
GLIS3: A Critical Transcription Factor in Islet β-Cell Generation


{kind=link}
{kind=link}
Abstract
:1. Introduction
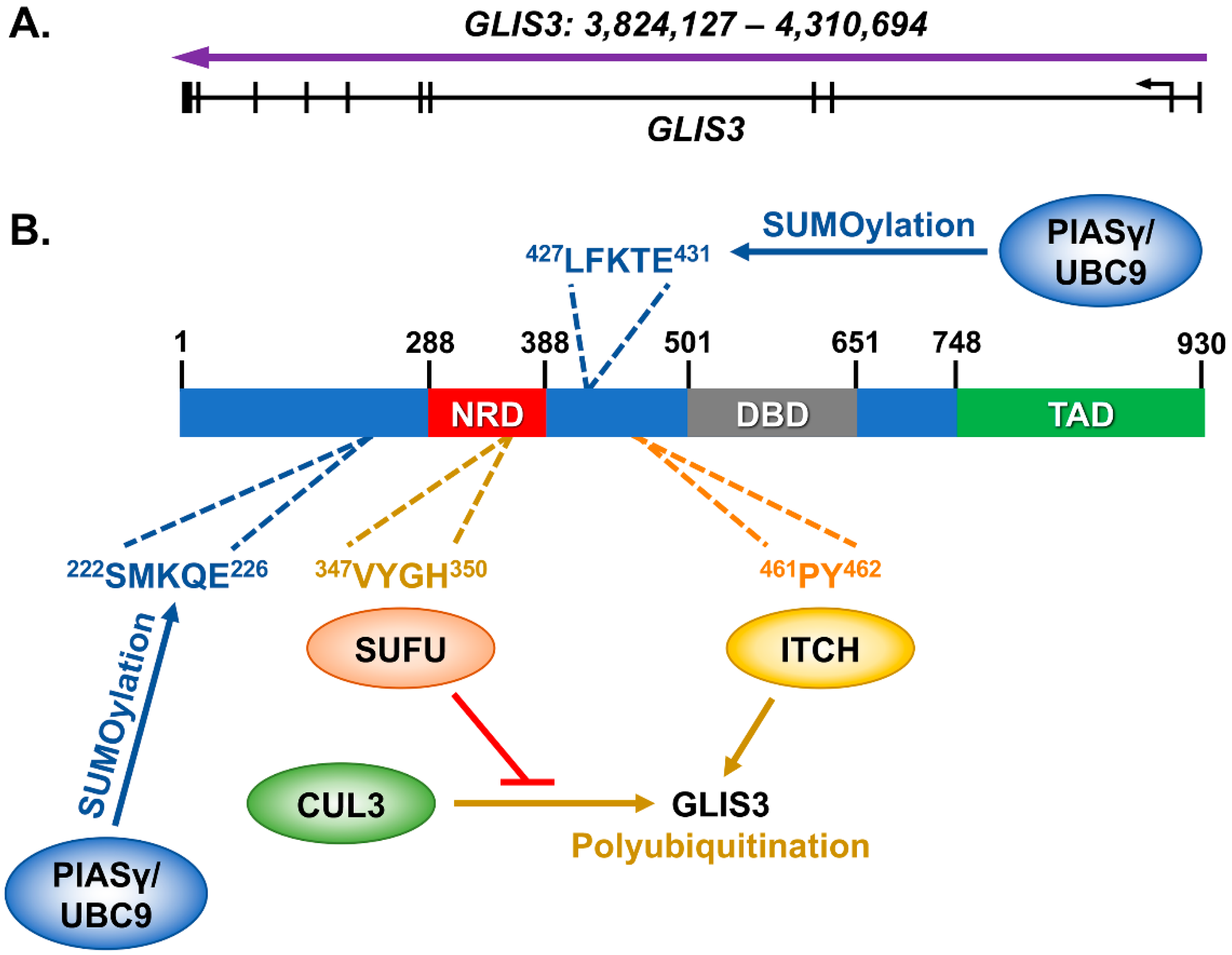
2. The GLIS3 Gene and Its Encoded Protein
3. Early Characterization of the GLIS3 Knockout Mice and Humans
4. Early Pancreas Development and Glis3 Expression in Mice
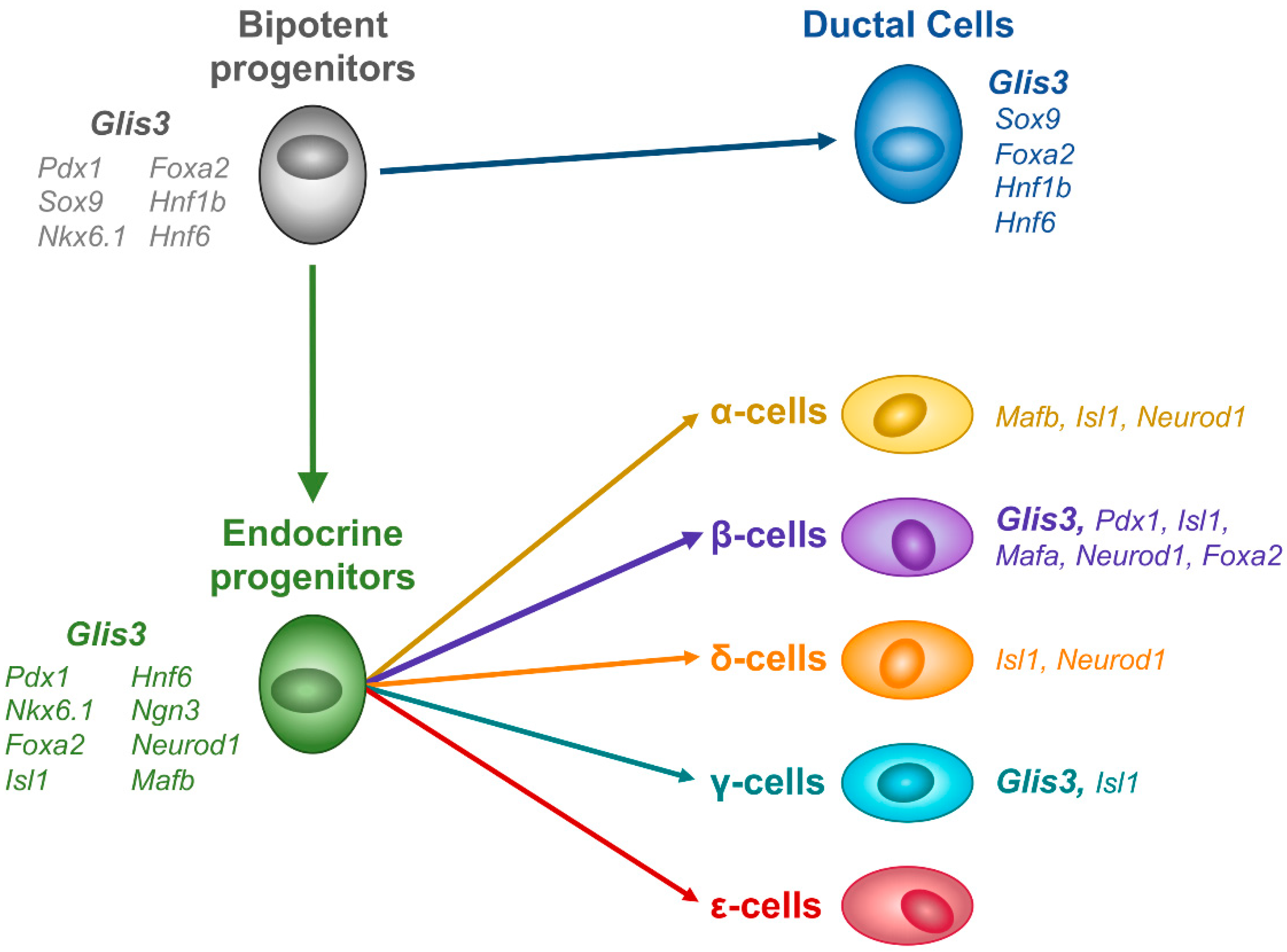
5. Glis3’s Role in the Secondary Transition and Ductal/Endocrine Lineage Determinations
6. Links between Glis3 and β-Cell Maturation
7. GLIS3 in Human β-Cell Development
8. Newly Identified Human Mutations within GLIS3
9. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Ionescu-Tirgoviste, C.; Gagniuc, P.A.; Gubceac, E.; Mardare, L.; Popescu, I.; Dima, S.; Militaru, M. A 3D map of the islet routes throughout the healthy human pancreas. Sci. Rep. 2015, 5, 14634. [Google Scholar] [CrossRef] [Green Version]
- Brissova, M.; Fowler, M.J.; Nicholson, W.E.; Chu, A.; Hirshberg, B.; Harlan, D.M.; Powers, A.C. Assessment of human pancreatic islet architecture and composition by laser scanning confocal microscopy. J. Histochem. Cytochem. 2005, 53, 1087–1097. [Google Scholar] [CrossRef] [PubMed]
- Murphy, R.; Ellard, S.; Hattersley, A.T. Clinical implications of a molecular genetic classification of monogenic beta-cell diabetes. Nat. Clin. Pract. Endocrinol. Metab. 2008, 4, 200–213. [Google Scholar] [CrossRef]
- Schneider, D.A.; von Herrath, M.G. Potential viral pathogenic mechanism in human type 1 diabetes. Diabetologia 2014, 57, 2009–2018. [Google Scholar] [CrossRef] [Green Version]
- Roivainen, M.; Klingel, K. Virus infections and type 1 diabetes risk. Curr. Diab. Rep. 2010, 10, 350–356. [Google Scholar] [CrossRef] [PubMed]
- Giongo, A.; Gano, K.A.; Crabb, D.B.; Mukherjee, N.; Novelo, L.L.; Casella, G.; Drew, J.C.; Ilonen, J.; Knip, M.; Hyoty, H.; et al. Toward defining the autoimmune microbiome for type 1 diabetes. ISME J. 2011, 5, 82–91. [Google Scholar] [CrossRef]
- Norris, J.M.; Barriga, K.; Klingensmith, G.; Hoffman, M.; Eisenbarth, G.S.; Erlich, H.A.; Rewers, M. Timing of initial cereal exposure in infancy and risk of islet autoimmunity. JAMA 2003, 290, 1713–1720. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, A.G.; Schmid, S.; Huber, D.; Hummel, M.; Bonifacio, E. Early infant feeding and risk of developing type 1 diabetes-associated autoantibodies. JAMA 2003, 290, 1721–1728. [Google Scholar] [CrossRef] [Green Version]
- Chan, J.M.; Rimm, E.B.; Colditz, G.A.; Stampfer, M.J.; Willett, W.C. Obesity, fat distribution, and weight gain as risk factors for clinical diabetes in men. Diabetes Care 1994, 17, 961–969. [Google Scholar] [CrossRef] [Green Version]
- Colditz, G.A.; Willett, W.C.; Rotnitzky, A.; Manson, J.E. Weight gain as a risk factor for clinical diabetes mellitus in women. Ann. Intern. Med. 1995, 122, 481–486. [Google Scholar] [CrossRef]
- Prasad, R.B.; Groop, L. Genetics of type 2 diabetes-pitfalls and possibilities. Genes 2015, 6, 87–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hattersley, A.T.; Greeley, S.A.W.; Polak, M.; Rubio-Cabezas, O.; Njolstad, P.R.; Mlynarski, W.; Castano, L.; Carlsson, A.; Raile, K.; Chi, D.V.; et al. ISPAD Clinical Practice Consensus Guidelines 2018: The diagnosis and management of monogenic diabetes in children and adolescents. Pediatr. Diabetes 2018, 19, 47–63. [Google Scholar] [CrossRef] [PubMed]
