
Pyroptosis: Mechanisms and Links with Fibrosis


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Inflammasomes
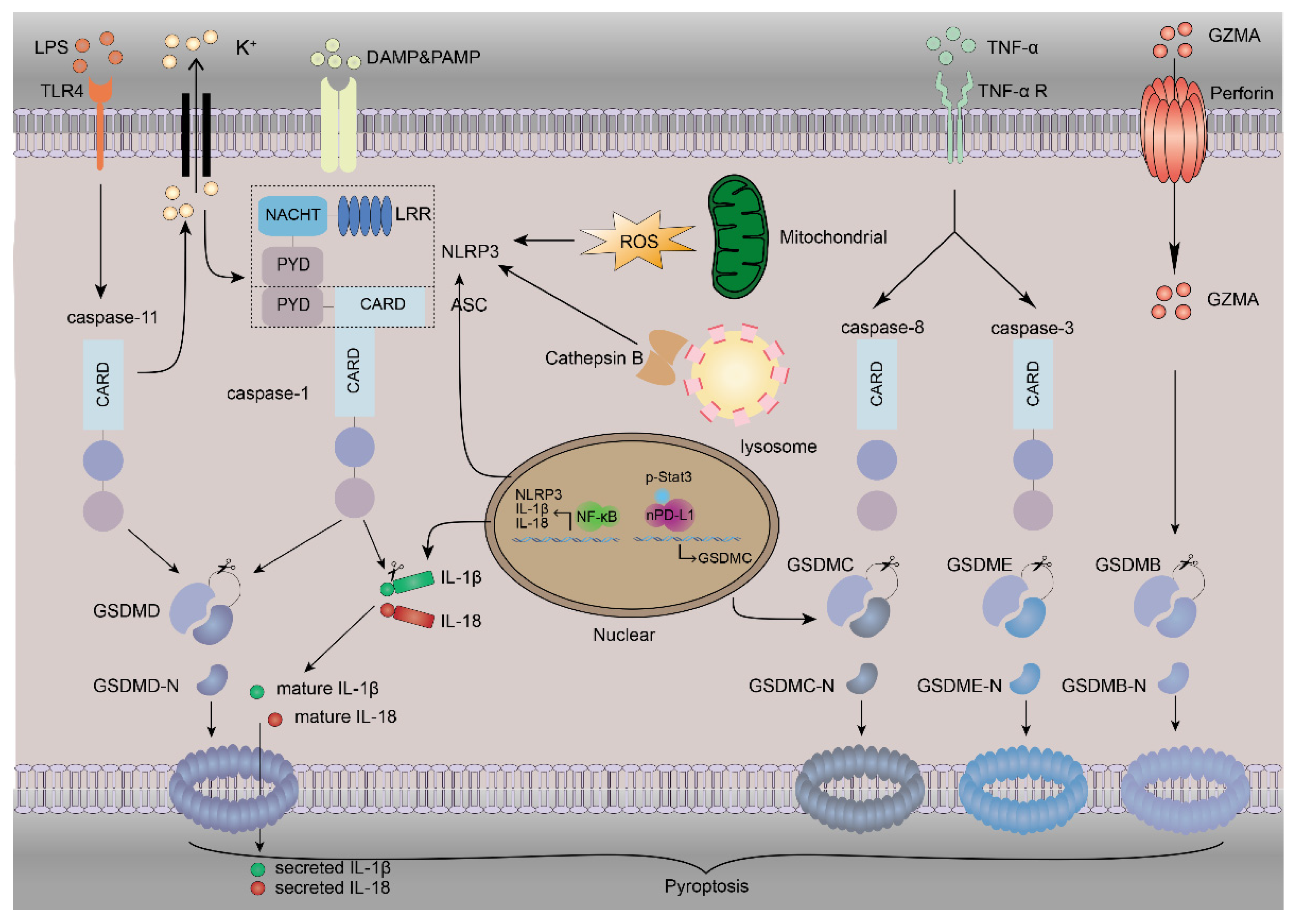
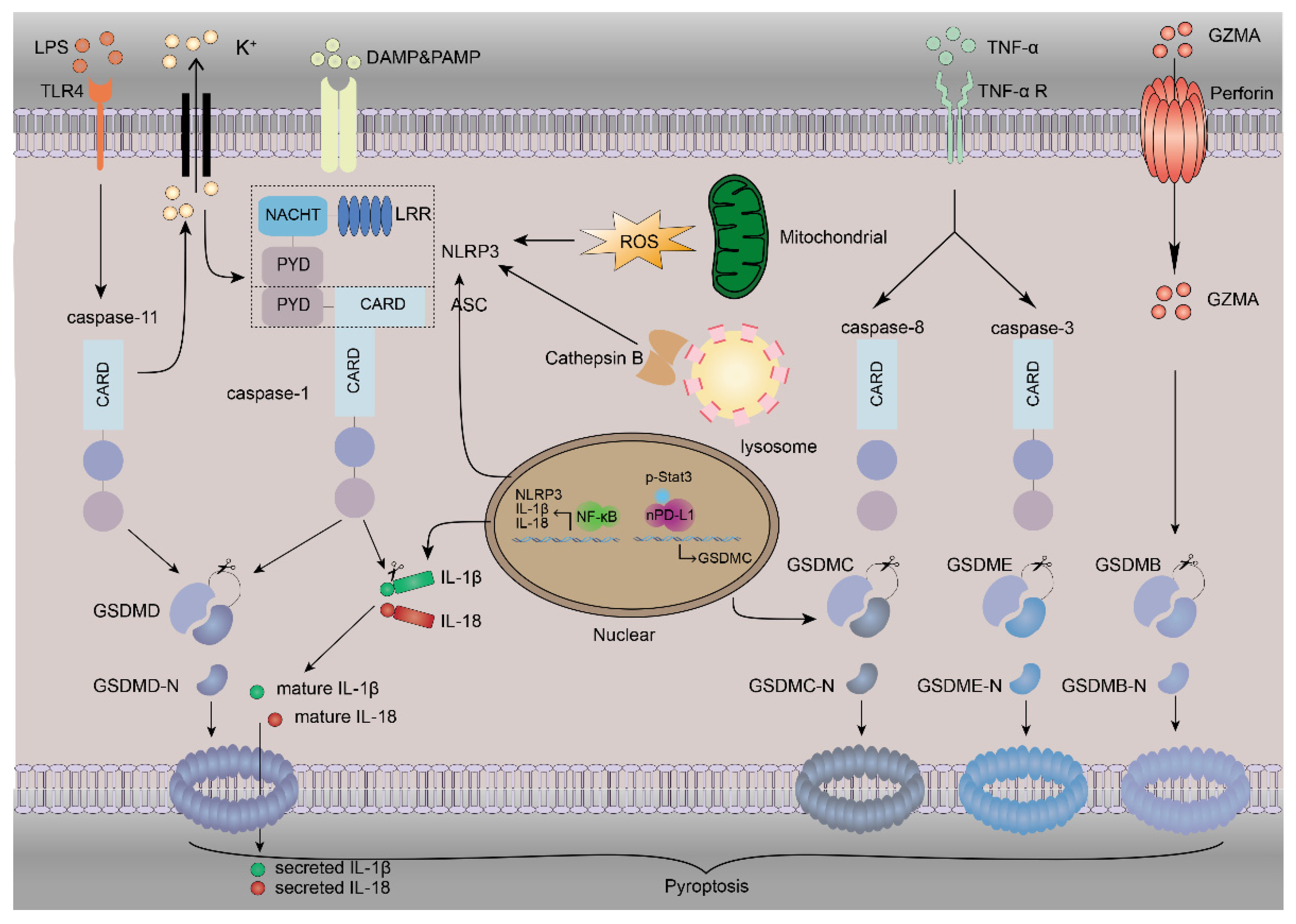
3. The Mechanism of Pyroptosis
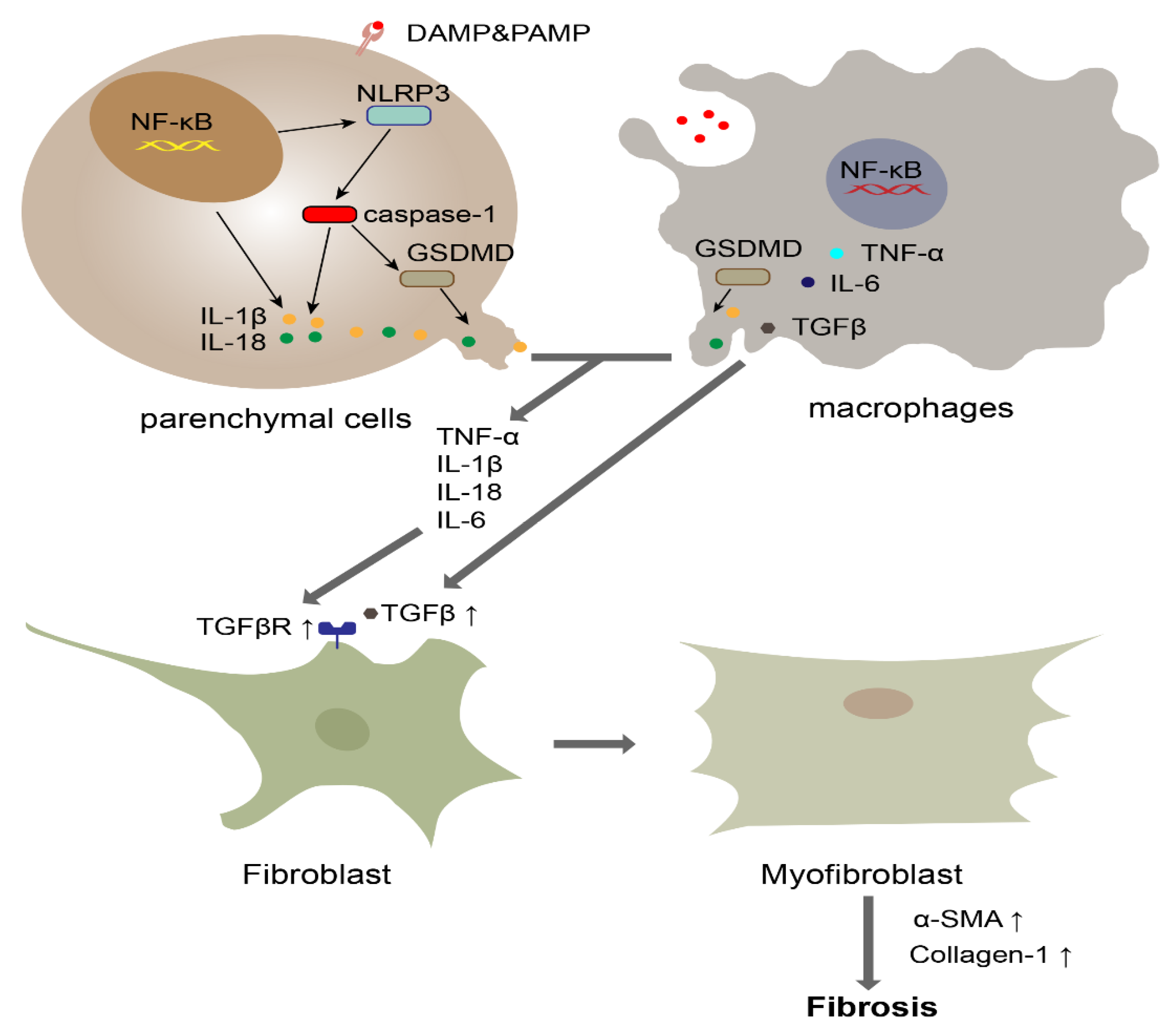
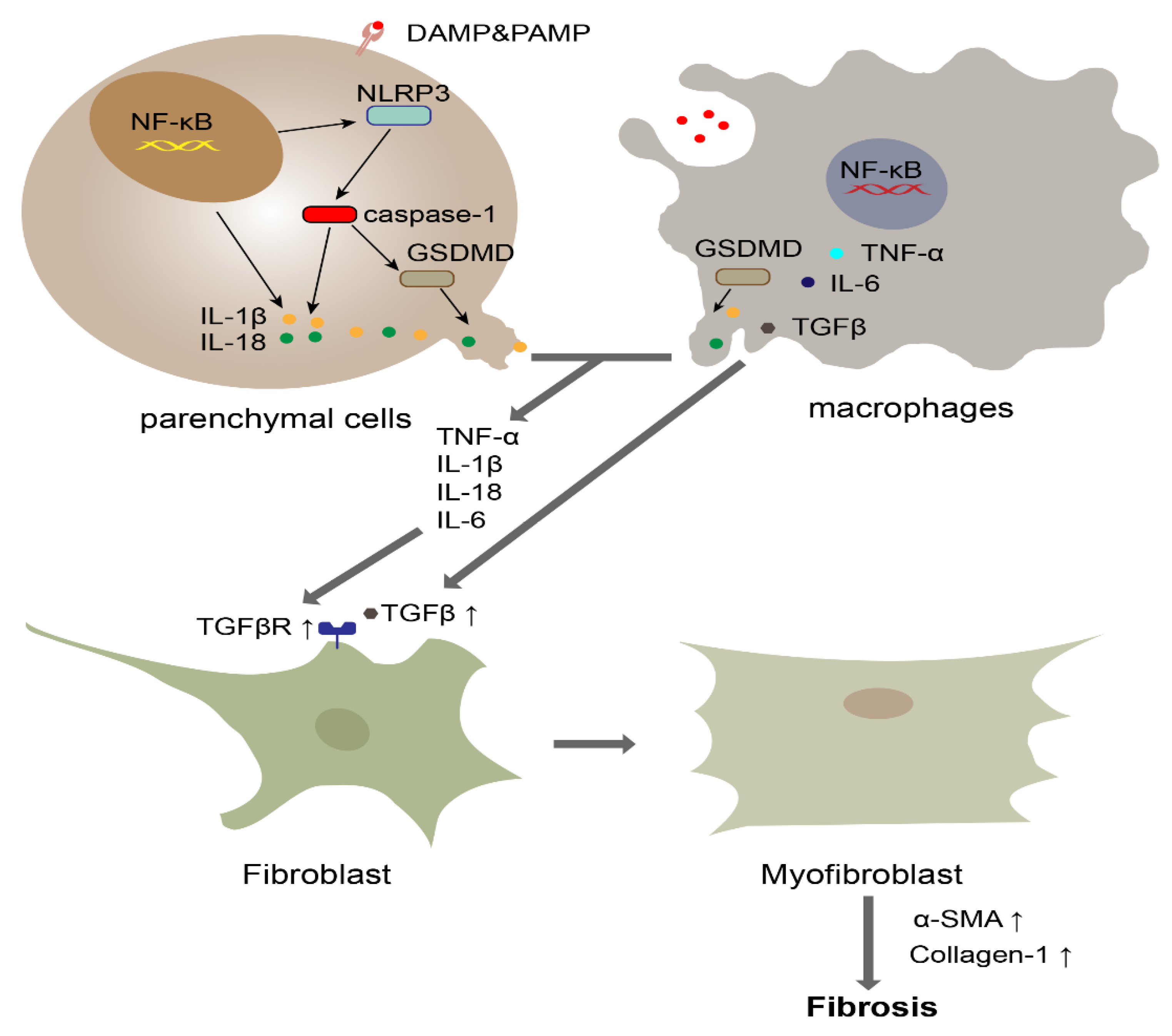
4. The Role of Pyroptosis in Fibrosis
4.1. Liver Fibrosis
4.2. Kidney Fibrosis
4.3. Lung Fibrosis
5. The Utility of Inflammasomes and Cytokines as Early Diagnostic Biomarkers of Fibrotic Disease and Therapeutic Targets
6. Conclusion and Perspective
Author Contributions
Funding
Conflicts of Interest
References
- Alegre, F.; Pelegrin, P.; Feldstein, A. Inflammasomes in Liver Fibrosis. Semin. Liver Dis. 2017, 37, 119–127. [Google Scholar] [CrossRef] [Green Version]
- Artlett, C. Inflammasomes in wound healing and fibrosis. J. Pathol. 2013, 229, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A. Cellular and molecular mechanisms of fibrosis. J. Pathol. 2008, 214, 199–210. [Google Scholar] [CrossRef] [Green Version]
- Van De Water, L.; Varney, S.; Tomasek, J.J. Mechanoregulation of the Myofibroblast in Wound Contraction, Scarring, and Fibrosis: Opportunities for New Therapeutic Intervention. Adv. Wound Care 2013, 2, 122–141. [Google Scholar] [CrossRef] [Green Version]
- Artlett, C.M.; Thacker, J.D. Molecular activation of the NLRP3 Inflammasome in fibrosis: Common threads linking divergent fibrogenic diseases. Antioxid. Redox Signal. 2015, 22, 1162–1175. [Google Scholar] [CrossRef] [PubMed]
- Henderson, N.; Rieder, F.; Wynn, T. Fibrosis: From mechanisms to medicines. Nature 2020, 587, 555–566. [Google Scholar] [CrossRef] [PubMed]
- Christgen, S.; Tweedell, R.E.; Kanneganti, T.D. Programming inflammatory cell death for therapy. Pharmacol. Ther. 2021, 108010. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.; Pandey, A.; Man, S. Gasdermins deliver a deadly punch to cancer. Cell Res. 2020, 30, 463–464. [Google Scholar] [CrossRef] [Green Version]
- von Moltke, J.; Ayres, J.S.; Kofoed, E.M.; Chavarría-Smith, J.; Vance, R.E. Recognition of bacteria by inflammasomes. Annu. Rev. Immunol. 2013, 31, 73–106. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Q.; Pan, J.; Zhou, Z.L.; Yin, F.; Xie, H.Y.; Chen, P.P.; Li, J.Y.; Zheng, P.Q.; Zhou, L.; Zhang, W.; et al. Caspase-11/4 and gasdermin D-mediated pyroptosis contributes to podocyte injury in mouse diabetic nephropathy. Acta Pharmacol. Sin. 2021, 42, 954–963. [Google Scholar] [CrossRef]
- Coutinho-Budd, J.; Broihier, H. Pyroptosis Takes Aim at Neurodevelopment. Dev. Cell 2020, 53, 498–499. [Google Scholar] [CrossRef]
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, J.; Gao, W.; Shao, F. Pyroptosis: Gasdermin-Mediated Programmed Necrotic Cell Death. Trends Biochem. Sci. 2017, 42, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Najafov, A.; Py, B.F. Roles of Caspases in Necrotic Cell Death. Cell 2016, 167, 1693–1704. [Google Scholar] [CrossRef] [Green Version]
- Byrne, N.; Soni, S.; Takahara, S.; Ferdaoussi, M.; Al Batran, R.; Darwesh, A.; Levasseur, J.; Beker, D.; Vos, D.; Schmidt, M.; et al. Chronically Elevating Circulating Ketones Can Reduce Cardiac Inflammation and Blunt the Development of Heart Failure. Circ. Heart Fail. 2020, 13, e006573. [Google Scholar] [CrossRef] [PubMed]
- Suetomi, T.; Willeford, A.; Brand, C.; Cho, Y.; Ross, R.; Miyamoto, S.; Brown, J. Inflammation and NLRP3 Inflammasome Activation Initiated in Response to Pressure Overload by Ca/Calmodulin-Dependent Protein Kinase II δ Signaling in Cardiomyocytes Are Essential for Adverse Cardiac Remodeling. Circulation 2018, 138, 2530–2544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef]
