
Progressive Liver Fibrosis in Non-Alcoholic Fatty Liver Disease


 , , ,
, , , 

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
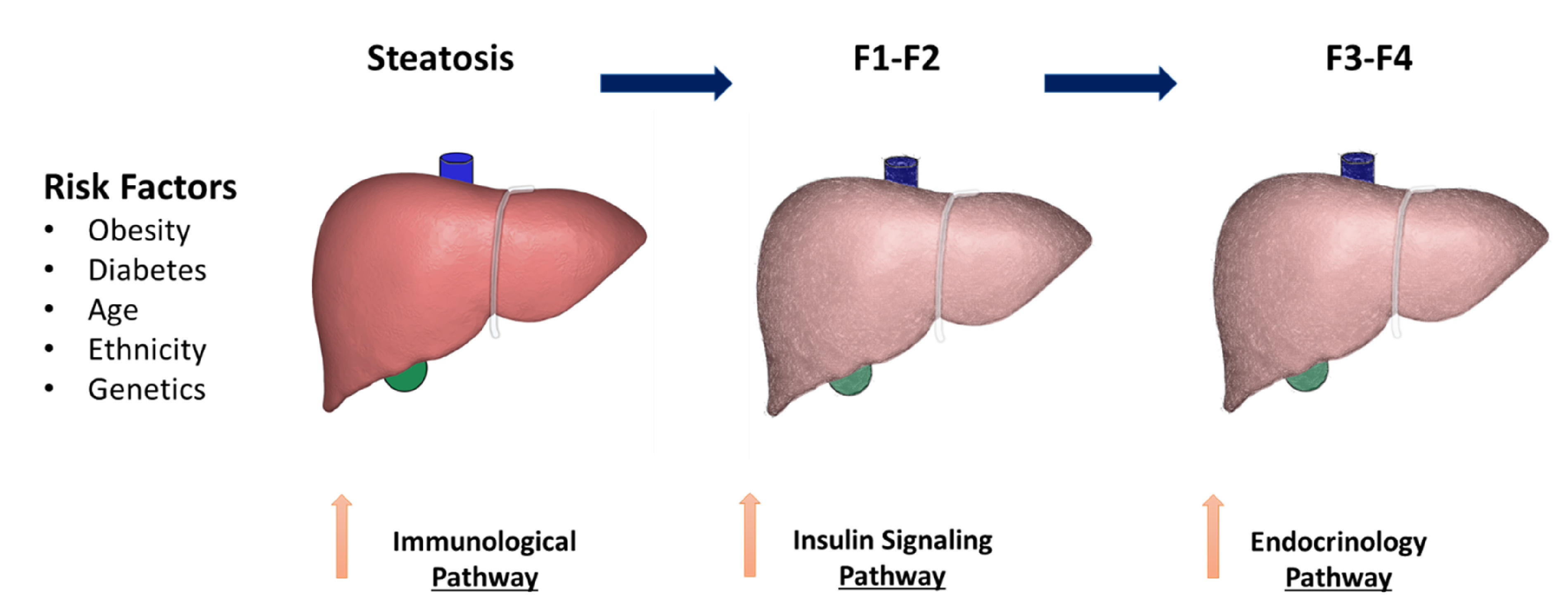
2. NAFLD Risk Factors
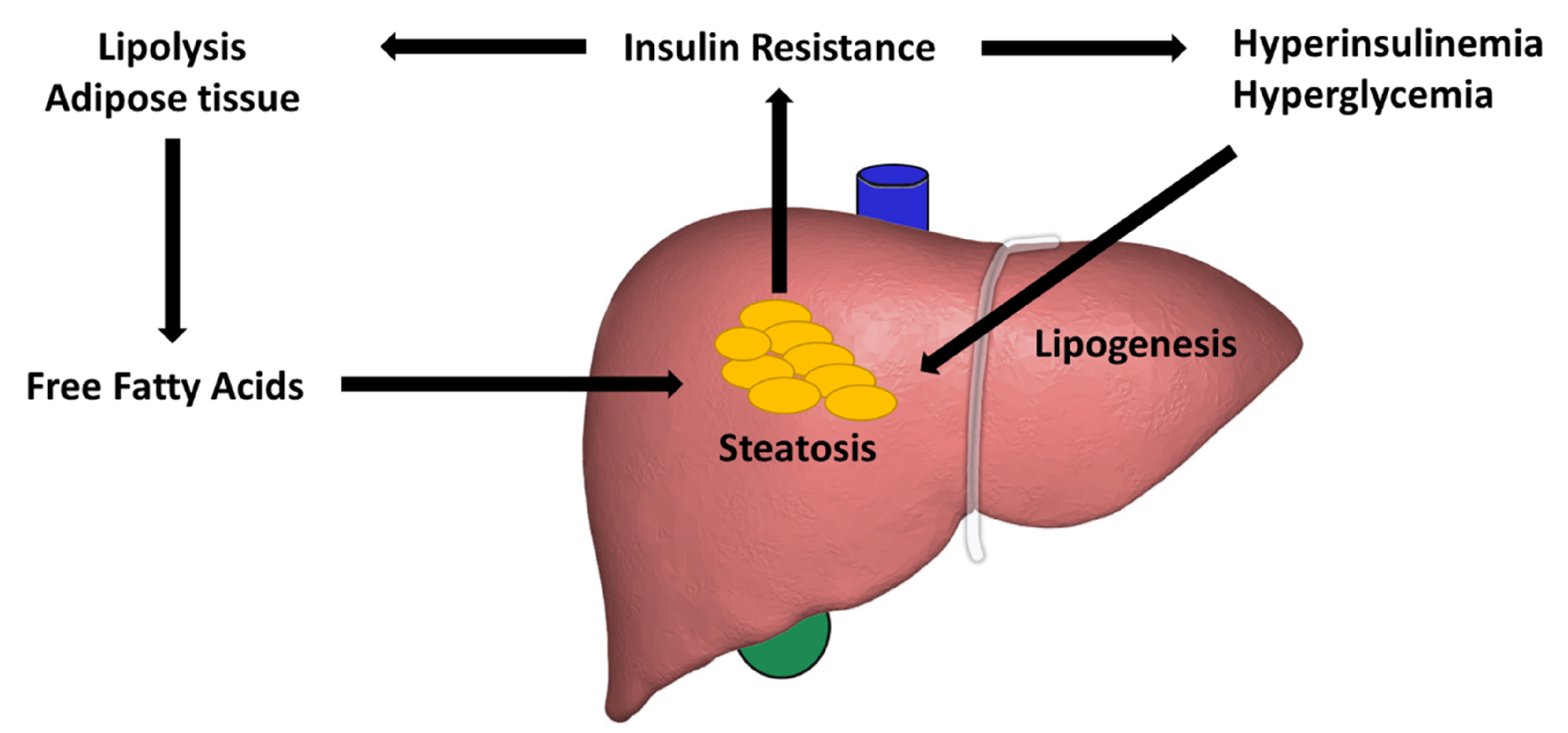
3. Lipid Accumulation
4. The Insulin Signaling Pathway
5. The Role of Autophagy
6. Immunologic and Genetic Factors
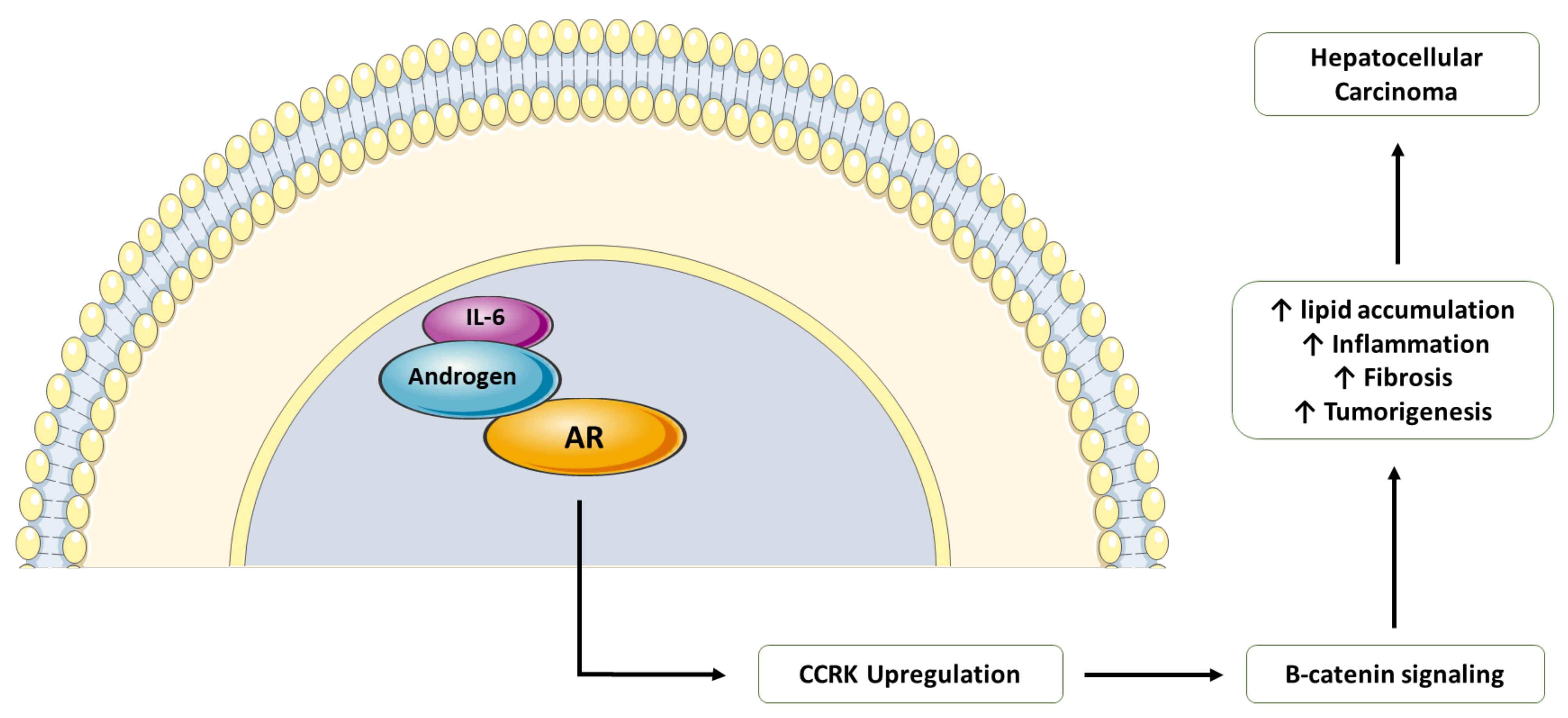
7. Endocrine Pathway
8. The Gut Microbiota
9. Biomarkers
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wree, A.; Broderick, L.; Canbay, A.; Hoffman, H.M.; Feldstein, A.E. From NAFLD to NASH to cirrhosis—New insights into disease mechanisms. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 627–636. [Google Scholar] [CrossRef] [PubMed]
- Wanless, I.R.; Lentz, J.S. Fatty liver hepatitis (steatohepatitis) and obesity: An autopsy study with analysis of risk factors. Hepatology 1990, 12, 1106–1110. [Google Scholar] [CrossRef]
- Gutiérrez-Cuevas, J.; Santos, A.; Armendariz-Borunda, J. Pathophysiological molecular mechanisms of obesity: A link between MAFLD and NASH with cardiovascular diseases. Int. J. Mol. Sci. 2021, 22, 11629. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Golabi, P.; de Avila, L.; Paik, J.M.; Srishord, M.; Fukui, N.; Qiu, Y.; Burns, L.; Afendy, A.; Nader, F. The global epidemiology of NAFLD and NASH in patients with type 2 diabetes: A systematic review and meta-analysis. J. Hepatol. 2019, 71, 793–801. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.J.; Fallon, M.B. Gender and racial differences in nonalcoholic fatty liver disease. World J. Hepatol. 2014, 6, 274–283. [Google Scholar] [CrossRef] [PubMed]
- Clark, J.M.; Brancati, F.L.; Diehl, A.M. The prevalence and etiology of elevated aminotransferase levels in the United States. Am. J. Gastroenterol. 2003, 98, 960–967. [Google Scholar] [CrossRef]
- Ioannou, G.N.; Boyko, E.J.; Lee, S.P. The prevalence and predictors of elevated serum aminotransferase activity in the United States in 1999–2002. Am. J. Gastroenterol. 2006, 101, 76–82. [Google Scholar] [CrossRef]
- Kanwal, F.; Kramer, J.R.; Duan, Z.; Yu, X.; White, D.; El-Serag, H.B. Trends in the Burden of Nonalcoholic Fatty Liver Disease in a United States Cohort of Veterans. Clin. Gastroenterol. Hepatol. 2016, 14, 301–308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, M.; Huang, J.; George, J.; Huang, J.; Leung, C.; Eslam, M.; Chan, H.; Ng, S.C. The changing epidemiology of liver diseases in the Asia-Pacific region. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 57–73. [Google Scholar] [CrossRef]
- Mitra, S.; De, A.; Chowdhury, A. Epidemiology of non-alcoholic and alcoholic fatty liver diseases. Transl. Gastroenterol. Hepatol. 2020, 5, 16. [Google Scholar] [CrossRef]
- Schneider, A.L.; Lazo, M.; Selvin, E.; Clark, J.M. Racial differences in nonalcoholic fatty liver disease in the U.S. population. Obesity 2014, 22, 292–299. [Google Scholar] [CrossRef] [Green Version]
- Romeo, S.; Kozlitina, J.; Xing, C.; Pertsemlidis, A.; Cox, D.; Pennacchio, L.A.; Boerwinkle, E.; Cohen, J.C.; Hobbs, H.H. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2008, 40, 1461–1465. [Google Scholar] [CrossRef] [Green Version]
