; Duan, W.; Guo, Y.; Jiang, H.; Li, W.; Li, C. Full-length TDP-43 and its C-terminal fragments activate mitophagy in NSC34 cell line. Neurosci. Lett. 2012, 530, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Jun, M.H.; Jang, J.W.; Jeon, P.; Lee, S.K.; Lee, S.H.; Choi, H.E.; Lee, Y.K.; Choi, H.; Park, S.W.; Kim, J.; et al. Nonmuscle myosin IIB regulates Parkin-mediated mitophagy associated with amyotrophic lateral sclerosis-linked TDP-43. Cell Death Dis. 2020, 11, 952. [Google Scholar] [CrossRef] [PubMed]
- Limanaqi, F.; Biagioni, F.; Gambardella, S.; Familiari, P.; Frati, A.; Fornai, F. Promiscuous Roles of Autophagy and Proteasome in Neurodegenerative Proteinopathies. Int. J. Mol. Sci. 2020, 21, 3028. [Google Scholar] [CrossRef]
- Budini, M.; Buratti, E.; Morselli, E.; Criollo, A. Autophagy and Its Impact on Neurodegenerative Diseases: New Roles for TDP-43 and C9orf72. Front. Mol. Neurosci. 2017, 10, 170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chua, J.P.; De Calbiac, H.; Kabashi, E.; Barmada, S.J. Autophagy and ALS: Mechanistic insights and therapeutic implications. Autophagy 2021, 1–29. [Google Scholar] [CrossRef]
- Leibiger, C.; Deisel, J.; Aufschnaiter, A.; Ambros, S.; Tereshchenko, M.; Verheijen, B.M.; Buttner, S.; Braun, R.J. TDP-43 controls lysosomal pathways thereby determining its own clearance and cytotoxicity. Hum. Mol. Genet. 2018, 27, 1593–1607. [Google Scholar] [CrossRef] [Green Version]
- Root, J.; Merino, P.; Nuckols, A.; Johnson, M.; Kukar, T. Lysosome dysfunction as a cause of neurodegenerative diseases: Lessons from frontotemporal dementia and amyotrophic lateral sclerosis. Neurobiol. Dis. 2021, 154, 105360. [Google Scholar] [CrossRef]
- Caccamo, A.; Majumder, S.; Oddo, S. Cognitive decline typical of frontotemporal lobar degeneration in transgenic mice expressing the 25-kDa C-terminal fragment of TDP-43. Am. J. Pathol. 2012, 180, 293–302. [Google Scholar] [CrossRef] [Green Version]
- Swadling, L.; Pallett, L.J.; Diniz, M.O.; Baker, J.M.; Amin, O.E.; Stegmann, K.A.; Burton, A.R.; Schmidt, N.M.; Jeffery-Smith, A.; Zakeri, N.; et al. Human Liver Memory CD8(+) T Cells Use Autophagy for Tissue Residence. Cell Rep. 2020, 30, 687–698.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onishi, M.; Yamano, K.; Sato, M.; Matsuda, N.; Okamoto, K. Molecular mechanisms and physiological functions of mitophagy. EMBO J. 2021, 40, e104705. [Google Scholar] [CrossRef]
- Nguyen, D.K.H.; Thombre, R.; Wang, J. Autophagy as a common pathway in amyotrophic lateral sclerosis. Neurosci. Lett. 2019, 697, 34–48. [Google Scholar] [CrossRef]
- Kumar, S.; Julien, J.P. TDP-43 triggers immune response via mitochondrial DNA release. Cell Res. 2021, 31, 379–380. [Google Scholar] [CrossRef]