- Py, B.F.; Kim, M.S.; Vakifahmetoglu-Norberg, H.; Yuan, J. Deubiquitination of NLRP3 by BRCC3 critically regulates inflammasome activity. Mol. Cell 2013, 49, 331–338. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Hu, D.; Wu, C.; Feng, Y.; Li, A.; Liu, W.; Wang, Y.; Chen, K.; Tian, M.; Xiao, F.; et al. STING promotes NLRP3 localization in ER and facilitates NLRP3 deubiquitination to activate the inflammasome upon HSV-1 infection. PLoS Pathog. 2020, 16, e1008335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, G.; Zhang, X.; Xiao, Y.; Zhang, W.; Wang, Y.; Ma, W.; Wang, X.; Song, P.; Lai, L.; Chen, H.; et al. ABRO1 promotes NLRP3 inflammasome activation through regulation of NLRP3 deubiquitination. EMBO J. 2019, 38. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Planillo, R.; Kuffa, P.; Martínez-Colón, G.; Smith, B.L.; Rajendiran, T.M.; Núñez, G. K+ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 2013, 38, 1142–1153. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Zhang, D.; Hu, D.; Zhou, X.; Zhou, Y. The role of mitochondria in NLRP3 inflammasome activation. Mol. Immunol. 2018, 103, 115–124. [Google Scholar] [CrossRef]
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 2012, 36, 401–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iyer, S.S.; He, Q.; Janczy, J.R.; Elliott, E.I.; Zhong, Z.; Olivier, A.K.; Sadler, J.J.; Knepper-Adrian, V.; Han, R.; Qiao, L.; et al. Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity 2013, 39, 311–323. [Google Scholar] [CrossRef] [Green Version]
- Hornung, V.; Bauernfeind, F.; Halle, A.; Samstad, E.O.; Kono, H.; Rock, K.L.; Fitzgerald, K.A.; Latz, E. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat. Immunol. 2008, 9, 847–856. [Google Scholar] [CrossRef]
- Strowig, T.; Henao-Mejia, J.; Elinav, E.; Flavell, R. Inflammasomes in health and disease. Nature 2012, 481, 278–286. [Google Scholar] [CrossRef]
- Sun, B.; Wang, X.; Liao, Y.; Ji, Z.; Chang, C.; Pokhrel, S.; Ku, J.; Liu, X.; Wang, M.; Dunphy, D.; et al. Repetitive Dosing of Fumed Silica Leads to Profibrogenic Effects through Unique Structure-Activity Relationships and Biopersistence in the Lung. ACS Nano 2016, 10, 8054–8066. [Google Scholar] [CrossRef] [Green Version]
- Pavan, C.; Rabolli, V.; Tomatis, M.; Fubini, B.; Lison, D. Why does the hemolytic activity of silica predict its pro-inflammatory activity? Part. Fibre Toxicol. 2014, 11, 76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koh, E.H.; Yoon, J.E.; Ko, M.S.; Leem, J.; Yun, J.Y.; Hong, C.H.; Cho, Y.K.; Lee, S.E.; Jang, J.E.; Baek, J.Y.; et al. Sphingomyelin synthase 1 mediates hepatocyte pyroptosis to trigger non-alcoholic steatohepatitis. Gut 2021, 70, 1954–1964. [Google Scholar] [CrossRef] [PubMed]
- Kadono, K.; Kageyama, S.; Nakamura, K.; Hirao, H.; Ito, T.; Kojima, H.; Dery, K.J.; Li, X.; Kupiec-Weglinski, J.W. Myeloid ikaros-SIRT1 signaling axis regulates hepatic inflammation and pyroptosis in ischemia-stressed mouse and human liver. J. Hepatol. 2021. in preprint. [Google Scholar] [CrossRef]
- Xu, W.; Che, Y.; Zhang, Q.; Huang, H.; Ding, C.; Wang, Y.; Wang, G.; Cao, L.; Hao, H. Apaf-1 Pyroptosome Senses Mitochondrial Permeability Transition. Cell Metab. 2021, 33, 424–436.e10. [Google Scholar] [CrossRef]