- Ramai, D.; Tai, W.; Rivera, M.; Facciorusso, A.; Tartaglia, N.; Pacilli, M.; Sacco, R. Natural Progression of Non-Alcoholic Steatohepatitis to Hepatocellular Carcinoma. Biomedicines 2021, 9, 184. [Google Scholar] [CrossRef] [PubMed]
- Lazarus, J.V.; Mark, H.E.; Anstee, Q.M.; Arab, J.P.; Batterham, R.L.; Castera, L.; Cortez-Pinto, H.; Crespo, J.; Cusi, K.; Dirac, M.A.; et al. Advancing the global public health agenda for NAFLD: A consensus statement. Nat. Rev. Gastroenterol. Hepatol. 2021. Epub ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, P.; Hellerbrand, C. Non-alcoholic fatty liver disease, obesity and the metabolic syndrome. Best Pract. Res. Clin. Gastroenterol. 2014, 28, 637–653. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, K.; Abrams, G.A. Metabolic liver disease of obesity and role of adipose tissue in the pathogenesis of nonalcoholic fatty liver disease. World J. Gastroenterol. 2007, 13, 3540–3553. [Google Scholar] [CrossRef] [Green Version]
- Van der Poorten, D.; Milner, K.-L.; Hui, J.; Hodge, A.; Trenell, M.I.; Kench, J.G.; London, R.; Peduto, T.; Chisholm, D.J.; George, J. Visceral fat: A key mediator of steatohepatitis in metabolic liver disease. Hepatology 2008, 48, 449–457. [Google Scholar] [CrossRef]
- Petta, S.; Amato, M.C.; Di Marco, V.; Cammà, C.; Pizzolanti, G.; Barcellona, M.R.; Cabibi, D.; Galluzzo, A.; Sinagra, D.; Giordano, C.; et al. Visceral adiposity index is associated with significant fibrosis in patients with non-alcoholic fatty liver disease. Aliment. Pharmacol. Ther. 2012, 35, 238–247. [Google Scholar] [CrossRef]
- Facciorusso, A. The influence of diabetes in the pathogenesis and the clinical course of hepatocellular carcinoma: Recent findings and new perspectives. Curr. Diabetes. Rev. 2013, 9, 382–386. [Google Scholar] [CrossRef] [PubMed]
- Garg, K.; Brackett, S.; Hirsch, I.; Garg, S. NAFLD/NASH and Diabetes. Diabetes Technol. Ther. 2020, 22, 174–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pang, Y.; Kartsonaki, C.; Turnbull, I.; Guo, Y.; Clarke, R.; Chen, Y.; Bragg, F.; Yang, L.; Bian, Z.; Millwood, I.; et al. Diabetes, Plasma Glucose, and Incidence of Fatty Liver, Cirrhosis, and Liver Cancer: A Prospective Study of 0.5 Million People. Hepatology 2018, 68, 1308–1318. [Google Scholar] [CrossRef] [PubMed]
- Lonardo, A.; Nascimbeni, F.; Mantovani, A.; Targher, G. Hypertension, diabetes, atherosclerosis and NASH: Cause or consequence? J. Hepatol. 2018, 68, 335–352. [Google Scholar] [CrossRef] [PubMed]
- Maciejewska-Markiewicz, D.; Stachowska, E.; Hawryłkowicz, V.; Stachowska, L.; Prowans, P. The role of resolvins, protectins and marensins in non-alcoholic fatty liver disease (NAFLD). Biomolecules 2021, 11, 937. [Google Scholar] [CrossRef]
- Schuppan, D.; Surabattula, R.; Wang, X.Y. Determinants of fibrosis progression and regression in NASH. J. Hepatol. 2018, 68, 238–250. [Google Scholar] [CrossRef] [PubMed]
- Charlton, M.; Krishnan, A.; Viker, K.; Sanderson, S.; Cazanave, S.; McConico, A.; Masuoko, H.; Gores, G. Fast food diet mouse: Novel small animal model of NASH with ballooning, pro413 gressive fibrosis, and high physiological fidelity to the human condition. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 301, G825–G834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neuschwander-Tetri, B.A. Lifestyle modification as the primary treatment of NASH. Clin. Liver. Dis. 2009, 13, 649–665. [Google Scholar] [CrossRef]