- Yu, C.H.; Davidson, S.; Harapas, C.R.; Hilton, J.B.; Mlodzianoski, M.J.; Laohamonthonkul, P.; Louis, C.; Low, R.R.J.; Moecking, J.; De Nardo, D.; et al. TDP-43 Triggers Mitochondrial DNA Release via mPTP to Activate cGAS/STING in ALS. Cell 2020, 183, 636–649.e18. [Google Scholar] [CrossRef]
- Tank, E.M.; Figueroa-Romero, C.; Hinder, L.M.; Bedi, K.; Archbold, H.C.; Li, X.; Weskamp, K.; Safren, N.; Paez-Colasante, X.; Pacut, C.; et al. Abnormal RNA stability in amyotrophic lateral sclerosis. Nat. Commun. 2018, 9, 2845. [Google Scholar] [CrossRef] [Green Version]
- Gautam, M.; Jara, J.H.; Kocak, N.; Rylaarsdam, L.E.; Kim, K.D.; Bigio, E.H.; Hande Ozdinler, P. Mitochondria, ER, and nuclear membrane defects reveal early mechanisms for upper motor neuron vulnerability with respect to TDP-43 pathology. Acta Neuropathol. 2019, 137, 47–69. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Deng, J.; Dong, J.; Liu, J.; Bigio, E.H.; Mesulam, M.; Wang, T.; Sun, L.; Wang, L.; Lee, A.Y.; et al. TDP-43 induces mitochondrial damage and activates the mitochondrial unfolded protein response. PLoS Genet. 2019, 15, e1007947. [Google Scholar] [CrossRef] [Green Version]
- Zuo, X.; Zhou, J.; Li, Y.; Wu, K.; Chen, Z.; Luo, Z.; Zhang, X.; Liang, Y.; Esteban, M.A.; Zhou, Y.; et al. TDP-43 aggregation induced by oxidative stress causes global mitochondrial imbalance in ALS. Nat. Struct. Mol. Biol. 2021, 28, 132–142. [Google Scholar] [CrossRef]
- Mackenzie, I.R.; Arzberger, T.; Kremmer, E.; Troost, D.; Lorenzl, S.; Mori, K.; Weng, S.M.; Haass, C.; Kretzschmar, H.A.; Edbauer, D.; et al. Dipeptide repeat protein pathology in C9ORF72 mutation cases: Clinico-pathological correlations. Acta Neuropathol. 2013, 126, 859–879. [Google Scholar] [CrossRef] [Green Version]
- Mackenzie, I.R. The role of dipeptide-repeat protein pathology in C9orf72 mutation cases. Neuropathol. Appl. Neurobiol. 2016, 42, 217–219. [Google Scholar] [CrossRef]
- Mayl, K.; Shaw, C.E.; Lee, Y.B. Disease Mechanisms and Therapeutic Approaches in C9orf72 ALS-FTD. Biomedicines 2021, 9, 601. [Google Scholar] [CrossRef]
- Dafinca, R.; Barbagallo, P.; Farrimond, L.; Candalija, A.; Scaber, J.; Ababneh, N.A.; Sathyaprakash, C.; Vowles, J.; Cowley, S.A.; Talbot, K. Impairment of Mitochondrial Calcium Buffering Links Mutations in C9ORF72 and TARDBP in iPS-Derived Motor Neurons from Patients with ALS/FTD. Stem Cell Rep. 2020, 14, 892–908. [Google Scholar] [CrossRef]
- Lynch, E.; Semrad, T.; Belsito, V.S.; FitzGibbons, C.; Reilly, M.; Hayakawa, K.; Suzuki, M. C9ORF72-related cellular pathology in skeletal myocytes derived from ALS-patient induced pluripotent stem cells. Dis. Models Mech. 2019, 12, dmm039552. [Google Scholar] [CrossRef] [Green Version]
- Debska-Vielhaber, G.; Miller, I.; Peeva, V.; Zuschratter, W.; Walczak, J.; Schreiber, S.; Petri, S.; Machts, J.; Vogt, S.; Szczepanowska, J.; et al. Impairment of mitochondrial oxidative phosphorylation in skin fibroblasts of SALS and FALS patients is rescued by in vitro treatment with ROS scavengers. Exp. Neurol. 2021, 339, 113620. [Google Scholar] [CrossRef]
