
NK Cells Armed with Chimeric Antigen Receptors (CAR): Roadblocks to Successful Development


 , , ,
, , ,  and
and 
Abstract
1. Introduction
2. NK Cells—A Promising Cellular Platform for CAR Engineering
3. Precise Design for a Functional CAR Structure
4. Established Targets for CAR NK Cells
4.1. CD19
4.2. EGFR
4.3. HER2
4.4. EpCAM (CD326)
4.5. GD2
4.6. Mesothelin
4.7. HSP70
5. CAR Transfer Methodology into NK Cells
5.1. Viral Vectors for CAR Transduction
5.2. Non-Viral Techniques for CAR Transfection
5.2.1. Electroporation-Mediated Delivery
5.2.2. Microfluidics-Based Cell Squeezing
5.2.3. Nanocarrier-Mediated Delivery
5.2.4. Transposon System
5.2.5. CRISPR-Cas9
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Laversanne, M.; Weiderpass, E.; Soerjomataram, I. The ever-increasing importance of cancer as a leading cause of premature death worldwide. Cancer 2021, 127, 3029–3030. [Google Scholar] [CrossRef]
- Multhoff, G.; Astner, S.T. Role of the Immune System in Cancer Development and Therapeutic Implications. In The Impact of Tumor Biology on Cancer Treatment and Multidisciplinary Strategies; Springer: Berlin/Heidelberg, Germany, 2009; pp. 129–145. [Google Scholar]
- Kobold, S.; Steffen, J.; Chaloupka, M.; Grassmann, S.; Henkel, J.; Castoldi, R.; Zeng, Y.; Chmielewski, M.; Schmollinger, J.C.; Schnurr, M. Selective bispecific T cell recruiting antibody and antitumor activity of adoptive T cell transfer. J. Natl. Cancer Inst. 2015, 107, 364. [Google Scholar] [CrossRef] [PubMed]
- Kobold, S.; Duewell, P.; Schnurr, M.; Subklewe, M.; Rothenfusser, S.; Endres, S. Immunotherapy in tumors: Activated T cells as a new treatment modality. Dtsch. Ärzteblatt Int. 2015, 112, 809. [Google Scholar]
- Multhoff, G.; Seier, S.; Stangl, S.; Sievert, W.; Shevtsov, M.; Werner, C.; Pockley, A.G.; Blankenstein, C.; Hildebrandt, M.; Offner, R. Targeted Natural Killer Cell–Based Adoptive Immunotherapy for the Treatment of Patients with NSCLC after Radiochemotherapy: A Randomized Phase II Clinical Trial. Clin. Cancer Res. 2020, 26, 5368–5379. [Google Scholar] [CrossRef] [PubMed]
- Milani, V.; Stangl, S.; Issels, R.; Gehrmann, M.; Wagner, B.; Hube, K.; Mayr, D.; Hiddemann, W.; Molls, M.; Multhoff, G. Anti-tumor activity of patient-derived NK cells after cell-based immunotherapy–a case report. J. Transl. Med. 2009, 7, 1–18. [Google Scholar] [CrossRef]
- Haslauer, T.; Greil, R.; Zaborsky, N.; Geisberger, R. CAR T-Cell Therapy in Hematological Malignancies. Int. J. Mol. Sci. 2021, 22, 8996. [Google Scholar] [CrossRef]
- Daher, M.; Melo Garcia, L.; Li, Y.; Rezvani, K. CAR-NK cells: The next wave of cellular therapy for cancer. Clin. Transl. Immunol. 2021, 10, e1274. [Google Scholar] [CrossRef]
- Tokarew, N.; Ogonek, J.; Endres, S.; von Bergwelt-Baildon, M.; Kobold, S. Teaching an old dog new tricks: Next-generation CAR T cells. Br. J. Cancer 2019, 120, 26–37. [Google Scholar] [CrossRef]
- Munshi, N.C.; Anderson, L.D., Jr.; Shah, N.; Madduri, D.; Berdeja, J.; Lonial, S.; Raje, N.; Lin, Y.; Siegel, D.; Oriol, A. Idecabtagene vicleucel in relapsed and refractory multiple myeloma. N. Engl. J. Med. 2021, 384, 705–716. [Google Scholar] [CrossRef]
- Lesch, S.; Benmebarek, M.-R.; Cadilha, B.L.; Stoiber, S.; Subklewe, M.; Endres, S.; Kobold, S. Determinants of response and resistance to CAR T cell therapy. Semin. Cancer Biol. 2020, 65, 80–90. [Google Scholar] [CrossRef]
- Sterner, R.C.; Sterner, R.M. CAR-T cell therapy: Current limitations and potential strategies. Blood Cancer J. 2021, 11, 1–11. [Google Scholar] [CrossRef]
- Zhang, C.; Hu, Y.; Xiao, W.; Tian, Z. Chimeric antigen receptor-and natural killer cell receptor-engineered innate killer cells in cancer immunotherapy. Cell. Mol. Immunol. 2021, 18, 2083–2100. [Google Scholar] [CrossRef]
- Liu, E.; Marin, D.; Banerjee, P.; Macapinlac, H.A.; Thompson, P.; Basar, R.; Nassif Kerbauy, L.; Overman, B.; Thall, P.; Kaplan, M. Use of CAR-transduced natural killer cells in CD19-positive lymphoid tumors. N. Engl. J. Med. 2020, 382, 545–553. [Google Scholar] [CrossRef]
- Quintarelli, C.; Sivori, S.; Caruso, S.; Carlomagno, S.; Boffa, I.; Orlando, D.; Guercio, M.; Cembrola, B.; Pitisci, A.; Di Cecca, S. CD19 redirected CAR NK cells are equally effective but less toxic than CAR T cells. Blood 2018, 132, 3491. [Google Scholar] [CrossRef]
- Morgan, M.A.; Büning, H.; Sauer, M.; Schambach, A. Use of cell and genome modification technologies to generate improved “off-the-shelf” CAR T and CAR NK cells. Front. Immunol. 2020, 11, 1965. [Google Scholar] [CrossRef]
- Rafei, H.; Daher, M.; Rezvani, K. Chimeric antigen receptor (CAR) natural killer (NK)-cell therapy: Leveraging the power of innate immunity. Br. J. Haematol. 2021, 193, 216–230. [Google Scholar] [CrossRef] [PubMed]
- Lamb, M.G.; Rangarajan, H.G.; Tullius, B.P.; Lee, D.A. Natural killer cell therapy for hematologic malignancies: Successes, challenges, and the future. Stem Cell Res. Ther. 2021, 12, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, M.; Mace, E.M.; Carisey, A.F.; Ahmed, N.; Orange, J.S. Quantitative imaging approaches to study the CAR immunological synapse. Mol. Ther. 2017, 25, 1757–1768. [Google Scholar] [CrossRef]
- Davey, A.S.; Call, M.E.; Call, M.J. The influence of chimeric antigen receptor structural domains on clinical outcomes and associated toxicities. Cancers 2021, 13, 38. [Google Scholar] [CrossRef]
- Schmidt, P.; Raftery, M.J.; Pecher, G. Engineering NK cells for CAR therapy—recent advances in gene transfer methodology. Front. Immunol. 2021, 11, 3404. [Google Scholar] [CrossRef]
- Vivier, E.; Raulet, D.H.; Moretta, A.; Caligiuri, M.A.; Zitvogel, L.; Lanier, L.L.; Yokoyama, W.M.; Ugolini, S. Innate or adaptive immunity? The example of natural killer cells. Science 2011, 331, 44–49. [Google Scholar] [CrossRef]
- Fehniger, T.A.; Cooper, M.A.; Nuovo, G.J.; Cella, M.; Facchetti, F.; Colonna, M.; Caligiuri, M.A. CD56bright natural killer cells are present in human lymph nodes and are activated by T cell–derived IL-2: A potential new link between adaptive and innate immunity. Blood J. Am. Soc. Hematol. 2003, 101, 3052–3057. [Google Scholar] [CrossRef]
- Cooper, M.A.; Fehniger, T.A.; Caligiuri, M.A. The biology of human natural killer-cell subsets. Trends Immunol. 2001, 22, 633–640. [Google Scholar] [CrossRef]
- Lanier, L.L. NK cell recognition. Annu. Rev. Immunol. 2005, 23, 225–274. [Google Scholar] [CrossRef]
- Talanian, R.V.; Yang, X.; Turbov, J.; Seth, P.; Ghayur, T.; Casiano, C.A.; Orth, K.; Froelich, C.J. Granule-mediated killing: Pathways for granzyme B–initiated apoptosis. J. Exp. Med. 1997, 186, 1323–1331. [Google Scholar] [CrossRef]
- Prager, I.; Watzl, C. Mechanisms of natural killer cell-mediated cellular cytotoxicity. J. Leukoc. Biol. 2019, 105, 1319–1329. [Google Scholar] [CrossRef]
- Abel, A.; Yang, C.; Thakar, M.; Malarkannan, S. Natural killer cells: Development, maturation, and clinical utilization. Front. Immunol. 2018, 9, 1869. [Google Scholar] [CrossRef]
- Konjević, G.M.; Vuletić, A.M.; Martinović, K.M.M.; Larsen, A.K.; Jurišić, V.B. The role of cytokines in the regulation of NK cells in the tumor environment. Cytokine 2019, 117, 30–40. [Google Scholar] [CrossRef]
- Penack, O.; Koenecke, C. Complications after CD19+ CAR T-cell therapy. Cancers 2020, 12, 3445. [Google Scholar] [CrossRef]
- Ghorashian, S.; Kramer, A.M.; Onuoha, S.; Wright, G.; Bartram, J.; Richardson, R.; Albon, S.J.; Casanovas-Company, J.; Castro, F.; Popova, B. Enhanced CAR T cell expansion and prolonged persistence in pediatric patients with ALL treated with a low-affinity CD19 CAR. Nat. Med. 2019, 25, 1408–1414. [Google Scholar] [CrossRef] [PubMed]
- DiTommaso, T.; Cole, J.M.; Cassereau, L.; Buggé, J.A.; Hanson, J.L.S.; Bridgen, D.T.; Stokes, B.D.; Loughhead, S.M.; Beutel, B.A.; Gilbert, J.B. Cell engineering with microfluidic squeezing preserves functionality of primary immune cells in vivo. Proc. Natl. Acad. Sci. USA 2018, 115, E10907–E10914. [Google Scholar] [CrossRef]
- Mamonkin, M.; Mukherjee, M.; Srinivasan, M.; Sharma, S.; Gomes-Silva, D.; Mo, F.; Krenciute, G.; Orange, J.S.; Brenner, M.K. Reversible transgene expression reduces fratricide and permits 4-1BB costimulation of CAR T cells directed to T-cell malignancies. Cancer Immunol. Res. 2018, 6, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Depil, S.; Duchateau, P.; Grupp, S.; Mufti, G.; Poirot, L. ‘Off-the-shelf’allogeneic CAR T cells: Development and challenges. Nat. Rev. Drug Discov. 2020, 19, 185–199. [Google Scholar] [CrossRef]