- Jensen, J. Gene regulatory factors in pancreatic development. Dev. Dyn. 2004, 229, 176–200. [Google Scholar] [CrossRef] [PubMed]
- Murtaugh, L.C.; Melton, D.A. Genes, signals, and lineages in pancreas development. Annu. Rev. Cell Dev. Biol. 2003, 19, 71–89. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Law, A.C.; Rajagopal, J.; Anderson, W.J.; Gray, P.A.; Melton, D.A. A multipotent progenitor domain guides pancreatic organogenesis. Dev. Cell 2007, 13, 103–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scoville, D.W.; Kang, H.S.; Jetten, A.M. Transcription factor GLIS3: Critical roles in thyroid hormone biosynthesis, hypothyroidism, pancreatic beta cells and diabetes. Pharmacol. Ther. 2020, 215, 107632. [Google Scholar] [CrossRef] [PubMed]
- Scoville, D.W.; Kang, H.S.; Jetten, A.M. GLIS1-3: Emerging roles in reprogramming, stem and progenitor cell differentiation and maintenance. Stem Cell Investig. 2017, 4, 80. [Google Scholar] [CrossRef] [Green Version]
- Jetten, A.M. GLIS1-3 transcription factors: Critical roles in the regulation of multiple physiological processes and diseases. Cell Mol. Life Sci. 2018, 75, 3473–3494. [Google Scholar] [CrossRef]
- Kim, Y.S.; Nakanishi, G.; Lewandoski, M.; Jetten, A.M. GLIS3, a novel member of the GLIS subfamily of Kruppel-like zinc finger proteins with repressor and activation functions. Nucleic Acids Res. 2003, 31, 5513–5525. [Google Scholar] [CrossRef]
- ZeRuth, G.T.; Yang, X.P.; Jetten, A.M. Modulation of the transactivation function and stability of Kruppel-like zinc finger protein Gli-similar 3 (Glis3) by Suppressor of Fused. J. Biol. Chem. 2011, 286, 22077–22089. [Google Scholar] [CrossRef] [Green Version]
- ZeRuth, G.T.; Williams, J.G.; Cole, Y.C.; Jetten, A.M. HECT E3 Ubiquitin Ligase Itch Functions as a Novel Negative Regulator of Gli-Similar 3 (Glis3) Transcriptional Activity. PLoS ONE 2015, 10, e0131303. [Google Scholar] [CrossRef]
- Hoard, T.M.; Yang, X.P.; Jetten, A.M.; ZeRuth, G.T. PIAS-family proteins negatively regulate Glis3 transactivation function through SUMO modification in pancreatic beta cells. Heliyon 2018, 4, e00709. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, N.; Hiramatsu, K.; Miyamoto, R.; Yasuda, K.; Suzuki, N.; Oshima, N.; Kiyonari, H.; Shiba, D.; Nishio, S.; Mochizuki, T.; et al. A murine model of neonatal diabetes mellitus in Glis3-deficient mice. FEBS Lett. 2009, 583, 2108–2113. [Google Scholar] [CrossRef] [Green Version]
- Kang, H.S.; Kim, Y.S.; ZeRuth, G.; Beak, J.Y.; Gerrish, K.; Kilic, G.; Sosa-Pineda, B.; Jensen, J.; Pierreux, C.E.; Lemaigre, F.P.; et al. Transcription factor Glis3, a novel critical player in the regulation of pancreatic beta-cell development and insulin gene expression. Mol. Cell. Biol. 2009, 29, 6366–6379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Chang, B.H.; Yechoor, V.; Chen, W.; Li, L.; Tsai, M.J.; Chan, L. The Kruppel-like zinc finger protein GLIS3 transactivates neurogenin 3 for proper fetal pancreatic islet differentiation in mice. Diabetologia 2011, 54, 2595–2605. [Google Scholar] [CrossRef] [Green Version]