- Wree, A.; Eguchi, A.; McGeough, M.; Pena, C.; Johnson, C.; Canbay, A.; Hoffman, H.; Feldstein, A. NLRP3 inflammasome activation results in hepatocyte pyroptosis, liver inflammation, and fibrosis in mice. Hepatoogy 2014, 59, 898–910. [Google Scholar] [CrossRef] [Green Version]
- Zychlinsky, A.; Prevost, M.C.; Sansonetti, P.J. Shigella flexneri induces apoptosis in infected macrophages. Nature 1992, 358, 167–169. [Google Scholar] [CrossRef] [PubMed]
- Weinrauch, Y.; Zychlinsky, A. The induction of apoptosis by bacterial pathogens. Annu. Rev. Microbiol. 1999, 53, 155–187. [Google Scholar] [CrossRef]
- Feng, S.; Fox, D.; Man, S. Mechanisms of Gasdermin Family Members in Inflammasome Signaling and Cell Death. J. Mol. Biol. 2018, 430, 3068–3080. [Google Scholar] [CrossRef]
- Ding, J.; Wang, K.; Liu, W.; She, Y.; Sun, Q.; Shi, J.; Sun, H.; Wang, D.C.; Shao, F. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 2016, 535, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, I.; Rayamajhi, M.; Miao, E.A. Programmed cell death as a defence against infection. Nat. Rev. Immunol. 2017, 17, 151–164. [Google Scholar] [CrossRef] [PubMed]
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T.; et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 2015, 526, 666–671. [Google Scholar] [CrossRef]
- Wang, Y.; Gao, W.; Shi, X.; Ding, J.; Liu, W.; He, H.; Wang, K.; Shao, F. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 2017, 547, 99–103. [Google Scholar] [CrossRef]
- Zhou, Z.; He, H.; Wang, K.; Shi, X.; Wang, Y.; Su, Y.; Wang, Y.; Li, D.; Liu, W.; Zhang, Y.; et al. Granzyme A from cytotoxic lymphocytes cleaves GSDMB to trigger pyroptosis in target cells. Science 2020, 368. [Google Scholar] [CrossRef]
- Hou, J.; Zhao, R.; Xia, W.; Chang, C.W.; You, Y.; Hsu, J.M.; Nie, L.; Chen, Y.; Wang, Y.C.; Liu, C.; et al. PD-L1-mediated gasdermin C expression switches apoptosis to pyroptosis in cancer cells and facilitates tumour necrosis. Nat. Cell Biol. 2020, 22, 1264–1275. [Google Scholar] [CrossRef]
- Wang, K.; Sun, Q.; Zhong, X.; Zeng, M.; Zeng, H.; Shi, X.; Li, Z.; Wang, Y.; Zhao, Q.; Shao, F.; et al. Structural Mechanism for GSDMD Targeting by Autoprocessed Caspases in Pyroptosis. Cell 2020, 180, 941–955.e20. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Zhao, Y.; Wang, Y.; Gao, W.; Ding, J.; Li, P.; Hu, L.; Shao, F. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 2014, 514, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; He, Y.; Muñoz-Planillo, R.; Liu, Q.; Núñez, G. Caspase-11 Requires the Pannexin-1 Channel and the Purinergic P2X7 Pore to Mediate Pyroptosis and Endotoxic Shock. Immunity 2015, 43, 923–932. [Google Scholar] [CrossRef] [Green Version]
- Han, X.; Song, J.; Lian, L.; Yao, Y.; Shao, D.; Fan, Y.; Hou, L.; Wang, G.; Zheng, S.; Wu, Y.; et al. Ginsenoside 25-OCH-PPD Promotes Activity of LXRs To Ameliorate P2X7R-Mediated NLRP3 Inflammasome in the Development of Hepatic Fibrosis. J. Agric. Food Chem. 2018, 66, 7023–7035. [Google Scholar] [CrossRef]
- Sasaki, R.; Devhare, P.; Steele, R.; Ray, R.; Ray, R. Hepatitis C virus-induced CCL5 secretion from macrophages activates hepatic stellate cells. Hepatology 2017, 66, 746–757. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Tu, K.; Liu, D.; Guo, L.; Chen, Y.; Li, Q.; Maiers, J.; Liu, Z.; Shah, V.; Dou, C.; et al. p300 Acetyltransferase Is a Cytoplasm-to-Nucleus Shuttle for SMAD2/3 and TAZ Nuclear Transport in Transforming Growth Factor β-Stimulated Hepatic Stellate Cells. Hepatology 2019, 70, 1409–1423. [Google Scholar] [CrossRef] [PubMed]
