
Development of Stem Cell-Derived Immune Cells for Off-the-Shelf Cancer Immunotherapies



Abstract
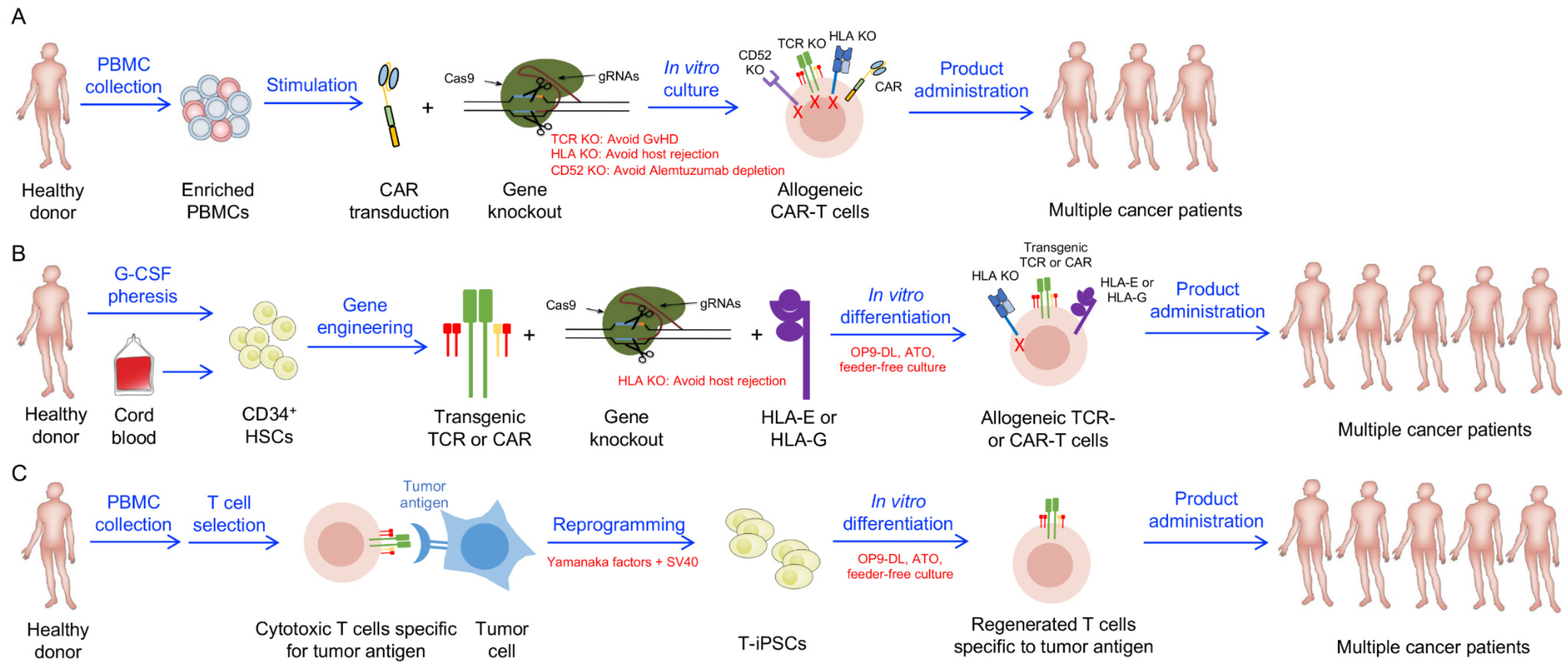
:1. Introduction
2. Stem Cell Resources and Culture Systems
3. Allogeneic Stem Cell-Engineered T Cell-Based Therapy
4. Allogeneic Stem Cell-Engineered Unconventional T Cell-Based Therapy
4.1. Allogeneic iNKT Cell-Based Therapy
4.2. Allogeneic MAIT Cell-Based Therapy
4.3. Allogeneic γδ T Cell-Based Therapy
5. Allogeneic Stem Cell-Engineered NK Cell-Based Therapy
5.1. CAR-NK Cells Derived from Umbilical Cord Blood (UCB)
5.2. CAR-NK Cells Derived from Other PSCs
6. Outlook
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Mohanty, R.; Chowdhury, C.R.; Arega, S.; Sen, P.; Ganguly, P.; Ganguly, N. CAR T cell therapy: A new era for cancer treatment (Review). Oncol. Rep. 2019, 42, 2183–2195. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Restifo, N.P. Adoptive cell transfer as personalized immunotherapy for human cancer. Science 2015, 348, 62–68. [Google Scholar] [CrossRef] [Green Version]
- Dudley, M.E.; Rosenberg, S.A. Adoptive-cell-transfer therapy for the treatment of patients with cancer. Nat. Rev. Cancer 2003, 3, 666–675. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenberg, S.A.; Yannelli, J.R.; Yang, J.C.; Topalian, S.L.; Schwartzentruber, D.J.; Weber, J.S.; Parkinson, D.R.; Seipp, C.A.; Einhorn, J.H.; White, D.E. Treatment of patients with metastatic melanoma with autologous tumor-infiltrating lymphocytes and interleukin 2. J. Natl. Cancer Inst. 1994, 86, 1159–1166. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Cao, Y.J. Engineered T Cell Therapy for Cancer in the Clinic. Front. Immunol. 2019, 10, 2250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadelain, M.; Brentjens, R.; Rivière, I. The basic principles of chimeric antigen receptor design. Cancer Discov. 2013, 3, 388–398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atrash, S.; Moyo, T.K. A Review of Chimeric Antigen Receptor T-Cell Therapy for Myeloma and Lymphoma. Onco. Targets. Ther. 2021, 14, 2185–2201. [Google Scholar] [CrossRef]
- Vormittag, P.; Gunn, R.; Ghorashian, S.; Veraitch, F.S. A guide to manufacturing CAR T cell therapies. Curr. Opin. Biotechnol. 2018, 53, 164–181. [Google Scholar] [CrossRef]
- Li, Y.-R.; Zhou, Y.; Kramer, A.; Yang, L. Engineering stem cells for cancer immunotherapy. Trends Cancer 2021, 7, 1059–1073. [Google Scholar] [CrossRef] [PubMed]
- Fraietta, J.A.; Lacey, S.F.; Orlando, E.J.; Pruteanu-Malinici, I.; Gohil, M.; Lundh, S.; Boesteanu, A.C.; Wang, Y.; O’Connor, R.S.; Hwang, W.-T.; et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat. Med. 2018, 24, 563–571. [Google Scholar] [CrossRef] [PubMed]
- Guedan, S.; Ruella, M.; June, C.H. Emerging Cellular Therapies for Cancer. Annu. Rev. Immunol. 2018, 37, 145–171. [Google Scholar] [CrossRef] [PubMed]
- Perez, C.; Gruber, I.; Arber, C. Off-the-Shelf Allogeneic T Cell Therapies for Cancer: Opportunities and Challenges Using Naturally Occurring “Universal” Donor T Cells. Front. Immunol. 2020, 11, 583716. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.K.; McGuirk, J.P. Allogeneic Stem Cell Transplantation: A Historical and Scientific Overview. Cancer Res. 2016, 76, 6445–6451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrara, J.L.M.; Levine, J.E.; Reddy, P.; Holler, E. Graft-versus-host disease. Lancet 2009, 373, 1550–1561. [Google Scholar] [CrossRef]
- Penack, O.; Marchetti, M.; Ruutu, T.; Aljurf, M.; Bacigalupo, A.; Bonifazi, F.; Ciceri, F.; Cornelissen, J.; Malladi, R.; Duarte, R.F.; et al. Prophylaxis and management of graft versus host disease after stem-cell transplantation for haematological malignancies: Updated consensus recommendations of the European Society for Blood and Marrow Transplantation. Lancet Haematol. 2020, 7, e157–e167. [Google Scholar] [CrossRef]
- Hill, G.R.; Betts, B.C.; Tkachev, V.; Kean, L.S.; Blazar, B.R. Current Concepts and Advances in Graft-Versus-Host Disease Immunology. Annu. Rev. Immunol. 2021, 39, 19–49. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Cibrian, N.; Zeiser, R.; Perez-Simon, J.A. Graft-versus-host disease prophylaxis: Pathophysiology-based review on current approaches and future directions. Blood Rev. 2021, 48, 100792. [Google Scholar] [CrossRef]
- Zeiser, R.; von Bubnoff, N.; Butler, J.; Mohty, M.; Niederwieser, D.; Or, R.; Szer, J.; Wagner, E.M.; Zuckerman, T.; Mahuzier, B.; et al. Ruxolitinib for Glucocorticoid-Refractory Acute Graft-versus-Host Disease. N. Engl. J. Med. 2020, 382, 1800–1810. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.-J.; Zhao, X.-Y.; Huang, X.-J. Strategies for Enhancing and Preserving Anti-leukemia Effects Without Aggravating Graft-Versus-Host Disease. Front. Immunol. 2018, 9, 3041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Souza, A.; Lee, S.; Zhu, X.; Pasquini, M. Current Use and Trends in Hematopoietic Cell Transplantation in the United States. Biol. Blood Marrow Transplant. 2017, 23, 1417–1421. [Google Scholar] [CrossRef] [Green Version]
- Depil, S.; Duchateau, P.; Grupp, S.A.; Mufti, G.; Poirot, L. “Off-the-shelf” allogeneic CAR T cells: Development and challenges. Nat. Rev. Drug Discov. 2020, 19, 185–199. [Google Scholar] [CrossRef] [PubMed]
- Ghaffari, S.; Khalili, N.; Rezaei, N. CRISPR/Cas9 revitalizes adoptive T-cell therapy for cancer immunotherapy. J. Exp. Clin. Cancer Res. 2021, 40, 269. [Google Scholar] [CrossRef] [PubMed]
