
The Novel Monoacylglycerol Lipase Inhibitor MJN110 Suppresses Neuroinflammation, Normalizes Synaptic Composition and Improves Behavioral Performance in the Repetitive Traumatic Brain Injury Mouse Model


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
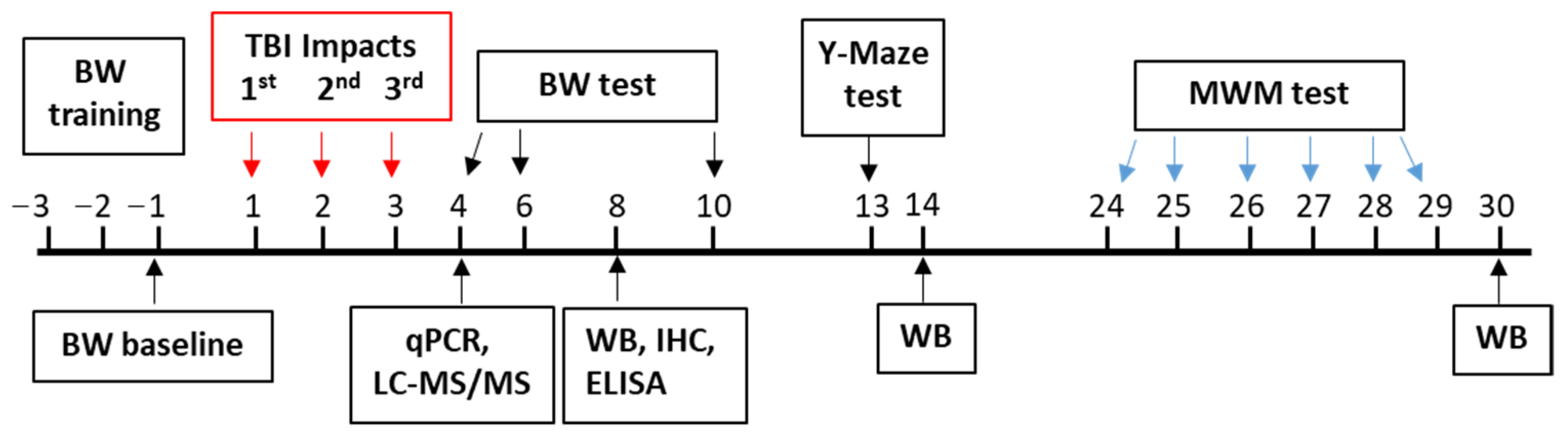
2.2. Animal Model and Drug Treatment
2.3. Behavioral Assay
2.3.1. Beam Walk Test
2.3.2. Spontaneous Alternation Y-Maze Test
2.3.3. Morris Water Maze Test
2.4. Quantitative Real-Time PCR
2.5. PGE2 Assay
2.6. Western Blot
2.7. LC-MS/MS Analysis
2.8. Immunohistochemistry
2.9. Fluoro-Jade C Staining
2.10. Statistical Analysis
3. Results
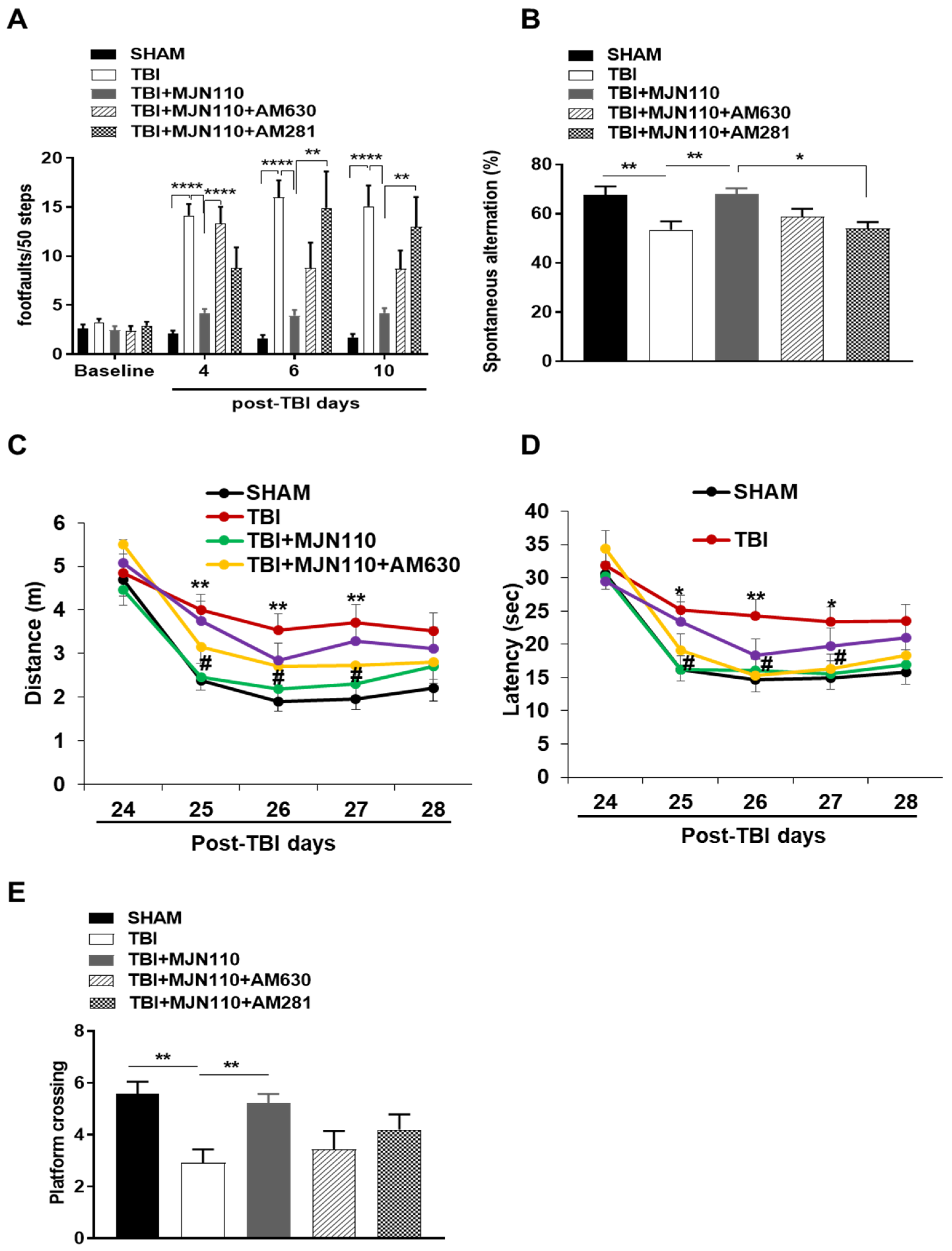
3.1. Locomotor and Working Memory Deficits Associated with Repetitive mTBI Were Mitigated by the MAGL Inhibition
3.2. MAGL Inhibition Selectively Elevated the Endogenous 2-AG Levels in the TBI Mouse Brain
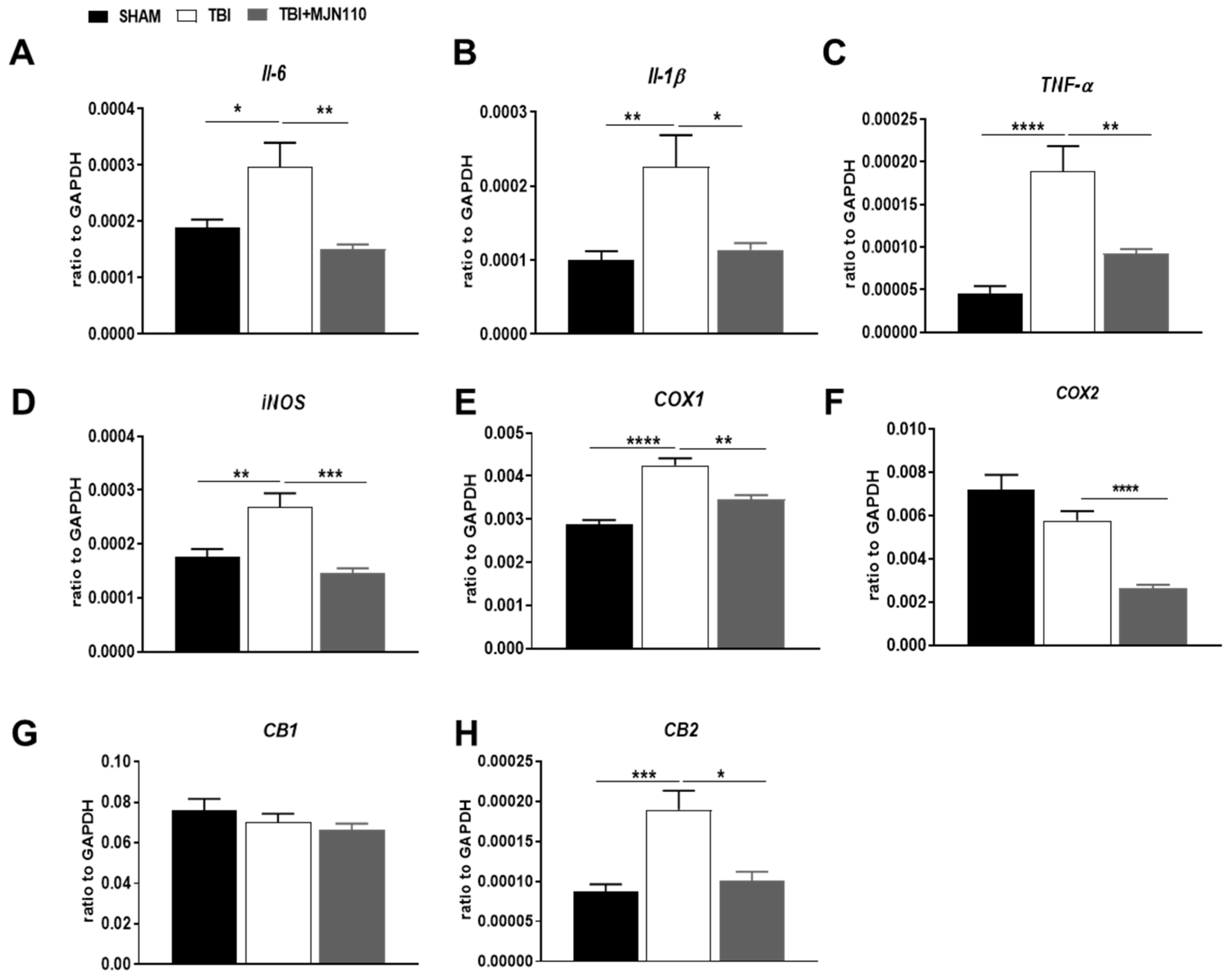
3.3. Inhibition of MAGL Attenuated TBI-Induced Neuroinflammation and Neuronal Cell Death
3.4. Alteration of the Ionotropic Glutamate and GABA Receptor Components and the Survival Signaling Molecules in the TBI Mouse Hippocampus Was Restored by MAGL Inhibition
3.5. Reversal of Locomotor and Short-Term Working Memory, but Not Long-Term Cognitive Function by MJN110 Was Mediated by the CB1 Receptor Activation
3.6. Inhibition of MAGL Normalized the Expression of Ionotropic Glutamate and GABA Receptors at 30 Days Post-TBI
3.7. The Therapeutic Effects of MJN110 Were Eliminated by Simultaneous Blockade of 2-AG Synthesis in the TBI Mouse Brain
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Reilly, P. The impact of neurotrauma on society: An international perspective. Prog. Brain Res. 2007, 161, 3–9. [Google Scholar] [CrossRef]
- Faul, M.; Coronado, V. Epidemiology of traumatic brain injury. Handb. Clin. Neurol. 2015, 127, 3–13. [Google Scholar] [CrossRef]
- Hobbs, J.G.; Young, J.S.; Bailes, J.E. Sports-related concussions: Diagnosis, complications, and current management strategies. Neurosurg. Focus 2016, 40, E5. [Google Scholar] [CrossRef]
- Lindberg, M.A.; Kiser, S.A.; Moy Martin, E.M. Mild TBI/Concussion Clinical Tools for Providers Used Within the Department of Defense and Defense Health Agency. Fed. Pract. 2020, 37, 410–419. [Google Scholar] [CrossRef] [PubMed]
- Johnson, V.E.; Stewart, W.; Arena, J.D.; Smith, D.H. Traumatic Brain Injury as a Trigger of Neurodegeneration. Adv. Neurobiol. 2017, 15, 383–400. [Google Scholar] [CrossRef]
- Pertwee, R.G. Elevating endocannabinoid levels: Pharmacological strategies and potential therapeutic applications. Proc. Nutr. Soc. 2014, 73, 96–105. [Google Scholar] [CrossRef] [Green Version]
- Hill, M.N.; Patel, S. Translational evidence for the involvement of the endocannabinoid system in stress-related psychiatric illnesses. Biol. Mood Anxiety Disord. 2013, 3, 19. [Google Scholar] [CrossRef]
- Piomelli, D. The molecular logic of endocannabinoid signalling. Nat. Rev. Neurosci. 2003, 4, 873–884. [Google Scholar] [CrossRef] [Green Version]