- Taha, D.; Barbar, M.; Kanaan, H.; Williamson Balfe, J. Neonatal diabetes mellitus, congenital hypothyroidism, hepatic fibrosis, polycystic kidneys, and congenital glaucoma: A new autosomal recessive syndrome? Am. J. Med. Genet. A 2003, 122, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Senee, V.; Chelala, C.; Duchatelet, S.; Feng, D.; Blanc, H.; Cossec, J.C.; Charon, C.; Nicolino, M.; Boileau, P.; Cavener, D.R.; et al. Mutations in GLIS3 are responsible for a rare syndrome with neonatal diabetes mellitus and congenital hypothyroidism. Nat. Genet. 2006, 38, 682–687. [Google Scholar] [CrossRef]
- Dimitri, P.; Warner, J.T.; Minton, J.A.; Patch, A.M.; Ellard, S.; Hattersley, A.T.; Barr, S.; Hawkes, D.; Wales, J.K.; Gregory, J.W. Novel GLIS3 mutations demonstrate an extended multisystem phenotype. Eur. J. Endocrinol. 2011, 164, 437–443. [Google Scholar] [CrossRef] [Green Version]
- Dimitri, P.; Habeb, A.M.; Gurbuz, F.; Millward, A.; Wallis, S.; Moussa, K.; Akcay, T.; Taha, D.; Hogue, J.; Slavotinek, A.; et al. Expanding the Clinical Spectrum Associated With GLIS3 Mutations. J. Clin. Endocrinol. Metab. 2015, 100, 1362–1369. [Google Scholar] [CrossRef] [Green Version]
- Dimitri, P.; De Franco, E.; Habeb, A.M.; Gurbuz, F.; Moussa, K.; Taha, D.; Wales, J.K.; Hogue, J.; Slavotinek, A.; Shetty, A.; et al. An emerging, recognizable facial phenotype in association with mutations in GLI-similar 3 (GLIS3). Am. J. Med. Genet. A 2016, 170, 1918–1923. [Google Scholar] [CrossRef] [Green Version]
- Fu, C.; Luo, S.; Long, X.; Li, Y.; She, S.; Hu, X.; Mo, M.; Wang, Z.; Chen, Y.; He, C.; et al. Mutation screening of the GLIS3 gene in a cohort of 592 Chinese patients with congenital hypothyroidism. Clin. Chim. Acta 2018, 476, 38–43. [Google Scholar] [CrossRef]
- Alghamdi, K.A.; Alsaedi, A.B.; Aljasser, A.; Altawil, A.; Kamal, N.M. Extended clinical features associated with novel Glis3 mutation: A case report. BMC Endocr. Disord. 2017, 17, 14. [Google Scholar] [CrossRef] [Green Version]
- Fu, C.; Luo, S.; Zhang, Y.; Fan, X.; D’Gama, A.M.; Zhang, X.; Zheng, H.; Su, J.; Li, C.; Luo, J.; et al. Chromosomal microarray and whole exome sequencing identify genetic causes of congenital hypothyroidism with extra-thyroidal congenital malformations. Clin. Chim. Acta 2019, 489, 103–108. [Google Scholar] [CrossRef]
- Gittes, G.K. Developmental biology of the pancreas: A comprehensive review. Dev. Biol. 2009, 326, 4–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, F.C.; Wright, C. Pancreas organogenesis: From bud to plexus to gland. Dev. Dyn. 2011, 240, 530–565. [Google Scholar] [CrossRef]
- Villasenor, A.; Chong, D.C.; Henkemeyer, M.; Cleaver, O. Epithelial dynamics of pancreatic branching morphogenesis. Development 2010, 137, 4295–4305. [Google Scholar] [CrossRef] [Green Version]
- Herrera, P.L. Adult insulin- and glucagon-producing cells differentiate from two independent cell lineages. Development 2000, 127, 2317–2322. [Google Scholar] [CrossRef] [PubMed]
- Gu, G.; Dubauskaite, J.; Melton, D.A. Direct evidence for the pancreatic lineage: NGN3+ cells are islet progenitors and are distinct from duct progenitors. Development 2002, 129, 2447–2457. [Google Scholar] [CrossRef]
- Doyle, M.J.; Sussel, L. Engineering islets: Lessons from stem cells and embryonic development. Endocrinol. Metab. Clin. N. Am. 2004, 33, 149–162. [Google Scholar] [CrossRef]