- Baldwin, K.R.; Godena, V.K.; Hewitt, V.L.; Whitworth, A.J. Axonal transport defects are a common phenotype in Drosophila models of ALS. Hum. Mol. Genet. 2016, 25, 2378–2392. [Google Scholar]
- Mehta, A.R.; Gregory, J.M.; Dando, O.; Carter, R.N.; Burr, K.; Nanda, J.; Story, D.; McDade, K.; Smith, C.; Morton, N.M.; et al. Mitochondrial bioenergetic deficits in C9orf72 amyotrophic lateral sclerosis motor neurons cause dysfunctional axonal homeostasis. Acta Neuropathol. 2021, 141, 257–279. [Google Scholar] [CrossRef]
- Onesto, E.; Colombrita, C.; Gumina, V.; Borghi, M.O.; Dusi, S.; Doretti, A.; Fagiolari, G.; Invernizzi, F.; Moggio, M.; Tiranti, V.; et al. Gene-specific mitochondria dysfunctions in human TARDBP and C9ORF72 fibroblasts. Acta Neuropathol. Commun. 2016, 4, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.; Liu, H.; Itoh, K.; Oh, S.; Zhao, L.; Murata, D.; Sesaki, H.; Hartung, T.; Na, C.H.; Wang, J. C9orf72 regulates energy homeostasis by stabilizing mitochondrial complex I assembly. Cell Metab. 2021, 33, 531–546.e9. [Google Scholar] [CrossRef]
- Allen, S.P.; Hall, B.; Woof, R.; Francis, L.; Gatto, N.; Shaw, A.C.; Myszczynska, M.; Hemingway, J.; Coldicott, I.; Willcock, A.; et al. C9orf72 expansion within astrocytes reduces metabolic flexibility in amyotrophic lateral sclerosis. Brain 2019, 142, 3771–3790. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.Y.; Lopez-Gonzalez, R.; Krishnan, G.; Phillips, H.L.; Li, A.N.; Seeley, W.W.; Yao, W.D.; Almeida, S.; Gao, F.B. C9ORF72-ALS/FTD-associated poly(GR) binds Atp5a1 and compromises mitochondrial function in vivo. Nat. Neurosci. 2019, 22, 851–862. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Gonzalez, R.; Lu, Y.; Gendron, T.F.; Karydas, A.; Tran, H.; Yang, D.; Petrucelli, L.; Miller, B.L.; Almeida, S.; Gao, F.B. Poly(GR) in C9ORF72-Related ALS/FTD Compromises Mitochondrial Function and Increases Oxidative Stress and DNA Damage in iPSC-Derived Motor Neurons. Neuron 2016, 92, 383–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Wu, Z.; Li, Y.; Tantray, I.; De Stefani, D.; Mattarei, A.; Krishnan, G.; Gao, F.B.; Vogel, H.; Lu, B. Altered MICOS Morphology and Mitochondrial Ion Homeostasis Contribute to Poly(GR) Toxicity Associated with C9-ALS/FTD. Cell Rep. 2020, 32, 107989. [Google Scholar] [CrossRef]
- Dafinca, R.; Scaber, J.; Ababneh, N.; Lalic, T.; Weir, G.; Christian, H.; Vowles, J.; Douglas, A.G.; Fletcher-Jones, A.; Browne, C.; et al. C9orf72 Hexanucleotide Expansions Are Associated with Altered Endoplasmic Reticulum Calcium Homeostasis and Stress Granule Formation in Induced Pluripotent Stem Cell-Derived Neurons from Patients with Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Stem Cells 2016, 34, 2063–2078. [Google Scholar]