- Kundu, S.; Gurney, M.; O’Dwyer, M. Generating natural killer cells for adoptive transfer: Expanding horizons. Cytotherapy 2021, 23, 559–566. [Google Scholar] [CrossRef]
- Stoiber, S.; Cadilha, B.L.; Benmebarek, M.-R.; Lesch, S.; Endres, S.; Kobold, S. Limitations in the design of chimeric antigen receptors for cancer therapy. Cells 2019, 8, 472. [Google Scholar] [CrossRef]
- Fujiwara, K.; Masutani, M.; Tachibana, M.; Okada, N. Impact of scFv structure in chimeric antigen receptor on receptor expression efficiency and antigen recognition properties. Biochem. Biophys. Res. Commun. 2020, 527, 350–357. [Google Scholar] [CrossRef] [PubMed]
- Genßler, S.; Burger, M.C.; Zhang, C.; Oelsner, S.; Mildenberger, I.; Wagner, M.; Steinbach, J.P.; Wels, W.S. Dual targeting of glioblastoma with chimeric antigen receptor-engineered natural killer cells overcomes heterogeneity of target antigen expression and enhances antitumor activity and survival. Oncoimmunology 2016, 5, e1119354. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; An, X.; Brown, R.D.; Ho, J.; Gibson, J.; Joshua, D.E.; Wels, W.; Sze, D.M. Development of Retargeted CD38-Specific NK-92 Cell Line for Potential Anti-Myeloma Immunotherapy. Am. Soc. Hematol. 2005, 106, 5104. [Google Scholar] [CrossRef]
- Chowdhury, P.S.; Vasmatzis, G. Engineering scFvs for improved stability. In Recombinant Antibodies for Cancer Therapy; Springer: Berlin/Heidelberg, Germany, 2003; pp. 237–254. [Google Scholar]
- Landoni, E.; Fucá, G.; Wang, J.; Chirasani, V.R.; Yao, Z.; Dukhovlinova, E.; Ferrone, S.; Savoldo, B.; Hong, L.K.; Shou, P. Modifications to the Framework Regions Eliminate Chimeric Antigen Receptor Tonic Signaling. Cancer Immunol. Res. 2021, 9, 441–453. [Google Scholar] [CrossRef]
- Alabanza, L.; Pegues, M.; Geldres, C.; Shi, V.; Wiltzius, J.J.; Sievers, S.A.; Yang, S.; Kochenderfer, J.N. Function of novel anti-CD19 chimeric antigen receptors with human variable regions is affected by hinge and transmembrane domains. Mol. Ther. 2017, 25, 2452–2465. [Google Scholar] [CrossRef]
- Kulemzin, S.V.; Matvienko, D.A.; Sabirov, A.H.; Sokratyan, A.M.; Chernikova, D.S.; Belovezhets, T.N.; Chikaev, A.N.; Taranin, A.V.; Gorchakov, A.A. Design and analysis of stably integrated reporters for inducible transgene expression in human T cells and CAR NK-cell lines. BMC Med. Genom. 2019, 12, 87–95. [Google Scholar] [CrossRef]
- Oelsner, S.; Friede, M.E.; Zhang, C.; Wagner, J.; Badura, S.; Bader, P.; Ullrich, E.; Ottmann, O.G.; Klingemann, H.; Tonn, T. Continuously expanding CAR NK-92 cells display selective cytotoxicity against B-cell leukemia and lymphoma. Cytotherapy 2017, 19, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Töpfer, K.; Cartellieri, M.; Michen, S.; Wiedemuth, R.; Müller, N.; Lindemann, D.; Bachmann, M.; Füssel, M.; Schackert, G.; Temme, A. DAP12-based activating chimeric antigen receptor for NK cell tumor immunotherapy. J. Immunol. 2015, 194, 3201–3212. [Google Scholar] [CrossRef]
- Watanabe, N.; Bajgain, P.; Sukumaran, S.; Ansari, S.; Heslop, H.E.; Rooney, C.M.; Brenner, M.K.; Leen, A.M.; Vera, J.F. Fine-tuning the CAR spacer improves T-cell potency. Oncoimmunology 2016, 5, e1253656. [Google Scholar] [CrossRef]
- Altvater, B.; Landmeier, S.; Pscherer, S.; Temme, J.; Schweer, K.; Kailayangiri, S.; Campana, D.; Juergens, H.; Pule, M.; Rossig, C. 2B4 (CD244) signaling by recombinant antigen-specific chimeric receptors costimulates natural killer cell activation to leukemia and neuroblastoma cells. Clin. Cancer Res. 2009, 15, 4857–4866. [Google Scholar] [CrossRef]
- Zhang, G.; Liu, R.; Zhu, X.; Wang, L.; Ma, J.; Han, H.; Wang, X.; Zhang, G.; He, W.; Wang, W. Retargeting NK-92 for anti-melanoma activity by a TCR-like single-domain antibody. Immunol. Cell Biol. 2013, 91, 615–624. [Google Scholar] [CrossRef]
- Müller, T.; Uherek, C.; Maki, G.; Chow, K.U.; Schimpf, A.; Klingemann, H.-G.; Tonn, T.; Wels, W.S. Expression of a CD20-specific chimeric antigen receptor enhances cytotoxic activity of NK cells and overcomes NK-resistance of lymphoma and leukemia cells. Cancer Immunol. Immunother. 2008, 57, 411–423. [Google Scholar] [CrossRef]
- Li, Y.; Hermanson, D.L.; Moriarity, B.S.; Kaufman, D.S. Human iPSC-derived natural killer cells engineered with chimeric antigen receptors enhance anti-tumor activity. Cell Stem Cell 2018, 23, 181–192.e5. [Google Scholar] [CrossRef]
- Kruschinski, A.; Moosmann, A.; Poschke, I.; Norell, H.; Chmielewski, M.; Seliger, B.; Kiessling, R.; Blankenstein, T.; Abken, H.; Charo, J. Engineering antigen-specific primary human NK cells against HER-2 positive carcinomas. Proc. Natl. Acad. Sci. USA 2008, 105, 17481–17486. [Google Scholar] [CrossRef]
- Fujiwara, K.; Tsunei, A.; Kusabuka, H.; Ogaki, E.; Tachibana, M.; Okada, N. Hinge and transmembrane domains of chimeric antigen receptor regulate receptor expression and signaling threshold. Cells 2020, 9, 1182. [Google Scholar] [CrossRef]
- Kasahara, Y.; Shin, C.; Kubo, N.; Mihara, K.; Iwabuchi, H.; Takachi, T.; Imamura, M.; Saitoh, A.; Imai, C. Development and characterisation of NKp44-based chimeric antigen receptors that confer T cells with NK cell-like specificity. Clin. Transl. Immunol. 2020, 9, e1147. [Google Scholar] [CrossRef]
- Han, J.; Chu, J.; Chan, W.K.; Zhang, J.; Wang, Y.; Cohen, J.B.; Victor, A.; Meisen, W.H.; Kim, S.-h.; Grandi, P. CAR-engineered NK cells targeting wild-type EGFR and EGFRvIII enhance killing of glioblastoma and patient-derived glioblastoma stem cells. Sci. Rep. 2015, 5, 1–13. [Google Scholar] [CrossRef]
- Imai, C.; Iwamoto, S.; Campana, D. Genetic modification of primary natural killer cells overcomes inhibitory signals and induces specific killing of leukemic cells. Blood 2005, 106, 376–383. [Google Scholar] [CrossRef] [PubMed]
- Romanski, A.; Uherek, C.; Bug, G.; Seifried, E.; Klingemann, H.; Wels, W.S.; Ottmann, O.G.; Tonn, T. CD 19-CAR engineered NK-92 cells are sufficient to overcome NK cell resistance in B-cell malignancies. J. Cell. Mol. Med. 2016, 20, 1287–1294. [Google Scholar] [CrossRef]
- Xu, D.; Jin, G.; Chai, D.; Zhou, X.; Gu, W.; Chong, Y.; Song, J.; Zheng, J. The development of CAR design for tumor CAR-T cell therapy. Oncotarget 2018, 9, 13991. [Google Scholar] [CrossRef]
- Savoldo, B.; Rooney, C.M.; Di Stasi, A.; Abken, H.; Hombach, A.; Foster, A.E.; Zhang, L.; Heslop, H.E.; Brenner, M.K.; Dotti, G. Epstein Barr virus–specific cytotoxic T lymphocytes expressing the anti-CD30ζ artificial chimeric T-cell receptor for immunotherapy of Hodgkin disease. Blood J. Am. Soc. Hematol. 2007, 110, 2620–2630. [Google Scholar] [CrossRef]
- Long, A.H.; Haso, W.M.; Shern, J.F.; Wanhainen, K.M.; Murgai, M.; Ingaramo, M.; Smith, J.P.; Walker, A.J.; Kohler, M.E.; Venkateshwara, V.R. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat. Med. 2015, 21, 581–590. [Google Scholar] [CrossRef]
- Xu, Y.; Liu, Q.; Zhong, M.; Wang, Z.; Chen, Z.; Zhang, Y.; Xing, H.; Tian, Z.; Tang, K.; Liao, X. 2B4 costimulatory domain enhancing cytotoxic ability of anti-CD5 chimeric antigen receptor engineered natural killer cells against T cell malignancies. J. Hematol. Oncol. 2019, 12, 1–13. [Google Scholar] [CrossRef]
- Upshaw, J.L.; Arneson, L.N.; Schoon, R.A.; Dick, C.J.; Billadeau, D.D.; Leibson, P.J. NKG2D-mediated signaling requires a DAP10-bound Grb2-Vav1 intermediate and phosphatidylinositol-3-kinase in human natural killer cells. Nat. Immunol. 2006, 7, 524–532. [Google Scholar] [CrossRef]
- Chang, Y.-H.; Connolly, J.; Shimasaki, N.; Mimura, K.; Kono, K.; Campana, D. A chimeric receptor with NKG2D specificity enhances natural killer cell activation and killing of tumor cells. Cancer Res. 2013, 73, 1777–1786. [Google Scholar] [CrossRef]
- Huang, Y.; Zeng, J.; Liu, T.; Xu, Q.; Song, X.; Zeng, J. DNAM1 and 2B4 costimulatory domains enhance the cytotoxicity of anti-GPC3 chimeric antigen receptor-modified natural killer cells against hepatocellular cancer cells in vitro. Cancer Manag. Res. 2020, 12, 3247. [Google Scholar] [CrossRef]