- Guney, M.A.; Gannon, M. Pancreas cell fate. Birth Defects Res. C Embryo Today 2009, 87, 232–248. [Google Scholar] [CrossRef]
- Offield, M.F.; Jetton, T.L.; Labosky, P.A.; Ray, M.; Stein, R.W.; Magnuson, M.A.; Hogan, B.L.; Wright, C.V. PDX-1 is required for pancreatic outgrowth and differentiation of the rostral duodenum. Development 1996, 122, 983–995. [Google Scholar] [CrossRef] [PubMed]
- Ohlsson, H.; Karlsson, K.; Edlund, T. IPF1, a homeodomain-containing transactivator of the insulin gene. EMBO J. 1993, 12, 4251–4259. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, Y.; Cooper, B.; Gannon, M.; Ray, M.; MacDonald, R.J.; Wright, C.V. The role of the transcriptional regulator Ptf1a in converting intestinal to pancreatic progenitors. Nat. Genet. 2002, 32, 128–134. [Google Scholar] [CrossRef]
- Jonsson, J.; Carlsson, L.; Edlund, T.; Edlund, H. Insulin-promoter-factor 1 is required for pancreas development in mice. Nature 1994, 371, 606–609. [Google Scholar] [CrossRef] [PubMed]
- Stoffers, D.A.; Zinkin, N.T.; Stanojevic, V.; Clarke, W.L.; Habener, J.F. Pancreatic agenesis attributable to a single nucleotide deletion in the human IPF1 gene coding sequence. Nat. Genet. 1997, 15, 106–110. [Google Scholar] [CrossRef]
- Schwitzgebel, V.M.; Mamin, A.; Brun, T.; Ritz-Laser, B.; Zaiko, M.; Maret, A.; Jornayvaz, F.R.; Theintz, G.E.; Michielin, O.; Melloul, D.; et al. Agenesis of human pancreas due to decreased half-life of insulin promoter factor 1. J. Clin. Endocrinol. Metab. 2003, 88, 4398–4406. [Google Scholar] [CrossRef] [Green Version]
- Thomas, I.H.; Saini, N.K.; Adhikari, A.; Lee, J.M.; Kasa-Vubu, J.Z.; Vazquez, D.M.; Menon, R.K.; Chen, M.; Fajans, S.S. Neonatal diabetes mellitus with pancreatic agenesis in an infant with homozygous IPF-1 Pro63fsX60 mutation. Pediatr. Diabetes 2009, 10, 492–496. [Google Scholar] [CrossRef] [Green Version]
- Nicolino, M.; Claiborn, K.C.; Senee, V.; Boland, A.; Stoffers, D.A.; Julier, C. A novel hypomorphic PDX1 mutation responsible for permanent neonatal diabetes with subclinical exocrine deficiency. Diabetes 2010, 59, 733–740. [Google Scholar] [CrossRef] [Green Version]
- Caetano, L.A.; Santana, L.S.; Costa-Riquetto, A.D.; Lerario, A.M.; Nery, M.; Nogueira, G.F.; Ortega, C.D.; Rocha, M.S.; Jorge, A.A.L.; Teles, M.G. PDX1 -MODY and dorsal pancreatic agenesis: New phenotype of a rare disease. Clin. Genet. 2018, 93, 382–386. [Google Scholar] [CrossRef] [PubMed]
- Rose, S.D.; Kruse, F.; Swift, G.H.; MacDonald, R.J.; Hammer, R.E. A single element of the elastase I enhancer is sufficient to direct transcription selectively to the pancreas and gut. Mol. Cell. Biol. 1994, 14, 2048–2057. [Google Scholar] [CrossRef]
- Krapp, A.; Knofler, M.; Frutiger, S.; Hughes, G.J.; Hagenbuchle, O.; Wellauer, P.K. The p48 DNA-binding subunit of transcription factor PTF1 is a new exocrine pancreas-specific basic helix-loop-helix protein. EMBO J. 1996, 15, 4317–4329. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, H.; Kim, J.E.; Nakashima, K.; Balmes, G.; Iwai, N.; Deng, J.M.; Zhang, Z.; Martin, J.F.; Behringer, R.R.; Nakamura, T.; et al. Osteo-chondroprogenitor cells are derived from Sox9 expressing precursors. Proc. Natl. Acad. Sci. USA 2005, 102, 14665–14670. [Google Scholar] [CrossRef] [Green Version]
