
From NSAIDs to Glucocorticoids and Beyond


{kind=link}
{kind=link}
Abstract
:1. Introduction
Inflammation and Pain
2. Anti-Inflammatory Pharmacology
2.1. NSAIDs
2.2. Glucocorticoids
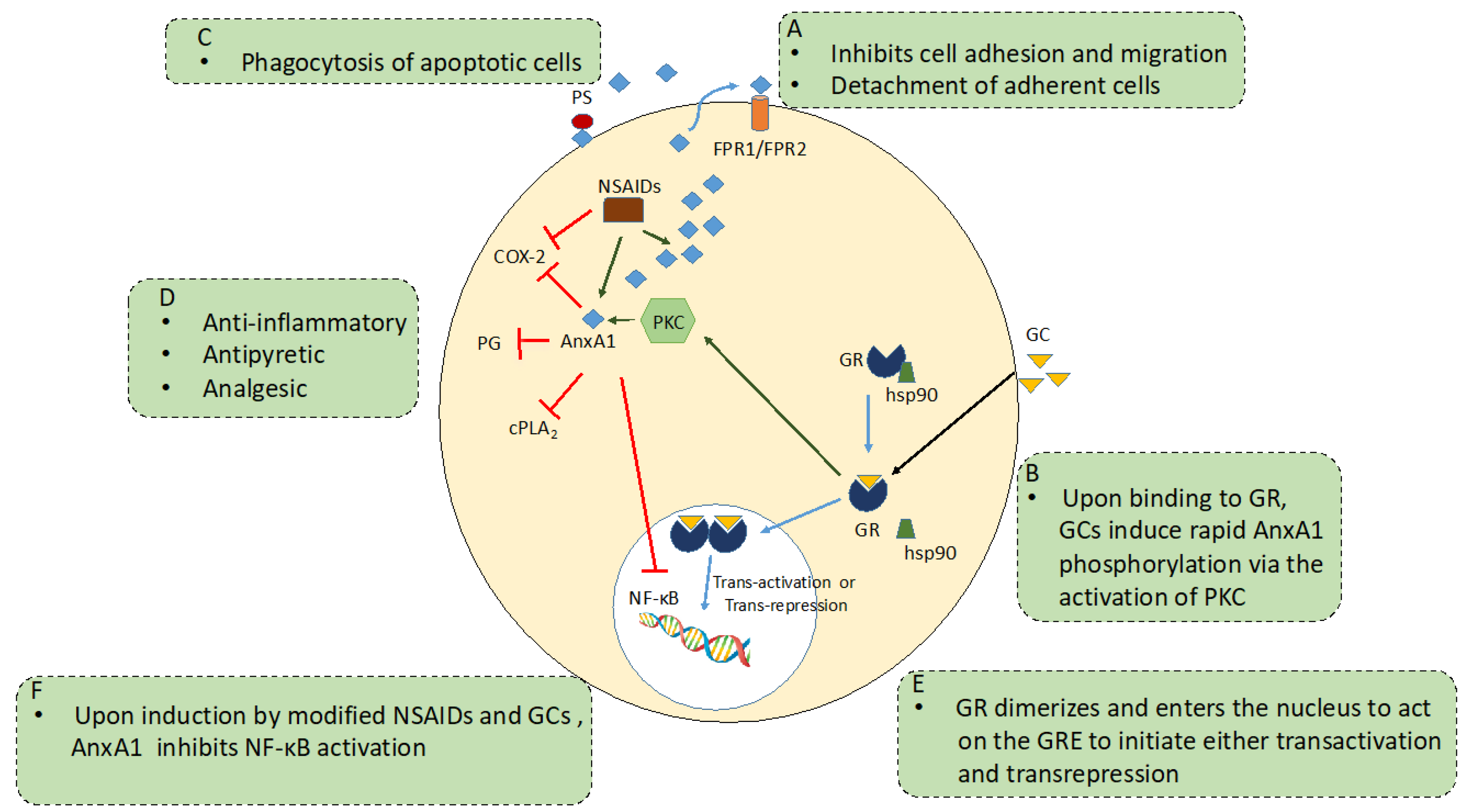
GC Receptor/Mechanism of Action
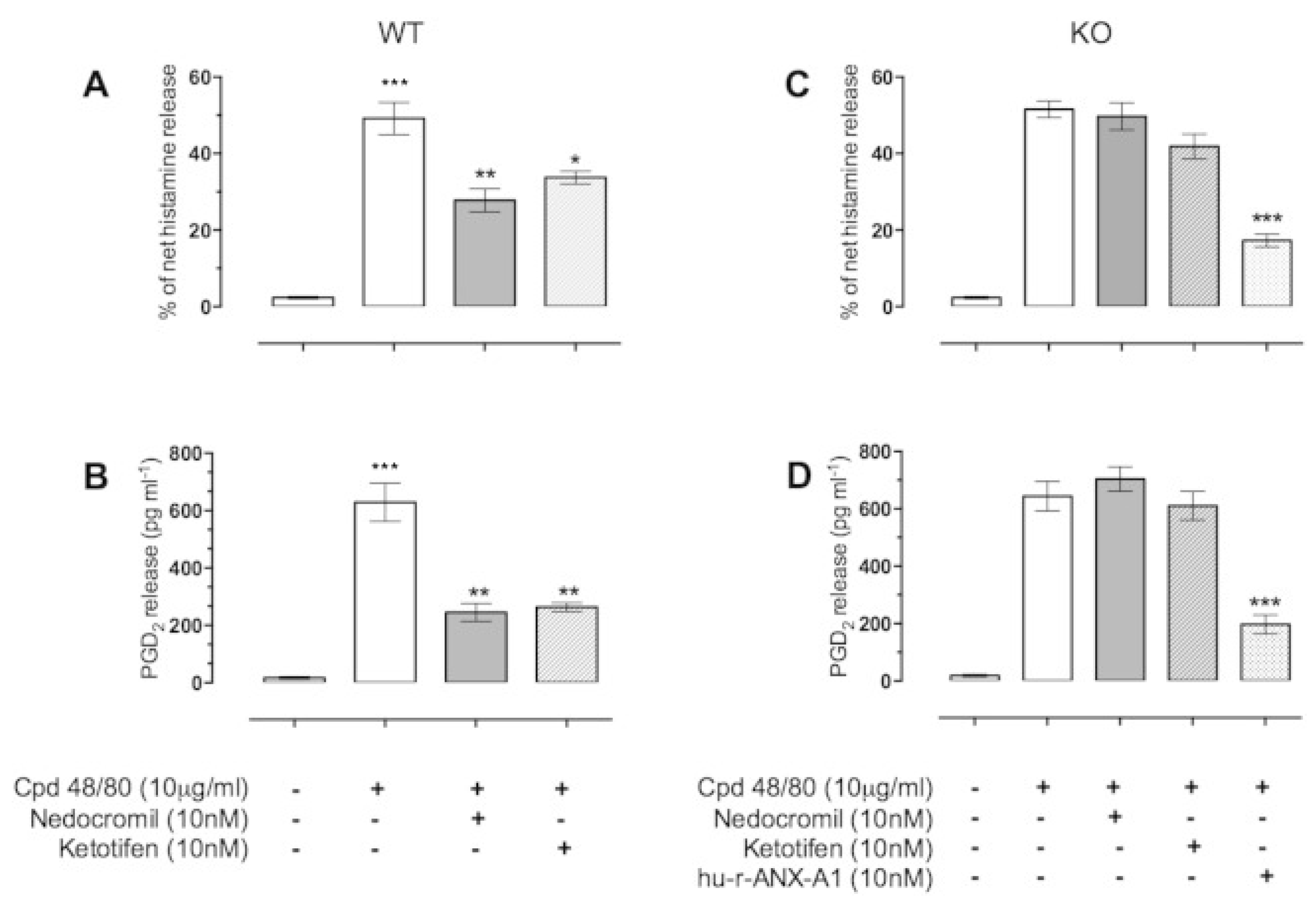
2.3. AnxA1 and Anti-Inflammation
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Köckerling, F.; Köckerling, D.; Lomas, C. Cornelius Celsus--ancient encyclopedist, surgeon-scientist, or master of surgery? Langenbeck’s Arch. Surg. 2013, 398, 609–916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, M.R.E. A brief survey of the history of inflammation. Inflamm. Res. 1994, 43, 86–90. [Google Scholar]
- Heidland, A.; Klassen, A.; Rutkowski, P.; Bahner, U. The contribution of Rudolf Virchow to the concept of inflammation: What is still of importance? J. Nephrol. 2006, 19 (Suppl. S10), 102–109. [Google Scholar]
- Chen, L.; Deng, H.; Cui, H.; Fang, J.; Zuo, Z.; Deng, J.; Li, Y.; Wang, X.; Zhao, L. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 2018, 9, 7204–7218. [Google Scholar] [CrossRef] [Green Version]
- Bennett, J.M.; Reeves, G.; Billman, G.E.; Sturmberg, J.P. Inflammation–Nature’s Way to Efficiently Respond to All Types of Challenges: Implications for Understanding and Managing “the Epidemic” of Chronic Diseases. Front. Med. 2018, 5, 316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubin, A.E.; Patapoutian, A. Nociceptors: The sensors of the pain pathway. J. Clin. Investig. 2010, 120, 3760–3772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinho-Ribeiro, F.A.; Verri, W.A., Jr.; Chiu, I.M. Nociceptor Sensory Neuron–Immune Interactions in Pain and Inflammation. Trends Immunol. 2017, 38, 5–19. [Google Scholar] [CrossRef] [Green Version]
- Netea, M.G.; Balkwill, F.; Chonchol, M.; Cominelli, F.; Donath, M.Y.; Giamarellos-Bourboulis, E.J.; Golenbock, D.; Gresnigt, M.S.; Heneka, M.T.; Hoffman, H.M.; et al. A guiding map for inflammation. Nat. Immunol. 2017, 18, 826–831. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Chen, Z.; Shen, W.; Huang, G.; Sedivy, J.M.; Wang, H.; Ju, Z. Inflammation, epigenetics, and metabolism converge to cell senescence and ageing: The regulation and intervention. Signal Transduct. Target. Ther. 2021, 6, 245. [Google Scholar] [CrossRef]
- Resolution Phase of Inflammation: Novel Endogenous Anti-Inflammatory and Proresolving Lipid Mediators and Pathways. Annu. Rev. Immunol. 2007, 25, 101–137. [CrossRef] [Green Version]
- Serhan, C.N. Pro-resolving lipid mediators are leads for resolution physiology. Nature 2014, 510, 92–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freire, M.O.; Van Dyke, T.E. Natural resolution of inflammation. Periodontology 2000 2013, 63, 149–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tompkins, D.A.; Hobelmann, J.G.; Compton, P. Providing chronic pain management in the “Fifth Vital Sign” Era: Historical and treatment perspectives on a modern-day medical dilemma. Drug Alcohol Depend. 2017, 173 (Suppl. S1), S11–S21. [Google Scholar] [CrossRef] [PubMed]