- Sordo-Bahamonde, C.; Vitale, M.; Lorenzo-Herrero, S.; López-Soto, A.; Gonzalez, S. Mechanisms of resistance to NK cell immunotherapy. Cancers 2020, 12, 893. [Google Scholar] [CrossRef]
- Tahmasebi, S.; Elahi, R.; Esmaeilzadeh, A. Solid tumors challenges and new insights of CAR T cell engineering. Stem Cell Rev. Rep. 2019, 15, 619–636. [Google Scholar] [CrossRef]
- Leko, V.; Rosenberg, S.A. Identifying and targeting human tumor antigens for T cell-based immunotherapy of solid tumors. Cancer Cell 2020, 38. [Google Scholar] [CrossRef]
- Jo, Y.; Ali, L.A.; Shim, J.A.; Lee, B.H.; Hong, C. Innovative CAR-T cell therapy for solid tumor; current duel between CAR-T spear and tumor shield. Cancers 2020, 12, 2087. [Google Scholar] [CrossRef]
- Liu, Y.; Zhou, Y.; Huang, K.H.; Fang, X.; Li, Y.; Wang, F.; An, L.; Chen, Q.; Zhang, Y.; Shi, A. Targeting epidermal growth factor-overexpressing triple-negative breast cancer by natural killer cells expressing a specific chimeric antigen receptor. Cell Prolif. 2020, 53, e12858. [Google Scholar] [CrossRef]
- Zhang, C.; Burger, M.C.; Jennewein, L.; Genßler, S.; Schönfeld, K.; Zeiner, P.; Hattingen, E.; Harter, P.N.; Mittelbronn, M.; Tonn, T. ErbB2/HER2-specific NK cells for targeted therapy of glioblastoma. J. Natl. Cancer Inst. 2016, 108, djv375. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhang, H.; Ding, J.; Liu, H.; Li, H.; Li, H.; Lu, M.; Miao, Y.; Li, L.; Zheng, J. Combination therapy with EpCAM-CAR-NK-92 cells and regorafenib against human colorectal cancer models. J. Immunol. Res. 2018, 2018, 4263520. [Google Scholar] [CrossRef]
- Kailayangiri, S.; Altvater, B.; Spurny, C.; Jamitzky, S.; Schelhaas, S.; Jacobs, A.H.; Wiek, C.; Roellecke, K.; Hanenberg, H.; Hartmann, W. Targeting Ewing sarcoma with activated and GD2-specific chimeric antigen receptor-engineered human NK cells induces upregulation of immune-inhibitory HLA-G. Oncoimmunology 2017, 6, e1250050. [Google Scholar] [CrossRef] [PubMed]
- Cao, B.; Liu, M.; Wang, L.; Liang, B.; Feng, Y.; Chen, X.; Shi, Y.; Zhang, J.; Ye, X.; Tian, Y. Use of chimeric antigen receptor NK-92 cells to target mesothelin in ovarian cancer. Biochem. Biophys. Res. Commun. 2020, 524, 96–102. [Google Scholar] [CrossRef]
- Tang, X.; Yang, L.; Li, Z.; Nalin, A.P.; Dai, H.; Xu, T.; Yin, J.; You, F.; Zhu, M.; Shen, W. First-in-man clinical trial of CAR NK-92 cells: Safety test of CD33-CAR NK-92 cells in patients with relapsed and refractory acute myeloid leukemia. Am. J. Cancer Res. 2018, 8, 1083. [Google Scholar]
- Morgan, M.A.; Kloos, A.; Lenz, D.; Kattre, N.; Nowak, J.; Bentele, M.; Keisker, M.; Dahlke, J.; Zimmermann, K.; Sauer, M. Improved Activity against Acute Myeloid Leukemia with Chimeric Antigen Receptor (CAR)-NK-92 Cells Designed to Target CD123. Viruses 2021, 13, 1365. [Google Scholar] [CrossRef] [PubMed]
- Chu, J.; Deng, Y.; Benson, D.M.; He, S.; Hughes, T.; Zhang, J.; Peng, Y.; Mao, H.; Yi, L.; Ghoshal, K. CS1-specific chimeric antigen receptor (CAR)-engineered natural killer cells enhance in vitro and in vivo antitumor activity against human multiple myeloma. Leukemia 2014, 28, 917–927. [Google Scholar] [CrossRef]
- You, F.; Wang, Y.; Jiang, L.; Zhu, X.; Chen, D.; Yuan, L.; An, G.; Meng, H.; Yang, L. A novel CD7 chimeric antigen receptor-modified NK-92MI cell line targeting T-cell acute lymphoblastic leukemia. Am. J. Cancer Res. 2019, 9, 64. [Google Scholar] [PubMed]
- Montagner, I.M.; Penna, A.; Fracasso, G.; Carpanese, D.; Dalla Pietà, A.; Barbieri, V.; Zuccolotto, G.; Rosato, A. Anti-PSMA CAR-engineered NK-92 cells: An off-the-shelf cell therapy for prostate cancer. Cells 2020, 9, 1382. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Yang, N.; Li, H.; Wang, Z. Robo1-specific chimeric antigen receptor natural killer cell therapy for pancreatic ductal adenocarcinoma with liver metastasis. J. Cancer Res. Ther. 2020, 16, 393. [Google Scholar] [PubMed]
- Ng, Y.Y.; Du, Z.; Zhang, X.; Chng, W.J.; Wang, S. CXCR4 and anti-BCMA CAR co-modified natural killer cells suppress multiple myeloma progression in a xenograft mouse model. Cancer Gene Ther. 2021, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Chu, Y.; Yahr, A.; Huang, B.; Ayello, J.; Barth, M.; Cairo, M.S. Romidepsin alone or in combination with anti-CD20 chimeric antigen receptor expanded natural killer cells targeting Burkitt lymphoma in vitro and in immunodeficient mice. Oncoimmunology 2017, 6, e1341031. [Google Scholar] [CrossRef]
- Matvienko, D.A.; Kulemzin, S.V.; Smagina, A.S.; Belovezhets, T.N.; Chikaev, A.N.; Volkova, O.Y.; Chikaev, N.A.; Koval, O.A.; Kuligina, E.V.; Taranin, A.V. Analysis of in vitro activity of PSCA-specific CARs in the context of human NK cell line YT. Cell Ther. Transplant. 2018, 7, 70–76. [Google Scholar] [CrossRef]
- Leivas, A.; Valeri, A.; Córdoba, L.; García-Ortiz, A.; Ortiz, A.; Sánchez-Vega, L.; Graña-Castro, O.; Fernández, L.; Carreño-Tarragona, G.; Pérez, M. NKG2D-CAR-transduced natural killer cells efficiently target multiple myeloma. Blood Cancer J. 2021, 11, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Lin, L.; Luo, Y.; Cai, Q.; Jiang, X.; Liao, C.; Wei, H. Optimization of GPC3-specific chimeric antigen receptor structure and its effect on killing hepatocellular carcinoma cells. Bioengineered 2021, 12, 3674–3683. [Google Scholar] [CrossRef] [PubMed]
- Ao, X.; Yang, Y.; Li, W.; Tan, Y.; Guo, W.; Ao, L.; He, X.; Wu, X.; Xia, J.; Xu, X. Anti-αFR CAR-engineered NK-92 cells display potent cytotoxicity against αFR-positive ovarian cancer. J. Immunother. 2019, 42, 284. [Google Scholar] [CrossRef]
- Grote, S.; Chan, K.C.H.; Baden, C.; Bösmüller, H.; Sulyok, M.; Frauenfeld, L.; Ebinger, M.; Handgretinger, R.; Schleicher, S. CD276 as a novel CAR NK-92 therapeutic target for neuroblastoma. Adv. Cell Gene Ther. 2021, 4, e105. [Google Scholar] [CrossRef]
- Oelsner, S.; Waldmann, A.; Billmeier, A.; Röder, J.; Lindner, A.; Ullrich, E.; Marschalek, R.; Dotti, G.; Jung, G.; Große-Hovest, L. Genetically engineered CAR NK cells display selective cytotoxicity against FLT3-positive B-ALL and inhibit in vivo leukemia growth. Int. J. Cancer 2019, 145, 1935–1945. [Google Scholar] [CrossRef]
- Robbins, Y.; Greene, S.; Friedman, J.; Clavijo, P.E.; Van Waes, C.; Fabian, K.P.; Padget, M.R.; Sater, H.A.; Lee, J.H.; Soon-Shiong, P. Tumor control via targeting PD-L1 with chimeric antigen receptor modified NK cells. Elife 2020, 9, e54854. [Google Scholar] [CrossRef]
- Hu, Z. Tissue factor as a new target for CAR-NK cell immunotherapy of triple-negative breast cancer. Sci. Rep. 2020, 10, 1–13. [Google Scholar] [CrossRef]
- Stikvoort, A.; van der Schans, J.; Sarkar, S.; Poels, R.; Ruiter, R.; Naik, J.; Yuan, H.; de Bruijn, J.D.; van de Donk, N.W.; Zweegman, S. CD38-specific Chimeric Antigen Receptor Expressing Natural Killer KHYG-1 Cells: A Proof of Concept for an “Off the Shelf” Therapy for Multiple Myeloma. HemaSphere 2021, 5, e596. [Google Scholar] [CrossRef]
- Shiozawa, M.; Chang, C.-H.; Huang, Y.-C.; Chen, Y.-C.; Chi, M.-S.; Hao, H.-C.; Chang, Y.-C.; Takeda, S.; Chi, K.-H.; Wang, Y.-S. Pharmacologically upregulated carcinoembryonic antigen-expression enhances the cytolytic activity of genetically-modified chimeric antigen receptor NK-92MI against colorectal cancer cells. BMC Immunol. 2018, 19, 1–13. [Google Scholar] [CrossRef]
- Tseng, H.-c.; Xiong, W.; Badeti, S.; Yang, Y.; Ma, M.; Liu, T.; Ramos, C.A.; Dotti, G.; Fritzky, L.; Jiang, J.-g. Efficacy of anti-CD147 chimeric antigen receptors targeting hepatocellular carcinoma. Nat. Commun. 2020, 11, 1–15. [Google Scholar] [CrossRef]
- Zhao, Q.; Ahmed, M.; Tassev, D.V.; Hasan, A.; Kuo, T.-Y.; Guo, H.; O’Reilly, R.J.; Cheung, N.V. Affinity maturation of T-cell receptor-like antibodies for Wilms tumor 1 peptide greatly enhances therapeutic potential. Leukemia 2015, 29, 2238–2247. [Google Scholar] [CrossRef]
- Salman, H.; Pinz, K.G.; Wada, M.; Shuai, X.; Yan, L.E.; Petrov, J.C.; Ma, Y. Preclinical targeting of human acute myeloid leukemia using CD4-specific chimeric antigen receptor (CAR) T cells and NK cells. J. Cancer 2019, 10, 4408. [Google Scholar] [CrossRef]
- Liu, B.; Liu, Z.Z.; Zhou, M.L.; Lin, J.W.; Chen, X.M.; Li, Z.; Gao, W.B.; Yu, Z.D.; Liu, T. Development of c-MET-specific chimeric antigen receptor-engineered natural killer cells with cytotoxic effects on human liver cancer HepG2 cells. Mol. Med. Rep. 2019, 20, 2823–2831. [Google Scholar] [CrossRef]