- Jacquemin, P.; Lemaigre, F.P.; Rousseau, G.G. The Onecut transcription factor HNF-6 (OC-1) is required for timely specification of the pancreas and acts upstream of Pdx-1 in the specification cascade. Dev. Biol. 2003, 258, 105–116. [Google Scholar] [CrossRef] [Green Version]
- Pierreux, C.E.; Poll, A.V.; Kemp, C.R.; Clotman, F.; Maestro, M.A.; Cordi, S.; Ferrer, J.; Leyns, L.; Rousseau, G.G.; Lemaigre, F.P. The transcription factor hepatocyte nuclear factor-6 controls the development of pancreatic ducts in the mouse. Gastroenterology 2006, 130, 532–541. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Ables, E.T.; Pope, C.F.; Washington, M.K.; Hipkens, S.; Means, A.L.; Path, G.; Seufert, J.; Costa, R.H.; Leiter, A.B.; et al. Multiple, temporal-specific roles for HNF6 in pancreatic endocrine and ductal differentiation. Mech. Dev. 2009, 126, 958–973. [Google Scholar] [CrossRef] [Green Version]
- Haumaitre, C.; Barbacci, E.; Jenny, M.; Ott, M.O.; Gradwohl, G.; Cereghini, S. Lack of TCF2/vHNF1 in mice leads to pancreas agenesis. Proc. Natl. Acad. Sci. USA 2005, 102, 1490–1495. [Google Scholar] [CrossRef] [Green Version]
- Seymour, P.A.; Freude, K.K.; Tran, M.N.; Mayes, E.E.; Jensen, J.; Kist, R.; Scherer, G.; Sander, M. SOX9 is required for maintenance of the pancreatic progenitor cell pool. Proc. Natl. Acad. Sci. USA 2007, 104, 1865–1870. [Google Scholar] [CrossRef] [Green Version]
- Haumaitre, C.; Fabre, M.; Cormier, S.; Baumann, C.; Delezoide, A.L.; Cereghini, S. Severe pancreas hypoplasia and multicystic renal dysplasia in two human fetuses carrying novel HNF1beta/MODY5 mutations. Hum. Mol. Genet. 2006, 15, 2363–2375. [Google Scholar] [CrossRef] [Green Version]
- Ang, S.L.; Rossant, J. HNF-3 beta is essential for node and notochord formation in mouse development. Cell 1994, 78, 561–574. [Google Scholar] [CrossRef]
- Dufort, D.; Schwartz, L.; Harpal, K.; Rossant, J. The transcription factor HNF3beta is required in visceral endoderm for normal primitive streak morphogenesis. Development 1998, 125, 3015–3025. [Google Scholar] [CrossRef]
- Weinstein, D.C.; Ruiz i Altaba, A.; Chen, W.S.; Hoodless, P.; Prezioso, V.R.; Jessell, T.M.; Darnell, J.E., Jr. The winged-helix transcription factor HNF-3 beta is required for notochord development in the mouse embryo. Cell 1994, 78, 575–588. [Google Scholar] [CrossRef]
- Kang, H.S.; Takeda, Y.; Jeon, K.; Jetten, A.M. The Spatiotemporal Pattern of Glis3 Expression Indicates a Regulatory Function in Bipotent and Endocrine Progenitors during Early Pancreatic Development and in Beta, PP and Ductal Cells. PLoS ONE 2016, 11, e0157138. [Google Scholar] [CrossRef] [PubMed]
- Norgaard, G.A.; Jensen, J.N.; Jensen, J. FGF10 signaling maintains the pancreatic progenitor cell state revealing a novel role of Notch in organ development. Dev. Biol. 2003, 264, 323–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hart, A.; Papadopoulou, S.; Edlund, H. Fgf10 maintains notch activation, stimulates proliferation, and blocks differentiation of pancreatic epithelial cells. Dev. Dyn. 2003, 228, 185–193. [Google Scholar] [CrossRef]