- Guerrero, R.; Vega, G.L.; Grundy, S.M.; Browning, J.D. Ethnic differences in hepatic steatosis: An insulin resistance paradox? Hepatology 2009, 49, 791–801. [Google Scholar] [CrossRef] [Green Version]
- Dongiovanni, P.; Anstee, Q.M.; Valenti, L. Genetic predisposition in NAFLD and NASH: Impact on severity of liver disease and response to treatment. Curr. Pharm. Des. 2013, 19, 5219–5238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dludla, P.V.; Nkambule, B.B.; Mazibuko-Mbeje, S.E.; Nyambuya, T.M.; Marcheggiani, F.; Cirilli, I.; Ziqubu, K.; Shabalala, S.C.; Johnson, R.; Louw, J.; et al. N-Acetyl Cysteine Targets Hepatic Lipid Accumulation to Curb Oxidative Stress and Inflammation in NAFLD: A Comprehensive Analysis of the Literature. Antioxidants 2020, 9, 1283. [Google Scholar] [CrossRef]
- Pierantonelli, I.; Svegliati-Baroni, G. Nonalcoholic Fatty Liver Disease. Transplantation 2019, 103, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Lara-Castro, C.; Garvey, W. Intracellular Lipid Accumulation in Liver and Muscle and the Insulin Resistance Syndrome. Endocrinol. Metabol. Clin. North. Am. 2008, 37, 841–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arner, P. The adipocyte in insulin resistance: Key molecules and the impact of the thiazolidinediones. Trends Endocrinol. Metab. 2003, 14, 137–145. [Google Scholar] [CrossRef]
- Samuel, V.T.; Shulman, G.I. Mechanisms for insulin resistance: Common threads and missing links. Cells 2012, 148, 852–871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aboubakr, A.; Stroud, A.; Kumar, S.; Newberry, C. Dietary approaches for management of non-alcoholic fatty liver disease: A clinician’s guide. Curr. Gastroenterol. Rep. 2021, 23, 21. [Google Scholar] [CrossRef] [PubMed]
- Al-Busafi, S.A.; Bhat, M.; Wong, P.; Ghali, P.; Deschenes, M. Antioxidant Therapy in Nonalcoholic Steatohepatitis. Hepat. Res. Treat. 2012, 2012, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Browning, J.D.; Horton, J.D. Molecular mediators of hepatic steatosis and liver injury. J. Clin. Investig. 2004, 114, 147–152. [Google Scholar] [CrossRef] [Green Version]
- Brunt, E.M.; Kleiner, D.E.; Wilson, L.A.; Belt, P.; Neuschwander-Tetri, B.A. For the NASH Clinical Research Network (CRN) Nonalcoholic fatty liver disease (NAFLD) activity score and the histopathologic diagnosis in NAFLD: Distinct clinicopathologic meanings. Hepatology 2010, 53, 810–820. [Google Scholar] [CrossRef] [Green Version]
- George, D.; Goldwurm, S.; Macdonald, G.A.; Cowley, L.L.; Walker, N.I.; Ward, P.J.; Jazwinska, E.C.; Powell, L.W. Increased hepatic iron concentration in nonalcoholic steatohepatitis is associated with increased fibrosis. Gastroenterology 1998, 114, 311–318. [Google Scholar] [CrossRef]
- Zangar, R.C.; Novak, R.F. Effects of Fatty Acids and Ketone Bodies on Cytochromes P450 2B, 4A, and 2E1 Expression in Primary Cultured Rat Hepatocytes. Arch. Biochem. Biophys. 1997, 337, 217–224. [Google Scholar] [CrossRef]
- Ockner, R.K.; Kaikus, R.M.; Bass, N.M. Fatty-acid metabolism and the pathogenesis of hepatocellular carcinoma: Review and hypothesis. Hepatology 1993, 18, 669–676. [Google Scholar] [CrossRef]