- Torikai, H.; Reik, A.; Liu, P.-Q.; Zhou, Y.; Zhang, L.; Maiti, S.; Huls, H.; Miller, J.C.; Kebriaei, P.; Rabinovich, B.; et al. A foundation for universal T-cell based immunotherapy: T cells engineered to express a CD19-specific chimeric-antigen-receptor and eliminate expression of endogenous TCR. Blood 2012, 119, 5697–5705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torikai, H.; Reik, A.; Soldner, F.; Warren, E.H.; Yuen, C.; Zhou, Y.; Crossland, D.L.; Huls, H.; Littman, N.; Zhang, Z.; et al. Toward eliminating HLA class I expression to generate universal cells from allogeneic donors. Blood 2013, 122, 1341–1349. [Google Scholar] [CrossRef]
- Benjamin, R.; Graham, C.; Yallop, D.; Jozwik, A.; Mirci-Danicar, O.C.; Lucchini, G.; Pinner, D.; Jain, N.; Kantarjian, H.; Boissel, N.; et al. Genome-edited, donor-derived allogeneic anti-CD19 chimeric antigen receptor T cells in paediatric and adult B-cell acute lymphoblastic leukaemia: Results of two phase 1 studies. Lancet 2020, 396, 1885–1894. [Google Scholar] [CrossRef]
- DiNofia, A.M.; Grupp, S.A. Will allogeneic CAR T cells for CD19(+) malignancies take autologous CAR T cells “off the shelf”? Nat. Rev. Clin. Oncol. 2021, 18, 195–196. [Google Scholar] [CrossRef] [PubMed]
- Lim, O.; Jung, M.Y.; Hwang, Y.K.; Shin, E.-C. Present and Future of Allogeneic Natural Killer Cell Therapy. Front. Immunol. 2015, 6, 286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hicklin, D.J.; Marincola, F.M.; Ferrone, S. HLA class I antigen downregulation in human cancers: T-cell immunotherapy revives an old story. Mol. Med. Today 1999, 5, 178–186. [Google Scholar] [CrossRef]
- Fang, F.; Xiao, W.; Tian, Z. NK cell-based immunotherapy for cancer. Semin. Immunol. 2017, 31, 37–54. [Google Scholar] [CrossRef]
- Romee, R.; Rosario, M.; Berrien-Elliott, M.M.; Wagner, J.A.; Jewell, B.A.; Schappe, T.; Leong, J.W.; Abdel-Latif, S.; Schneider, S.E.; Willey, S.; et al. Cytokine-induced memory-like natural killer cells exhibit enhanced responses against myeloid leukemia. Sci. Transl. Med. 2016, 8, 357ra123. [Google Scholar] [CrossRef] [Green Version]
- Liu, E.; Marin, D.; Banerjee, P.; Macapinlac, H.A.; Thompson, P.; Basar, R.; Nassif Kerbauy, L.; Overman, B.; Thall, P.; Kaplan, M.; et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N. Engl. J. Med. 2020, 382, 545–553. [Google Scholar] [CrossRef]
- Godfrey, D.I.; Le Nours, J.; Andrews, D.M.; Uldrich, A.P.; Rossjohn, J. Unconventional T Cell Targets for Cancer Immunotherapy. Immunity 2018, 48, 453–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Godfrey, D.I.; Uldrich, A.P.; McCluskey, J.; Rossjohn, J.; Moody, D.B. The burgeoning family of unconventional T cells. Nat. Immunol. 2015, 16, 1114–1123. [Google Scholar] [CrossRef] [PubMed]
- Heczey, A.; Courtney, A.N.; Montalbano, A.; Robinson, S.; Liu, K.; Li, M.; Ghatwai, N.; Dakhova, O.; Liu, B.; Raveh-Sadka, T.; et al. Anti-GD2 CAR-NKT cells in patients with relapsed or refractory neuroblastoma: An interim analysis. Nat. Med. 2020, 26, 1686–1690. [Google Scholar] [CrossRef]
- Xu, Y.; Xiang, Z.; Alnaggar, M.; Kouakanou, L.; Li, J.; He, J.; Yang, J.; Hu, Y.; Chen, Y.; Lin, L.; et al. Allogeneic Vγ9Vδ2 T-cell immunotherapy exhibits promising clinical safety and prolongs the survival of patients with late-stage lung or liver cancer. Cell. Mol. Immunol. 2021, 18, 427–439. [Google Scholar] [CrossRef] [PubMed]
- Qasim, W.; Zhan, H.; Samarasinghe, S.; Adams, S.; Amrolia, P.; Stafford, S.; Butler, K.; Rivat, C.; Wright, G.; Somana, K.; et al. Molecular remission of infant B-ALL after infusion of universal TALEN gene-edited CAR T cells. Sci. Transl. Med. 2017, 9, 374. [Google Scholar] [CrossRef]
- MacLeod, D.T.; Antony, J.; Martin, A.J.; Moser, R.J.; Hekele, A.; Wetzel, K.J.; Brown, A.E.; Triggiano, M.A.; Hux, J.A.; Pham, C.D.; et al. Integration of a CD19 CAR into the TCR Alpha Chain Locus Streamlines Production of Allogeneic Gene-Edited CAR T Cells. Mol. Ther. 2017, 25, 949–961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Themeli, M.; Kloss, C.C.; Ciriello, G.; Fedorov, V.D.; Perna, F.; Gonen, M.; Sadelain, M. Generation of tumor-targeted human T lymphocytes from induced pluripotent stem cells for cancer therapy. Nat. Biotechnol. 2013, 31, 928–933. [Google Scholar] [CrossRef]
- Snauwaert, S.; Verstichel, G.; Bonte, S.; Goetgeluk, G.; Vanhee, S.; Van Caeneghem, Y.; De Mulder, K.; Heirman, C.; Stauss, H.; Heemskerk, M.H.M.; et al. In vitro generation of mature, naive antigen-specific CD8+ T cells with a single T-cell receptor by agonist selection. Leukemia 2014, 28, 830–841. [Google Scholar] [CrossRef] [PubMed]
- Montel-Hagen, A.; Seet, C.S.; Li, S.; Chick, B.; Zhu, Y.; Chang, P.; Tsai, S.; Sun, V.; Lopez, S.; Chen, H.C.; et al. Organoid-Induced Differentiation of Conventional T Cells from Human Pluripotent Stem Cells. Cell Stem Cell 2019, 24, 376–389.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seet, C.S.; He, C.; Bethune, M.T.; Li, S.; Chick, B.; Gschweng, E.H.; Zhu, Y.; Kim, K.; Kohn, D.B.; Baltimore, D.; et al. Generation of mature T cells from human hematopoietic stem and progenitor cells in artificial thymic organoids. Nat. Methods 2017, 14, 521–530. [Google Scholar] [CrossRef]
- Minagawa, A.; Yoshikawa, T.; Yasukawa, M.; Hotta, A.; Kunitomo, M.; Iriguchi, S.; Takiguchi, M.; Kassai, Y.; Imai, E.; Yasui, Y.; et al. Enhancing T Cell Receptor Stability in Rejuvenated iPSC-Derived T Cells Improves Their Use in Cancer Immunotherapy. Cell Stem Cell 2018, 23, 850–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishimura, T.; Kaneko, S.; Kawana-Tachikawa, A.; Tajima, Y.; Goto, H.; Zhu, D.; Nakayama-Hosoya, K.; Iriguchi, S.; Uemura, Y.; Shimizu, T.; et al. Generation of rejuvenated antigen-specific T cells by reprogramming to pluripotency and redifferentiation. Cell Stem Cell 2013, 12, 114–126.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Honda, T.; Ando, M.; Ando, J.; Ishii, M.; Sakiyama, Y.; Ohara, K.; Toyota, T.; Ohtaka, M.; Masuda, A.; Terao, Y.; et al. Sustainable Tumor-Suppressive Effect of iPSC-Derived Rejuvenated T Cells Targeting Cervical Cancers. Mol. Ther. 2020, 28, 2394–2405. [Google Scholar] [CrossRef] [PubMed]