- Gradwohl, G.; Dierich, A.; LeMeur, M.; Guillemot, F. neurogenin3 is required for the development of the four endocrine cell lineages of the pancreas. Proc. Natl. Acad. Sci. USA 2000, 97, 1607–1611. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Jensen, J.N.; Seymour, P.A.; Hsu, W.; Dor, Y.; Sander, M.; Magnuson, M.A.; Serup, P.; Gu, G. Sustained Neurog3 expression in hormone-expressing islet cells is required for endocrine maturation and function. Proc. Natl. Acad. Sci. USA 2009, 106, 9715–9720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, J.; Asa, S.L.; Drucker, D.J. Islet cell and extrapancreatic expression of the LIM domain homeobox gene isl-1. Mol. Endocrinol. 1991, 5, 1633–1641. [Google Scholar] [CrossRef] [Green Version]
- Naya, F.J.; Huang, H.P.; Qiu, Y.; Mutoh, H.; DeMayo, F.J.; Leiter, A.B.; Tsai, M.J. Diabetes, defective pancreatic morphogenesis, and abnormal enteroendocrine differentiation in BETA2/neuroD-deficient mice. Genes Dev. 1997, 11, 2323–2334. [Google Scholar] [CrossRef] [Green Version]
- Du, A.; Hunter, C.S.; Murray, J.; Noble, D.; Cai, C.L.; Evans, S.M.; Stein, R.; May, C.L. Islet-1 is required for the maturation, proliferation, and survival of the endocrine pancreas. Diabetes 2009, 58, 2059–2069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scoville, D.; Lichti-Kaiser, K.; Grimm, S.; Jetten, A. GLIS3 binds pancreatic beta cell regulatory regions alongside other islet transcription factors. J. Endocrinol. 2019, 243, 1–14. [Google Scholar] [CrossRef]
- Wortham, M.; Sander, M. Transcriptional mechanisms of pancreatic beta-cell maturation and functional adaptation. Trends Endocrinol. Metab. 2021, 32, 474–487. [Google Scholar] [CrossRef] [PubMed]
- Blum, B.; Hrvatin, S.; Schuetz, C.; Bonal, C.; Rezania, A.; Melton, D.A. Functional beta-cell maturation is marked by an increased glucose threshold and by expression of urocortin 3. Nat. Biotechnol. 2012, 30, 261–264. [Google Scholar] [CrossRef] [Green Version]
- Hang, Y.; Stein, R. MafA and MafB activity in pancreatic beta cells. Trends Endocrinol. Metab. 2011, 22, 364–373. [Google Scholar] [CrossRef] [Green Version]
- Akerman, I.; Maestro, M.A.; De Franco, E.; Grau, V.; Flanagan, S.; Garcia-Hurtado, J.; Mittler, G.; Ravassard, P.; Piemonti, L.; Ellard, S.; et al. Neonatal diabetes mutations disrupt a chromatin pioneering function that activates the human insulin gene. Cell Rep. 2021, 35, 108981. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Moriguchi, T.; Kajihara, M.; Esaki, R.; Harada, A.; Shimohata, H.; Oishi, H.; Hamada, M.; Morito, N.; Hasegawa, K.; et al. MafA is a key regulator of glucose-stimulated insulin secretion. Mol. Cell. Biol. 2005, 25, 4969–4976. [Google Scholar] [CrossRef] [Green Version]
- Hang, Y.; Yamamoto, T.; Benninger, R.K.; Brissova, M.; Guo, M.; Bush, W.; Piston, D.W.; Powers, A.C.; Magnuson, M.; Thurmond, D.C.; et al. The MafA transcription factor becomes essential to islet beta-cells soon after birth. Diabetes 2014, 63, 1994–2005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scoville, D.W.; Cyphert, H.A.; Liao, L.; Xu, J.; Reynolds, A.; Guo, S.; Stein, R. MLL3 and MLL4 Methyltransferases Bind to the MAFA and MAFB Transcription Factors to Regulate Islet beta-Cell Function. Diabetes 2015, 64, 3772–3783. [Google Scholar] [CrossRef] [Green Version]
- Conrad, E.; Dai, C.; Spaeth, J.; Guo, M.; Cyphert, H.A.; Scoville, D.; Carroll, J.; Yu, W.M.; Goodrich, L.V.; Harlan, D.M.; et al. The MAFB transcription factor impacts islet alpha-cell function in rodents and represents a unique signature of primate islet beta-cells. Am. J. Physiol. Endocrinol. Metab. 2016, 310, 91–102. [Google Scholar] [CrossRef] [Green Version]