- Luanpitpong, S.; Poohadsuan, J.; Klaihmon, P.; Issaragrisil, S. Selective Cytotoxicity of Single and Dual Anti-CD19 and Anti-CD138 Chimeric Antigen Receptor-Natural Killer Cells against Hematologic Malignancies. J. Immunol. Res. 2021, 2021. [Google Scholar] [CrossRef]
- Chen, K.H.; Wada, M.; Firor, A.E.; Pinz, K.G.; Jares, A.; Liu, H.; Salman, H.; Golightly, M.; Lan, F.; Jiang, X. Novel anti-CD3 chimeric antigen receptor targeting of aggressive T cell malignancies. Oncotarget 2016, 7, 56219. [Google Scholar] [CrossRef]
- Maude, S.L.; Laetsch, T.W.; Buechner, J.; Rives, S.; Boyer, M.; Bittencourt, H.; Bader, P.; Verneris, M.R.; Stefanski, H.E.; Myers, G.D. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N. Engl. J. Med. 2018, 378, 439–448. [Google Scholar] [CrossRef]
- Ravi, D.; Sarkar, S.; Purvey, S.; Passero, F.; Beheshti, A.; Chen, Y.; Mokhtar, M.; David, K.; Konry, T.; Evens, A.M. Interaction kinetics with transcriptomic and secretory responses of CD19-CAR natural killer-cell therapy in CD20 resistant non-hodgkin lymphoma. Leukemia 2020, 34, 1291–1304. [Google Scholar] [CrossRef]
- Braendstrup, P.; Levine, B.L.; Ruella, M. The long road to the first FDA-approved gene therapy: Chimeric antigen receptor T cells targeting CD19. Cytotherapy 2020, 22, 57–69. [Google Scholar] [CrossRef]
- Xu, X.; Sun, Q.; Liang, X.; Chen, Z.; Zhang, X.; Zhou, X.; Li, M.; Tu, H.; Liu, Y.; Tu, S. Mechanisms of relapse after CD19 CAR T-cell therapy for acute lymphoblastic leukemia and its prevention and treatment strategies. Front. Immunol. 2019, 10, 2664. [Google Scholar] [CrossRef] [PubMed]
- Spiegel, J.Y.; Patel, S.; Muffly, L.; Hossain, N.M.; Oak, J.; Baird, J.H.; Frank, M.J.; Shiraz, P.; Sahaf, B.; Craig, J. CAR T cells with dual targeting of CD19 and CD22 in adult patients with recurrent or refractory B cell malignancies: A phase 1 trial. Nat. Med. 2021, 27, 1419–1431. [Google Scholar] [CrossRef]
- Ruella, M.; Barrett, D.M.; Kenderian, S.S.; Shestova, O.; Hofmann, T.J.; Perazzelli, J.; Klichinsky, M.; Aikawa, V.; Nazimuddin, F.; Kozlowski, M. Dual CD19 and CD123 targeting prevents antigen-loss relapses after CD19-directed immunotherapies. J. Clin. Investig. 2016, 126, 3814–3826. [Google Scholar] [CrossRef]
- Ko, A.H.; Jordan, A.C.; Tooker, E.; Lacey, S.F.; Chang, R.B.; Li, Y.; Venook, A.P.; Tempero, M.; Damon, L.; Fong, L. Dual targeting of mesothelin and CD19 with chimeric antigen receptor-modified T cells in patients with metastatic pancreatic cancer. Mol. Ther. 2020, 28, 2367–2378. [Google Scholar] [CrossRef] [PubMed]
- Fousek, K.; Watanabe, J.; George, A.; An, X.; Samaha, H.; Navai, S.A.; Byrd, T.T.; Jang, A.; Kim, H.N.; Joseph, S. Targeting primary pre-B cell acute lymphoblastic leukemia and CD19-negative relapses using trivalent CAR T cells. Blood 2017, 130, 4614. [Google Scholar]
- Liu, E.; Tong, Y.; Dotti, G.; Shaim, H.; Savoldo, B.; Mukherjee, M.; Orange, J.; Wan, X.; Lu, X.; Reynolds, A. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia 2018, 32, 520–531. [Google Scholar] [CrossRef] [PubMed]
- Müller, S.; Bexte, T.; Gebel, V.; Kalensee, F.; Stolzenberg, E.; Hartmann, J.; Koehl, U.; Schambach, A.; Wels, W.S.; Modlich, U. High cytotoxic efficiency of lentivirally and alpharetrovirally engineered CD19-specific chimeric antigen receptor natural killer cells against acute lymphoblastic leukemia. Front. Immunol. 2020, 10, 3123. [Google Scholar] [CrossRef] [PubMed]
- Mendelsohn, J.; Baselga, J. Epidermal growth factor receptor targeting in cancer. Semin. Oncol. 2006, 33, 369–385. [Google Scholar] [CrossRef]
- Shah, R.R.; Shah, D.R. Safety and tolerability of epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors in oncology. Drug Saf. 2019, 42, 181–198. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, Z.; Ding, Y.; Fang, Y.; Wang, P.; Chu, W.; Jin, Z.; Yang, X.; Wang, J.; Lou, J. Phase I clinical trial of EGFR-specific CAR-T cells generated by the piggyBac transposon system in advanced relapsed/refractory non-small cell lung cancer patients. J. Cancer Res. Clin. Oncol. 2021, 147, 3725–3734. [Google Scholar] [CrossRef]
- Chen, X.; Han, J.; Chu, J.; Zhang, L.; Zhang, J.; Chen, C.; Chen, L.; Wang, Y.; Wang, H.; Yi, L. A combinational therapy of EGFR-CAR NK cells and oncolytic herpes simplex virus 1 for breast cancer brain metastases. Oncotarget 2016, 7, 27764. [Google Scholar] [CrossRef]
- Zhang, Q.; Tian, K.; Xu, J.; Zhang, H.; Li, L.; Fu, Q.; Chai, D.; Li, H.; Zheng, J. Synergistic effects of cabozantinib and EGFR-specific CAR-NK-92 cells in renal cell carcinoma. J. Immunol. Res. 2017, 2017, 6915912. [Google Scholar] [CrossRef] [PubMed]
- Ma, R.; Lu, T.; Li, Z.; Teng, K.-Y.; Mansour, A.G.; Yu, M.; Tian, L.; Xu, B.; Ma, S.; Zhang, J. An oncolytic virus expressing IL-15/IL-15Rα combined with off-the-shelf EGFR-CAR NK cells targets glioblastoma. Cancer Res. 2021, 81, 3635–3648. [Google Scholar] [CrossRef]
- Murakami, T.; Nakazawa, T.; Natsume, A.; Nishimura, F.; Nakamura, M.; Matsuda, R.; Omoto, K.; Tanaka, Y.; Shida, Y.; Park, Y.-S. Novel human NK cell line carrying CAR targeting EGFRvIII induces antitumor effects in glioblastoma cells. Anticancer Res. 2018, 38, 5049–5056. [Google Scholar] [CrossRef]
- O’Rourke, D.M.; Nasrallah, M.P.; Desai, A.; Melenhorst, J.J.; Mansfield, K.; Morrissette, J.J.; Martinez-Lage, M.; Brem, S.; Maloney, E.; Shen, A. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 2017, 9, eaaa0984. [Google Scholar] [CrossRef]
- Li, K.; Qian, S.; Huang, M.; Chen, M.; Peng, L.; Liu, J.; Xu, W.; Xu, J. Development of GPC3 and EGFR-dual-targeting chimeric antigen receptor-T cells for adoptive T cell therapy. Am. J. Transl. Res. 2021, 13, 156. [Google Scholar]
- Nakazawa, T.; Natsume, A.; Nishimura, F.; Morimoto, T.; Matsuda, R.; Nakamura, M.; Yamada, S.; Nakagawa, I.; Motoyama, Y.; Park, Y.-S. Effect of CRISPR/Cas9-mediated PD-1-disrupted primary human third-generation CAR-T cells targeting EGFRvIII on in vitro human glioblastoma cell growth. Cells 2020, 9, 998. [Google Scholar] [CrossRef] [PubMed]
- Xia, L.; Zheng, Z.; Liu, J.-y.; Chen, Y.-j.; Ding, J.; Hu, G.-s.; Hu, Y.-h.; Liu, S.; Luo, W.-x.; Xia, N.-s. Targeting Triple-Negative Breast Cancer with Combination Therapy of EGFR CAR T Cells and CDK7 Inhibition. Cancer Immunol. Res. 2021, 9, 707–722. [Google Scholar] [CrossRef]
- Meric-Bernstam, F.; Johnson, A.M.; Dumbrava, E.E.I.; Raghav, K.; Balaji, K.; Bhatt, M.; Murthy, R.K.; Rodon, J.; Piha-Paul, S.A. Advances in HER2-targeted therapy: Novel agents and opportunities beyond breast and gastric cancer. Clin. Cancer Res. 2019, 25, 2033–2041. [Google Scholar] [CrossRef]
- Ni, J.; Zhang, L. Progress in Treatment of Non-Small Cell Lung Cancer Harboring HER2 Aberrations. OncoTargets Ther. 2021, 14, 4087. [Google Scholar] [CrossRef]
- Scaltriti, M.; Rojo, F.; Ocaña, A.; Anido, J.; Guzman, M.; Cortes, J.; Di Cosimo, S.; Matias-Guiu, X.; Ramon y Cajal, S.; Arribas, J. Expression of p95HER2, a truncated form of the HER2 receptor, and response to anti-HER2 therapies in breast cancer. J. Natl. Cancer Inst. 2007, 99, 628–638. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, N.; Shi, H. Driving better and safer HER2-specific CARs for cancer therapy. Oncotarget 2017, 8, 62730. [Google Scholar] [CrossRef]
- Dreyer, T.F.; Kuhn, S.; Stange, C.; Heithorst, N.; Schilling, D.; Jelsma, J.; Sievert, W.; Seitz, S.; Stangl, S.; Hapfelmeier, A. The Chemokine CX3CL1 Improves Trastuzumab Efficacy in HER2 Low–Expressing Cancer In Vitro and In Vivo. Cancer Immunol. Res. 2021, 9, 779–789. [Google Scholar] [CrossRef]
- Szöőr, Á.; Tóth, G.; Zsebik, B.; Szabó, V.; Eshhar, Z.; Abken, H.; Vereb, G. Trastuzumab derived HER2-specific CARs for the treatment of trastuzumab-resistant breast cancer: CAR T cells penetrate and eradicate tumors that are not accessible to antibodies. Cancer Lett. 2020, 484, 1–8. [Google Scholar] [CrossRef]
- Portillo, A.L.; Hogg, R.; Poznanski, S.M.; Rojas, E.A.; Cashell, N.J.; Hammill, J.A.; Chew, M.V.; Shenouda, M.M.; Ritchie, T.M.; Cao, Q.T. Expanded human NK cells armed with CAR uncouple potent anti-tumor activity from off-tumor toxicity against solid tumors. Iscience 2021, 24, 102619. [Google Scholar] [CrossRef]