- Mackie, K. Cannabinoid receptors as therapeutic targets. Annu. Rev. Pharmacol. Toxicol. 2006, 46, 101–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Marzo, V. The endocannabinoid system: Its general strategy of action, tools for its pharmacological manipulation and potential therapeutic exploitation. Pharmacol. Res. 2009, 60, 77–84. [Google Scholar] [CrossRef]
- Hwang, J.; Adamson, C.; Butler, D.; Janero, D.R.; Makriyannis, A.; Bahr, B.A. Enhancement of endocannabinoid signaling by fatty acid amide hydrolase inhibition: A neuroprotective therapeutic modality. Life Sci. 2010, 86, 615–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ueda, N.; Tsuboi, K.; Uyama, T.; Ohnishi, T. Biosynthesis and degradation of the endocannabinoid 2-arachidonoylglycerol. Biofactors 2011, 37, 1–7. [Google Scholar] [CrossRef]
- Tchantchou, F.; Tucker, L.B.; Fu, A.H.; Bluett, R.J.; McCabe, J.T.; Patel, S.; Zhang, Y. The fatty acid amide hydrolase inhibitor PF-3845 promotes neuronal survival, attenuates inflammation and improves functional recovery in mice with traumatic brain injury. Neuropharmacology 2014, 85, 427–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tchantchou, F.; Zhang, Y. Selective inhibition of alpha/beta-hydrolase domain 6 attenuates neurodegeneration, alleviates blood brain barrier breakdown, and improves functional recovery in a mouse model of traumatic brain injury. J. Neurotrauma 2013, 30, 565–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Teng, Z.; Song, Y.; Hu, M.; Chen, C. Inhibition of monoacylglycerol lipase prevents chronic traumatic encephalopathy-like neuropathology in a mouse model of repetitive mild closed head injury. J. Cereb. Blood Flow Metab. 2015, 35, 443–453. [Google Scholar] [CrossRef]
- Katz, P.S.; Sulzer, J.K.; Impastato, R.A.; Teng, S.X.; Rogers, E.K.; Molina, P.E. Endocannabinoid degradation inhibition improves neurobehavioral function, blood-brain barrier integrity, and neuroinflammation following mild traumatic brain injury. J. Neurotrauma 2015, 32, 297–306. [Google Scholar] [CrossRef] [Green Version]
- Mayeux, J.; Katz, P.; Edwards, S.; Middleton, J.W.; Molina, P.E. Inhibition of Endocannabinoid Degradation Improves Outcomes from Mild Traumatic Brain Injury: A Mechanistic Role for Synaptic Hyperexcitability. J. Neurotrauma 2017, 34, 436–443. [Google Scholar] [CrossRef]
- Selvaraj, P.; Wen, J.; Tanaka, M.; Zhang, Y. Therapeutic Effect of a Novel Fatty Acid Amide Hydrolase Inhibitor PF04457845 in the Repetitive Closed Head Injury Mouse Model. J. Neurotrauma 2019, 36, 1655–1669. [Google Scholar] [CrossRef] [PubMed]
- Schurman, L.D.; Lichtman, A.H. Endocannabinoids: A Promising Impact for Traumatic Brain Injury. Front. Pharmacol. 2017, 8, 69. [Google Scholar] [CrossRef] [Green Version]
- Panikashvili, D.; Simeonidou, C.; Ben-Shabat, S.; Hanus, L.; Breuer, A.; Mechoulam, R.; Shohami, E. An endogenous cannabinoid (2-AG) is neuroprotective after brain injury. Nature 2001, 413, 527–531. [Google Scholar] [CrossRef]
- Panikashvili, D.; Shein, N.A.; Mechoulam, R.; Trembovler, V.; Kohen, R.; Alexandrovich, A.; Shohami, E. The endocannabinoid 2-AG protects the blood-brain barrier after closed head injury and inhibits mRNA expression of proinflammatory cytokines. Neurobiol. Dis. 2006, 22, 257–264. [Google Scholar] [CrossRef]