- Rutter, G.A.; Pullen, T.J.; Hodson, D.J.; Martinez-Sanchez, A. Pancreatic beta-cell identity, glucose sensing and the control of insulin secretion. Biochem. J. 2015, 466, 203–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pullen, T.J.; Rutter, G.A. When less is more: The forbidden fruits of gene repression in the adult beta-cell. Diabetes Obes. Metab. 2013, 15, 503–512. [Google Scholar] [CrossRef]
- Talchai, C.; Xuan, S.; Lin, H.V.; Sussel, L.; Accili, D. Pancreatic beta cell dedifferentiation as a mechanism of diabetic beta cell failure. Cell 2012, 150, 1223–1234. [Google Scholar] [CrossRef] [Green Version]
- Jennings, R.E.; Berry, A.A.; Strutt, J.P.; Gerrard, D.T.; Hanley, N.A. Human pancreas development. Development 2015, 142, 3126–3137. [Google Scholar] [CrossRef] [Green Version]
- Jennings, R.E.; Berry, A.A.; Kirkwood-Wilson, R.; Roberts, N.A.; Hearn, T.; Salisbury, R.J.; Blaylock, J.; Piper Hanley, K.; Hanley, N.A. Development of the human pancreas from foregut to endocrine commitment. Diabetes 2013, 62, 3514–3522. [Google Scholar] [CrossRef] [Green Version]
- Sussel, L.; Kalamaras, J.; Hartigan-O’Connor, D.J.; Meneses, J.J.; Pedersen, R.A.; Rubenstein, J.L.; German, M.S. Mice lacking the homeodomain transcription factor Nkx2.2 have diabetes due to arrested differentiation of pancreatic beta cells. Development 1998, 125, 2213–2221. [Google Scholar] [CrossRef]
- Pagliuca, F.W.; Melton, D.A. How to make a functional beta-cell. Development 2013, 140, 2472–2483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez, F.; Zhu, Z.; Shi, Z.D.; Lelli, K.; Verma, N.; Li, Q.V.; Huangfu, D. An iCRISPR platform for rapid, multiplexable, and inducible genome editing in human pluripotent stem cells. Cell Stem Cell 2014, 15, 215–226. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Z.; Li, Q.V.; Lee, K.; Rosen, B.P.; Gonzalez, F.; Soh, C.L.; Huangfu, D. Genome Editing of Lineage Determinants in Human Pluripotent Stem Cells Reveals Mechanisms of Pancreatic Development and Diabetes. Cell Stem Cell 2016, 18, 755–768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amin, S.; Cook, B.; Zhou, T.; Ghazizadeh, Z.; Lis, R.; Zhang, T.; Khalaj, M.; Crespo, M.; Perera, M.; Xiang, J.Z.; et al. Discovery of a drug candidate for GLIS3-associated diabetes. Nat. Commun. 2018, 9, 2681. [Google Scholar] [CrossRef]
- Nogueira, T.C.; Paula, F.M.; Villate, O.; Colli, M.L.; Moura, R.F.; Cunha, D.A.; Marselli, L.; Marchetti, P.; Cnop, M.; Julier, C.; et al. GLIS3, a susceptibility gene for type 1 and type 2 diabetes, modulates pancreatic beta cell apoptosis via regulation of a splice variant of the BH3-only protein Bim. PLoS Genet. 2013, 9, e1003532. [Google Scholar] [CrossRef]
- Ediger, B.N.; Lim, H.W.; Juliana, C.; Groff, D.N.; Williams, L.T.; Dominguez, G.; Liu, J.H.; Taylor, B.L.; Walp, E.R.; Kameswaran, V.; et al. LIM domain-binding 1 maintains the terminally differentiated state of pancreatic beta cells. J. Clin. Investig. 2017, 127, 215–229. [Google Scholar] [CrossRef] [PubMed]