- Gossel, L.D.; Heim, C.; Pfeffermann, L.-M.; Moser, L.M.; Bönig, H.B.; Klingebiel, T.E.; Bader, P.; Wels, W.S.; Merker, M.; Rettinger, E. Retargeting of NK-92 Cells against High-Risk Rhabdomyosarcomas by Means of an ERBB2 (HER2/Neu)-Specific Chimeric Antigen Receptor. Cancers 2021, 13, 1443. [Google Scholar] [CrossRef]
- Wilkie, S.; van Schalkwyk, M.C.; Hobbs, S.; Davies, D.M.; van der Stegen, S.J.; Pereira, A.C.P.; Burbridge, S.E.; Box, C.; Eccles, S.A.; Maher, J. Dual targeting of ErbB2 and MUC1 in breast cancer using chimeric antigen receptors engineered to provide complementary signaling. J. Clin. Immunol. 2012, 32, 1059–1070. [Google Scholar] [CrossRef]
- Shaw, A.R.; Porter, C.E.; Watanabe, N.; Tanoue, K.; Sikora, A.; Gottschalk, S.; Brenner, M.K.; Suzuki, M. Adenovirotherapy delivering cytokine and checkpoint inhibitor augments CAR T cells against metastatic head and neck cancer. Mol. Ther. 2017, 25, 2440–2451. [Google Scholar] [CrossRef]
- Gires, O.; Pan, M.; Schinke, H.; Canis, M.; Baeuerle, P.A. Expression and function of epithelial cell adhesion molecule EpCAM: Where are we after 40 years? Cancer Metastasis Rev. 2020, 39, 969–987. [Google Scholar] [CrossRef]
- Mohtar, M.A.; Syafruddin, S.E.; Nasir, S.N.; Low, T.Y. Revisiting the roles of pro-metastatic EpCAM in cancer. Biomolecules 2020, 10, 255. [Google Scholar] [CrossRef]
- Münz, M.; Murr, A.; Kvesic, M.; Rau, D.; Mangold, S.; Pflanz, S.; Lumsden, J.; Volkland, J.; Fagerberg, J.; Riethmüller, G. Side-by-side analysis of five clinically tested anti-EpCAM monoclonal antibodies. Cancer Cell Int. 2010, 10, 1–12. [Google Scholar] [CrossRef]
- Sahm, C.; Schönfeld, K.; Wels, W.S. Expression of IL-15 in NK cells results in rapid enrichment and selective cytotoxicity of gene-modified effectors that carry a tumor-specific antigen receptor. Cancer Immunol. Immunother. 2012, 61, 1451–1461. [Google Scholar] [CrossRef]
- Zhou, Y.; Wen, P.; Li, M.; Li, Y.; Li, X.A. Construction of chimeric antigen receptor-modified T cells targeting EpCAM and assessment of their anti-tumor effect on cancer cells. Mol. Med. Rep. 2019, 20, 2355–2364. [Google Scholar] [CrossRef]
- Qin, D.; Li, D.; Zhang, B.; Chen, Y.; Liao, X.; Li, X.; Alexander, P.B.; Wang, Y.; Li, Q.-J. Potential lung attack and lethality generated by EpCAM-specific CAR-T cells in immunocompetent mouse models. Oncoimmunology 2020, 9, 1806009. [Google Scholar] [CrossRef]
- Nazha, B.; Inal, C.; Owonikoko, T.K. Disialoganglioside GD2 expression in solid tumors and role as a target for cancer therapy. Front. Oncol. 2020, 10, 1000. [Google Scholar] [CrossRef] [PubMed]
- Cavdarli, S.; Groux-Degroote, S.; Delannoy, P. Gangliosides: The double-edge sword of neuro-ectodermal derived tumors. Biomolecules 2019, 9, 311. [Google Scholar] [CrossRef] [PubMed]
- Barry, W.E.; Jackson, J.R.; Asuelime, G.E.; Wu, H.-W.; Sun, J.; Wan, Z.; Malvar, J.; Sheard, M.A.; Wang, L.; Seeger, R.C. Activated natural killer cells in combination with anti-GD2 antibody dinutuximab improve survival of mice after surgical resection of primary neuroblastoma. Clin. Cancer Res. 2019, 25, 325–333. [Google Scholar] [CrossRef]
- Seitz, C.M.; Schroeder, S.; Knopf, P.; Krahl, A.-C.; Hau, J.; Schleicher, S.; Martella, M.; Quintanilla-Martinez, L.; Kneilling, M.; Pichler, B. GD2-targeted chimeric antigen receptor T cells prevent metastasis formation by elimination of breast cancer stem-like cells. Oncoimmunology 2020, 9, 1683345. [Google Scholar] [CrossRef]
- Esser, R.; Müller, T.; Stefes, D.; Kloess, S.; Seidel, D.; Gillies, S.D.; Aperlo-Iffland, C.; Huston, J.S.; Uherek, C.; Schönfeld, K. NK cells engineered to express a GD2-specific antigen receptor display built-in ADCC-like activity against tumour cells of neuroectodermal origin. J. Cell. Mol. Med. 2012, 16, 569–581. [Google Scholar] [CrossRef] [PubMed]
- Heczey, A.; Courtney, A.N.; Montalbano, A.; Robinson, S.; Liu, K.; Li, M.; Ghatwai, N.; Dakhova, O.; Liu, B.; Raveh-Sadka, T. Anti-GD2 CAR-NKT cells in patients with relapsed or refractory neuroblastoma: An interim analysis. Nat. Med. 2020, 26, 1686–1690. [Google Scholar] [CrossRef]
- Caforio, M.; Sorino, C.; Caruana, I.; Weber, G.; Camera, A.; Cifaldi, L.; De Angelis, B.; Del Bufalo, F.; Vitale, A.; Goffredo, B.M. GD2 redirected CAR T and activated NK-cell-mediated secretion of IFNγ overcomes MYCN-dependent IDO1 inhibition, contributing to neuroblastoma cell immune escape. J. Immunother. Cancer 2021, 9, e001502. [Google Scholar] [CrossRef]
- Xu, X.; Huang, W.; Heczey, A.; Liu, D.; Guo, L.; Wood, M.; Jin, J.; Courtney, A.N.; Liu, B.; Di Pierro, E.J. NKT cells coexpressing a GD2-specific chimeric antigen receptor and IL15 show enhanced in vivo persistence and antitumor activity against neuroblastoma. Clin. Cancer Res. 2019, 25, 7126–7138. [Google Scholar] [CrossRef]
- Klampatsa, A.; Dimou, V.; Albelda, S.M. Mesothelin-targeted CAR-T cell therapy for solid tumors. Expert Opin. Biol. Ther. 2021, 21, 473–486. [Google Scholar] [CrossRef]
- Le, Q.; Castro, S.; Tang, T.; Loeb, A.M.; Hylkema, T.; McKay, C.N.; Perkins, L.; Srivastava, S.; Call, L.; Smith, J. Therapeutic Targeting of Mesothelin with Chimeric Antigen Receptor T Cells in Acute Myeloid Leukemia. Clin. Cancer Res. 2021, 27, 5718–5730. [Google Scholar] [CrossRef]
- Lv, J.; Li, P. Mesothelin as a biomarker for targeted therapy. Biomark. Res. 2019, 7, 1–18. [Google Scholar] [CrossRef]
- Cao, B.; Liu, M.; Huang, J.; Zhou, J.; Li, J.; Lian, H.; Huang, W.; Guo, Y.; Yang, S.; Lin, L. Development of mesothelin-specific CAR NK-92 cells for the treatment of gastric cancer. Int. J. Biol. Sci. 2021, 17, 3850. [Google Scholar] [CrossRef]
- Batchu, R.B.; Gruzdyn, O.V.; Tavva, P.S.; Kolli, B.K.; Dachepalli, R.; Weaver, D.W.; Gruber, S.A. Engraftment of mesothelin chimeric antigen receptor using a hybrid Sleeping Beauty/minicircle vector into NK-92MI cells for treatment of pancreatic cancer. Surgery 2019, 166, 503–508. [Google Scholar] [CrossRef]
- Watanabe, K.; Luo, Y.; Da, T.; Guedan, S.; Ruella, M.; Scholler, J.; Keith, B.; Young, R.M.; Engels, B.; Sorsa, S. Pancreatic cancer therapy with combined mesothelin-redirected chimeric antigen receptor T cells and cytokine-armed oncolytic adenoviruses. JCI Insight 2018, 3, 499573. [Google Scholar] [CrossRef]
- Cinay, G.E. Cell-based cancer immunotherapy using chimeric antigen receptor (CAR)-engineered natural killer (NK) cells surface-modified with drug-loaded nanoparticles targeting adenosine receptors. Am. Assoc. Immnol. 2020, 204 (Suppl. S1), 170.10. [Google Scholar]
- Adusumilli, P.S.; Zauderer, M.G.; Rivière, I.; Solomon, S.B.; Rusch, V.W.; O’Cearbhaill, R.E.; Zhu, A.; Cheema, W.; Chintala, N.K.; Halton, E. A Phase I Trial of Regional Mesothelin-Targeted CAR T-cell Therapy in Patients with Malignant Pleural Disease, in Combination with the Anti–PD-1 Agent Pembrolizumab. Cancer Discov. 2021, 11, 2748–2763. [Google Scholar] [CrossRef]
- Vaupel, P.; Multhoff, G. Revisiting the Warburg effect: Historical dogma versus current understanding. J. Physiol. 2021, 599, 1745–1757. [Google Scholar] [CrossRef] [PubMed]
- Multhoff, G.; Botzler, C.; Wiesnet, M.; Müller, E.; Meier, T.; Wilmanns, W.; Issels, R.D. A stress-inducible 72-kDa heat-shock protein (HSP72) is expressed on the surface of human tumor cells, but not on normal cells. Int. J. Cancer 1995, 61, 272–279. [Google Scholar] [CrossRef]
- Gehrmann, M.; Liebisch, G.; Schmitz, G.; Anderson, R.; Steinem, C.; De Maio, A.; Pockley, G.; Multhoff, G. Tumor-specific Hsp70 plasma membrane localization is enabled by the glycosphingolipid Gb3. PLoS ONE 2008, 3, e1925. [Google Scholar] [CrossRef]
- Stangl, S.; Gehrmann, M.; Riegger, J.; Kuhs, K.; Riederer, I.; Sievert, W.; Hube, K.; Mocikat, R.; Dressel, R.; Kremmer, E. Targeting membrane heat-shock protein 70 (Hsp70) on tumors by cmHsp70. 1 antibody. Proc. Natl. Acad. Sci. USA 2011, 108, 733–738. [Google Scholar] [CrossRef] [PubMed]
- Botzler, C.; Schmidt, J.; Luz, A.; Jennen, L.; Issels, R.; Multhoff, G. Differential Hsp70 plasma-membrane expression on primary human tumors and metastases in mice with severe combined immunodeficiency. Int. J. Cancer 1998, 77, 942–948. [Google Scholar] [CrossRef]
- Multhoff, G.; Pockley, A.G.; Schmid, T.E.; Schilling, D. The role of heat shock protein 70 (Hsp70) in radiation-induced immunomodulation. Cancer Lett. 2015, 368, 179–184. [Google Scholar] [CrossRef]