- Shohami, E.; Cohen-Yeshurun, A.; Magid, L.; Algali, M.; Mechoulam, R. Endocannabinoids and traumatic brain injury. Br. J. Pharmacol. 2011, 163, 1402–1410. [Google Scholar] [CrossRef] [Green Version]
- Nomura, D.K.; Morrison, B.E.; Blankman, J.L.; Long, J.Z.; Kinsey, S.G.; Marcondes, M.C.; Ward, A.M.; Hahn, Y.K.; Lichtman, A.H.; Conti, B.; et al. Endocannabinoid hydrolysis generates brain prostaglandins that promote neuroinflammation. Science 2011, 334, 809–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niphakis, M.J.; Cognetta, A.B., 3rd; Chang, J.W.; Buczynski, M.W.; Parsons, L.H.; Byrne, F.; Burston, J.J.; Chapman, V.; Cravatt, B.F. Evaluation of NHS carbamates as a potent and selective class of endocannabinoid hydrolase inhibitors. ACS Chem. Neurosci. 2013, 4, 1322–1332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ignatowska-Jankowska, B.; Wilkerson, J.L.; Mustafa, M.; Abdullah, R.; Niphakis, M.; Wiley, J.L.; Cravatt, B.F.; Lichtman, A.H. Selective monoacylglycerol lipase inhibitors: Antinociceptive versus cannabimimetic effects in mice. J. Pharmacol. Exp. Ther. 2015, 353, 424–432. [Google Scholar] [CrossRef] [Green Version]
- Serrano, A.; Pavon, F.J.; Buczynski, M.W.; Schlosburg, J.; Natividad, L.A.; Polis, I.Y.; Stouffer, D.G.; Zorrilla, E.P.; Roberto, M.; Cravatt, B.F.; et al. Deficient endocannabinoid signaling in the central amygdala contributes to alcohol dependence-related anxiety-like behavior and excessive alcohol intake. Neuropsychopharmacology 2018, 43, 1840–1850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, S.H.; Arai, A.L.; Mou, Y.; Kang, B.; Yen, C.C.; Hallenbeck, J.; Silva, A.C. Neuroprotective Effects of MAGL (Monoacylglycerol Lipase) Inhibitors in Experimental Ischemic Stroke. Stroke 2018, 49, 718–726. [Google Scholar] [CrossRef]
- Fucich, E.A.; Stielper, Z.F.; Cancienne, H.L.; Edwards, S.; Gilpin, N.W.; Molina, P.E.; Middleton, J.W. Endocannabinoid degradation inhibitors ameliorate neuronal and synaptic alterations following traumatic brain injury. J. Neurophysiol. 2020, 123, 707–717. [Google Scholar] [CrossRef]
- Ogasawara, D.; Deng, H.; Viader, A.; Baggelaar, M.P.; Breman, A.; den Dulk, H.; van den Nieuwendijk, A.M.; Soethoudt, M.; van der Wel, T.; Zhou, J.; et al. Rapid and profound rewiring of brain lipid signaling networks by acute diacylglycerol lipase inhibition. Proc. Natl. Acad. Sci. USA 2016, 113, 26–33. [Google Scholar] [CrossRef] [Green Version]
- Yu, F.; Wang, Z.; Tchantchou, F.; Chiu, C.T.; Zhang, Y.; Chuang, D.M. Lithium ameliorates neurodegeneration, suppresses neuroinflammation, and improves behavioral performance in a mouse model of traumatic brain injury. J. Neurotrauma 2012, 29, 362–374. [Google Scholar] [CrossRef] [Green Version]
- Velosky, A.G.; Tucker, L.B.; Fu, A.H.; Liu, J.; McCabe, J.T. Cognitive performance of male and female C57BL/6J mice after repetitive concussive brain injuries. Behav. Brain Res. 2017, 324, 115–124. [Google Scholar] [CrossRef]