- Ore, A.; Akinloye, O.A. Oxidative stress and antioxidant biomarkers in clinical and experimental models of non-alcoholic fatty liver disease. Medicina 2019, 55, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gentric, G.; Maillet, V.; Paradis, V.; Couton, D.; L’Hermitte, A.; Panasyuk, G.; Fromenty, B.; Celton-Morizur, S.; Desdouets, C. Oxidative stress promotes pathologic polyploidization in nonalcoholic fatty liver disease. J. Clin. Investig. 2015, 125, 981–992. [Google Scholar] [CrossRef] [Green Version]
- Feldstein, A.E.; Werneburg, N.W.; Canbay, A.; Guicciardi, M.E.; Bronk, S.F.; Rydzewski, R.; Burgart, L.J.; Gores, G.J. Free fatty acids promote hepatic lipotoxicity by stimulating TNF-alpha expression via a lysosomal pathway. Hepatology 2004, 40, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Boucher, J.; Kleinridders, A.; Kahn, C.R. Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb. Perspect. Biol. 2014, 6, a009191. [Google Scholar] [CrossRef] [Green Version]
- Lu, M.; Wan, M.; Leavens, K.F.; Chu, Q.; Monks, B.R.; Fernandez, S.; Ahima, R.S.; Ueki, K.; Kahn, C.R.; Birnbaum, M.J. Insulin regulates liver metabolism in vivo in the absence of hepatic Akt and Foxo1. Nat. Med. 2012, 18, 388–395. [Google Scholar] [CrossRef] [PubMed]
- Russell, J.O.; Monga, S.P. Wnt/β-Catenin Signaling in Liver Development, Homeostasis, and Pathobiology. Annu. Rev. Pathol. 2018, 13, 351–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elsayed, H.R.H.; El-Nablaway, M.; Khattab, B.A.; Sherif, R.N.; Elkashef, W.F.; Abdalla, A.M.; El Nashar, E.M.; Abd-Elmonem, M.M.; El-Gamal, R. Independent of Calorie Intake, Short-term Alternate-day Fasting Alleviates NASH, With Modulation of Markers of Lipogenesis, Autophagy, Apoptosis, and Inflammation in Rats. J. Histochem. Cytochem. 2021, 69, 575–596. [Google Scholar] [CrossRef]
- Liu, L.; Liao, J.Z.; He, X.X.; Li, P.Y. The role of autophagy in hepatocellular carcinoma: Friend or foe. Oncotarget 2017, 8, 57707–57722. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, Y.; Yu, F.; Wang, J.; Guo, C.; Fan, X. Autophagy: A new target for nonalcoholic fatty liver disease therapy. Hepat. Med. 2016, 8, 27–37. [Google Scholar] [CrossRef] [Green Version]
- Park, E.J.; Lee, J.H.; Yu, G.Y.; He, G.; Ali, S.R.; Holzer, R.G.; Karin, M. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell 2010, 140, 197–208. [Google Scholar] [CrossRef] [Green Version]
- Highton, A.J.; Schuster, I.S.; Degli-Esposti, M.A.; Altfeld, M. The role of natural killer cells in liver inflammation. Semin. Immunopathol. 2021, 43, 1–15. [Google Scholar] [CrossRef]
- Tosello-Trampont, A.C.; Krueger, P.; Narayanan, S.; Landes, S.G.; Leitinger, N.; Hahn, Y.S. NKp46(+) natural killer cells attenuate metabolism-induced hepatic fibrosis by regulating macrophage activation in mice. Hepatology 2016, 63, 799–812. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Chantar, M.L.; Delgado, T.C.; Beraza, N. Revisiting the Role of Natural Killer Cells in Non-Alcoholic Fatty Liver Disease. Front. Immunol. 2021, 12, 640869. [Google Scholar] [CrossRef]
- Martin-Murphy, B.V.; You, Q.; Wang, H.; Becky, A.; Reilly, T.P.; Friedman, J.E.; Ju, C. Mice lacking natural killer T cells are more susceptible to metabolic alterations following high fat diet feeding. PLoS ONE 2014, 9, e80949. [Google Scholar]