- Rice, A.S.; Smith, B.H.; Blyth, F.M. Pain and the global burden of disease. Pain 2016, 157, 791–796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldberg, D.S.; McGee, S.J. Pain as a global public health priority. BMC Public Health 2011, 11, 770. [Google Scholar] [CrossRef] [Green Version]
- Burke, R.E. Sir Charles Sherrington’s The integrative action of the nervous system: A centenary appreciation. Brain 2006, 130, 887–894. [Google Scholar] [CrossRef]
- Bonica, J.J. The management of pain of cancer. J. Mich. State Med. Soc. 1953, 52, 284–290. [Google Scholar] [PubMed]
- Renn, C.L.; Dorsey, S.G. The physiology and processing of pain: A review. AACN Clin. Issues 2005, 16, 277–290. [Google Scholar] [CrossRef] [Green Version]
- Meyr, A.J.; Steinberg, J.S. The Physiology of the Acute Pain Pathway. Clin. Podiatr. Med. Surg. 2008, 25, 305–326. [Google Scholar] [CrossRef]
- Mifflin, K.; Kerr, B.J. The transition from acute to chronic pain: Understanding how different biological systems interact. Can. J. Anesth./J. Can. D’anesthésie 2013, 61, 112–122. [Google Scholar] [CrossRef]
- Kean, W.; Forestier, F.; Kassam, Y.; Buchanan, W.; Rooney, P. The history of gold therapy in rheumatoid disease. Semin. Arthritis Rheum. 1985, 14, 180–186. [Google Scholar] [CrossRef]
- Suarez-Almazor, M.E.; Belseck, E.; Spooner, C. Penicillamine for treating rheumatoid arthritis. Cochrane Database Syst. Rev. 2000, 2011, CD001460. [Google Scholar] [CrossRef]
- Rynes, R.I. Antimalarial drugs in the treatment of rheumatological diseases. Rheumatology 1997, 36, 799–805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinblatt, M.E. Methotrexate in Rheumatoid Arthritis: A Quarter Century of Development. Trans. Am. Clin. Clim. Assoc. 2013, 124, 16–25. [Google Scholar]
- Cronstein, B.N.; Aune, T.M. Methotrexate and its mechanisms of action in inflammatory arthritis. Nat. Rev. Rheumatol. 2020, 16, 145–154. [Google Scholar] [CrossRef]
- Chan, E.S.; Cronstein, B.N. Methotrexate--how does it really work? Nat. Rev. Rheumatol. 2010, 6, 175–178. [Google Scholar] [CrossRef]
- Cavaillon, J.-M. Once upon a time, inflammation. J. Venom. Anim. Toxins Incl. Trop. Dis. 2021, 27, 20200147. [Google Scholar] [CrossRef]
- Jack, D.B. One hundred years of aspirin. Lancet 1997, 350, 437–439. [Google Scholar] [CrossRef]
- Desborough, M.J.R.; Keeling, D.M. The aspirin story—From willow to wonder drug. Br. J. Haematol. 2017, 177, 674–683. [Google Scholar] [CrossRef] [Green Version]
- Sneader, W. The discovery of aspirin: A reappraisal. BMJ 2000, 321, 1591–1594. [Google Scholar] [CrossRef] [Green Version]
- Flower, R.J. The development of COX2 inhibitors. Nat. Rev. Drug Discov. 2003, 2, 179–191. [Google Scholar] [CrossRef]
- Vane, J.R. Inhibition of Prostaglandin Synthesis as a Mechanism of Action for Aspirin-like Drugs. Nat. New Biol. 1971, 231, 232–235. [Google Scholar] [CrossRef] [PubMed]
- Botting, R.M. Vane’s discovery of the mechanism of action of aspirin changed our understanding of its clinical pharmacology. Pharmacol. Rep. 2010, 62, 518–525. [Google Scholar] [CrossRef]
- Vane, J.R. Adventures and Excursions in Bioassay: The Stepping Stones to Prostacylin. Biosci. Rep. 2004, 24, 254–279. [Google Scholar] [CrossRef]