- Tanaka, K.; Yagi, T.; Shimakoshi, R.; Azuma, K.; Nanba, T.; Ogo, H.; Tamura, A.; Asanuma, M. Effects of galantamine on L-NAME-induced behavioral impairment in Y-maze task in mice. Neurosci. Lett. 2009, 462, 235–238. [Google Scholar] [CrossRef]
- Jassam, Y.N.; Izzy, S.; Whalen, M.; McGavern, D.B.; El Khoury, J. Neuroimmunology of Traumatic Brain Injury: Time for a Paradigm Shift. Neuron 2017, 95, 1246–1265. [Google Scholar] [CrossRef] [Green Version]
- Pearn, M.L.; Niesman, I.R.; Egawa, J.; Sawada, A.; Almenar-Queralt, A.; Shah, S.B.; Duckworth, J.L.; Head, B.P. Pathophysiology Associated with Traumatic Brain Injury: Current Treatments and Potential Novel Therapeutics. Cell Mol. Neurobiol. 2017, 37, 571–585. [Google Scholar] [CrossRef]
- Carvajal, F.J.; Mattison, H.A.; Cerpa, W. Role of NMDA Receptor-Mediated Glutamatergic Signaling in Chronic and Acute Neuropathologies. Neural Plast. 2016, 2016, 2701526. [Google Scholar] [CrossRef]
- Wu, C.; Sun, D. GABA receptors in brain development, function, and injury. Metab. Brain Dis. 2015, 30, 367–379. [Google Scholar] [CrossRef] [Green Version]
- Simon, D.W.; McGeachy, M.J.; Bayir, H.; Clark, R.S.; Loane, D.J.; Kochanek, P.M. The far-reaching scope of neuroinflammation after traumatic brain injury. Nat. Rev. Neurol. 2017, 13, 171–191. [Google Scholar] [CrossRef] [Green Version]
- Mechoulam, R.; Spatz, M.; Shohami, E. Endocannabinoids and neuroprotection. Sci. STKE 2002, 2002, re5. [Google Scholar] [CrossRef]
- Dash, P.K.; Mach, S.A.; Moore, A.N. The role of extracellular signal-regulated kinase in cognitive and motor deficits following experimental traumatic brain injury. Neuroscience 2002, 114, 755–767. [Google Scholar] [CrossRef]
- Zhang, C.; Zhu, J.; Zhang, J.; Li, H.; Zhao, Z.; Liao, Y.; Wang, X.; Su, J.; Sang, S.; Yuan, X.; et al. Neuroprotective and anti-apoptotic effects of valproic acid on adult rat cerebral cortex through ERK and Akt signaling pathway at acute phase of traumatic brain injury. Brain Res. 2014, 1555, 1–9. [Google Scholar] [CrossRef]
- Walker, C.L.; Liu, N.K.; Xu, X.M. PTEN/PI3K and MAPK signaling in protection and pathology following CNS injuries. Front. Biol. 2013, 8, 421–433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, M.; Chang, E.; Igarashi, T.; Noble, L.J. Neuronal injury and loss after traumatic brain injury: Time course and regional variability. Brain Res. 2001, 917, 45–54. [Google Scholar] [CrossRef]
- Hemphill, M.A.; Dauth, S.; Yu, C.J.; Dabiri, B.E.; Parker, K.K. Traumatic brain injury and the neuronal microenvironment: A potential role for neuropathological mechanotransduction. Neuron 2015, 85, 1177–1192. [Google Scholar] [CrossRef] [Green Version]
- Gao, X.; Chen, J. Mild traumatic brain injury results in extensive neuronal degeneration in the cerebral cortex. J. Neuropathol. Exp. Neurol. 2011, 70, 183–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barria, A.; Malinow, R. NMDA receptor subunit composition controls synaptic plasticity by regulating binding to CaMKII. Neuron 2005, 48, 289–301. [Google Scholar] [CrossRef] [Green Version]