- Ando, M.; Nakauchi, H. “Off-the-shelf” immunotherapy with iPSC-derived rejuvenated cytotoxic T lymphocytes. Exp. Hematol. 2017, 47, 2–12. [Google Scholar] [CrossRef]
- Vizcardo, R.; Masuda, K.; Yamada, D.; Ikawa, T.; Shimizu, K.; Fujii, S.I.; Koseki, H.; Kawamoto, H. Regeneration of human tumor antigen-specific T cells from iPSCs derived from mature CD8+ T cells. Cell Stem Cell 2013, 12, 31–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawamoto, H.; Masuda, K.; Nagano, S. Regeneration of antigen-specific T cells by using induced pluripotent stem cell (iPSC) technology. Int. Immunol. 2021. [Google Scholar] [CrossRef]
- Yamada, D.; Iyoda, T.; Vizcardo, R.; Shimizu, K.; Sato, Y.; Endo, T.A.; Kitahara, G.; Okoshi, M.; Kobayashi, M.; Sakurai, M.; et al. Efficient Regeneration of Human Vα24(+) Invariant Natural Killer T Cells and Their Anti-Tumor Activity In Vivo. Stem Cells 2016, 34, 2852–2860. [Google Scholar] [CrossRef]
- Kitayama, S.; Zhang, R.; Liu, T.Y.; Ueda, N.; Iriguchi, S.; Yasui, Y.; Kawai, Y.; Tatsumi, M.; Hirai, N.; Mizoro, Y.; et al. Cellular Adjuvant Properties, Direct Cytotoxicity of Re-differentiated Vα24 Invariant NKT-like Cells from Human Induced Pluripotent Stem Cells. Stem Cell Rep. 2016, 6, 213–227. [Google Scholar] [CrossRef] [Green Version]
- Wakao, H.; Yoshikiyo, K.; Koshimizu, U.; Furukawa, T.; Enomoto, K.; Matsunaga, T.; Tanaka, T.; Yasutomi, Y.; Yamada, T.; Minakami, H.; et al. Expansion of functional human mucosal-associated invariant T cells via reprogramming to pluripotency and redifferentiation. Cell Stem Cell 2013, 12, 546–558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wakao, H. Reprogramming of MAIT Cells to Pluripotency and Redifferentiation. Methods Mol. Biol. 2020, 2098, 237–257. [Google Scholar] [CrossRef] [PubMed]
- Wakao, H.; Fujita, H. Toward the realization of cell therapy: The advent of MAIT cells from iPSCs. Cell Cycle 2013, 12, 2341–2342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, D.; Koyanagi-Aoi, M.; Taniguchi-Ikeda, M.; Yoshida, Y.; Azuma, T.; Aoi, T. The Generation of Human γδT Cell-Derived Induced Pluripotent Stem Cells from Whole Peripheral Blood Mononuclear Cell Culture. Stem Cells Transl. Med. 2018, 7, 34–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, E.; Tong, Y.; Dotti, G.; Shaim, H.; Savoldo, B.; Mukherjee, M.; Orange, J.; Wan, X.; Lu, X.; Reynolds, A.; et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia 2018, 32, 520–531. [Google Scholar] [CrossRef] [PubMed]
- Hermanson, D.L.; Bendzick, L.; Pribyl, L.; McCullar, V.; Vogel, R.I.; Miller, J.S.; Geller, M.A.; Kaufman, D.S. Induced Pluripotent Stem Cell-Derived Natural Killer Cells for Treatment of Ovarian Cancer. Stem Cells 2016, 34, 93–101.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Hermanson, D.L.; Moriarity, B.S.; Kaufman, D.S. Human iPSC-Derived Natural Killer Cells Engineered with Chimeric Antigen Receptors Enhance Anti-tumor Activity. Cell Stem Cell 2018, 23, 181–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woll, P.S.; Martin, C.H.; Miller, J.S.; Kaufman, D.S. Human embryonic stem cell-derived NK cells acquire functional receptors and cytolytic activity. J. Immunol. 2005, 175, 5095–5103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cichocki, F.; Bjordahl, R.; Gaidarova, S.; Mahmood, S.; Abujarour, R.; Wang, H.; Tuininga, K.; Felices, M.; Davis, Z.B.; Bendzick, L.; et al. iPSC-derived NK cells maintain high cytotoxicity and enhance In Vivo tumor control in concert with T cells and anti–PD-1 therapy. Sci. Transl. Med. 2020, 12, eaaz5618. [Google Scholar] [CrossRef]
- Zhu, H.; Blum, R.H.; Bjordahl, R.; Gaidarova, S.; Rogers, P.; Lee, T.T.; Abujarour, R.; Bonello, G.B.; Wu, J.; Tsai, P.-F.; et al. Pluripotent stem cell-derived NK cells with high-affinity noncleavable CD16a mediate improved antitumor activity. Blood 2020, 135, 399–410. [Google Scholar] [CrossRef]
- Goldenson, B.H.; Zhu, H.; Wang, Y.M.; Heragu, N.; Bernareggi, D.; Ruiz-Cisneros, A.; Bahena, A.; Ask, E.H.; Hoel, H.J.; Malmberg, K.-J.; et al. Umbilical Cord Blood and iPSC-Derived Natural Killer Cells Demonstrate Key Differences in Cytotoxic Activity and KIR Profiles. Front. Immunol. 2020, 11, 561553. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Kaufman, D.S. An Improved Method to Produce Clinical-Scale Natural Killer Cells from Human Pluripotent Stem Cells. Methods Mol. Biol. 2019, 2048, 107–119. [Google Scholar] [CrossRef]
- Gumperz, J.E.; Miyake, S.; Yamamura, T.; Brenner, M.B. Functionally distinct subsets of CD1d-restricted natural killer T cells revealed by CD1d tetramer staining. J. Exp. Med. 2002, 195, 625–636. [Google Scholar] [CrossRef] [PubMed]
- Montoya, C.J.; Pollard, D.; Martinson, J.; Kumari, K.; Wasserfall, C.; Mulder, C.B.; Rugeles, M.T.; Atkinson, M.A.; Landay, A.L.; Wilson, S.B. Characterization of human invariant natural killer T subsets in health and disease using a novel invariant natural killer T cell-clonotypic monoclonal antibody, 6B11. Immunology 2007, 122, 1–14. [Google Scholar] [CrossRef]
- Zhu, Y.; Smith, D.J.; Zhou, Y.; Li, Y.R.; Yu, J.; Lee, D.; Wang, Y.C.; Di Biase, S.; Wang, X.; Hardoy, C.; et al. Development of Hematopoietic Stem Cell-Engineered Invariant Natural Killer T Cell Therapy for Cancer. Cell Stem Cell 2019, 25, 542–557.e9. [Google Scholar] [CrossRef]