- Fan, Y.; Zhang, W.; Wei, H.; Sun, R.; Tian, Z.; Chen, Y. Hepatic NK cells attenuate fibrosis progression of non-alcoholic steatohepatitis in dependent of CXCL10-mediated recruitment. Liver Int. 2020, 40, 598–608. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Zeng, W.; Gai, X.; Xu, Q.; Li, C.; Liang, Z.; Liu, Q. Role of the Hedgehog pathway in hepatocellular carcinoma. Oncol. Rep. 2013, 30, 2020–2026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czaja, A.J. Incorporating mucosal-associated invariant T cells into the pathogenesis of chronic liver disease. World J. Gastroenterol. 2021, 27, 3705–3733. [Google Scholar] [CrossRef]
- Bolte, F.J.; Rehermann, B. Mucosal-Associated Invariant T Cells in Chronic Inflammatory Liver Disease. Liver. Dis. 2018, 38, 60–65. [Google Scholar] [CrossRef]
- Li, Y.; Huang, B.; Jiang, X.; Chen, W.; Zhang, J.; Wei, Y.; Chen, Y.; Lian, M.; Bian, Z.; Miao, Q.; et al. Mucosal-Associated Invariant T Cells Improve Nonalcoholic Fatty Liver Disease Through Regulating Macrophage Polarization. Front. Immunol. 2018, 9, 1994. [Google Scholar] [CrossRef]
- Hegde, P.; Weiss, E.; Paradis, V.; Wan, J.; Mabire, M.; Sukriti, S.; Rautou, P.E.; Albuquerque, M.; Picq, O.; Gupta, A.C.; et al. Mucosal-associated invariant T cells are a profibrogenic immune cell population in the liver. Nat. Commun. 2018, 9, 2146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singal, A.G.; Manjunath, H.; Yopp, A.C.; Beg, M.S.; Marrero, J.A.; Gopal, P.; Waljee, A.K. The Effect of PNPLA3 on Fibrosis Progression and Development of Hepatocellular Carcinoma: A Meta-analysis. Am. J. Gastroenterol. 2014, 109, 325–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sookoian, S.; Pirola, C.J. Meta-analysis of the influence of I148M variant of patatin-like phospholipase domain containing 3 gene (PNPLA3) on the susceptibility and histological severity of nonalcoholic fatty liver disease. Hepatology 2011, 53, 1883–1894. [Google Scholar] [CrossRef] [PubMed]
- Bruschi, F.; Tardelli, M.; Claudel, T.; Trauner, M. PNPLA3 expression and its impact on the liver: Current perspectives. Hepatic Med. Evid. Res. 2017, 9, 55–66. [Google Scholar] [CrossRef] [Green Version]
- Pingitore, P.; Romeo, S. The role of PNPLA3 in health and disease. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2019, 1864, 900–906. [Google Scholar] [CrossRef]
- Baulande, S.; Lasnier, F.; Lucas, M.; Pairault, J. Adiponutrin, a Transmembrane Protein Corresponding to a Novel Dietary- and Obesity-linked mRNA Specifically Expressed in the Adipose Lineage. J. Biol. Chem. 2001, 276, 33336–33344. [Google Scholar] [CrossRef] [Green Version]
- Hao, L.; Ito, K.; Huang, K.H.; Sae-tan, S.; Lambert, J.D.; Ross, A.C. Shifts in dietary carbohydrate-lipid exposure regulate expression of the non-alcoholic fatty liver disease-associated gene PNPLA3/adiponutrin in mouse liver and HepG2 human liver cells. Metabolism 2014, 63, 1352–1362. [Google Scholar] [CrossRef] [Green Version]
- Moldes, M.; Beauregard, G.; Faraj, M.; Peretti, N.; Ducluzeau, P.-H.; Laville, M.; Rabasa-Lhoret, R.; Vidal, H.; Clément, K. Adiponutrin gene is regulated by insulin and glucose in human adipose tissue. Eur. J. Endocrinol. 2006, 155, 461–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; He, S.; Li, J.Z.; Seo, Y.-K.; Osborne, T.F.; Cohen, J.C.; Hobbs, H.H. A feed-forward loop amplifies nutritional regulation of PNPLA3. Proc. Natl. Acad. Sci. USA 2010, 107, 7892–7897. [Google Scholar] [CrossRef] [Green Version]