- Moser, C.; Schmidbauer, C.; Gürtler, U.; Gross, C.; Gehrmann, M.; Thonigs, G.; Pfister, K.; Multhoff, G. Inhibition of tumor growth in mice with severe combined immunodeficiency is mediated by heat shock protein 70 (Hsp70)-peptide–activated, CD94 positive natural killer cells. Cell Stress Chaperones 2002, 7, 365. [Google Scholar] [CrossRef]
- Stangl, S.; Tei, L.; De Rose, F.; Reder, S.; Martinelli, J.; Sievert, W.; Shevtsov, M.; Öllinger, R.; Rad, R.; Schwaiger, M. Preclinical evaluation of the Hsp70 peptide tracer TPP-PEG24-DFO [89Zr] for tumor-specific PET/CT imaging. Cancer Res. 2018, 78, 6268–6281. [Google Scholar] [CrossRef]
- Kokowski, K.; Stangl, S.; Seier, S.; Hildebrandt, M.; Vaupel, P.; Multhoff, G. Radiochemotherapy combined with NK cell transfer followed by second-line PD-1 inhibition in a patient with NSCLC stage IIIb inducing long-term tumor control: A case study. Strahlenther. Und Onkol. 2019, 195, 352–361. [Google Scholar] [CrossRef] [PubMed]
- Krause, S.W.; Gastpar, R.; Andreesen, R.; Gross, C.; Ullrich, H.; Thonigs, G.; Pfister, K.; Multhoff, G. Treatment of colon and lung cancer patients with ex vivo heat shock protein 70-peptide-activated, autologous natural killer cells: A clinical phase I trial. Clin. Cancer Res. 2004, 10, 3699–3707. [Google Scholar] [CrossRef]
- Matosevic, S. Viral and nonviral engineering of natural killer cells as emerging adoptive cancer immunotherapies. J. Immunol. Res. 2018, 2018, 4054815. [Google Scholar] [CrossRef]
- Wagner, E. Converging paths of viral and non-viral vector engineering. Mol. Ther. 2008, 16, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Lundstrom, K. Viral vectors in gene therapy. Diseases 2018, 6, 42. [Google Scholar] [CrossRef]
- Morgan, M.A.; Galla, M.; Grez, M.; Fehse, B.; Schambach, A. Retroviral gene therapy in Germany with a view on previous experience and future perspectives. Gene Ther. 2021, 28, 494–512. [Google Scholar] [CrossRef] [PubMed]
- Streltsova, M.A.; Barsov, E.; Erokhina, S.A.; Kovalenko, E.I. Retroviral gene transfer into primary human NK cells activated by IL-2 and K562 feeder cells expressing membrane-bound IL-21. J. Immunol. Methods 2017, 450, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Suerth, J.D.; Morgan, M.A.; Kloess, S.; Heckl, D.; Neudörfl, C.; Falk, C.S.; Koehl, U.; Schambach, A. Efficient generation of gene-modified human natural killer cells via alpharetroviral vectors. J. Mol. Med. 2016, 94, 83–93. [Google Scholar] [CrossRef]
- Montini, E.; Cesana, D.; Schmidt, M.; Sanvito, F.; Ponzoni, M.; Bartholomae, C.; Sergi, L.S.; Benedicenti, F.; Ambrosi, A.; Di Serio, C. Hematopoietic stem cell gene transfer in a tumor-prone mouse model uncovers low genotoxicity of lentiviral vector integration. Nat. Biotechnol. 2006, 24, 687–696. [Google Scholar] [CrossRef] [PubMed]
- Milone, M.C.; O’Doherty, U. Clinical use of lentiviral vectors. Leukemia 2018, 32, 1529–1541. [Google Scholar] [CrossRef]
- Micucci, F.; Zingoni, A.; Piccoli, M.; Frati, L.; Santoni, A.; Galandrini, R. High-efficient lentiviral vector-mediated gene transfer into primary human NK cells. Exp. Hematol. 2006, 34, 1344–1352. [Google Scholar] [CrossRef]
- Becknell, B.; Trotta, R.; Yu, J.; Ding, W.; Mao, H.C.; Hughes, T.; Marburger, T.; Caligiuri, M.A. Efficient infection of human natural killer cells with an EBV/retroviral hybrid vector. J. Immunol. Methods 2005, 296, 115–123. [Google Scholar] [CrossRef]
- Fraietta, J.A.; Nobles, C.L.; Sammons, M.A.; Lundh, S.; Carty, S.A.; Reich, T.J.; Cogdill, A.P.; Morrissette, J.J.; DeNizio, J.E.; Reddy, S. Disruption of TET2 promotes the therapeutic efficacy of CD19-targeted T cells. Nature 2018, 558, 307–312. [Google Scholar] [CrossRef] [PubMed]
- Sutlu, T.; Nyström, S.; Gilljam, M.; Stellan, B.; Applequist, S.E.; Alici, E. Inhibition of intracellular antiviral defense mechanisms augments lentiviral transduction of human natural killer cells: Implications for gene therapy. Hum. Gene Ther. 2012, 23, 1090–1100. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Guerrero, A.; Cosset, F.-L.; Verhoeyen, E. Lentiviral Vector Pseudotypes: Precious Tools to Improve Gene Modification of Hematopoietic Cells for Research and Gene Therapy. Viruses 2020, 12, 1016. [Google Scholar] [CrossRef]
- Levine, B.L.; Humeau, L.M.; Boyer, J.; MacGregor, R.-R.; Rebello, T.; Lu, X.; Binder, G.K.; Slepushkin, V.; Lemiale, F.; Mascola, J.R. Gene transfer in humans using a conditionally replicating lentiviral vector. Proc. Natl. Acad. Sci. USA 2006, 103, 17372–17377. [Google Scholar] [CrossRef]
- Jamali, A.; Hadjati, J.; Madjd, Z.; Mirzaei, H.R.; Thalheimer, F.B.; Agarwal, S.; Bonig, H.; Ullrich, E.; Hartmann, J. Highly efficient generation of transgenically augmented CAR NK cells overexpressing CXCR4. Front. Immunol. 2020, 11, 2028. [Google Scholar] [CrossRef]
- Gong, Y.; Wolterink, R.G.K.; Janssen, I.; Groot, A.J.; Bos, G.M.; Germeraad, W.T. Rosuvastatin enhances VSV-G lentiviral transduction of NK cells via upregulation of the low-density lipoprotein receptor. Mol. Ther. -Methods Clin. Dev. 2020, 17, 634–646. [Google Scholar] [CrossRef]
- Colamartino, A.B.; Lemieux, W.; Bifsha, P.; Nicoletti, S.; Chakravarti, N.; Sanz, J.; Roméro, H.; Selleri, S.; Béland, K.; Guiot, M. Efficient and robust NK-cell transduction with baboon envelope pseudotyped lentivector. Front. Immunol. 2019, 10, 2873. [Google Scholar] [CrossRef]
- Bari, R.; Granzin, M.; Tsang, K.S.; Roy, A.; Krueger, W.; Orentas, R.; Schneider, D.; Pfeifer, R.; Moeker, N.; Verhoeyen, E. A distinct subset of highly proliferative and lentiviral vector (LV)-transducible NK cells define a readily engineered subset for adoptive cellular therapy. Front. Immunol. 2019, 10, 2001. [Google Scholar] [CrossRef]
- Bell Jr, A.J.; Fegen, D.; Ward, M.; Bank, A. RD114 envelope proteins provide an effective and versatile approach to pseudotype lentiviral vectors. Exp. Biol. Med. 2010, 235, 1269–1276. [Google Scholar] [CrossRef] [PubMed]
- Klöß, S.; Oberschmidt, O.; Morgan, M.; Dahlke, J.; Arseniev, L.; Huppert, V.; Granzin, M.; Gardlowski, T.; Matthies, N.; Soltenborn, S. Optimization of human NK cell manufacturing: Fully automated separation, improved ex vivo expansion using IL-21 with autologous feeder cells, and generation of anti-CD123-CAR-expressing effector cells. Hum. Gene Ther. 2017, 28, 897–913. [Google Scholar] [CrossRef] [PubMed]
- Davis, H.E.; Morgan, J.R.; Yarmush, M.L. Polybrene increases retrovirus gene transfer efficiency by enhancing receptor-independent virus adsorption on target cell membranes. Biophys. Chem. 2002, 97, 159–172. [Google Scholar] [CrossRef]
- Littwitz, E.; Francois, S.; Dittmer, U.; Gibbert, K. Distinct roles of NK cells in viral immunity during different phases of acute Friend retrovirus infection. Retrovirology 2013, 10, 1–7. [Google Scholar] [CrossRef]
- Robbins, G.M.; Wang, M.; Pomeroy, E.J.; Moriarity, B.S. Nonviral genome engineering of natural killer cells. Stem Cell Res. Ther. 2021, 12, 1–9. [Google Scholar] [CrossRef]
- Ingegnere, T.; Mariotti, F.R.; Pelosi, A.; Quintarelli, C.; De Angelis, B.; Tumino, N.; Besi, F.; Cantoni, C.; Locatelli, F.; Vacca, P. Human CAR NK cells: A new non-viral method allowing high efficient transfection and strong tumor cell killing. Front. Immunol. 2019, 10, 957. [Google Scholar] [CrossRef]
- Liu, H.; Yang, B.; Sun, T.; Lin, L.; Hu, Y.; Deng, M.; Yang, J.; Liu, T.; Li, J.; Sun, S. Specific growth inhibition of ErbB2-expressing human breast cancer cells by genetically modified NK-92 cells. Oncol. Rep. 2015, 33, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Boissel, L.; Betancur, M.; Wels, W.S.; Tuncer, H.; Klingemann, H. Transfection with mRNA for CD19 specific chimeric antigen receptor restores NK cell mediated killing of CLL cells. Leuk. Res. 2009, 33, 1255–1259. [Google Scholar] [CrossRef] [PubMed]
- Strobel, I.; Berchtold, S.; Götze, A.; Schulze, U.; Schuler, G.; Steinkasserer, A. Human dendritic cells transfected with either RNA or DNA encoding influenza matrix protein M1 differ in their ability to stimulate cytotoxic T lymphocytes. Gene Ther. 2000, 7, 2028–2035. [Google Scholar] [CrossRef]