- Zhou, Y.; Li, Y.-R.; Zeng, S.; Yang, L. Methods for Studying Mouse and Human Invariant Natural Killer T Cells. Methods Mol. Biol. 2021, 2388, 35–57. [Google Scholar] [CrossRef]
- Groh, V.; Porcelli, S.; Fabbi, M.; Lanier, L.L.; Picker, L.J.; Anderson, T.; Warnke, R.A.; Bhan, A.K.; Strominger, J.L.; Brenner, M.B. Human lymphocytes bearing T cell receptor gamma/delta are phenotypically diverse and evenly distributed throughout the lymphoid system. J. Exp. Med. 1989, 169, 1277–1294. [Google Scholar] [CrossRef] [PubMed]
- Notta, F.; Doulatov, S.; Laurenti, E.; Poeppl, A.; Jurisica, I.; Dick, J.E. Isolation of single human hematopoietic stem cells capable of long-term multilineage engraftment. Science 2011, 333, 218–221. [Google Scholar] [CrossRef]
- Larson, S.; De Oliveira, S.N. Gene-modified hematopoietic stem cells for cancer immunotherapy. Hum. Vaccines Immunother. 2014, 10, 982–985. [Google Scholar] [CrossRef] [Green Version]
- Saetersmoen, M.L.; Hammer, Q.; Valamehr, B.; Kaufman, D.S.; Malmberg, K.J. Off-the-shelf cell therapy with induced pluripotent stem cell-derived natural killer cells. Semin. Immunopathol. 2019, 41, 59–68. [Google Scholar] [CrossRef] [Green Version]
- Nishimura, T.; Nakauchi, H. Generation of antigen-specific T cells from human induced pluripotent stem cells. Methods Mol. Biol. 2019, 1899, 25–40. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, T.M.; Zúñiga-Pflücker, J.C. Induction of T cell development from hematopoietic progenitor cells by delta-like-1 In Vitro. Immunity 2002, 17, 749–756. [Google Scholar] [CrossRef] [Green Version]
- Ciofani, M.; Zúñiga-Pflücker, J.C. Notch promotes survival of pre-T cells at the β-selection checkpoint by regulating cellular metabolism. Nat. Immunol. 2005, 6, 881–888. [Google Scholar] [CrossRef]
- Ciofani, M.; Knowles, G.C.; Wiest, D.L.; von Boehmer, H.; Zúñiga-Pflücker, J.C. Stage-Specific and Differential Notch Dependency at the αβ and γδ T Lineage Bifurcation. Immunity 2006, 25, 105–116. [Google Scholar] [CrossRef] [Green Version]
- Besseyrias, V.; Fiorini, E.; Strobl, L.J.; Zimber-Strobl, U.; Dumortier, A.; Koch, U.; Arcangeli, M.L.; Ezine, S.; MacDonald, H.R.; Radtke, F. Hierarchy of Notch-Delta interactions promoting T cell lineage commitment and maturation. J. Exp. Med. 2007, 204, 331–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Awong, G.; La Motte-Mohs, R.N.; Zúñiga-Pflücker, J.C. Generation of pro-T cells In Vitro: Potential for immune reconstitution. Semin. Immunol. 2007, 19, 341–349. [Google Scholar] [CrossRef] [PubMed]
- Vodyanik, M.A.; Bork, J.A.; Thomson, J.A.; Slukvin, I.I. Human embryonic stem cell-derived CD34+ cells: Efficient production in the coculture with OP9 stromal cells and analysis of lymphohematopoietic potential. Blood 2005, 105, 617–626. [Google Scholar] [CrossRef] [Green Version]
- Takayama, N.; Nishikii, H.; Usui, J.; Tsukui, H.; Sawaguchi, A.; Hiroyama, T.; Eto, K.; Nakauchi, H. Generation of functional platelets from human embryonic stem cells In Vitro via ES-sacs, VEGF-promoted structures that concentrate hematopoietic progenitors. Blood 2008, 111, 5298–5306. [Google Scholar] [CrossRef] [Green Version]
- Yamasaki, K.; Horiguchi, S.; Kurosaki, M.; Kunii, N.; Nagato, K.; Hanaoka, H.; Shimizu, N.; Ueno, N.; Yamamoto, S.; Taniguchi, M.; et al. Induction of NKT cell-specific immune responses in cancer tissues after NKT cell-targeted adoptive immunotherapy. Clin. Immunol. 2011, 138, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knorr, P. Rotocols and Manufacturing for Cell -Based Therapies Clinical-Scale Derivation of Natural Killer Cells From Human Pluripotent Stem Cells for Cancer Therapy. Stem Cells Transl. Med. 2013, 2, 274–283. [Google Scholar] [CrossRef] [PubMed]
- Shimasaki, N.; Jain, A.; Campana, D. NK cells for cancer immunotherapy. Nat. Rev. Drug Discov. 2020, 19, 200–218. [Google Scholar] [CrossRef] [PubMed]
- Woll, P.S.; Grzywacz, B.; Tian, X.; Marcus, R.K.; Knorr, D.A.; Verneris, M.R.; Kaufman, D.S. Human embryonic stem cells differentiate into a homogeneous population of natural killer cells with potent In Vivo antitumor activity. Blood 2009, 113, 6094–6101. [Google Scholar] [CrossRef] [PubMed]
- Delaney, C.; Heimfeld, S.; Brashem-Stein, C.; Voorhies, H.; Manger, R.L.; Bernstein, I.D. Notch-mediated expansion of human cord blood progenitor cells capable of rapid myeloid reconstitution. Nat. Med. 2010, 16, 232–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melkus, M.W.; Estes, J.D.; Padgett-Thomas, A.; Gatlin, J.; Denton, P.W.; Othieno, F.A.; Wege, A.K.; Haase, A.T.; Garcia, J.V. Humanized mice mount specific adaptive and innate immune responses to EBV and TSST-1. Nat. Med. 2006, 12, 1316–1322. [Google Scholar] [CrossRef] [PubMed]
- Lan, P.; Tonomura, N.; Shimizu, A.; Wang, S.; Yang, Y.G. Reconstitution of a functional human immune system in immunodeficient mice through combined human fetal thymus/liver and CD34+ cell transplantation. Blood 2006, 108, 487–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kitchen, S.G.; Bennett, M.; Galić, Z.; Kim, J.; Xu, Q.; Young, A.; Lieberman, A.; Joseph, A.; Goldstein, H.; Ng, H.; et al. Engineering antigen-specific T cells from genetically modified human hematopoietic stem cells in immunodeficient mice. PLoS ONE 2009, 4, e8208. [Google Scholar] [CrossRef] [PubMed]
- Vatakis, D.N.; Arumugam, B.; Kim, S.G.; Bristol, G.; Yang, O.; Zack, J.A. Introduction of exogenous T-cell receptors into human hematopoietic progenitors results in exclusion of endogenous T-cell receptor expression. Mol. Ther. 2013, 21, 1055–1063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, D.J.; Liu, S.; Ji, S.; Li, B.; McLaughlin, J.; Cheng, D.; Witte, O.N.; Yang, L. Genetic engineering of hematopoietic stem cells to generate invariant natural killer T cells. Proc. Natl. Acad. Sci. USA 2015, 112, 1523–1528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenblatt, M.B.; Vbranac, V.; Tivey, T.; Tsang, K.; Tager, A.M.; Aliprantis, A.O. Graft versus Host Disease in the Bone Marrow, Liver and Thymus Humanized Mouse Model. PLoS ONE 2012, 7, e44664. [Google Scholar] [CrossRef] [PubMed]