- Dubuquoy, C.; Robichon, C.; Lasnier, F.; Langlois, C.; Dugail, I.; Foufelle, F.; Girard, J.; Burnol, A.-F.; Postic, C.; Moldes, M. Distinct regulation of adiponutrin/PNPLA3 gene expression by the transcription factors ChREBP and SREBP1c in mouse and human hepatocytes. J. Hepatol. 2011, 55, 145–153. [Google Scholar] [CrossRef]
- Perttilä, J.; Huaman-Samanez, C.; Caron, S.; Tanhuanpää, K.; Staels, B.; Yki-Järvinen, H.; Olkkonen, V.M. PNPLA3 is regulated by glucose in human hepatocytes, and its I148M mutant slows down triglyceride hydrolysis. Am. J. Physiol. Endocrinol. Metab. 2012, 302, E1063–E1069. [Google Scholar] [CrossRef] [PubMed]
- Loria, P.; Carulli, L.; Bertolotti, M.; Lonardo, A. Endocrine and liver interaction: The role of endocrine pathways in NASH. Nat. Rev. Gastroenterol. Hepatol. 2009, 6, 236–247. [Google Scholar] [CrossRef]
- Sun, H.; Yang, W.; Tian, Y.; Zeng, X.; Zhou, J.; Mok, M.T.; Cheng, A.S. An inflammatory-CCRK circuitry drives mTORC1-dependent metabolic and immunosuppressive reprogramming in obesity-associated hepatocellular carcinoma. Nat. Commun. 2018, 9, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Yu, Z.; Tian, Y.; Lee, Y.Y.; Li, M.S.; Go Cheng, M.Y.; Cheung, Y.S.; Lai, P.B.; Chan, A.M.; To, K.F.; et al. A CCRK-EZH2 epigenetic circuitry drives hepatocarcinogenesis and associates with tumor recurrence and poor survival of patients. J. Hepatol. 2015, 62, 1100–1111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagasue, N.; Yu, L.; Yukaya, H.; Kohno, H.; Nakamura, T. Androgen and oestrogen receptors in hepatocellular carcinoma and surrounding liver parenchyma: Impact on intrahepatic recurrence after hepatic resection. Br. J. Surg. 1995, 82, 542–547. [Google Scholar] [CrossRef] [PubMed]
- Awuah, P.K.; Monga, S.P. Cell cycle–related kinase links androgen receptor and β-catenin signaling in hepatocellular carcinoma: Why are men at a loss? Hepatology 2012, 55, 970–974. [Google Scholar] [CrossRef] [Green Version]
- Arslan, N. Obesity, fatty liver disease and intestinal microbiota. World J. Gastroenterol. 2014, 20, 16452–16463. [Google Scholar] [CrossRef] [PubMed]
- Machado, M.V.; Cortez-Pinto, H. Gut microbiota and nonalcoholic fatty liver disease. Ann. Hepatol. 2012, 11, 440–449. [Google Scholar] [CrossRef]
- Albhaisi, S.A.M.; Bajaj, J.S.; Sanyal, A.J. Role of gut microbiota in liver disease. Am. J. Physiol. Gastrointest. Liver. Physiol. 2019, 318, G84–G98. [Google Scholar] [CrossRef] [Green Version]
- Bajaj, J.S.; Betrapally, N.S.; Hylemon, P.B.; Thacker, L.R.; Daita, K.; Kang, D.J.; White, M.B.; Unser, A.B.; Fagan, A.; Gavis, E.A.; et al. Gut microbiota alterations can predict hospitalizations in cirrhosis independent of diabetes mellitus. Sci. Rep. 2015, 5, 18559. [Google Scholar] [CrossRef] [Green Version]
- Albhaisi, S.A.M.; Bajaj, J.S. The Influence of the Microbiome on NAFLD and NASH. Clin. Liver. Dis. 2021, 17, 15–18. [Google Scholar] [CrossRef] [PubMed]
- Neuman, M.G.; Cohen, L.B.; Nanau, R.M. Biomarkers in nonalcoholic fatty liver disease. Can. J. Gastroenterol. Hepatol. 2014, 28, 607–618. [Google Scholar] [CrossRef] [PubMed]