- Gautheron, J.; Gores, G.J.; Rodrigues, C.M.P. Lytic cell death in metabolic liver disease. J. Hepatol. 2020, 73, 394–408. [Google Scholar] [CrossRef] [PubMed]
- Cai, S.; Yang, R.; Li, Y.; Ning, Z.; Zhang, L.; Zhou, G.; Luo, W.; Li, D.; Chen, Y.; Pan, M.; et al. Angiotensin-(1-7) Improves Liver Fibrosis by Regulating the NLRP3 Inflammasome via Redox Balance Modulation. Antioxid. Redox Signal. 2016, 24, 795–812. [Google Scholar] [CrossRef]
- Gaul, S.; Leszczynska, A.; Alegre, F.; Kaufmann, B.; Johnson, C.D.; Adams, L.A.; Wree, A.; Damm, G.; Seehofer, D.; Calvente, C.J.; et al. Hepatocyte pyroptosis and release of inflammasome particles induce stellate cell activation and liver fibrosis. J. Hepatol. 2021, 74, 156–167. [Google Scholar] [CrossRef] [PubMed]
- Petrasek, J.; Bala, S.; Csak, T.; Lippai, D.; Kodys, K.; Menashy, V.; Barrieau, M.; Min, S.; Kurt-Jones, E.; Szabo, G. IL-1 receptor antagonist ameliorates inflammasome-dependent alcoholic steatohepatitis in mice. J. Clin. Investig. 2012, 122, 3476–3489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Li, Y.; Siraj, S.; Jin, H.; Fan, Y.; Yang, X.; Huang, X.; Wang, X.; Wang, J.; Liu, L.; et al. FUN14 Domain-Containing 1-Mediated Mitophagy Suppresses Hepatocarcinogenesis by Inhibition of Inflammasome Activation in Mice. Hepatology 2019, 69, 604–621. [Google Scholar] [CrossRef]
- Inzaugarat, M.; Johnson, C.; Holtmann, T.; McGeough, M.; Trautwein, C.; Papouchado, B.; Schwabe, R.; Hoffman, H.; Wree, A.; Feldstein, A. NLR Family Pyrin Domain-Containing 3 Inflammasome Activation in Hepatic Stellate Cells Induces Liver Fibrosis in Mice. Hepatology 2019, 69, 845–859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schuster-Gaul, S.; Geisler, L.; McGeough, M.; Johnson, C.; Zagorska, A.; Li, L.; Wree, A.; Barry, V.; Mikaelian, I.; Jih, L.; et al. ASK1 inhibition reduces cell death and hepatic fibrosis in an Nlrp3 mutant liver injury model. JCI Insight 2020, 5, e123294. [Google Scholar] [CrossRef]
- Mridha, A.; Wree, A.; Robertson, A.; Yeh, M.; Johnson, C.; Van Rooyen, D.; Haczeyni, F.; Teoh, N.; Savard, C.; Ioannou, G.; et al. NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. J. Hepatol. 2017, 66, 1037–1046. [Google Scholar] [CrossRef]
- Kim, S.; Kim, G.; Han, D.; Lee, M.; Kim, I.; Kim, B.; Kim, K.; Song, Y.; Yoo, J.; Wang, H.; et al. Ezetimibe ameliorates steatohepatitis via AMP activated protein kinase-TFEB-mediated activation of autophagy and NLRP3 inflammasome inhibition. Autophagy 2017, 13, 1767–1781. [Google Scholar] [CrossRef]
- Zhao, Y.; Wang, Z.; Feng, D.; Zhao, H.; Lin, M.; Hu, Y.; Zhang, N.; Lv, L.; Gao, Z.; Zhai, X.; et al. p66Shc Contributes to Liver Fibrosis through the Regulation of Mitochondrial Reactive Oxygen Species. Theranostics 2019, 9, 1510–1522. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaka, T.; Atsumi, A.; Matsumori, R.; Nie, T.; Shinozaki, H.; Suzuki-Kemuriyama, N.; Kuba, M.; Nakagawa, Y.; Ishii, K.; Shimada, M.; et al. Elovl6 promotes nonalcoholic steatohepatitis. Hepatology 2012, 56, 2199–2208. [Google Scholar] [CrossRef]
- Oakley, F.; Teoh, V.; Ching-A-Sue, G.; Bataller, R.; Colmenero, J.; Jonsson, J.; Eliopoulos, A.; Watson, M.; Manas, D.; Mann, D. Angiotensin II activates I kappaB kinase phosphorylation of RelA at Ser 536 to promote myofibroblast survival and liver fibrosis. Gastroenterology 2009, 136, 2334–2344.e1. [Google Scholar] [CrossRef]