- Eizirik, D.L.; Sammeth, M.; Bouckenooghe, T.; Bottu, G.; Sisino, G.; Igoillo-Esteve, M.; Ortis, F.; Santin, I.; Colli, M.L.; Barthson, J.; et al. The human pancreatic islet transcriptome: Expression of candidate genes for type 1 diabetes and the impact of pro-inflammatory cytokines. PLoS Genet. 2012, 8, e1002552. [Google Scholar] [CrossRef] [PubMed]
- Alvelos, M.I.; Bruggemann, M.; Sutandy, F.R.; Juan-Mateu, J.; Colli, M.L.; Busch, A.; Lopes, M.; Castela, A.; Aartsma-Rus, A.; Konig, J.; et al. The RNA-binding profile of the splicing factor SRSF6 in immortalized human pancreatic beta-cells. Life Sci. Alliance 2021, 4, e202000825. [Google Scholar] [CrossRef]
- Santin, I.; Moore, F.; Colli, M.L.; Gurzov, E.N.; Marselli, L.; Marchetti, P.; Eizirik, D.L. PTPN2, a candidate gene for type 1 diabetes, modulates pancreatic beta-cell apoptosis via regulation of the BH3-only protein Bim. Diabetes 2011, 60, 3279–3288. [Google Scholar] [CrossRef] [Green Version]
- Barthson, J.; Germano, C.M.; Moore, F.; Maida, A.; Drucker, D.J.; Marchetti, P.; Gysemans, C.; Mathieu, C.; Nunez, G.; Jurisicova, A.; et al. Cytokines tumor necrosis factor-alpha and interferon-gamma induce pancreatic beta-cell apoptosis through STAT1-mediated Bim protein activation. J. Biol. Chem. 2011, 286, 39632–39643. [Google Scholar] [CrossRef] [Green Version]
- Moore, F.; Santin, I.; Nogueira, T.C.; Gurzov, E.N.; Marselli, L.; Marchetti, P.; Eizirik, D.L. The transcription factor C/EBP delta has anti-apoptotic and anti-inflammatory roles in pancreatic beta cells. PLoS ONE 2012, 7, e31062. [Google Scholar] [CrossRef] [Green Version]
- Dooley, J.; Tian, L.; Schonefeldt, S.; Delghingaro-Augusto, V.; Garcia-Perez, J.E.; Pasciuto, E.; Di Marino, D.; Carr, E.J.; Oskolkov, N.; Lyssenko, V.; et al. Genetic predisposition for beta cell fragility underlies type 1 and type 2 diabetes. Nat. Genet. 2016, 48, 519–527. [Google Scholar] [CrossRef] [PubMed]
- London, S.; De Franco, E.; Elias-Assad, G.; Barhoum, M.N.; Felszer, C.; Paniakov, M.; Weiner, S.A.; Tenenbaum-Rakover, Y. Case Report: Neonatal Diabetes Mellitus Caused by a Novel GLIS3 Mutation in Twins. Front. Endocrinol. 2021, 12, 673755. [Google Scholar] [CrossRef]
- ZeRuth, G.T.; Takeda, Y.; Jetten, A.M. The Kruppel-like protein Gli-similar 3 (Glis3) functions as a key regulator of insulin transcription. Mol. Endocrinol. 2013, 27, 1692–1705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yalcintepe, S.; Ozguc Comlek, F.; Gurkan, H.; Demir, S.; Atli, E.I.; Atli, E.; Eker, D.; Tutunculer Kokenli, F. The Application of Next Generation Sequencing Maturity Onset Diabetes of the Young Gene Panel in Turkish Patients from Trakya Region. J. Clin. Res. Pediatr. Endocrinol. 2021, 13, 320–331. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scoville, D.W.; Jetten, A.M. GLIS3: A Critical Transcription Factor in Islet β-Cell Generation. Cells 2021, 10, 3471. https://doi.org/10.3390/cells10123471
Scoville DW, Jetten AM. GLIS3: A Critical Transcription Factor in Islet β-Cell Generation. Cells. 2021; 10(12):3471. https://doi.org/10.3390/cells10123471
Chicago/Turabian StyleScoville, David W., and Anton M. Jetten. 2021. "GLIS3: A Critical Transcription Factor in Islet β-Cell Generation" Cells 10, no. 12: 3471. https://doi.org/10.3390/cells10123471
APA StyleScoville, D. W., & Jetten, A. M. (2021). GLIS3: A Critical Transcription Factor in Islet β-Cell Generation. Cells, 10(12), 3471. https://doi.org/10.3390/cells10123471