- Hawkey, C.J. COX-2 chronology. Gut 2005, 54, 1509–1514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitchell, J.A.; Akarasereenont, P.; Thiemermann, C.; Flower, R.J.; Vane, J.R. Selectivity of nonsteroidal antiinflammatory drugs as inhibitors of constitutive and inducible cyclooxygenase. Proc. Natl. Acad. Sci. USA 1993, 90, 11693–11697. [Google Scholar] [CrossRef] [Green Version]
- Cairns, J.A. The coxibs and traditional nonsteroidal anti-inflammatory drugs: A current perspective on cardiovascular risks. Can. J. Cardiol. 2007, 23, 125–131. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Stark, L.A. Aspirin Prevention of Colorectal Cancer: Focus on NF-κB Signalling and the Nucleolus. Biomedicines 2017, 5, 43. [Google Scholar] [CrossRef] [Green Version]
- Dachineni, R.; Ai, G.; Kumar, D.R.; Sadhu, S.S.; Tummala, H.; Bhat, G.J. Cyclin A2 and CDK2 as Novel Targets of Aspirin and Salicylic Acid: A Potential Role in Cancer Prevention. Mol. Cancer Res. 2016, 14, 241–252. [Google Scholar] [CrossRef] [Green Version]
- Ishihara, T.; Yoshida, M.; Arita, M. Omega-3 fatty acid-derived mediators that control inflammation and tissue homeostasis. Int. Immunol. 2019, 31, 559–567. [Google Scholar] [CrossRef] [Green Version]
- Weng, J.; Zhao, G.; Weng, L.; Guan, J.; for the Alzheimer’s Disease Neuroimaging Initiative. Aspirin using was associated with slower cognitive decline in patients with Alzheimer’s disease. PLoS ONE 2021, 16, e0252969. [Google Scholar] [CrossRef] [PubMed]
- Jorda, A.; Aldasoro, M.; Aldasoro, C.; Guerra-Ojeda, S.; Iradi, A.; Vila, J.M.; Campos-Campos, J.; Valles, S.L. Action of low doses of Aspirin in Inflammation and Oxidative Stress induced by aβ. Int. J. Med. Sci. 2020, 17, 834–843. [Google Scholar] [CrossRef]
- Flower, R. What are all the things that aspirin does? BMJ 2003, 327, 572–573. [Google Scholar] [CrossRef]
- Wallace, J.L.; Pittman, Q.; Cirino, G. Nitric Oxide-Releasing Nsaids: A Novel Class of Gi-Sparing Anti-Inflammatory Drugs. Nov. Mol. Approaches Anti-Inflamm. Ther. 1995, 46, 121–129. [Google Scholar]
- Vane, J.R.; Botting, R.M. Mechanism of Action of Nonsteroidal Anti-inflammatory Drugs. Am. J. Med. 1998, 104, 2S–8S. [Google Scholar] [CrossRef] [PubMed]
- Brune, K.; Renner, B.; Tiegs, G. Acetaminophen/paracetamol: A history of errors, failures and false decisions. Eur. J. Pain 2015, 19, 953–965. [Google Scholar] [CrossRef] [PubMed]
- Graham, G.G.; Scott, K. Mechanism of Action of Paracetamol. Am. J. Ther. 2005, 12, 46–55. [Google Scholar] [CrossRef]
- Prescott, L.F. Paracetamol: Past, present, and future. Am. J. Ther. 2000, 7, 143–147. [Google Scholar] [CrossRef]
- Flower, R.J.; Vane, J.R. Inhibition of Prostaglandin Synthetase in Brain explains the Anti-pyretic Activity of Paracetamol (4-Acetamidophenol). Nat. Cell Biol. 1972, 240, 410–411. [Google Scholar] [CrossRef] [PubMed]
- Dinchuk, J.E.; Liu, R.Q.; Trzaskos, J.M. COX-3: In the wrong frame in mind. Immunol. Lett. 2003, 86, 121. [Google Scholar] [CrossRef]