- Kwok, R.; Tse, Y.K.; Wong, G.L.; Ha, Y.; Lee, A.U.; Ngu, M.C.; Chan, H.L.; Wong, V.W. Systematic review with meta-analysis: Non-invasive assessment of non-alcoholic fatty liver disease--the role of transient elastography and plasma cytokeratin-18 fragments. Aliment. Pharmacol. Ther. 2014, 39, 254–269. [Google Scholar] [CrossRef]
- Manousou, P.; Kalambokis, G.; Grillo, F.; Watkins, J.; Xirouchakis, E.; Pleguezuelo, M.; Leandro, G.; Arvaniti, V.; Germani, G.; Patch, D.; et al. Serum ferritin is a discriminant marker for both fibrosis and inflammation in histologically proven non-alcoholic fatty liver disease patients. Liver Int. 2011, 31, 730–739. [Google Scholar] [CrossRef] [PubMed]
- Roskams, T.; Yang, S.Q.; Koteish, A.; Durnez, A.; DeVos, R.; Huang, X.; Achten, R.; Verslype, C.; Diehl, A.M. Oxidative stress and oval cell accumulation in mice and humans with alcoholic and nonalcoholic fatty liver disease. Am. J. Pathol. 2003, 163, 1301–1311. [Google Scholar] [CrossRef] [Green Version]
- Puri, P.; Wiest, M.M.; Cheung, O.; Mirshahi, F.; Sargeant, C.; Min, H.K.; Contos, M.J.; Sterling, R.K.; Fuchs, M.; Zhou, H. The plasma lipidomic signature of nonalcoholic steatohepatitis. Hepatology 2009, 50, 1827–1838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lkhouri, N.; Berk, M.; Yerian, L.; Lopez, R.; Chung, Y.M.; Zhang, R.; McIntyre, T.M.; Feldstein, A.E.; Hazen, S.L. OxNASH score correlates with histologic features and severity of nonalcoholic fatty liver disease. Dig. Dis. Sci. 2014, 59, 1617–1624. [Google Scholar] [CrossRef] [PubMed]
- Armutcu, F.; Akyol, S.; Ucar, F.; Erdogan, S.; Akyol, O. Markers in nonalcoholic steatohepatitis. Adv. Clin. Chem. 2013, 61, 67–125. [Google Scholar] [PubMed]
- Polyzos, S.A.; Toulis, K.A.; Goulis, D.G.; Zavos, C.; Kountouras, J. Serum total adiponectin in nonalcoholic fatty liver disease: A systematic review and meta-analysis. Metabolism 2011, 60, 313–326. [Google Scholar] [CrossRef]
- Ozcelik, F.; Yuksel, C.; Arslan, E.; Genc, S.; Omer, B.; Serdar, M.A. Relationship between visceral adipose tissue and adiponectin, inflammatory markers and thyroid hormones in obese males with hepatosteatosis and insulin resistance. Arch. Med. Res. 2013, 44, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Sheth, S.G.; Flamm, S.L.; Gordon, F.D.; Chopra, S. AST/ALT ratio predicts cirrhosis in patients with chronic hepatitis C virus infection. Am. J. Gastroenterol. 1998, 93, 44–48. [Google Scholar] [CrossRef] [PubMed]
- Facciorusso, A.; Del Prete, V.; Turco, A.; Buccino, R.V.; Nacchiero, M.C.; Muscatiello, N. Long-term liver stiffness assessment in hepatitis C virus patients undergoing antiviral therapy: Results from a 5-year cohort study. J. Gastroenterol. Hepatol. 2018, 33, 942–949. [Google Scholar] [CrossRef] [PubMed]
- Facciorusso, A.; Garcia Perdomo, H.A.; Muscatiello, N.; Buccino, R.V.; Wong, V.W.; Singh, S. Systematic review with meta-analysis: Change in liver stiffness during anti-viral therapy in patients with hepatitis B. Dig. Liver Dis. 2018, 50, 787–794. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Facciorusso, A.; Loomba, R.; Falck-Ytter, Y.T. Magnitude and Kinetics of Decrease in Liver Stiffness After Antiviral Therapy in Patients with Chronic Hepatitis C: A Systematic Review and Meta-analysis. Clin. Gastroenterol. Hepatol. 2018, 16, 27–38. [Google Scholar] [CrossRef] [Green Version]