- Kreiter, N.; Pal, A.; Lojewski, X.; Corcia, P.; Naujock, M.; Reinhardt, P.; Sterneckert, J.; Petri, S.; Wegner, F.; Storch, A.; et al. Age-dependent neurodegeneration and organelle transport deficiencies in mutant TDP43 patient-derived neurons are independent of TDP43 aggregation. Neurobiol. Dis. 2018, 115, 167–181. [Google Scholar] [CrossRef] [PubMed]
- Allen, S.P. Understanding metabolic flexibility: A potential key to unlocking metabolic therapies in amyotrophic lateral sclerosis? Neural Regen. Res. 2020, 15, 1654–1655. [Google Scholar] [CrossRef] [PubMed]
- Grunewald, A.; Kumar, K.R.; Sue, C.M. New insights into the complex role of mitochondria in Parkinson’s disease. Prog. Neurobiol. 2019, 177, 73–93. [Google Scholar] [CrossRef]
- Chang, M.C.; Srinivasan, K.; Friedman, B.A.; Suto, E.; Modrusan, Z.; Lee, W.P.; Kaminker, J.S.; Hansen, D.V.; Sheng, M. Progranulin deficiency causes impairment of autophagy and TDP-43 accumulation. J. Exp. Med. 2017, 214, 2611–2628. [Google Scholar] [CrossRef] [Green Version]
- Seelaar, H.; Rohrer, J.D.; Pijnenburg, Y.A.; Fox, N.C.; van Swieten, J.C. Clinical, genetic and pathological heterogeneity of frontotemporal dementia: A review. J. Neurol. Neurosurg. Psychiatry 2011, 82, 476–486. [Google Scholar] [CrossRef] [Green Version]
- Rosen, E.Y.; Wexler, E.M.; Versano, R.; Coppola, G.; Gao, F.; Winden, K.D.; Oldham, M.C.; Martens, L.H.; Zhou, P.; Farese, R.V., Jr.; et al. Functional genomic analyses identify pathways dysregulated by progranulin deficiency, implicating Wnt signaling. Neuron 2011, 71, 1030–1042. [Google Scholar] [CrossRef] [Green Version]
- Alquezar, C.; Esteras, N.; Alzualde, A.; Moreno, F.; Ayuso, M.S.; Lopez de Munain, A.; Martin-Requero, A. Inactivation of CDK/pRb pathway normalizes survival pattern of lymphoblasts expressing the FTLD-progranulin mutation c.709-1G>A. PLoS ONE 2012, 7, e37057. [Google Scholar]
- Evers, B.M.; Rodriguez-Navas, C.; Tesla, R.J.; Prange-Kiel, J.; Wasser, C.R.; Yoo, K.S.; McDonald, J.; Cenik, B.; Ravenscroft, T.A.; Plattner, F.; et al. Lipidomic and Transcriptomic Basis of Lysosomal Dysfunction in Progranulin Deficiency. Cell Rep. 2017, 20, 2565–2574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, D.; Zhou, M.; Wang, Z.; Fu, Y.; Jia, M.; Wang, X.; Liu, M.; Zhang, Y.; Sun, Y.; Lu, Y.; et al. PGRN acts as a novel regulator of mitochondrial homeostasis by facilitating mitophagy and mitochondrial biogenesis to prevent podocyte injury in diabetic nephropathy. Cell Death Dis. 2019, 10, 524. [Google Scholar] [CrossRef] [Green Version]
- Koentjoro, B.; Park, J.S.; Sue, C.M. Nix restores mitophagy and mitochondrial function to protect against PINK1/Parkin-related Parkinson’s disease. Sci. Rep. 2017, 7, 44373. [Google Scholar] [CrossRef] [PubMed]