- Krebs, E.E.; Gravely, A.; Nugent, S.; Jensen, A.C.; DeRonne, B.; Goldsmith, E.S.; Kroenke, K.; Bair, M.J.; Noorbaloochi, S. Effect of Opioid vs. Nonopioid Medications on Pain-Related Function in Patients with Chronic Back Pain or Hip or Knee Osteoarthritis Pain: The SPACE Randomized Clinical Trial. JAMA 2018, 319, 872–882. [Google Scholar] [CrossRef] [PubMed]
- Schug, S. Do NSAIDs Really Interfere with Healing after Surgery? J. Clin. Med. 2021, 10, 2359. [Google Scholar] [CrossRef]
- Chen, M.R.; Dragoo, J.L. The effect of nonsteroidal anti-inflammatory drugs on tissue healing. Knee Surg. Sports Traumatol. Arthrosc. 2012, 21, 540–549. [Google Scholar] [CrossRef] [PubMed]
- Zhao-Fleming, H.; Hand, A.; Zhang, K.; Polak, R.; Northcut, A.; Jacob, D.; Dissanaike, S.; Rumbaugh, K.P. Effect of non-steroidal anti-inflammatory drugs on post-surgical complications against the backdrop of the opioid crisis. Burn. Trauma 2018, 6, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pearce, J.M. Thomas Addison (1793–1860). J. R. Soc. Med. 2004, 97, 297–300. [Google Scholar] [CrossRef]
- Edwards, C. Sixty years after Hench--corticosteroids and chronic inflammatory disease. J. Clin. Endocrinol. Metab. 2012, 97, 1443–1451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glyn, J.H. The discovery of cortisone: A personal memory. BMJ 1998, 317, 822. [Google Scholar] [CrossRef] [Green Version]
- Hench, P.S.; Kendall, E.C.; Slocumb, C.H.; Polley, H.F. The effect of a hormone of the adrenal cortex (17-hydroxy-11-dehydrocorticosterone: Compound E) and of pituitary adrenocortical hormone in arthritis: Preliminary report. Ann. Rheum. Dis. 1949, 8, 97–104. [Google Scholar] [CrossRef] [Green Version]
- Karsh, J.; Hetényi, G., Jr. An historical review of rheumatoid arthritis treatment:1948 to 1952. Semin. Arthritis Rheum. 1997, 27, 57–65. [Google Scholar] [CrossRef]
- Burns, C.M. The History of Cortisone Discovery and Development. Rheum. Dis. Clin. N. Am. 2016, 42, 1–14. [Google Scholar] [CrossRef]
- Ramamoorthy, S.; Cidlowski, J.A. Corticosteroids: Mechanisms of Action in Health and Disease. Rheum. Dis. Clin. N. Am. 2016, 42, 15–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scherholz, M.L.; Schlesinger, N.; Androulakis, I.P. Chronopharmacology of glucocorticoids. Adv. Drug Deliv. Rev. 2019, 151–152, 245–261. [Google Scholar] [CrossRef]
- Uyl, D.D.; Bultink, I.E.M.; Lems, W.F. Advances in Glucocorticoid-Induced Osteoporosis. Curr. Rheumatol. Rep. 2011, 13, 233–240. [Google Scholar]
- Barnes, P.J.; Adcock, I. Glucocorticoid resistance in inflammatory diseases. Lancet 2009, 373, 1905–1917. [Google Scholar] [CrossRef]
- Barnes, P.J. Glucocorticosteroids: Current and future directions. Br. J. Pharmacol. 2010, 163, 29–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kino, T.; Burd, I.; Segars, J.H. Dexamethasone for Severe COVID-19: How Does It Work at Cellular and Molecular Levels? Int. J. Mol. Sci. 2021, 22, 6764. [Google Scholar] [CrossRef]
- Croxtall, J.D.; Van Hal, P.T.W.; Choudhury, Q.; Gilroy, D.; Flower, R.J. Different glucocorticoids vary in their genomic and non-genomic mechanism of action in A549 cells. Br. J. Pharmacol. 2002, 135, 511–519. [Google Scholar] [CrossRef] [Green Version]