- Gardoni, F.; Polli, F.; Cattabeni, F.; Di Luca, M. Calcium-calmodulin-dependent protein kinase II phosphorylation modulates PSD-95 binding to NMDA receptors. Eur. J. Neurosci. 2006, 24, 2694–2704. [Google Scholar] [CrossRef]
- Fan, X.; Jin, W.Y.; Wang, Y.T. The NMDA receptor complex: A multifunctional machine at the glutamatergic synapse. Front. Cell. Neurosci. 2014, 8, 160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hynd, M.R.; Scott, H.L.; Dodd, P.R. Glutamate-mediated excitotoxicity and neurodegeneration in Alzheimer’s disease. Neurochem. Int. 2004, 45, 583–595. [Google Scholar] [CrossRef] [PubMed]
- Bliss, T.V.; Collingridge, G.L. A synaptic model of memory: Long-term potentiation in the hippocampus. Nature 1993, 361, 31–39. [Google Scholar] [CrossRef]
- Collingridge, G.L.; Bliss, T.V. Memories of NMDA receptors and LTP. Trends Neurosci. 1995, 18, 54–56. [Google Scholar] [CrossRef]
- Mannix, R.; Berkner, J.; Mei, Z.; Alcon, S.; Hashim, J.; Robinson, S.; Jantzie, L.; Meehan, W.P., 3rd; Qiu, J. Adolescent Mice Demonstrate a Distinct Pattern of Injury after Repetitive Mild Traumatic Brain Injury. J. Neurotrauma 2017, 34, 495–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sze, C.; Bi, H.; Kleinschmidt-DeMasters, B.K.; Filley, C.M.; Martin, L.J. N-Methyl-D-aspartate receptor subunit proteins and their phosphorylation status are altered selectively in Alzheimer’s disease. J. Neurol. Sci. 2001, 182, 151–159. [Google Scholar] [CrossRef]
- Hynd, M.R.; Scott, H.L.; Dodd, P.R. Differential expression of N-methyl-d-aspartate receptor NR2 isoforms in Alzheimer’s disease. J. Neurochem. 2004, 90, 913–919. [Google Scholar] [CrossRef]
- Sakimura, K.; Kutsuwada, T.; Ito, I.; Manabe, T.; Takayama, C.; Kushiya, E.; Yagi, T.; Aizawa, S.; Inoue, Y.; Sugiyama, H.; et al. Reduced hippocampal LTP and spatial learning in mice lacking NMDA receptor epsilon 1 subunit. Nature 1995, 373, 151–155. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.P.; Shimizu, E.; Dube, G.R.; Rampon, C.; Kerchner, G.A.; Zhuo, M.; Liu, G.; Tsien, J.Z. Genetic enhancement of learning and memory in mice. Nature 1999, 401, 63–69. [Google Scholar] [CrossRef]
- Gascon, S.; Sobrado, M.; Roda, J.M.; Rodriguez-Pena, A.; Diaz-Guerra, M. Excitotoxicity and focal cerebral ischemia induce truncation of the NR2A and NR2B subunits of the NMDA receptor and cleavage of the scaffolding protein PSD-95. Mol. Psychiatry 2008, 13, 99–114. [Google Scholar] [CrossRef] [PubMed]
- Cui, Z.; Feng, R.; Jacobs, S.; Duan, Y.; Wang, H.; Cao, X.; Tsien, J.Z. Increased NR2A:NR2B ratio compresses long-term depression range and constrains long-term memory. Sci. Rep. 2013, 3, 1036. [Google Scholar] [CrossRef] [Green Version]
- Traynelis, S.F.; Wollmuth, L.P.; McBain, C.J.; Menniti, F.S.; Vance, K.M.; Ogden, K.K.; Hansen, K.B.; Yuan, H.; Myers, S.J.; Dingledine, R. Glutamate receptor ion channels: Structure, regulation, and function. Pharmacol. Rev. 2010, 62, 405–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwak, S.; Weiss, J.H. Calcium-permeable AMPA channels in neurodegenerative disease and ischemia. Curr. Opin. Neurobiol. 2006, 16, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Liao, M.; Mielke, J.G.; Ning, K.; Chen, Y.; Li, L.; El-Hayek, Y.H.; Gomez, E.; Zukin, R.S.; Fehlings, M.G.; et al. Ischemic insults direct glutamate receptor subunit 2-lacking AMPA receptors to synaptic sites. J. Neurosci. 2006, 26, 5309–5319. [Google Scholar] [CrossRef] [PubMed]