- Lonskaya, I.; Hebron, M.L.; Desforges, N.M.; Franjie, A.; Moussa, C.E. Tyrosine kinase inhibition increases functional parkin-Beclin-1 interaction and enhances amyloid clearance and cognitive performance. EMBO Mol. Med. 2013, 5, 1247–1262. [Google Scholar] [CrossRef] [PubMed]
- Lonskaya, I.; Hebron, M.L.; Desforges, N.M.; Schachter, J.B.; Moussa, C.E. Nilotinib-induced autophagic changes increase endogenous parkin level and ubiquitination, leading to amyloid clearance. J. Mol. Med. 2014, 92, 373–386. [Google Scholar] [CrossRef]
- Perera, N.D.; Tomas, D.; Wanniarachchillage, N.; Cuic, B.; Luikinga, S.J.; Rytova, V.; Turner, B.J. Stimulation of mTOR-independent autophagy and mitophagy by rilmenidine exacerbates the phenotype of transgenic TDP-43 mice. Neurobiol. Dis. 2021, 154, 105359. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Modeste, E.; Dammer, E.; Merino, P.; Taylor, G.; Duong, D.M.; Deng, Q.; Holler, C.J.; Gearing, M.; Dickson, D.; et al. Network analysis of the progranulin-deficient mouse brain proteome reveals pathogenic mechanisms shared in human frontotemporal dementia caused by GRN mutations. Acta Neuropathol. Commun. 2020, 8, 163. [Google Scholar] [CrossRef]
- Paushter, D.H.; Du, H.; Feng, T.; Hu, F. The lysosomal function of progranulin, a guardian against neurodegeneration. Acta Neuropathol. 2018, 136, 1–17. [Google Scholar] [CrossRef]
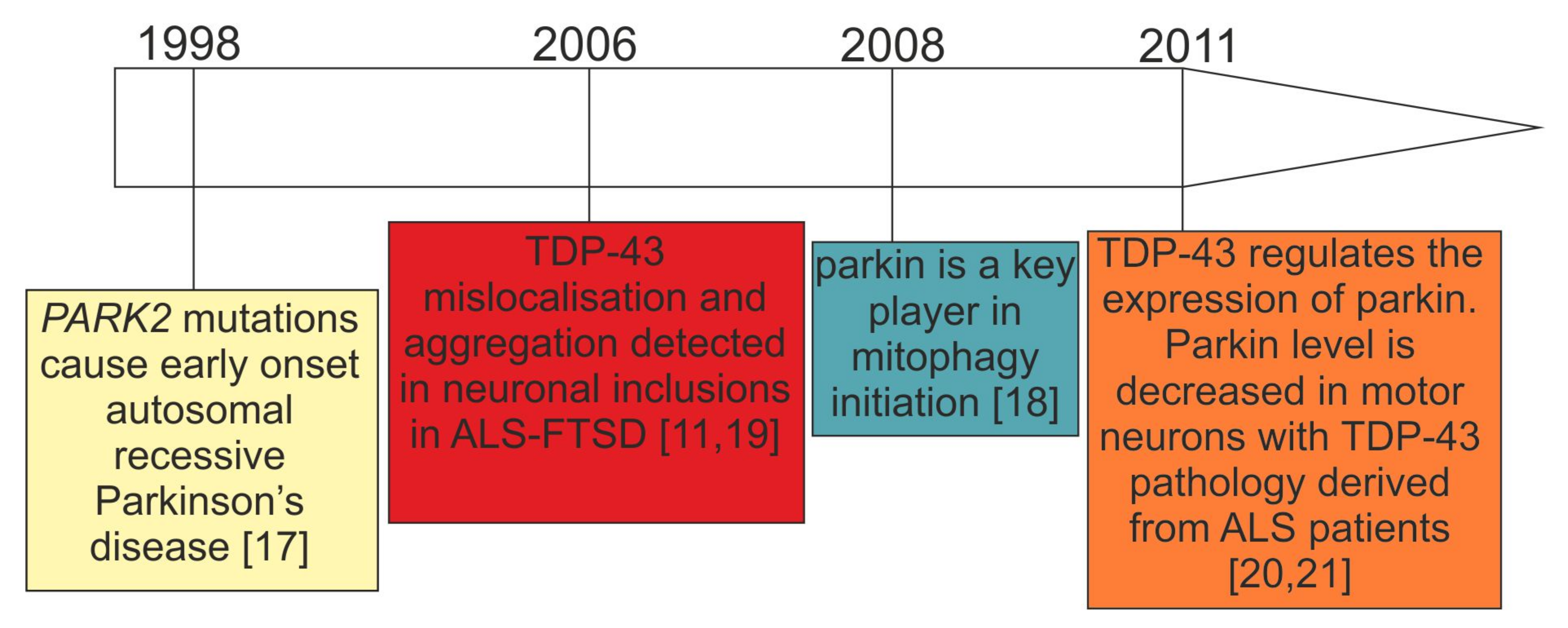


{kind=link}
{kind=link}
| TDP43-Proteinopathy Model | Cell Type/Treatment Length | Parkin mRNA/Protein | Accompanying Changes | References |
|---|---|---|---|---|
| Patients with sporadic ALS (n = 12) vs. control subjects | ca. 1000 motor neurons/- | Trend for decreased mRNA (microarray) | - | [95] |
| Patients with sporadic ALS (n = 11) vs. control subjects (n = 3) | Spinal cord motor neurons—only those with TDP-43 inclusions/- | Decreased protein (IF) | - | [21] |
| Carriers of PGRN mutations from families with FTLD | Human primary skin fibroblasts with PGRN mutations | Decreased mRNA/protein by ca. 60% (qRT-PCR) | Unchanged MFN2 and VDAC1 mRNA and protein | [94] |
| Mouse TDP-43 knockdown | Striatum injection of antisense oligonucleotides/2 weeks | Decreased mRNA by ca. 70% (RNAseq) | - | [20] |
| Mouse TDP-43 knockdown | Brain and spinal-cord injection of antisense oligonucleotides/2 weeks | Decreased mRNA by ca. 80% (qRT-PCR) | - | [21] |
| TDP-43 knockdown in human neurons (TDP-43 expression reduction by 60–75%) | Human neurons (iPSC-derived and HUES6 line) lentiviral shRNA constructs/na | Decreased mRNA by ca. 25% (qRT-PCR) | - | [21] |
| TDP-43 silencing (siRNA) in HEK293T | Human HEK293T (DMSO vs. mitochondrial uncoupler CCCP; siTDP-43 or si CTRL)/na | Decreased protein cytoplasmic localization (IF) | Decreased prohibitin 2 (PHB2) | [91] |
| TDP-43 silencing (siRNA) in skin fibroblasts derived from patients with FTLD | Human primary skin fibroblasts with PGRN mutations and control fibroblasts (siTDP-43 or siCTRL)/48 h | Decreased protein by ca. 40% (WB) | - | [94] |