- Shultz, L.D.; Saito, Y.; Najima, Y.; Tanaka, S.; Ochi, T.; Tomizawa, M.; Doi, T.; Sone, A.; Suzuki, N.; Fujiwara, H.; et al. Generation of functional human T-cell subsets with HLA-restricted immune responses in HLA class I expressing NOD/SCID/IL2rγnull humanized mice. Proc. Natl. Acad. Sci. USA 2010, 107, 13022–13027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishimura, K.; Sano, M.; Ohtaka, M.; Furuta, B.; Umemura, Y.; Nakajima, Y.; Ikehara, Y.; Kobayashi, T.; Segawa, H.; Takayasu, S.; et al. Development of defective and persistent Sendai virus vector: A unique gene delivery/expression system ideal for cell reprogramming. J. Biol. Chem. 2011, 286, 4760–4771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, J.; Tang, S.Y.; Toh, L.L.; Wang, S. Generation of “Off-the-Shelf” Natural Killer Cells from Peripheral Blood Cell-Derived Induced Pluripotent Stem Cells. Stem Cell Rep. 2017, 9, 1796–1812. [Google Scholar] [CrossRef] [Green Version]
- Choi, B.D.; Yu, X.; Castano, A.P.; Darr, H.; Henderson, D.B.; Bouffard, A.A.; Larson, R.C.; Scarfò, I.; Bailey, S.R.; Gerhard, G.M.; et al. CRISPR-Cas9 disruption of PD-1 enhances activity of universal EGFRvIII CAR T cells in a preclinical model of human glioblastoma. J. Immunother. Cancer 2019, 7, 304. [Google Scholar] [CrossRef] [PubMed]
- Webber, B.R.; Lonetree, C.-L.; Kluesner, M.G.; Johnson, M.J.; Pomeroy, E.J.; Diers, M.D.; Lahr, W.S.; Draper, G.M.; Slipek, N.J.; Smeester, B.A.; et al. Highly efficient multiplex human T cell engineering without double-strand breaks using Cas9 base editors. Nat. Commun. 2019, 10, 5222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benjamin, R.; Graham, C.; Yallop, D.; Jozwik, A.; Ciocarlie, O.; Jain, N.; Jabbour, E.J.; Maus, M.V.; Frigault, M.; Boissel, N.; et al. Preliminary Data on Safety, Cellular Kinetics and Anti-Leukemic Activity of UCART19, an Allogeneic Anti-CD19 CAR T-Cell Product, in a Pool of Adult and Pediatric Patients with High-Risk CD19+ Relapsed/Refractory B-Cell Acute Lymphoblastic Leukemia. Blood 2018, 132, 896. [Google Scholar] [CrossRef]
- Van Caeneghem, Y.; De Munter, S.; Tieppo, P.; Goetgeluk, G.; Weening, K.; Verstichel, G.; Bonte, S.; Taghon, T.; Leclercq, G.; Kerre, T.; et al. Antigen receptor-redirected T cells derived from hematopoietic precursor cells lack expression of the endogenous TCR/CD3 receptor and exhibit specific antitumor capacities. Oncoimmunology 2017, 6, e1283460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giannoni, F.; Hardee, C.L.; Wherley, J.; Gschweng, E.; Senadheera, S.; Kaufman, M.L.; Chan, R.; Bahner, I.; Gersuk, V.; Wang, X.; et al. Allelic exclusion and peripheral reconstitution by TCR transgenic T cells arising from transduced human hematopoietic stem/progenitor cells. Mol. Ther. 2013, 21, 1044–1054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, M.A.; Büning, H.; Sauer, M.; Schambach, A. Use of Cell and Genome Modification Technologies to Generate Improved “Off-the-Shelf” CAR T and CAR NK Cells. Front. Immunol. 2020, 11, 1965. [Google Scholar] [CrossRef] [PubMed]
- Braud, V.; Calan, C.A.O.; So, K.; Andrea, A.D.; Ogg, G.S.; Lazetic, S.; Young, N.T.; Bell, J.I.; Phillips, J.H.; Lanier, L.L.; et al. HLA-E binds to natural killer cell receptors CD94/NKG2A, B and C. Nature 1998, 391, 795–799. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.; Llano, M.; Carretero, M.; Ishitani, A.; Navarro, F.; López-Botet, M.; Geraghty, D.E. HLA-E is a major ligand for the natural killer inhibitory receptor CD94/NKG2A. Proc. Natl. Acad. Sci. USA 1998, 95, 5199–5204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rouas-Freiss, N.; Marchal, R.E.; Kirszenbaum, M.; Dausset, J.; Carosella, E.D. The α1 domain of HLA-G1 and HLA-G2 inhibits cytotoxicity induced by natural killer cells: Is HLA-G the public ligand for natural killer cell inhibitory receptors? Proc. Natl. Acad. Sci. USA 1997, 94, 5249–5254. [Google Scholar] [CrossRef] [Green Version]
- Gornalusse, G.G.; Hirata, R.K.; Funk, S.E.; Riolobos, L.; Lopes, V.S.; Manske, G.; Prunkard, D.; Colunga, A.G.; Hanafi, L.-A.; Clegg, D.O.; et al. HLA-E-expressing pluripotent stem cells escape allogeneic responses and lysis by NK cells. Nat. Biotechnol. 2017, 35, 765–772. [Google Scholar] [CrossRef] [Green Version]
- Bendelac, A.; Savage, P.B.; Teyton, L. The biology of NKT cells. Annu. Rev. Immunol. 2007, 25, 297–336. [Google Scholar] [CrossRef] [Green Version]
- Bae, E.-A.; Seo, H.; Kim, I.-K.; Jeon, I.; Kang, C.-Y. Roles of NKT cells in cancer immunotherapy. Arch. Pharm. Res. 2019, 42, 543–548. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Basabe, A.; Strati, F.; Facciotti, F. License to Kill: When iNKT Cells Are Granted the Use of Lethal Cytotoxicity. Int. J. Mol. Sci. 2020, 21, 3909. [Google Scholar] [CrossRef] [PubMed]
- Krijgsman, D.; Hokland, M.; Kuppen, P.J.K. The Role of Natural Killer T Cells in Cancer-A Phenotypical and Functional Approach. Front. Immunol. 2018, 9, 367. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Oh, S.; Lim, S.; Shin, J.H.; Yoon, M.S.; Park, S.-H. Invariant NKT cells facilitate cytotoxic T-cell activation via direct recognition of CD1d on T cells. Exp. Mol. Med. 2019, 51, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Santo, C.; Arscott, R.; Booth, S.; Karydis, I.; Jones, M.; Asher, R.; Salio, M.; Middleton, M.; Cerundolo, V. Invariant NKT cells modulate the suppressive activity of IL-10-secreting neutrophils differentiated with serum amyloid A. Nat. Immunol. 2010, 11, 1039–1046. [Google Scholar] [CrossRef] [Green Version]