- Youn, H.; Chung, J.-K. Modified mRNA as an alternative to plasmid DNA (pDNA) for transcript replacement and vaccination therapy. Expert Opin. Biol. Ther. 2015, 15, 1337–1348. [Google Scholar] [CrossRef]
- Carlsten, M.; Levy, E.; Karambelkar, A.; Li, L.; Reger, R.; Berg, M.; Peshwa, M.V.; Childs, R.W. Efficient mRNA-based genetic engineering of human NK cells with high-affinity CD16 and CCR7 augments rituximab-induced ADCC against lymphoma and targets NK cell migration toward the lymph node-associated chemokine CCL19. Front. Immunol. 2016, 7, 105. [Google Scholar] [CrossRef]
- Li, L.; Liu, L.N.; Feller, S.; Allen, C.; Shivakumar, R.; Fratantoni, J.; Wolfraim, L.A.; Fujisaki, H.; Campana, D.; Chopas, N. Expression of chimeric antigen receptors in natural killer cells with a regulatory-compliant non-viral method. Cancer Gene Ther. 2010, 17, 147–154. [Google Scholar] [CrossRef]
- Boissel, L.; Betancur, M.; Lu, W.; Wels, W.S.; Marino, T.; Van Etten, R.A.; Klingemann, H. Comparison of mRNA and lentiviral based transfection of natural killer cells with chimeric antigen receptors recognizing lymphoid antigens. Leuk. Lymphoma 2012, 53, 958–965. [Google Scholar] [CrossRef]
- Shimasaki, N.; Fujisaki, H.; Cho, D.; Masselli, M.; Lockey, T.; Eldridge, P.; Leung, W.; Campana, D. A clinically adaptable method to enhance the cytotoxicity of natural killer cells against B-cell malignancies. Cytotherapy 2012, 14, 830–840. [Google Scholar] [CrossRef]
- Xiao, L.; Cen, D.; Gan, H.; Sun, Y.; Huang, N.; Xiong, H.; Jin, Q.; Su, L.; Liu, X.; Wang, K. Adoptive transfer of NKG2D CAR mRNA-engineered natural killer cells in colorectal cancer patients. Mol. Ther. 2019, 27, 1114–1125. [Google Scholar] [CrossRef]
- Brunner, S.; Fürtbauer, E.; Sauer, T.; Kursa, M.; Wagner, E. Overcoming the nuclear barrier: Cell cycle independent nonviral gene transfer with linear polyethylenimine or electroporation. Mol. Ther. 2002, 5, 80–86. [Google Scholar] [CrossRef]
- Grund, E.M.; Muise-Helmericks, R.C. Cost efficient and effective gene transfer into the human natural killer cell line, NK92. J. Immunol. Methods 2005, 296, 31–36. [Google Scholar] [CrossRef]
- Radons, J.; Gross, C.; Stangl, S.; Multhoff, G. Nucleofection of non-B cells with mini-Epstein-Barr virus DNA. J. Immunol. Methods 2005, 303, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Trompeter, H.-I.; Weinhold, S.; Thiel, C.; Wernet, P.; Uhrberg, M. Rapid and highly efficient gene transfer into natural killer cells by nucleofection. J. Immunol. Methods 2003, 274, 245–256. [Google Scholar] [CrossRef]
- Flanagan, M.; Gimble, J.M.; Yu, G.; Wu, X.; Xia, X.; Hu, J.; Yao, S.; Li, S. Competitive electroporation formulation for cell therapy. Cancer Gene Ther. 2011, 18, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Simon, B.; Harrer, D.C.; Schuler-Thurner, B.; Schaft, N.; Schuler, G.; Dörrie, J.; Uslu, U. The si RNA-mediated downregulation of PD-1 alone or simultaneously with CTLA-4 shows enhanced in vitro CAR-T-cell functionality for further clinical development towards the potential use in immunotherapy of melanoma. Exp. Dermatol. 2018, 27, 769–778. [Google Scholar] [CrossRef]
- Sharei, A.; Zoldan, J.; Adamo, A.; Sim, W.Y.; Cho, N.; Jackson, E.; Mao, S.; Schneider, S.; Han, M.-J.; Lytton-Jean, A. A vector-free microfluidic platform for intracellular delivery. Proc. Natl. Acad. Sci. USA 2013, 110, 2082–2087. [Google Scholar] [CrossRef] [PubMed]
- Freitag, F.; Wagner, E. Optimizing synthetic nucleic acid and protein nanocarriers: The chemical evolution approach. Adv. Drug Deliv. Rev. 2021, 168, 30–54. [Google Scholar] [CrossRef]
- Wang, Y.; Wagner, E. Non-viral targeted nucleic acid delivery: Apply sequences for optimization. Pharmaceutics 2020, 12, 888. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Wagner, E. Polymeric carriers for nucleic acid delivery: Current designs and future directions. Biomacromolecules 2019, 20, 3613–3626. [Google Scholar] [CrossRef]
- Wagner, E. Polymers for nucleic acid transfer—An overview. Adv. Genet. 2014, 88, 231–261. [Google Scholar]
- Abdalla, A.M.; Xiao, L.; Miao, Y.; Huang, L.; Fadlallah, G.M.; Gauthier, M.; Ouyang, C.; Yang, G. Nanotechnology promotes genetic and functional modifications of therapeutic T cells against cancer. Adv. Sci. 2020, 7, 1903164. [Google Scholar] [CrossRef]
- Wilk, A.J.; Weidenbacher, N.L.-B.; Vergara, R.; Haabeth, O.A.; Levy, R.; Waymouth, R.M.; Wender, P.A.; Blish, C.A. Charge-altering releasable transporters enable phenotypic manipulation of natural killer cells for cancer immunotherapy. Blood Adv. 2020, 4, 4244–4255. [Google Scholar] [CrossRef]
- Moffett, H.; Coon, M.; Radtke, S.; Stephan, S.; McKnight, L.; Lambert, A.; Stoddard, B.; Kiem, H.; Stephan, M. Hit-and-run programming of therapeutic cytoreagents using mRNA nanocarriers. Nat. Commun. 2017, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Billingsley, M.M.; Singh, N.; Ravikumar, P.; Zhang, R.; June, C.H.; Mitchell, M.J. Ionizable lipid nanoparticle-mediated mRNA delivery for human CAR T cell engineering. Nano Lett. 2020, 20, 1578–1589. [Google Scholar] [CrossRef]
- Kim, K.-S.; Han, J.-H.; Park, J.-H.; Kim, H.-K.; Choi, S.H.; Kim, G.R.; Song, H.; An, H.J.; Han, D.K.; Park, W. Multifunctional nanoparticles for genetic engineering and bioimaging of natural killer (NK) cell therapeutics. Biomaterials 2019, 221, 119418. [Google Scholar] [CrossRef]
- Huang, J.; Yuen, D.; Mintern, J.D.; Johnston, A.P. Opportunities for Innovation: Building on the success of lipid nanoparticle vaccines. Curr. Opin. Colloid Interface Sci. 2021, 55, 101468. [Google Scholar] [CrossRef]
- Ogris, M.; Wagner, E. To be targeted: Is the magic bullet concept a viable option for synthetic nucleic acid therapeutics? Hum. Gene Ther. 2011, 22, 799–807. [Google Scholar] [CrossRef]
- Frank, A.M.; Braun, A.H.; Scheib, L.; Agarwal, S.; Schneider, I.C.; Fusil, F.; Perian, S.; Sahin, U.; Thalheimer, F.B.; Verhoeyen, E. Combining T-cell–specific activation and in vivo gene delivery through CD3-targeted lentiviral vectors. Blood Adv. 2020, 4, 5702–5715. [Google Scholar]
- Smith, T.T.; Stephan, S.B.; Moffett, H.F.; McKnight, L.E.; Ji, W.; Reiman, D.; Bonagofski, E.; Wohlfahrt, M.E.; Pillai, S.P.; Stephan, M.T. In situ programming of leukaemia-specific T cells using synthetic DNA nanocarriers. Nat. Nanotechnol. 2017, 12, 813–820. [Google Scholar] [CrossRef]
- Lächelt, U.; Wagner, E. Nucleic acid therapeutics using polyplexes: A journey of 50 years (and beyond). Chem. Rev. 2015, 115, 11043–11078. [Google Scholar]
- Liu, M.A. A comparison of plasmid DNA and mRNA as vaccine technologies. Vaccines 2019, 7, 37. [Google Scholar] [CrossRef]
- Levacic, A.K.; Morys, S.; Kempter, S.; Lächelt, U.; Wagner, E. Minicircle versus plasmid DNA delivery by receptor-targeted polyplexes. Hum. Gene Ther. 2017, 28, 862–874. [Google Scholar] [CrossRef]
- Vargas, J.E.; Chicaybam, L.; Stein, R.T.; Tanuri, A.; Delgado-Cañedo, A.; Bonamino, M.H. Retroviral vectors and transposons for stable gene therapy: Advances, current challenges and perspectives. J. Transl. Med. 2016, 14, 1–15. [Google Scholar] [CrossRef]
- Tipanee, J.; Chai, Y.C.; VandenDriessche, T.; Chuah, M.K. Preclinical and clinical advances in transposon-based gene therapy. Biosci. Rep. 2017, 37, BSR20160614. [Google Scholar] [CrossRef]
- Magnani, C.F.; Mezzanotte, C.; Cappuzzello, C.; Bardini, M.; Tettamanti, S.; Fazio, G.; Cooper, L.J.; Dastoli, G.; Cazzaniga, G.; Biondi, A. Preclinical efficacy and safety of CD19CAR cytokine-induced killer cells transfected with sleeping beauty transposon for the treatment of acute lymphoblastic leukemia. Hum. Gene Ther. 2018, 29, 602–613. [Google Scholar] [CrossRef]
- Magnani, C.F.; Tettamanti, S.; Alberti, G.; Pisani, I.; Biondi, A.; Serafini, M.; Gaipa, G. Transposon-based CAR T cells in acute leukemias: Where are we going? Cells 2020, 9, 1337. [Google Scholar] [CrossRef]
- Kebriaei, P.; Singh, H.; Huls, M.H.; Figliola, M.J.; Bassett, R.; Olivares, S.; Jena, B.; Dawson, M.J.; Kumaresan, P.R.; Su, S. Phase I trials using Sleeping Beauty to generate CD19-specific CAR T cells. J. Clin. Investig. 2016, 126, 3363–3376. [Google Scholar] [CrossRef]