- Ning, Z.; Luo, X.; Wang, G.; Li, Y.; Pan, M.; Yang, R.; Ling, X.; Huang, S.; Ma, X.; Jin, S.; et al. MicroRNA-21 Mediates Angiotensin II-Induced Liver Fibrosis by Activating NLRP3 Inflammasome/IL-1β Axis via Targeting Smad7 and Spry1. Antioxid. Redox Signal. 2017, 27, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Jimenez Calvente, C.; Del Pilar, H.; Tameda, M.; Johnson, C.; Feldstein, A. MicroRNA 223 3p Negatively Regulates the NLRP3 Inflammasome in Acute and Chronic Liver Injury. Mol. Ther. J. Am. Soc. Gene Ther. 2020, 28, 653–663. [Google Scholar] [CrossRef]
- Lebeaupin, C.; Vallée, D.; Rousseau, D.; Patouraux, S.; Bonnafous, S.; Adam, G.; Luciano, F.; Luci, C.; Anty, R.; Iannelli, A.; et al. Bax inhibitor-1 protects from nonalcoholic steatohepatitis by limiting inositol-requiring enzyme 1 alpha signaling in mice. Hepatology 2018, 68, 515–532. [Google Scholar] [CrossRef] [Green Version]
- Lozano-Ruiz, B.; Bachiller, V.; García-Martínez, I.; Zapater, P.; Gómez-Hurtado, I.; Moratalla, A.; Giménez, P.; Bellot, P.; Francés, R.; Such, J.; et al. Absent in melanoma 2 triggers a heightened inflammasome response in ascitic fluid macrophages of patients with cirrhosis. J. Hepatol. 2015, 62, 64–71. [Google Scholar] [CrossRef]
- De Minicis, S.; Rychlicki, C.; Agostinelli, L.; Saccomanno, S.; Candelaresi, C.; Trozzi, L.; Mingarelli, E.; Facinelli, B.; Magi, G.; Palmieri, C.; et al. Dysbiosis contributes to fibrogenesis in the course of chronic liver injury in mice. Hepatology 2014, 59, 1738–1749. [Google Scholar] [CrossRef]
- Kämpfer, H.; Paulukat, J.; Mühl, H.; Wetzler, C.; Pfeilschifter, J.; Frank, S. Lack of interferon-gamma production despite the presence of interleukin-18 during cutaneous wound healing. Mol. Med. 2000, 6, 1016–1027. [Google Scholar] [CrossRef] [Green Version]
- Fix, C.; Bingham, K.; Carver, W. Effects of interleukin-18 on cardiac fibroblast function and gene expression. Cytokine 2011, 53, 19–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruscitti, P.; Masedu, F.; Alvaro, S.; Airò, P.; Battafarano, N.; Cantarini, L.; Cantatore, F.; Carlino, G.; D’Abrosca, V.; Frassi, M.; et al. Anti-interleukin-1 treatment in patients with rheumatoid arthritis and type 2 diabetes (TRACK): A multicentre, open-label, randomised controlled trial. PLoS Med. 2019, 16, e1002901. [Google Scholar] [CrossRef] [PubMed]
- Kone-Paut, I.; Cimaz, R.; Herberg, J.; Bates, O.; Carbasse, A.; Saulnier, J.; Maggio, M.; Anton, J.; Piram, M. The use of interleukin 1 receptor antagonist (anakinra) in Kawasaki disease: A retrospective cases series. Autoimmun. Rev. 2018, 17, 768–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Tassell, B.; Canada, J.; Carbone, S.; Trankle, C.; Buckley, L.; Oddi Erdle, C.; Abouzaki, N.; Dixon, D.; Kadariya, D.; Christopher, S.; et al. Interleukin-1 Blockade in Recently Decompensated Systolic Heart Failure: Results from REDHART (Recently Decompensated Heart Failure Anakinra Response Trial). Circ. Heart Fail. 2017, 10, 4373. [Google Scholar] [CrossRef]
- Kooistra, E.; Waalders, N.; Grondman, I.; Janssen, N.; de Nooijer, A.; Netea, M.; van de Veerdonk, F.; Ewalds, E.; van der Hoeven, J.; Kox, M.; et al. Anakinra treatment in critically ill COVID-19 patients: A prospective cohort study. Crit. Care 2020, 24, 688. [Google Scholar] [CrossRef]
- van Poppel, P.; van Asseldonk, E.; Holst, J.; Vilsbøll, T.; Netea, M.; Tack, C. The interleukin-1 receptor antagonist anakinra improves first-phase insulin secretion and insulinogenic index in subjects with impaired glucose tolerance. Diabetes Obes. Metab. 2014, 16, 1269–1273. [Google Scholar] [CrossRef]