- Panettieri, R.A.; Schaafsma, D.; Amrani, Y.; Koziol-White, C.; Ostrom, R.; Tliba, O. Non-genomic Effects of Glucocorticoids: An Updated View. Trends Pharmacol. Sci. 2019, 40, 38–49. [Google Scholar] [CrossRef]
- Encío, I.J.; Detera-Wadleigh, S.D. The genomic structure of the human glucocorticoid receptor. J. Biol. Chem. 1991, 266, 7182–7188. [Google Scholar] [CrossRef]
- Kadmiel, M.; Cidlowski, J.A. Glucocorticoid receptor signaling in health and disease. Trends Pharmacol. Sci. 2013, 34, 518–530. [Google Scholar] [CrossRef] [Green Version]
- Renoir, J.M.; Radanyi, C.; Faber, L.E.; Baulieu, E.E. The non-DNA-binding heterooligomeric form of mammalian steroid hormone receptors contains a hsp90-bound 59-kilodalton protein. J. Biol. Chem. 1990, 265, 10740–10745. [Google Scholar] [CrossRef]
- Perretti, M.; D’Acquisto, F. Annexin A1 and glucocorticoids as effectors of the resolution of inflammation. Nat. Rev. Immunol. 2009, 9, 62–70. [Google Scholar] [CrossRef]
- Bodwell, J.E.; Ortí, E.; Coull, J.M.; Pappin, D.J.; Smith, L.I.; Swift, F. Identification of phosphorylated sites in the mouse glucocorticoid receptor. J. Biol. Chem. 1991, 266, 7549–7555. [Google Scholar] [CrossRef]
- Clark, A.R.; Lasa, M. Crosstalk between glucocorticoids and mitogen-activated protein kinase signalling pathways. Curr. Opin. Pharmacol. 2003, 3, 404–411. [Google Scholar] [CrossRef]
- Escoter-Torres, L.; Caratti, G.; Mechtidou, A.; Tuckermann, J.; Uhlenhaut, N.H.; Vettorazzi, S. Fighting the Fire: Mechanisms of Inflammatory Gene Regulation by the Glucocorticoid Receptor. Front. Immunol. 2019, 10, 1859. [Google Scholar] [CrossRef] [PubMed]
- Vago, J.P.; Tavares, L.P.; Garcia, C.C.; Lima, K.M.; Perucci, L.O.; Vieira, É.L.; Nogueira, C.R.C.; Soriani, F.M.; Martins, J.O.; Silva, P.M.R.; et al. The Role and Effects of Glucocorticoid-Induced Leucine Zipper in the Context of Inflammation Resolution. J. Immunol. 2015, 194, 4940–4950. [Google Scholar] [CrossRef] [Green Version]
- Song, I.-H.; Buttgereit, F. Non-genomic glucocorticoid effects to provide the basis for new drug developments. Mol. Cell. Endocrinol. 2006, 246, 142–146. [Google Scholar] [CrossRef]
- Reichardt, H.M.; Kellendonk, C.; Tronche, F.; Schütz, G. The Cre/loxP system—A versatile tool to study glucocorticoid signalling in mice. Biochem. Soc. Trans. 1999, 27, 78–83. [Google Scholar] [CrossRef] [Green Version]
- Flower, R.J.; Blackwell, G.J. Anti-inflammatory steroids induce biosynthesis of a phospholipase A2 inhibitor which prevents prostaglandin generation. Nat. Cell Biol. 1979, 278, 456–459. [Google Scholar] [CrossRef]
- Pepinsky, R.B.; Sinclair, L.K.; Browning, J.L.; Mattaliano, R.J.; Smart, J.E.; Chow, E.P.; Falbel, T.; Ribolini, A.; Garwin, J.L.; Wallner, B.P. Purification and partial sequence analysis of a 37-kDa protein that inhibits phospholipase A2 activity from rat peritoneal exudates. J. Biol. Chem. 1986, 261, 4239–4346. [Google Scholar] [CrossRef]
- Wallner, B.P.; Mattaliano, R.J.; Hession, C.; Cate, R.L.; Tizard, R.; Sinclair, L.K.; Foeller, C.; Chow, E.P.; Browing, J.L.; Ramachandran, K.L.; et al. Cloning and expression of human lipocortin, a phospholipase A2 inhibitor with potential anti-inflammatory activity. Nature 1986, 320, 77–81. [Google Scholar] [CrossRef]