| Overexpression of wild-type TDP-43-HA or mutant TDP-43-Q331K | Primary mouse neurons/motor cortex and human HEK293T cells/48 h | Decreased endogenous parkin mRNA and protein by c.a. 50% (qRT-PCR, WB) | Increased PINK1 protein | [92] |
| Exogenous co-expression of wild-type TDP-43-HA and intron-free human parkin or intron-free PINK1 | Human HEK293T cells/48 h | Decreased intron-free parkin mRNA and protein by c.a. 50% (qRT-PCR) | Increased cleaved PINK1 protein forms insoluble cytoplasmic aggregates | [92] |
| Transgenic Drosophila knock-in of wild-type human TDP-43-H | Fly heads/na | Decreased mRNA and protein by c.a. 45% (qRT-PCR, WB) | - | [92] |
| Wild-type TDP-43 overexpression | Human HEK293T (DMSO vs. mitochondrial uncoupler CCCP; wild-type pLX-TDP-43-v5 vector/na) | Increased protein cytoplasmic localization (IF) | Increased prohibitin 2 (PHB2) | [91] |
| Wild-type TDP-43 overexpression | Human primary skin fibroblasts with transiently silenced PGRN (48 h) overexpressing wild-type flag-TDP-43 (24 h) | Increased protein by c.a. 40% (WB) | Increased PGRN protein | [94] |
| Wild-type TDP-43 overexpression | Human skin fibroblasts with PGRN mutations overexpressing wild-type flag-TDP-43 (48 h) | Decreased protein by c.a. 50% (WB) | - | [94] |
| Transgenic mouse with heterozygous knock-in of human mutant TDP-43 (A315T) | Whole-brain tissue | mRNA and protein reduced by 70% compared to wild-type controls (qRT-PCR, WB) | Abnormal neuronal mitochondrial cristae, fusion and fission defects; | [96] |
| Overexpressed wild-type TDP-43 | Human M17 neuroblastoma cells | Increased protein by c.a. 50% (WB) | - | [93] |
| Transgenic mouse with knock-in of human mutant TDP-43A315T | Increased mRNA by c.a. 50% (qRT-PCR) | - | [93] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gaweda-Walerych, K.; Sitek, E.J.; Narożańska, E.; Buratti, E. Parkin beyond Parkinson’s Disease—A Functional Meaning of Parkin Downregulation in TDP-43 Proteinopathies. Cells 2021, 10, 3389. https://doi.org/10.3390/cells10123389
Gaweda-Walerych K, Sitek EJ, Narożańska E, Buratti E. Parkin beyond Parkinson’s Disease—A Functional Meaning of Parkin Downregulation in TDP-43 Proteinopathies. Cells. 2021; 10(12):3389. https://doi.org/10.3390/cells10123389
Chicago/Turabian StyleGaweda-Walerych, Katarzyna, Emilia Jadwiga Sitek, Ewa Narożańska, and Emanuele Buratti. 2021. "Parkin beyond Parkinson’s Disease—A Functional Meaning of Parkin Downregulation in TDP-43 Proteinopathies" Cells 10, no. 12: 3389. https://doi.org/10.3390/cells10123389
APA StyleGaweda-Walerych, K., Sitek, E. J., Narożańska, E., & Buratti, E. (2021). Parkin beyond Parkinson’s Disease—A Functional Meaning of Parkin Downregulation in TDP-43 Proteinopathies. Cells, 10(12), 3389. https://doi.org/10.3390/cells10123389