- Ambade, A.; Lowe, P.; Kodys, K.; Catalano, D.; Gyongyosi, B.; Cho, Y.; Iracheta-Vellve, A.; Adejumo, A.; Saha, B.; Calenda, C.; et al. Pharmacological Inhibition of CCR2/5 Signaling Prevents and Reverses Alcohol-Induced Liver Damage, Steatosis, and Inflammation in Mice. Hepatology 2019, 69, 1105–1121. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Jiang, M.; Chu, Y.; Wang, W.; Chen, D.; Li, X.; Zhang, Z.; Zhang, D.; Fan, D.; Nie, Y.; et al. Gasdermin D plays a key role as a pyroptosis executor of non-alcoholic steatohepatitis in humans and mice. J. Hepatol. 2018, 68, 773–782. [Google Scholar] [CrossRef]
- Komada, T.; Muruve, D. The role of inflammasomes in kidney disease. Nat. Rev. Nephrol. 2019, 15, 501–520. [Google Scholar] [CrossRef]
- Vilaysane, A.; Chun, J.; Seamone, M.; Wang, W.; Chin, R.; Hirota, S.; Li, Y.; Clark, S.; Tschopp, J.; Trpkov, K.; et al. The NLRP3 inflammasome promotes renal inflammation and contributes to CKD. J. Am. Soc. Nephrol. JASN 2010, 21, 1732–1744. [Google Scholar] [CrossRef] [Green Version]
- Komada, T.; Chung, H.; Lau, A.; Platnich, J.; Beck, P.; Benediktsson, H.; Duff, H.; Jenne, C.; Muruve, D. Macrophage Uptake of Necrotic Cell DNA Activates the AIM2 Inflammasome to Regulate a Proinflammatory Phenotype in CKD. J. Am. Soc. Nephrol. JASN 2018, 29, 1165–1181. [Google Scholar] [CrossRef] [PubMed]
- Ludwig-Portugall, I.; Bartok, E.; Dhana, E.; Evers, B.; Primiano, M.; Hall, J.; Franklin, B.; Knolle, P.; Hornung, V.; Hartmann, G.; et al. An NLRP3-specific inflammasome inhibitor attenuates crystal-induced kidney fibrosis in mice. Kidney Int. 2016, 90, 525–539. [Google Scholar] [CrossRef] [Green Version]
- Anders, H.; Suarez-Alvarez, B.; Grigorescu, M.; Foresto-Neto, O.; Steiger, S.; Desai, J.; Marschner, J.; Honarpisheh, M.; Shi, C.; Jordan, J.; et al. The macrophage phenotype and inflammasome component NLRP3 contributes to nephrocalcinosis-related chronic kidney disease independent from IL-1-mediated tissue injury. Kidney Int. 2018, 93, 656–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakker, P.; Butter, L.; Kors, L.; Teske, G.; Aten, J.; Sutterwala, F.; Florquin, S.; Leemans, J. Nlrp3 is a key modulator of diet-induced nephropathy and renal cholesterol accumulation. Kidney Int. 2014, 85, 1112–1122. [Google Scholar] [CrossRef] [Green Version]
- Song, W.; Wei, L.; Du, Y.; Wang, Y.; Jiang, S. Protective effect of ginsenoside metabolite compound K against diabetic nephropathy by inhibiting NLRP3 inflammasome activation and NF-κB/p38 signaling pathway in high-fat diet/streptozotocin-induced diabetic mice. Int. Immunopharmacol. 2018, 63, 227–238. [Google Scholar] [CrossRef]
- Luan, P.; Zhuang, J.; Zou, J.; Li, H.; Shuai, P.; Xu, X.; Zhao, Y.; Kou, W.; Ji, S.; Peng, A.; et al. NLRC5 deficiency ameliorates diabetic nephropathy through alleviating inflammation. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2018, 32, 1070–1084. [Google Scholar] [CrossRef] [Green Version]
- Schmidt-Lauber, C.; Bossaller, L.; Abujudeh, H.; Vladimer, G.; Christ, A.; Fitzgerald, K.; Latz, E.; Gravallese, E.; Marshak-Rothstein, A.; Kay, J. Gadolinium-based compounds induce NLRP3-dependent IL-1β production and peritoneal inflammation. Ann. Rheum. Dis. 2015, 74, 2062–2069. [Google Scholar] [CrossRef] [Green Version]