- Raynal, P.; Pollard, H.B. Annexins: The problem of assessing the biological role for a gene family of multifunctional calcium- and phospholipid-binding proteins. Biochim. Biophys. Acta (BBA)-Rev. Biomembr. 1994, 1197, 63–93. [Google Scholar] [CrossRef]
- Rosengarth, A.; Gerke, V.; Luecke, H. X-ray structure of full-length annexin 1 and implications for membrane aggregation. J. Mol. Biol. 2001, 306, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Perretti, M.; Flower, R.J. Measurement of lipocortin 1 levels in murine peripheral blood leukocytes by flow cytometry: Modulation by glucocorticoids and inflammation. Br. J. Pharmacol. 1996, 118, 605–610. [Google Scholar] [CrossRef] [Green Version]
- Solito, E.; de Coupade, C.; Parente, L.; Flower, R.J.; Russo-Marie, F. IL-6 stimulates annexin 1 expression and translocation and suggests a new biological role as class II acute phase protein. Cytokine 1998, 10, 514–521. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.D.; Cowell, A.-M.; Flower, J.; Buckingham, J.C. Lipocortin 1 Mediates an Early Inhibitory Action of Glucocorticoids on the Secretion of ACTH by the Rat Anterior Pituitary Gland in vitro. Neuroendocrinology 1993, 58, 430–439. [Google Scholar] [CrossRef]
- Loxley, H.D.; Cowell, A.-M.; Flower, R.J.; Buckingham, J.C. Effects of Lipocortin 1 and Dexamethasone on the Secretion of Corticotrophin-Releasing Factors in the Rat: In vitro and in vivo Studies. J. Neuroendocr. 1993, 5, 51–61. [Google Scholar] [CrossRef]
- Croxtall, J.D.; Choudhury, Q.; Flower, R.J. Glucocorticoids act within minutes to inhibit recruitment of signalling factors to activated EGF receptors through a receptor-dependent, transcription-independent mechanism. Br. J. Pharmacol. 2000, 130, 289–298. [Google Scholar] [CrossRef] [Green Version]
- Masferrer, J.L.; Seibert, K.; Zweifel, B.; Needleman, P. Endogenous glucocorticoids regulate an inducible cyclooxygenase enzyme. Proc. Natl. Acad. Sci. USA 1992, 89, 3917–3921. [Google Scholar] [CrossRef] [Green Version]
- Maridonneau-Parini, I.; Errasfa, M.; Russo-Marie, F. Inhibition of O2-generation by dexamethasone is mimicked by lipocortin I in alveolar macrophages. J. Clin. Investig. 1989, 83, 1936–1940. [Google Scholar] [CrossRef]
- Sheikh, M.H.; Solito, E. Annexin A1: Uncovering the Many Talents of an Old Protein. Int. J. Mol. Sci. 2018, 19, 1045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gold, R.; Pepinsky, R.B.; Zettl, U.K.; Toyka, K.V.; Hartung, H.P. Lipocortin-1 (annexin-1) suppresses activation of autoimmune T cell lines in the Lewis rat. J. Neuroimmunol. 1996, 69, 157–164. [Google Scholar] [CrossRef]
- Davidson, J.; Flower, R.; Milton, A.; Peers, S.; Rotondo, D. Antipyretic actions of human recombinant lipocortin-1. Br. J. Pharmacol. 1991, 102, 7–9. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, S.H.; Cunha, F.Q.; Lorenzetti, B.B.; Michelin, M.A.; Perretti, M.; Flower, R.J.; Poole, S. Role of lipocortin-1 in the anti-hyperalgesic actions of dexamethasone. Br. J. Pharmacol. 1997, 121, 883–888. [Google Scholar] [CrossRef] [Green Version]