- Mavers, M.; Maas-Bauer, K.; Negrin, R.S. Invariant Natural Killer T Cells As Suppressors of Graft-versus-Host Disease in Allogeneic Hematopoietic Stem Cell Transplantation. Front. Immunol. 2017, 8, 900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fereidouni, M.; Derakhshani, A.; Exley, M.A. iNKT cells and hematopoietic stem cell transplantation: Two-phase activation of iNKT cells may improve outcome. Clin. Immunol. 2019, 207, 43–48. [Google Scholar] [CrossRef]
- Guan, P.; Bassiri, H.; Patel, N.P.; Nichols, K.E.; Das, R. Invariant natural killer T cells in hematopoietic stem cell transplantation: Killer choice for natural suppression. Bone Marrow Transplant. 2016, 51, 629–637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chaidos, A.; Patterson, S.; Szydlo, R.; Chaudhry, M.S.; Dazzi, F.; Kanfer, E.; McDonald, D.; Marin, D.; Milojkovic, D.; Pavlu, J.; et al. Graft invariant natural killer T-cell dose predicts risk of acute graft-versus-host disease in allogeneic hematopoietic stem cell transplantation. Blood 2012, 119, 5030–5036. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Springfield, R.; Chen, S.; Li, X.; Feng, X.; Moshirian, R.; Yang, R.; Yuan, W. α-GalCer and iNKT Cell-Based Cancer Immunotherapy: Realizing the Therapeutic Potentials. Front. Immunol. 2019, 10, 1126. [Google Scholar] [CrossRef] [Green Version]
- Exley, M.A.; Friedlander, P.; Alatrakchi, N.; Vriend, L.; Yue, S.; Sasada, T.; Zeng, W.; Mizukami, Y.; Clark, J.; Nemer, D.; et al. Adoptive Transfer of Invariant NKT Cells as Immunotherapy for Advanced Melanoma: A Phase I Clinical Trial. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 3510–3519. [Google Scholar] [CrossRef] [Green Version]
- Motohashi, S.; Ishikawa, A.; Ishikawa, E.; Otsuji, M.; Iizasa, T.; Hanaoka, H.; Shimizu, N.; Horiguchi, S.; Okamoto, Y.; Fujii, S.; et al. A phase I study of In Vitro expanded natural killer T cells in patients with advanced and recurrent non-small cell lung cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2006, 12, 6079–6086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heczey, A.; Liu, D.; Tian, G.; Courtney, A.N.; Wei, J.; Marinova, E.; Gao, X.; Guo, L.; Yvon, E.; Hicks, J.; et al. Invariant NKT cells with chimeric antigen receptor provide a novel platform for safe and effective cancer immunotherapy. Blood 2014, 124, 2824–2833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ngai, H.; Tian, G.; Courtney, A.N.; Ravari, S.B.; Guo, L.; Liu, B.; Jin, J.; Shen, E.T.; Di Pierro, E.J.; Metelitsa, L.S. IL-21 Selectively Protects CD62L(+) NKT Cells and Enhances Their Effector Functions for Adoptive Immunotherapy. J. Immunol. 2018, 201, 2141–2153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, G.; Courtney, A.N.; Jena, B.; Heczey, A.; Liu, D.; Marinova, E.; Guo, L.; Xu, X.; Torikai, H.; Mo, Q.; et al. CD62L+ NKT cells have prolonged persistence and antitumor activity In Vivo. J. Clin. Investig. 2016, 126, 2341–2355. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Huang, W.; Heczey, A.; Liu, D.; Guo, L.; Wood, M.; Jin, J.; Courtney, A.N.; Liu, B.; Di Pierro, E.J.; et al. NKT Cells Coexpressing a GD2-Specific Chimeric Antigen Receptor and IL15 Show Enhanced In Vivo Persistence and Antitumor Activity against Neuroblastoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 7126–7138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujii, S.I.; Shimizu, K.; Okamoto, Y.; Kunii, N.; Nakayama, T.; Motohashi, S.; Taniguchi, M. NKT cells as an ideal anti-tumor immunotherapeutic. Front. Immunol. 2013, 4, 409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurioka, A.; Jahun, A.S.; Hannaway, R.F.; Walker, L.J.; Fergusson, J.R.; Sverremark-Ekström, E.; Corbett, A.J.; Ussher, J.E.; Willberg, C.B.; Klenerman, P. Shared and distinct phenotypes and functions of human cD161++ Vα7.2+ T cell subsets. Front. Immunol. 2017, 8, 1031. [Google Scholar] [CrossRef]
- Godfrey, D.I.; Koay, H.-F.; McCluskey, J.; Gherardin, N.A. The biology and functional importance of MAIT cells. Nat. Immunol. 2019, 20, 1110–1128. [Google Scholar] [CrossRef] [PubMed]
- Gherardin, N.A.; Loh, L.; Admojo, L.; Davenport, A.J.; Richardson, K.; Rogers, A.; Darcy, P.K.; Jenkins, M.R.; Prince, H.M.; Harrison, S.J.; et al. Enumeration, functional responses and cytotoxic capacity of MAIT cells in newly diagnosed and relapsed multiple myeloma. Sci. Rep. 2018, 8, 4159. [Google Scholar] [CrossRef] [Green Version]
- Sundström, P.; Szeponik, L.; Ahlmanner, F.; Sundquist, M.; Wong, J.S.B.; Lindskog, E.B.; Gustafsson, B.; Quiding-Järbrink, M. Tumor-infiltrating mucosal-associated invariant T (MAIT) cells retain expression of cytotoxic effector molecules. Oncotarget 2019, 10, 2810–2823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zabijak, L.; Attencourt, C.; Guignant, C.; Chatelain, D.; Marcelo, P.; Marolleau, J.-P.; Treiner, E. Increased tumor infiltration by mucosal-associated invariant T cells correlates with poor survival in colorectal cancer patients. Cancer Immunol. Immunother. 2015, 64, 1601–1608. [Google Scholar] [CrossRef]