- Noh, K.M.; Yokota, H.; Mashiko, T.; Castillo, P.E.; Zukin, R.S.; Bennett, M.V. Blockade of calcium-permeable AMPA receptors protects hippocampal neurons against global ischemia-induced death. Proc. Natl. Acad. Sci. USA 2005, 102, 12230–12235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rakhade, S.N.; Zhou, C.; Aujla, P.K.; Fishman, R.; Sucher, N.J.; Jensen, F.E. Early alterations of AMPA receptors mediate synaptic potentiation induced by neonatal seizures. J. Neurosci. 2008, 28, 7979–7990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell, J.D.; Ai, J.; Chen, Y.; Baker, A.J. Mild in vitro trauma induces rapid Glur2 endocytosis, robustly augments calcium permeability and enhances susceptibility to secondary excitotoxic insult in cultured Purkinje cells. Brain 2007, 130, 2528–2542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spaethling, J.M.; Klein, D.M.; Singh, P.; Meaney, D.F. Calcium-permeable AMPA receptors appear in cortical neurons after traumatic mechanical injury and contribute to neuronal fate. J. Neurotrauma 2008, 25, 1207–1216. [Google Scholar] [CrossRef] [PubMed]
- Noga, J.T.; Hyde, T.M.; Herman, M.M.; Spurney, C.F.; Bigelow, L.B.; Weinberger, D.R.; Kleinman, J.E. Glutamate receptors in the postmortem striatum of schizophrenic, suicide, and control brains. Synapse 1997, 27, 168–176. [Google Scholar] [CrossRef]
- Sokolov, B.P. Expression of NMDAR1, GluR1, GluR7, and KA1 glutamate receptor mRNAs is decreased in frontal cortex of “neuroleptic-free” schizophrenics: Evidence on reversible up-regulation by typical neuroleptics. J. Neurochem. 1998, 71, 2454–2464. [Google Scholar] [CrossRef]
- Klausberger, T.; Somogyi, P. Neuronal diversity and temporal dynamics: The unity of hippocampal circuit operations. Science 2008, 321, 53–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, D.A.; Cho, R.Y.; Carter, C.S.; Eklund, K.; Forster, S.; Kelly, M.A.; Montrose, D. Subunit-selective modulation of GABA type A receptor neurotransmission and cognition in schizophrenia. Am. J. Psychiatry 2008, 165, 1585–1593. [Google Scholar] [CrossRef] [Green Version]
- Benes, F.M.; Berretta, S. GABAergic interneurons: Implications for understanding schizophrenia and bipolar disorder. Neuropsychopharmacology 2001, 25, 1–27. [Google Scholar] [CrossRef]
- Zhang, Z.J.; Reynolds, G.P. A selective decrease in the relative density of parvalbumin-immunoreactive neurons in the hippocampus in schizophrenia. Schizophr. Res. 2002, 55, 1–10. [Google Scholar] [CrossRef]
- Sperk, G.; Furtinger, S.; Schwarzer, C.; Pirker, S. GABA and its receptors in epilepsy. Adv. Exp. Med. Biol. 2004, 548, 92–103. [Google Scholar] [CrossRef] [PubMed]