- Prommersberger, S.; Reiser, M.; Beckmann, J.; Danhof, S.; Amberger, M.; Quade-Lyssy, P.; Einsele, H.; Hudecek, M.; Bonig, H.; Ivics, Z. CARAMBA: A first-in-human clinical trial with SLAMF7 CAR-T cells prepared by virus-free Sleeping Beauty gene transfer to treat multiple myeloma. Gene Ther. 2021, 28, 560–571. [Google Scholar] [CrossRef]
- Wang, J.; Lupo, K.B.; Chambers, A.M.; Matosevic, S. Purinergic targeting enhances immunotherapy of CD73+ solid tumors with piggyBac-engineered chimeric antigen receptor natural killer cells. J. Immunother. Cancer 2018, 6, 1–14. [Google Scholar] [CrossRef]
- Huang, X.; Haley, K.; Wong, M.; Guo, H.; Lu, C.; Wilber, A.; Zhou, X. Unexpectedly high copy number of random integration but low frequency of persistent expression of the Sleeping Beauty transposase after trans delivery in primary human T cells. Hum. Gene Ther. 2010, 21, 1577–1590. [Google Scholar] [CrossRef]
- Monjezi, R.; Miskey, C.; Gogishvili, T.; Schleef, M.; Schmeer, M.; Einsele, H.; Ivics, Z.; Hudecek, M. Enhanced CAR T-cell engineering using non-viral Sleeping Beauty transposition from minicircle vectors. Leukemia 2017, 31, 186–194. [Google Scholar] [CrossRef]
- Querques, I.; Mades, A.; Zuliani, C.; Miskey, C.; Alb, M.; Grueso, E.; Machwirth, M.; Rausch, T.; Einsele, H.; Ivics, Z. A highly soluble Sleeping Beauty transposase improves control of gene insertion. Nat. Biotechnol. 2019, 37, 1502–1512. [Google Scholar] [CrossRef]
- Zheng, Y.; Li, Z.-R.; Yue, R.; Fu, Y.-L.; Liu, Z.-Y.; Feng, H.-Y.; Li, J.-G.; Han, S.-Y. PiggyBac transposon system with polymeric gene carrier transfected into human T cells. Am. J. Transl. Res. 2019, 11, 7126. [Google Scholar]
- Bishop, D.C.; Clancy, L.E.; Simms, R.; Burgess, J.; Mathew, G.; Moezzi, L.; Street, J.A.; Sutrave, G.; Atkins, E.; McGuire, H.M. Development of CAR T-cell lymphoma in two of ten patients effectively treated with piggyBac modified CD19 CAR T-cells. Blood 2021, 138, 1504–1509. [Google Scholar] [CrossRef]
- Afolabi, L.O.; Adeshakin, A.O.; Sani, M.M.; Bi, J.; Wan, X. Genetic reprogramming for NK cell cancer immunotherapy with CRISPR/Cas9. Immunology 2019, 158, 63–69. [Google Scholar] [CrossRef]
- Mollanoori, H.; Shahraki, H.; Rahmati, Y.; Teimourian, S. CRISPR/Cas9 and CAR-T cell, collaboration of two revolutionary technologies in cancer immunotherapy, an instruction for successful cancer treatment. Hum. Immunol. 2018, 79, 876–882. [Google Scholar] [CrossRef]
- Huang, R.-S.; Lai, M.-C.; Shih, H.-A.; Lin, S. A robust platform for expansion and genome editing of primary human natural killer cells. J. Exp. Med. 2021, 218, e20201529. [Google Scholar] [CrossRef] [PubMed]
- van Haasteren, J.; Li, J.; Scheideler, O.J.; Murthy, N.; Schaffer, D.V. The delivery challenge: Fulfilling the promise of therapeutic genome editing. Nat. Biotechnol. 2020, 38, 845–855. [Google Scholar] [CrossRef]
- Kuhn, J.; Lin, Y.; Krhac Levacic, A.; Al Danaf, N.; Peng, L.; Höhn, M.; Lamb, D.C.; Wagner, E.; Lächelt, U. Delivery of Cas9/sgRNA ribonucleoprotein complexes via hydroxystearyl oligoamino amides. Bioconjugate Chem. 2020, 31, 729–742. [Google Scholar] [CrossRef] [PubMed]
- Eyquem, J.; Mansilla-Soto, J.; Giavridis, T.; van der Stegen, S.J.; Hamieh, M.; Cunanan, K.M.; Odak, A.; Gönen, M.; Sadelain, M. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature 2017, 543, 113–117. [Google Scholar] [CrossRef]
- Shevtsov, M.; Pitkin, E.; Ischenko, A.; Stangl, S.; Khachatryan, W.; Galibin, O.; Edmond, S.; Lobinger, D.; Multhoff, G. Ex Vivo Hsp70-activated NK cells in combination with PD-1 inhibition significantly increase overall survival in preclinical models of glioblastoma and lung cancer. Front. Immunol. 2019, 10, 454. [Google Scholar] [CrossRef] [PubMed]
- Daher, M.; Basar, R.; Gokdemir, E.; Baran, N.; Uprety, N.; Nunez Cortes, A.K.; Mendt, M.; Kerbauy, L.N.; Banerjee, P.P.; Shanley, M. Targeting a cytokine checkpoint enhances the fitness of armored cord blood CAR-NK cells. Blood J. Am. Soc. Hematol. 2021, 137, 624–636. [Google Scholar] [CrossRef]



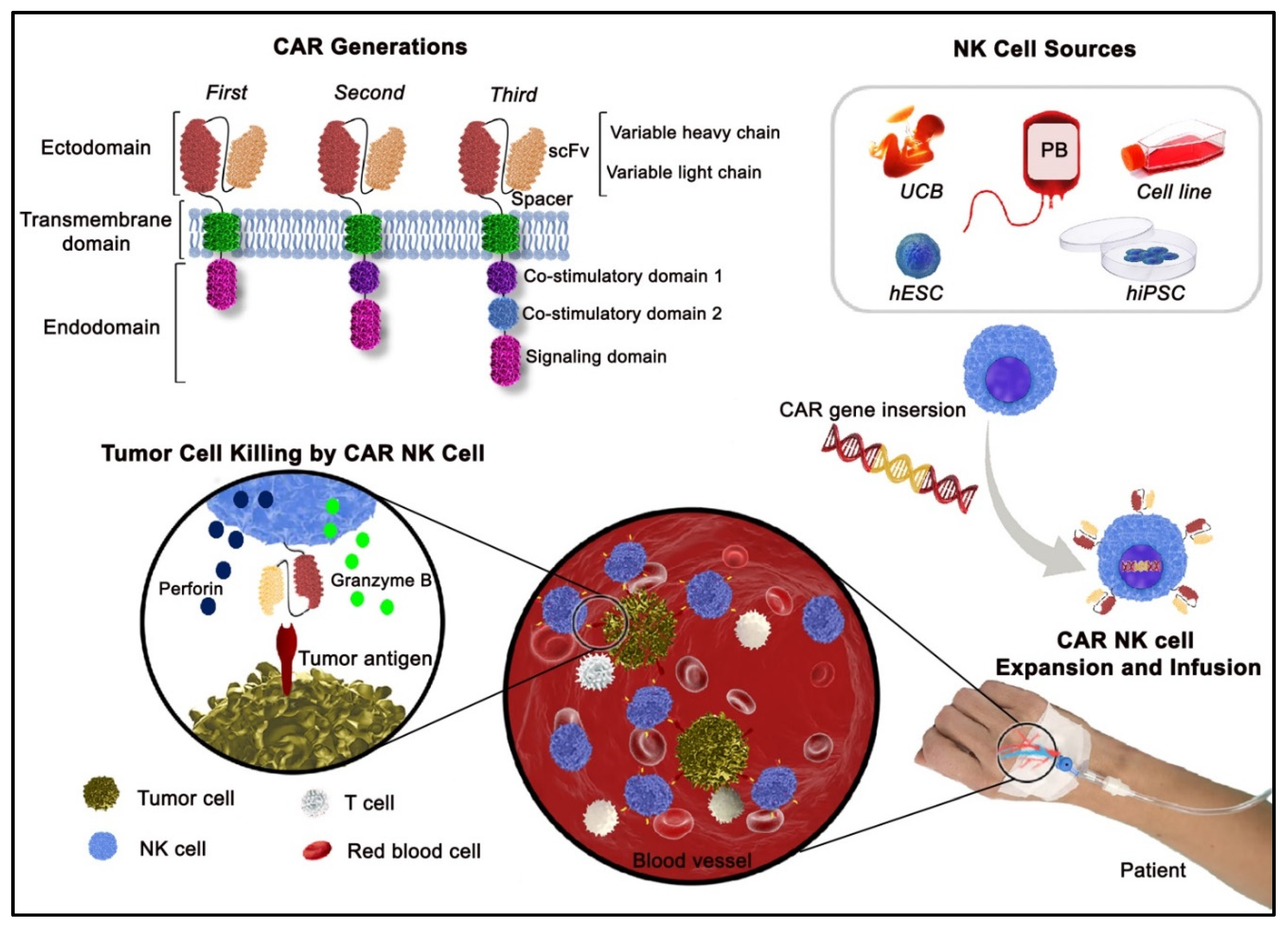{kind=link}
{kind=link}
| Tumor Antigen | Tumor | Source | Delivery Method | Transmembrane + Endodomain | State | Ref |
|---|---|---|---|---|---|---|
| CD19 | Non-Hodgkin’s lymphoma Chronic lymphocytic leukemia | CB NK | Retroviral | CD28, CD3ζ, iC9, IL-15 | Phase I and II trial | [14] |
| EGFR | Triple-negative breast cancer | PB NK | Lentiviral | CD8, CD28, 4-1BB, CD3ζ | Preclinical | [68] |
| HER2 | Glioblastoma | NK-92 | Lentiviral | CD28, CD3ζ | Preclinical | [69] |
| EpCAM | Colorectal cancer | NK-92 | Lentiviral | CD8, 4-1BB, CD3ζ | Preclinical | [70] |
| GD2 | Ewing sarcoma | PB NK | Retroviral | CD28, 4-1BB, CD3ζ | Preclinical | [71] |
| Mesothelin | Ovarian carcinoma | NK-92 | Lentiviral | CD8, CD28, 4-1BB, CD3ζ | Preclinical | [72] |
| CD33 | Acute myeloid leukemia | NK-92 | Lentiviral | CD28, 4-1BB, CD3ζ | Phase I and II trial | [73] |
| CD123 | Acute myeloid leukemia | NK-92 | Retroviral | CD28, 4-1BB, CD3ζ | Preclinical | [74] |
| CS1 | Multiple myeloma | NK-92 | Lentiviral | CD28, CD3ζ | Preclinical | [75] |
| CD7 | Acute T lymphoblastic leukemia | NK-92MI | Electroporation | CD28, 4-1BB, CD3ζ | Preclinical | [76] |
| PSMA | Prostate carcinoma | NK-92 | Lentiviral | CD28, CD3ζ | Preclinical | [77] |
| ROBO1 | Pancreatic ductal adenocarcinoma | NK-92 | Lentiviral | CD8, 4-1BB, CD3ζ | Phase I and II trial | [78] |
| BCMA | Multiple myeloma | PB NK | Electroporation | CD8, DAP12|CD3ζ | Preclinical | [79] |
| CD20 | Burkitt lymphoma | PB NK | Electroporation | 4-1BB, CD3ζ | Preclinical | [80] |
| PSCA | Prostate adenocarcinoma | YT NK | Lentiviral | CD8, CD28, CD3ζ | Preclinical | [81] |
| GPA7 | Melanoma | NK-92MI | Electroporation | HLA-A2, CD3ζ | Preclinical | [48] |
| NKG2DL | Multiple myeloma | PB NK | Lentiviral | 4-1BB, CD3ζ | Preclinical | [82] |
| GPC3 | Hepatocellular carcinoma | PB NK | Lentiviral | CD8, 4-1BB, CD3ζ | Preclinical | [83] |
| FR α | Ovarian adenocarcinoma | NK-92 | Lentiviral | CD8, CD28, 4-1BB, CD3ζ | Preclinical | [84] |
| CD276 | Neuroblastoma | NK-92 | Lentiviral | CD8, CD28, CD3ζ | Preclinical | [85] |
| CD135 | Acute B lymphoblastic leukemia | NK-92 | Lentiviral | CD28, CD3ζ | Preclinical | [86] |
| CD5 | Acute T lymphoblastic leukemia | NK-92 | Lentiviral | CD8, 2B4, CD3ζ | Preclinical | [60] |
| PDL1 | Head and neck squamous cell carcinoma | haNK | Electroporation | CD8, CD28, FcεR1γ | Preclinical | [87] |
| TF | Triple-negative breast cancer | NK92MI | Lentivirus | CD28, 4-1BB, CD3ζ | Preclinical | [88] |
| CD38 | Multiple myeloma | KHYG-1 | Retroviral | CD8, CD28, 4-1BB, CD3ζ | Preclinical | [89] |
| CEA | Colorectal carcinoma | NK92MI | Retroviral | CD8, CD3ζ | Preclinical | [90] |
| CD147 | Hepatocellular carcinoma | NK92MI | Retroviral | CD28, 4-1BB, CD3ζ | Preclinical | [91] |
| WT-1 | Leukemia | NK92MI | Retroviral | 4-1BB, CD3ζ | Preclinical | [92] |
| CD4 | Acute myeloid leukemia | NK-92 | Lentivirus | CD28, 4-1BB, CD3ζ | Preclinical | [93] |
| c-MET | Hepatocellular carcinoma | PB NK | Lentiviral | CD8, 4-1BB, DAP12 | Preclinical | [94] |
| CD138 | Hematologic malignancies | NK-92 | Lentiviral | CD8, CD28, 4-1BB, CD3ζ | Preclinical | [95] |
| CD3 | T-Cell lymphoma | NK-92 | Lentiviral | CD28, 4-1BB, CD3ζ | Preclinical | [96] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bashiri Dezfouli, A.; Yazdi, M.; Pockley, A.G.; Khosravi, M.; Kobold, S.; Wagner, E.; Multhoff, G. NK Cells Armed with Chimeric Antigen Receptors (CAR): Roadblocks to Successful Development. Cells 2021, 10, 3390. https://doi.org/10.3390/cells10123390
Bashiri Dezfouli A, Yazdi M, Pockley AG, Khosravi M, Kobold S, Wagner E, Multhoff G. NK Cells Armed with Chimeric Antigen Receptors (CAR): Roadblocks to Successful Development. Cells. 2021; 10(12):3390. https://doi.org/10.3390/cells10123390
Chicago/Turabian StyleBashiri Dezfouli, Ali, Mina Yazdi, Alan Graham Pockley, Mohammad Khosravi, Sebastian Kobold, Ernst Wagner, and Gabriele Multhoff. 2021. "NK Cells Armed with Chimeric Antigen Receptors (CAR): Roadblocks to Successful Development" Cells 10, no. 12: 3390. https://doi.org/10.3390/cells10123390
APA StyleBashiri Dezfouli, A., Yazdi, M., Pockley, A. G., Khosravi, M., Kobold, S., Wagner, E., & Multhoff, G. (2021). NK Cells Armed with Chimeric Antigen Receptors (CAR): Roadblocks to Successful Development. Cells, 10(12), 3390. https://doi.org/10.3390/cells10123390