- Chi, H.; Hua, K.; Lin, Y.; Chu, C.; Hsieh, C.; Hsu, Y.; Ka, S.; Tsai, Y.; Liu, F.; Chen, A. IL-36 Signaling Facilitates Activation of the NLRP3 Inflammasome and IL-23/IL-17 Axis in Renal Inflammation and Fibrosis. J. Am. Soc. Nephrol. JASN 2017, 28, 2022–2037. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Zhong, D.; Chen, H.; Jin, J.; Liu, Q.; Li, G. NLRP3 inflammasome activates interleukin-23/interleukin-17 axis during ischaemia-reperfusion injury in cerebral ischaemia in mice. Life Sci. 2019, 227, 101–113. [Google Scholar] [CrossRef]
- Li, L.; Dai, B.; Sun, Y.; Zhang, T. The activation of IL-17 signaling pathway promotes pyroptosis in pneumonia-induced sepsis. Ann. Transl. Med. 2020, 8, 674. [Google Scholar] [CrossRef]
- Duckles, S.P.; Krause, D.N. Mechanisms of cerebrovascular protection: Oestrogen, inflammation and mitochondria. Acta Physiol. 2011, 203, 149–154. [Google Scholar] [CrossRef]
- Nishi, Y.; Satoh, M.; Nagasu, H.; Kadoya, H.; Ihoriya, C.; Kidokoro, K.; Sasaki, T.; Kashihara, N. Selective estrogen receptor modulation attenuates proteinuria-induced renal tubular damage by modulating mitochondrial oxidative status. Kidney Int. 2013, 83, 662–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Homsi, E.; Janino, P.; de Faria, J.B. Role of caspases on cell death, inflammation, and cell cycle in glycerol-induced acute renal failure. Kidney Int. 2006, 69, 1385–1392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, L.; Zhang, J.; Yuan, X.; Tang, J.; Qiu, S.; Peng, Z.; Yuan, Q.; Xie, Y.; Mei, W.; Tang, Y.; et al. Fluorofenidone attenuates interleukin-1β production by interacting with NLRP3 inflammasome in unilateral ureteral obstruction. Nephrology 2018, 23, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Yuan, Y.; Huang, Z.; Chen, H.; Lan, R.; Wang, Z.; Lai, K.; Chen, H.; Chen, Z.; Zou, Z.; et al. GSDME-mediated pyroptosis promotes inflammation and fibrosis in obstructive nephropathy. Cell Death Differ. 2021, 28, 2333–2350. [Google Scholar] [CrossRef] [PubMed]
- Brusselle, G.G.; Provoost, S.; Bracke, K.R.; Kuchmiy, A.; Lamkanfi, M. Inflammasomes in respiratory disease: From bench to bedside. Chest 2014, 145, 1121–1133. [Google Scholar] [CrossRef]
- Cantin, A.M. Cystic Fibrosis Lung Disease and Immunometabolism. Targeting the NLRP3 Inflammasome. Am. J. Respir. Crit. Care Med. 2019, 200, 1335–1337. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.; Moon, J.; Nikahira, K.; Yun, H.; Harris, R.; Hong, K.; Huang, H.; Choi, A.; Stout-Delgado, H. GLUT1-dependent glycolysis regulates exacerbation of fibrosis via AIM2 inflammasome activation. Thorax 2020, 75, 227–236. [Google Scholar] [CrossRef] [Green Version]
- Lv, Z.; Wang, Y.; Liu, Y.; Mao, Y.; Dong, W.; Ding, Z.; Meng, G.; Jiang, L.; Zhu, X. NLRP3 Inflammasome Activation Contributes to Mechanical Stretch-Induced Endothelial-Mesenchymal Transition and Pulmonary Fibrosis. Crit. Care Med. 2018, 46, e49–e58. [Google Scholar] [CrossRef]
- Hussain, S.; Sangtian, S.; Anderson, S.; Snyder, R.; Marshburn, J.; Rice, A.; Bonner, J.; Garantziotis, S. Inflammasome activation in airway epithelial cells after multi-walled carbon nanotube exposure mediates a profibrotic response in lung fibroblasts. Part. Fibre Toxicol. 2014, 11, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef]
- Sun, B.; Wang, X.; Ji, Z.; Wang, M.; Liao, Y.; Chang, C.; Li, R.; Zhang, H.; Nel, A.; Xia, T. NADPH Oxidase-Dependent NLRP3 Inflammasome Activation and its Important Role in Lung Fibrosis by Multiwalled Carbon Nanotubes. Small 2015, 11, 2087–2097. [Google Scholar] [CrossRef] [Green Version]