- Kobori, N.; Dash, P.K. Reversal of brain injury-induced prefrontal glutamic acid decarboxylase expression and working memory deficits by D1 receptor antagonism. J. Neurosci. 2006, 26, 4236–4246. [Google Scholar] [CrossRef]
- Almeida-Suhett, C.P.; Prager, E.M.; Pidoplichko, V.; Figueiredo, T.H.; Marini, A.M.; Li, Z.; Eiden, L.E.; Braga, M.F. Reduced GABAergic inhibition in the basolateral amygdala and the development of anxiety-like behaviors after mild traumatic brain injury. PLoS ONE 2014, 9, e102627. [Google Scholar] [CrossRef] [Green Version]
- Almeida-Suhett, C.P.; Prager, E.M.; Pidoplichko, V.; Figueiredo, T.H.; Marini, A.M.; Li, Z.; Eiden, L.E.; Braga, M.F. GABAergic interneuronal loss and reduced inhibitory synaptic transmission in the hippocampal CA1 region after mild traumatic brain injury. Exp. Neurol. 2015, 273, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Gibson, C.J.; Meyer, R.C.; Hamm, R.J. Traumatic brain injury and the effects of diazepam, diltiazem, and MK-801 on GABA-A receptor subunit expression in rat hippocampus. J. Biomed. Sci. 2010, 17, 38. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Picon, F.; Snellman, A.; Shatillo, O.; Lehtiniemi, P.; Gronroos, T.J.; Marjamaki, P.; Trigg, W.; Jones, P.A.; Solin, O.; Pitkanen, A.; et al. Ex Vivo Tracing of NMDA and GABA-A Receptors in Rat Brain After Traumatic Brain Injury Using 18F-GE-179 and 18F-GE-194 Autoradiography. J. Nucl. Med. 2016, 57, 1442–1447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drexel, M.; Puhakka, N.; Kirchmair, E.; Hortnagl, H.; Pitkanen, A.; Sperk, G. Expression of GABA receptor subunits in the hippocampus and thalamus after experimental traumatic brain injury. Neuropharmacology 2015, 88, 122–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, Q.A.; Nicoll, R.A. The GABAA Receptor beta Subunit Is Required for Inhibitory Transmission. Neuron 2018, 98, 718–725 e713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sigel, E.; Baur, R.; Racz, I.; Marazzi, J.; Smart, T.G.; Zimmer, A.; Gertsch, J. The major central endocannabinoid directly acts at GABA(A) receptors. Proc. Natl. Acad. Sci. USA 2011, 108, 18150–18155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Y.; Vasilyev, D.V.; Goncalves, M.B.; Howell, F.V.; Hobbs, C.; Reisenberg, M.; Shen, R.; Zhang, M.Y.; Strassle, B.W.; Lu, P.; et al. Loss of retrograde endocannabinoid signaling and reduced adult neurogenesis in diacylglycerol lipase knock-out mice. J. Neurosci. 2010, 30, 2017–2024. [Google Scholar] [CrossRef]
- Tanimura, A.; Yamazaki, M.; Hashimotodani, Y.; Uchigashima, M.; Kawata, S.; Abe, M.; Kita, Y.; Hashimoto, K.; Shimizu, T.; Watanabe, M.; et al. The endocannabinoid 2-arachidonoylglycerol produced by diacylglycerol lipase alpha mediates retrograde suppression of synaptic transmission. Neuron 2010, 65, 320–327. [Google Scholar] [CrossRef] [Green Version]