- Ling, L.; Lin, Y.; Zheng, W.; Hong, S.; Tang, X.; Zhao, P.; Li, M.; Ni, J.; Li, C.; Wang, L.; et al. Circulating and tumor-infiltrating mucosal associated invariant T (MAIT) cells in colorectal cancer patients. Sci. Rep. 2016, 6, 20358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, M.; Goswami, S.; Shi, J.-Y.; Wu, L.-J.; Wang, X.-Y.; Ma, J.-Q.; Zhang, Z.; Shi, Y.; Ma, L.-J.; Zhang, S.; et al. Activated and Exhausted MAIT Cells Foster Disease Progression and Indicate Poor Outcome in Hepatocellular Carcinoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 3304–3316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, J.; Allen, S.; McDonald, E.; Das, I.; Mak, J.Y.W.; Liu, L.; Fairlie, D.P.; Meehan, B.S.; Chen, Z.; Corbett, A.J.; et al. MAIT Cells Promote Tumor Initiation, Growth, and Metastases via Tumor MR1. Cancer Discov. 2020, 10, 124–141. [Google Scholar] [CrossRef] [PubMed]
- Iriguchi, S.; Kaneko, S. Toward the development of true “off-the-shelf” synthetic T-cell immunotherapy. Cancer Sci. 2019, 110, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Trotman-Grant, A.C.; Mohtashami, M.; De Sousa Casal, J.; Martinez, E.C.; Lee, D.; Teichman, S.; Brauer, P.M.; Han, J.; Anderson, M.K.; Zúñiga-Pflücker, J.C. DL4-μbeads induce T cell lineage differentiation from stem cells in a stromal cell-free system. Nat. Commun. 2021, 12, 5023. [Google Scholar] [CrossRef]
- Legut, M.; Cole, D.K.; Sewell, A.K. The promise of γδ T cells and the γδ T cell receptor for cancer immunotherapy. Cell. Mol. Immunol. 2015, 12, 656–668. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, T.; Fichtner, A.S.; Karunakaran, M.M. An Update on the Molecular Basis of Phosphoantigen Recognition by Vγ9Vδ2 T Cells. Cells 2020, 9, 1433. [Google Scholar] [CrossRef]
- Blazquez, J.-L.; Benyamine, A.; Pasero, C.; Olive, D. New Insights Into the Regulation of γδ T Cells by BTN3A and Other BTN/BTNL in Tumor Immunity. Front. Immunol. 2018, 9, 1601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sebestyen, Z.; Prinz, I.; Déchanet-Merville, J.; Silva-Santos, B.; Kuball, J. Translating gammadelta (γδ) T cells and their receptors into cancer cell therapies. Nat. Rev. Drug Discov. 2020, 19, 169–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandes, M.; Willimann, K.; Bioley, G.; Lévy, N.; Eberl, M.; Luo, M.; Tampé, R.; Lévy, F.; Romero, P.; Moser, B. Cross-presenting human gammadelta T cells induce robust CD8+ alphabeta T cell responses. Proc. Natl. Acad. Sci. USA 2009, 106, 2307–2312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Wu, W.; Wong, W.M.; Ward, E.; Thrasher, A.J.; Goldblatt, D.; Osman, M.; Digard, P.; Canaday, D.H.; Gustafsson, K. Human gamma delta T cells: A lymphoid lineage cell capable of professional phagocytosis. J. Immunol. 2009, 183, 5622–5629. [Google Scholar] [CrossRef]
- Pistoia, V.; Tumino, N.; Vacca, P.; Veneziani, I.; Moretta, A.; Locatelli, F.; Moretta, L. Human γδ T-Cells: From Surface Receptors to the Therapy of High-Risk Leukemias. Front. Immunol. 2018, 9, 984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raverdeau, M.; Cunningham, S.P.; Harmon, C.; Lynch, L. γδ T cells in cancer: A small population of lymphocytes with big implications. Clin. Transl. Immunol. 2019, 8, e01080. [Google Scholar] [CrossRef] [PubMed]
- Kabelitz, D.; Serrano, R.; Kouakanou, L.; Peters, C.; Kalyan, S. Cancer immunotherapy with γδ T cells: Many paths ahead of us. Cell. Mol. Immunol. 2020, 17, 925–939. [Google Scholar] [CrossRef] [PubMed]
- Elahi, R.; Heidary, A.H.; Hadiloo, K.; Esmaeilzadeh, A. Chimeric Antigen Receptor-Engineered Natural Killer (CAR NK) Cells in Cancer Treatment; Recent Advances and Future Prospects. Stem Cell Rev. Rep. 2021, 17, 2081–2106. [Google Scholar] [CrossRef]
- Euchner, J.; Sprissler, J.; Cathomen, T.; Fürst, D.; Schrezenmeier, H.; Debatin, K.-M.; Schwarz, K.; Felgentreff, K. Natural Killer Cells Generated From Human Induced Pluripotent Stem Cells Mature to CD56(bright)CD16(+)NKp80(+/-) In-Vitro and Express KIR2DL2/DL3 and KIR3DL1. Front. Immunol. 2021, 12, 640672. [Google Scholar] [CrossRef] [PubMed]
- Biederstädt, A.; Rezvani, K. Engineering the next generation of CAR-NK immunotherapies. Int. J. Hematol. 2021, 114, 554–571. [Google Scholar] [CrossRef]
- Basar, R.; Daher, M.; Rezvani, K. Next-generation cell therapies: The emerging role of CAR-NK cells. Blood Adv. 2020, 4, 5868–5876. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.-J.; Feng, Q. CAR-NK cells from engineered pluripotent stem cells: Off-the-shelf therapeutics for all patients. Stem Cells Transl. Med. 2021, 10 (Suppl. 2), S10–S17. [Google Scholar] [CrossRef] [PubMed]
- Ni, Z.; Knorr, D.A.; Clouser, C.L.; Hexum, M.K.; Southern, P.; Mansky, L.M.; Park, I.-H.; Kaufman, D.S. Human pluripotent stem cells produce natural killer cells that mediate anti-HIV-1 activity by utilizing diverse cellular mechanisms. J. Virol. 2011, 85, 43–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shankar, K.; Capitini, C.M.; Saha, K. Genome engineering of induced pluripotent stem cells to manufacture natural killer cell therapies. Stem Cell Res. Ther. 2020, 11, 234. [Google Scholar] [CrossRef]
- Bansal, R.; Reshef, R. Revving the CAR–Combination strategies to enhance CAR T cell effectiveness. Blood Rev. 2021, 45, 100695. [Google Scholar] [CrossRef]
- Wu, R.; Forget, M.-A.; Chacon, J.; Bernatchez, C.; Haymaker, C.; Chen, J.Q.; Hwu, P.; Radvanyi, L.G. Adoptive T-cell therapy using autologous tumor-infiltrating lymphocytes for metastatic melanoma: Current status and future outlook. Cancer J. 2012, 18, 160–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.; Watkins, R.; Vilgelm, A.E. Cell Therapy With TILs: Training and Taming T Cells to Fight Cancer. Front. Immunol. 2021, 12, 690499. [Google Scholar] [CrossRef]
- Robbins, P.F.; Kassim, S.H.; Tran, T.L.N.; Crystal, J.S.; Morgan, R.A.; Feldman, S.A.; Yang, J.C.; Dudley, M.E.; Wunderlich, J.R.; Sherry, R.M.; et al. A pilot trial using lymphocytes genetically engineered with an NY-ESO-1-reactive T-cell receptor: Long-term follow-up and correlates with response. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 1019–1027. [Google Scholar] [CrossRef] [Green Version]