- Meng, Y.; Li, T.; Zhou, G.S.; Chen, Y.; Yu, C.H.; Pang, M.X.; Li, W.; Li, Y.; Zhang, W.Y.; Li, X. The angiotensin-converting enzyme 2/angiotensin (1-7)/Mas axis protects against lung fibroblast migration and lung fibrosis by inhibiting the NOX4-derived ROS-mediated RhoA/Rho kinase pathway. Antioxid. Redox Signal. 2015, 22, 241–258. [Google Scholar] [CrossRef]
- Meng, Y.; Pan, M.; Zheng, B.; Chen, Y.; Li, W.; Yang, Q.; Zheng, Z.; Sun, N.; Zhang, Y.; Li, X. Autophagy Attenuates Angiotensin II-Induced Pulmonary Fibrosis by Inhibiting Redox Imbalance-Mediated NOD-Like Receptor Family Pyrin Domain Containing 3 Inflammasome Activation. Antioxid. Redox Signal. 2019, 30, 520–541. [Google Scholar] [CrossRef] [PubMed]
- McElvaney, O.; Zaslona, Z.; Becker-Flegler, K.; Palsson-McDermott, E.; Boland, F.; Gunaratnam, C.; Gulbins, E.; O’Neill, L.; Reeves, E.; McElvaney, N. Specific Inhibition of the NLRP3 Inflammasome as an Antiinflammatory Strategy in Cystic Fibrosis. Am. J. Respir. Crit. Care Med. 2019, 200, 1381–1391. [Google Scholar] [CrossRef] [PubMed]
- Lasithiotaki, I.; Giannarakis, I.; Tsitoura, E.; Samara, K.; Margaritopoulos, G.; Choulaki, C.; Vasarmidi, E.; Tzanakis, N.; Voloudaki, A.; Sidiropoulos, P.; et al. NLRP3 inflammasome expression in idiopathic pulmonary fibrosis and rheumatoid lung. Eur. Respir. J. 2016, 47, 910–918. [Google Scholar] [CrossRef] [Green Version]
- Xiao, H.; Li, H.; Wang, J.; Zhang, J.; Shen, J.; An, X.; Zhang, C.; Wu, J.; Song, Y.; Wang, X.; et al. IL-18 cleavage triggers cardiac inflammation and fibrosis upon β-adrenergic insult. Eur. Heart J. 2018, 39, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Usui, F.; Shirasuna, K.; Kimura, H.; Tatsumi, K.; Kawashima, A.; Karasawa, T.; Yoshimura, K.; Aoki, H.; Tsutsui, H.; Noda, T.; et al. Inflammasome activation by mitochondrial oxidative stress in macrophages leads to the development of angiotensin II-induced aortic aneurysm. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 127–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hillegass, J.; Miller, J.; MacPherson, M.; Westbom, C.; Sayan, M.; Thompson, J.; Macura, S.; Perkins, T.; Beuschel, S.; Alexeeva, V.; et al. Asbestos and erionite prime and activate the NLRP3 inflammasome that stimulates autocrine cytokine release in human mesothelial cells. Part. Fibre Toxicol. 2013, 10, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheibe, K.; Kersten, C.; Schmied, A.; Vieth, M.; Primbs, T.; Carlé, B.; Knieling, F.; Claussen, J.; Klimowicz, A.; Zheng, J.; et al. Inhibiting Interleukin 36 Receptor Signaling Reduces Fibrosis in Mice with Chronic Intestinal Inflammation. Gastroenterology 2019, 156, 1082–1097.e11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pulskens, W.; Butter, L.; Teske, G.; Claessen, N.; Dessing, M.; Flavell, R.; Sutterwala, F.; Florquin, S.; Leemans, J. Nlrp3 prevents early renal interstitial edema and vascular permeability in unilateral ureteral obstruction. PLoS ONE 2014, 9, e85775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, Z.; Gong, Q.; Guo, J. Pyroptosis: Mechanisms and Links with Fibrosis. Cells 2021, 10, 3509. https://doi.org/10.3390/cells10123509
Song Z, Gong Q, Guo J. Pyroptosis: Mechanisms and Links with Fibrosis. Cells. 2021; 10(12):3509. https://doi.org/10.3390/cells10123509
Chicago/Turabian StyleSong, Zihao, Quan Gong, and Jiawei Guo. 2021. "Pyroptosis: Mechanisms and Links with Fibrosis" Cells 10, no. 12: 3509. https://doi.org/10.3390/cells10123509
APA StyleSong, Z., Gong, Q., & Guo, J. (2021). Pyroptosis: Mechanisms and Links with Fibrosis. Cells, 10(12), 3509. https://doi.org/10.3390/cells10123509