- Bellanti, F.; Villani, R.; Tamborra, R.; Blonda, M.; Iannelli, G.; di Bello, G.; Facciorusso, A.; Poli, G.; Iuliano, L.; Avolio, C.; et al. Synergistic interaction of fatty acids and oxysterols impairs mitochondrial function and limits liver adaptation during nafld progression. Redox Biol. 2018, 15, 86–96. [Google Scholar] [CrossRef] [PubMed]
- Sterling, R.K.; Lissen, E.; Clumeck, N.; Sola, R.; Correa, M.C.; Montaner, J.S.; Sulkowski, M.; Torriani, F.J.; Dieterich, D.T.; Thomas, D.L.; et al. APRICOT Clinical Investigators. Development of a simple noninvasive index to predict significant fibrosis in patients with HIV/HCV coinfection. Hepatology 2006, 43, 1317–1325. [Google Scholar] [CrossRef] [PubMed]
- Mózes, F.E.; Lee, J.A.; Selvaraj, E.A.; Jayaswal, A.; Trauner, M.; Boursier, J.; Fournier, C.; Staufer, K.; Stauber, R.E.; Bugianesi, E.; et al. Diagnostic accuracy of non-invasive tests for advanced fibrosis in patients with NAFLD: An individual patient data meta-analysis. Gut 2021. Online ahead of print. [Google Scholar] [CrossRef]
- Loomba, R. Role of imaging-based biomarkers in NAFLD: Recent advances in clinical application and future research directions. J. Hepatol. 2018, 68, 296–304. [Google Scholar] [CrossRef] [PubMed]
- Neuman, M.G.; Cohen, L.B.; Nanau, R.M. Hyaluronic acid as a non-invasive biomarker of liver fibrosis. Clin. Biochem. 2016, 49, 302–315. [Google Scholar] [CrossRef]
- Poynard, T.; Ratziu, V.; Charlotte, F.; Messous, D.; Munteanu, M.; Imbert-Bismut, F.; Massard, J.; Bonyhay, L.; Tahiri, M.; Thabut, D.; et al. Diagnostic value of biochemical markers (NashTest) for the prediction of non alcoholo steato hepatitis in patients with non-alcoholic fatty liver disease. BMC Gastroenterol. 2006, 6, 34. [Google Scholar] [CrossRef] [Green Version]
- Younossi, Z.M.; Jarrar, M.; Nugen, C.; Randhawa, M.; Afendy, M.; Stepanova, M.; Rafiq, N.; Goodman, Z.; Chandhoke, V.; Baranova, A. A novel diagnostic biomarker panel for obesity-related nonalcoholic steatohepatitis (NASH). Obes. Surg. 2008, 18, 1430–1437. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramai, D.; Facciorusso, A.; Vigandt, E.; Schaf, B.; Saadedeen, W.; Chauhan, A.; di Nunzio, S.; Shah, A.; Giacomelli, L.; Sacco, R. Progressive Liver Fibrosis in Non-Alcoholic Fatty Liver Disease. Cells 2021, 10, 3401. https://doi.org/10.3390/cells10123401
Ramai D, Facciorusso A, Vigandt E, Schaf B, Saadedeen W, Chauhan A, di Nunzio S, Shah A, Giacomelli L, Sacco R. Progressive Liver Fibrosis in Non-Alcoholic Fatty Liver Disease. Cells. 2021; 10(12):3401. https://doi.org/10.3390/cells10123401
Chicago/Turabian StyleRamai, Daryl, Antonio Facciorusso, Erika Vigandt, Bryan Schaf, Waleed Saadedeen, Aditya Chauhan, Sara di Nunzio, Aashni Shah, Luca Giacomelli, and Rodolfo Sacco. 2021. "Progressive Liver Fibrosis in Non-Alcoholic Fatty Liver Disease" Cells 10, no. 12: 3401. https://doi.org/10.3390/cells10123401
APA StyleRamai, D., Facciorusso, A., Vigandt, E., Schaf, B., Saadedeen, W., Chauhan, A., di Nunzio, S., Shah, A., Giacomelli, L., & Sacco, R. (2021). Progressive Liver Fibrosis in Non-Alcoholic Fatty Liver Disease. Cells, 10(12), 3401. https://doi.org/10.3390/cells10123401