- Hay, A.E.; Cheung, M.C. CAR T-cells: Costs, comparisons, and commentary. J. Med. Econ. 2019, 22, 613–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartmann, J.; Schüßler-Lenz, M.; Bondanza, A.; Buchholz, C.J. Clinical development of CAR T cells-challenges and opportunities in translating innovative treatment concepts. EMBO Mol. Med. 2017, 9, 1183–1197. [Google Scholar] [CrossRef]
- Ruella, M.; Kenderian, S.S. Next-Generation Chimeric Antigen Receptor T-Cell Therapy: Going off the Shelf. BioDrugs 2017, 31, 473–481. [Google Scholar] [CrossRef]
- Hmadcha, A.; Martin-Montalvo, A.; Gauthier, B.R.; Soria, B.; Capilla-Gonzalez, V. Therapeutic Potential of Mesenchymal Stem Cells for Cancer Therapy. Front. Bioeng. Biotechnol. 2020, 8, 43. [Google Scholar] [CrossRef]
- Aravindhan, S.; Ejam, S.S.; Lafta, M.H.; Markov, A.; Yumashev, A.V.; Ahmadi, M. Mesenchymal stem cells and cancer therapy: Insights into targeting the tumour vasculature. Cancer Cell Int. 2021, 21, 158. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef] [Green Version]
- Majzner, R.G.; Mackall, C.L. Tumor Antigen Escape from CAR T-cell Therapy. Cancer Discov. 2018, 8, 1219–1226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, E.; Turcotte, S.; Gros, A.; Robbins, P.F.; Lu, Y.-C.; Dudley, M.E.; Wunderlich, J.R.; Somerville, R.P.; Hogan, K.; Hinrichs, C.S.; et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science 2014, 344, 641–645. [Google Scholar] [CrossRef] [PubMed]
- Ghassemi, S.; Nunez-Cruz, S.; O’Connor, R.S.; Fraietta, J.A.; Patel, P.R.; Scholler, J.; Barrett, D.M.; Lundh, S.M.; Davis, M.M.; Bedoya, F.; et al. Reducing Ex Vivo Culture Improves the Antileukemic Activity of Chimeric Antigen Receptor (CAR) T Cells. Cancer Immunol. Res. 2018, 6, 1100–1109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bankoti, R.; Akbal, H.P.; Adorno, M.; Robilant, B. Di 830 Targeting cellular senescence to increase CAR-T cell fitness. J. Immunother. Cancer 2020, 8, A497. [Google Scholar] [CrossRef]
- Wang, B.; Iriguchi, S.; Waseda, M.; Ueda, N.; Ueda, T.; Xu, H.; Minagawa, A.; Ishikawa, A.; Yano, H.; Ishi, T.; et al. Generation of hypoimmunogenic T cells from genetically engineered allogeneic human induced pluripotent stem cells. Nat. Biomed. Eng. 2021, 5, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Yeku, O.O.; Brentjens, R.J. Armored CAR T-cells: Utilizing cytokines and pro-inflammatory ligands to enhance CAR T-cell anti-tumour efficacy. Biochem. Soc. Trans. 2016, 44, 412–418. [Google Scholar] [CrossRef] [PubMed]
- Rezvani, K. Adoptive cell therapy using engineered natural killer cells. Bone Marrow Transplant. 2019, 54, 785–788. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, E.R.; D’Souza, R.R.; Klampatsa, A. Armored CAR T-Cells: The Next Chapter in T-Cell Cancer Immunotherapy. Biologics 2021, 15, 95–105. [Google Scholar] [CrossRef]
- Li, Y.-R.; Zhou, Y.; Kim, Y.J.; Zhu, Y.; Ma, F.; Yu, J.; Wang, Y.-C.; Chen, X.; Li, Z.; Zeng, S.; et al. Development of allogeneic HSC-engineered iNKT cells for off-the-shelf cancer immunotherapy. Cell Rep. Med. 2021, 2, 100449. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Immune Cell Types | Tumor Recognition Receptors | Restriction Reactivity | Staining Markers | GvHDRisk | Allogeneic Cell Products |
|---|---|---|---|---|---|
| Conventional αβ T cells | Highly diverse αβ TCRs | MHC-I and MHC-II | CD3+ TCR αβ+ | High | Genome-edited, donor-derived UCART19 [25,36] |
| CD19 CAR-T cells with CAR integrated into the TCR α chain [37] | |||||
| iPSC-derived CD19 CAR-T cells [38] | |||||
| In vitro generation in OP9-DL1 cultures [39] | |||||
| In vitro generation in ATO cultures [40,41] | |||||
| Rejuvenated iPSC-Derived T Cells [42,43,44,45,46,47] | |||||
| Invariant natural killer T (iNKT) cells | Invariant TCR α-chain (Vα14-Jα18 in mice or Vα24-Jα18 in humans), restricted diverse TCR β-chain | CD1d | CD3+TCR αβ+6B11(iNKT TCR)+ | Low | iPSC-derived iNKT cells [48,49] |
| Mucosal associated invariant T (MAIT) cells | Semi-invariant TCR α-chain (Vα19-Jα33 in mice or Vα7.2-Jα33 in humans), restricted diverse TCR β-chain | MR1 | CD3+TCR αβ+Vα7.2+ | Low | iPSC-derived MAIT cells [50,51,52] |
| Gamma delta (γδ) T cells | Restricted diverse γδ TCRs | Butyrophilin 3A1, CD1d | CD3+ TCR γδ+ | Low | iPSC-derived γδ T cells [53] |
| Natural killer (NK) cells | NK activation and inhibition receptors (e.g., NKG2D, DNAM-1, KIR) | e.g., MIC-A/B, ULBP, CD155, CD112 | CD3-CD56+ | Low | Cord blood-derived CD19 CAR-NK cells [31,54] |
| PSC-derived NK cells [55,56,57,58,59,60,61] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.-R.; Dunn, Z.S.; Zhou, Y.; Lee, D.; Yang, L. Development of Stem Cell-Derived Immune Cells for Off-the-Shelf Cancer Immunotherapies. Cells 2021, 10, 3497. https://doi.org/10.3390/cells10123497
Li Y-R, Dunn ZS, Zhou Y, Lee D, Yang L. Development of Stem Cell-Derived Immune Cells for Off-the-Shelf Cancer Immunotherapies. Cells. 2021; 10(12):3497. https://doi.org/10.3390/cells10123497
Chicago/Turabian StyleLi, Yan-Ruide, Zachary Spencer Dunn, Yang Zhou, Derek Lee, and Lili Yang. 2021. "Development of Stem Cell-Derived Immune Cells for Off-the-Shelf Cancer Immunotherapies" Cells 10, no. 12: 3497. https://doi.org/10.3390/cells10123497
APA StyleLi, Y.-R., Dunn, Z. S., Zhou, Y., Lee, D., & Yang, L. (2021). Development of Stem Cell-Derived Immune Cells for Off-the-Shelf Cancer Immunotherapies. Cells, 10(12), 3497. https://doi.org/10.3390/cells10123497