- Mulla, A.; Leroux, C.; Solito, E.; Buckingham, J.C. Correlation between the Antiinflammatory Protein Annexin 1 (Lipocortin 1) and Serum Cortisol in Subjects with Normal and Dysregulated Adrenal Function. J. Clin. Endocrinol. Metab. 2005, 90, 557–562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hannon, R.; Croxtall, J.D.; Getting, S.J.; Roviezzo, F.; Yona, S.; Paul-Clark, M.J.; Gavins, F.N.E.; Perretti, M.; Morris, J.F.; Buckingham, J.C.; et al. Aberrant inflammation and resistance to glucocorticoids in annexin 1−/− mouse. FASEB J. 2003, 17, 253–255. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Huang, L.; Zhao, W.; Rigas, B. Annexin 1 induced by anti-inflammatory drugs binds to NF-kappaB and inhibits its activation: Anticancer effects in vitro and in vivo. Cancer Res. 2010, 70, 2379–2388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walther, A.; Riehemann, K.; Gerke, V. A novel ligand of the formyl peptide receptor: Annexin I regulates neutrophil extravasation by interacting with the FPR. Mol. Cell 2000, 5, 831–840. [Google Scholar] [CrossRef]
- Cooray, S.N.; Gobbetti, T.; Montero-Melendez, T.; McArthur, S.; Thompson, D.; Clark, A.J.L.; Flower, R.J.; Perretti, M. Ligand-specific conformational change of the G-protein-coupled receptor ALX/FPR2 determines proresolving functional responses. Proc. Natl. Acad. Sci. USA 2013, 110, 18232–18237. [Google Scholar] [CrossRef] [Green Version]
- Brancaleone, V.; Dalli, J.; Bena, S.; Flower, R.J.; Cirino, G.; Perretti, M. Evidence for an anti-inflammatory loop centered on polymorphonuclear leukocyte formyl peptide receptor 2/lipoxin A4 receptor and operative in the inflamed microvasculature. J. Immunol. 2011, 186, 4905–4914. [Google Scholar] [CrossRef] [Green Version]
- Sinniah, A.; Yazid, S.; Flower, R.J. The Anti-allergic Cromones: Past, Present, and Future. Front. Pharmacol. 2017, 8, 827. [Google Scholar] [CrossRef] [Green Version]
- Yazid, S.; Norling, L.V.; Flower, R.J. Anti-inflammatory drugs, eicosanoids and the annexin A1/FPR2 anti-inflammatory system. Prostaglandins Other Lipid Mediat. 2012, 98, 94–100. [Google Scholar] [CrossRef]
- Sinniah, A.; Yazid, S.; Bena, S.; Oliani, S.M.; Perretti, M.; Flower, R.J. Endogenous Annexin-A1 Negatively Regulates Mast Cell-Mediated Allergic Reactions. Front. Pharmacol. 2019, 10, 1313. [Google Scholar] [CrossRef] [Green Version]
- Sinniah, A.; Yazid, S.; Perretti, M.; Solito, E.; Flower, R.J. The role of the Annexin-A1/FPR2 system in the regulation of mast cell degranulation provoked by compound 48/80 and in the inhibitory action of nedocromil. Int. Immunopharmacol. 2016, 32, 87–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yazid, S.; Sinniah, A.; Solito, E.; Calder, V.; Flower, R.J. Anti-allergic cromones inhibit histamine and eicosanoid release from activated human and murine mast cells by releasing Annexin A1. PLoS ONE 2013, 8, e58963. [Google Scholar]