- Cavener, V.S.; Gaulden, A.; Pennipede, D.; Jagasia, P.; Uddin, J.; Marnett, L.J.; Patel, S. Inhibition of Diacylglycerol Lipase Impairs Fear Extinction in Mice. Front. Neurosci. 2018, 12, 479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schurman, L.D.; Carper, M.C.; Moncayo, L.V.; Ogasawara, D.; Richardson, K.; Yu, L.; Liu, X.; Poklis, J.L.; Liu, Q.S.; Cravatt, B.F.; et al. Diacylglycerol Lipase-Alpha Regulates Hippocampal-Dependent Learning and Memory Processes in Mice. J. Neurosci. 2019, 39, 5949–5965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, K.L.; Tsuboi, K.; Adibekian, A.; Pugh, H.; Masuda, K.; Cravatt, B.F. DAGLbeta inhibition perturbs a lipid network involved in macrophage inflammatory responses. Nat. Chem. Biol. 2012, 8, 999–1007. [Google Scholar] [CrossRef]
- Viader, A.; Ogasawara, D.; Joslyn, C.M.; Sanchez-Alavez, M.; Mori, S.; Nguyen, W.; Conti, B.; Cravatt, B.F. A chemical proteomic atlas of brain serine hydrolases identifies cell type-specific pathways regulating neuroinflammation. Elife 2016, 5, e12345. [Google Scholar] [CrossRef] [PubMed]
- Wilkerson, J.L.; Ghosh, S.; Bagdas, D.; Mason, B.L.; Crowe, M.S.; Hsu, K.L.; Wise, L.E.; Kinsey, S.G.; Damaj, M.I.; Cravatt, B.F.; et al. Diacylglycerol lipase beta inhibition reverses nociceptive behaviour in mouse models of inflammatory and neuropathic pain. Br. J. Pharmacol. 2016, 173, 1678–1692. [Google Scholar] [CrossRef] [PubMed]
- Wilkerson, J.L.; Donvito, G.; Grim, T.W.; Abdullah, R.A.; Ogasawara, D.; Cravatt, B.F.; Lichtman, A.H. Investigation of Diacylglycerol Lipase Alpha Inhibition in the Mouse Lipopolysaccharide Inflammatory Pain Model. J. Pharmacol. Exp. Ther. 2017, 363, 394–401. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, L.D.; Smith, T.L.; Donvito, G.; Cravatt, B.F.; Newton, J.; Spiegel, S.; Reeves, T.M.; Phillips, L.L.; Lichtman, A.H. Diacylglycerol Lipase-beta Knockout Mice Display a Sex-Dependent Attenuation of Traumatic Brain Injury-Induced Mortality with No Impact on Memory or Other Functional Consequences. Cannabis Cannabinoid Res. 2021. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Selvaraj, P.; Tanaka, M.; Wen, J.; Zhang, Y. The Novel Monoacylglycerol Lipase Inhibitor MJN110 Suppresses Neuroinflammation, Normalizes Synaptic Composition and Improves Behavioral Performance in the Repetitive Traumatic Brain Injury Mouse Model. Cells 2021, 10, 3454. https://doi.org/10.3390/cells10123454
Selvaraj P, Tanaka M, Wen J, Zhang Y. The Novel Monoacylglycerol Lipase Inhibitor MJN110 Suppresses Neuroinflammation, Normalizes Synaptic Composition and Improves Behavioral Performance in the Repetitive Traumatic Brain Injury Mouse Model. Cells. 2021; 10(12):3454. https://doi.org/10.3390/cells10123454
Chicago/Turabian StyleSelvaraj, Prabhuanand, Mikiei Tanaka, Jie Wen, and Yumin Zhang. 2021. "The Novel Monoacylglycerol Lipase Inhibitor MJN110 Suppresses Neuroinflammation, Normalizes Synaptic Composition and Improves Behavioral Performance in the Repetitive Traumatic Brain Injury Mouse Model" Cells 10, no. 12: 3454. https://doi.org/10.3390/cells10123454
APA StyleSelvaraj, P., Tanaka, M., Wen, J., & Zhang, Y. (2021). The Novel Monoacylglycerol Lipase Inhibitor MJN110 Suppresses Neuroinflammation, Normalizes Synaptic Composition and Improves Behavioral Performance in the Repetitive Traumatic Brain Injury Mouse Model. Cells, 10(12), 3454. https://doi.org/10.3390/cells10123454