- Yazid, S.; Leoni, G.; Getting, S.J.; Cooper, D.; Solito, E.; Perretti, M.; Flower, R.J. Antiallergic Cromones Inhibit Neutrophil Recruitment Onto Vascular Endothelium via Annexin-A1 Mobilization. Arter. Thromb. Vasc. Biol. 2010, 30, 1718–1724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yazid, S.; Solito, E.; Christian, H.; McArthur, S.; Goulding, N.; Flower, R. Cromoglycate drugs suppress eicosanoid generation in U937 cells by promoting the release of Anx-A1. Biochem. Pharmacol. 2009, 77, 1814–1826. [Google Scholar] [CrossRef]
- Maderna, P.; Cottell, D.C.; Toivonen, T.; Dufton, N.; Dalli, J.; Perretti, M.; Godson, C. FPR2/ALX receptor expression and internalization are critical for lipoxin A4 and annexin-derived peptide-stimulated phagocytosis. FASEB J. 2010, 24, 4240–4249. [Google Scholar] [CrossRef] [Green Version]
- Lim, L.; Pervaiz, S. Annexin 1: The new face of an old molecule. FASEB J. 2007, 21, 968–975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnes, P.J. Glucocorticoids. Chem. Immunol. Allergy 2014, 100, 311–316. [Google Scholar]
- Cheuk, B.L.; Cheng, S.W. Annexin A1 expression in atherosclerotic carotid plaques and its relationship with plaque characteristics. Eur. J. Vasc. Endovasc. Surg. 2011, 41, 364–371. [Google Scholar] [CrossRef] [Green Version]
- Sena, A.; Grishina, I.; Thai, A.; Goulart, L.; Macal, M.; Fenton, A.; Li, J.; Prindiville, T.; Oliani, S.M.; Dandekar, S.; et al. Dysregulation of Anti-Inflammatory Annexin A1 Expression in Progressive Crohns Disease. PLoS ONE 2013, 8, e76969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sena, A.A.; Pedrotti, L.P.; Barrios, B.E.; Cejas, H.; Balderramo, D.; Diller, A.; Correa, S.G. Lack of TNFRI signaling enhances annexin A1 biological activity in intestinal inflammation. Biochem. Pharmacol. 2015, 98, 422–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, X.; Zhao, J.X.; Yao, L.; Huang, J.J.; Fan, Y.Y.; Yu, L.S. Expression of Annexin A1 during Skin Incised Wound Healing in Mice. Fa Yi Xue Za Zhi 2019, 35, 5–10. [Google Scholar] [PubMed]
- McArthur, S.; Loiola, R.A.; Maggioli, E.; Errede, M.; Virgintino, D.; Solito, E. The restorative role of annexin A1 at the blood–brain barrier. Fluids Barriers CNS 2016, 13, 17. [Google Scholar] [CrossRef] [Green Version]
- Ganesan, T.; Sinniah, A.; Ibrahim, Z.A.; Chik, Z.; Alshawsh, M.A. Annexin A1: A Bane or a Boon in Cancer? A Systematic Review. Molecules 2020, 25, 3700. [Google Scholar] [CrossRef]
- Shao, G.; Zhou, H.; Zhang, Q.; Jin, Y.; Fu, C. Advancements of Annexin A1 in inflammation and tumorigenesis. OncoTargets Ther. 2019, 12, 3245–3254. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sinniah, A.; Yazid, S.; Flower, R.J. From NSAIDs to Glucocorticoids and Beyond. Cells 2021, 10, 3524. https://doi.org/10.3390/cells10123524
Sinniah A, Yazid S, Flower RJ. From NSAIDs to Glucocorticoids and Beyond. Cells. 2021; 10(12):3524. https://doi.org/10.3390/cells10123524
Chicago/Turabian StyleSinniah, Ajantha, Samia Yazid, and Rod J. Flower. 2021. "From NSAIDs to Glucocorticoids and Beyond" Cells 10, no. 12: 3524. https://doi.org/10.3390/cells10123524
APA StyleSinniah, A., Yazid, S., & Flower, R. J. (2021). From NSAIDs to Glucocorticoids and Beyond. Cells, 10(12), 3524. https://doi.org/10.3390/cells10123524