
Emerging Molecular Dependencies of Mutant EGFR-Driven Non-Small Cell Lung Cancer

 ,
,
Abstract
:1. Introduction
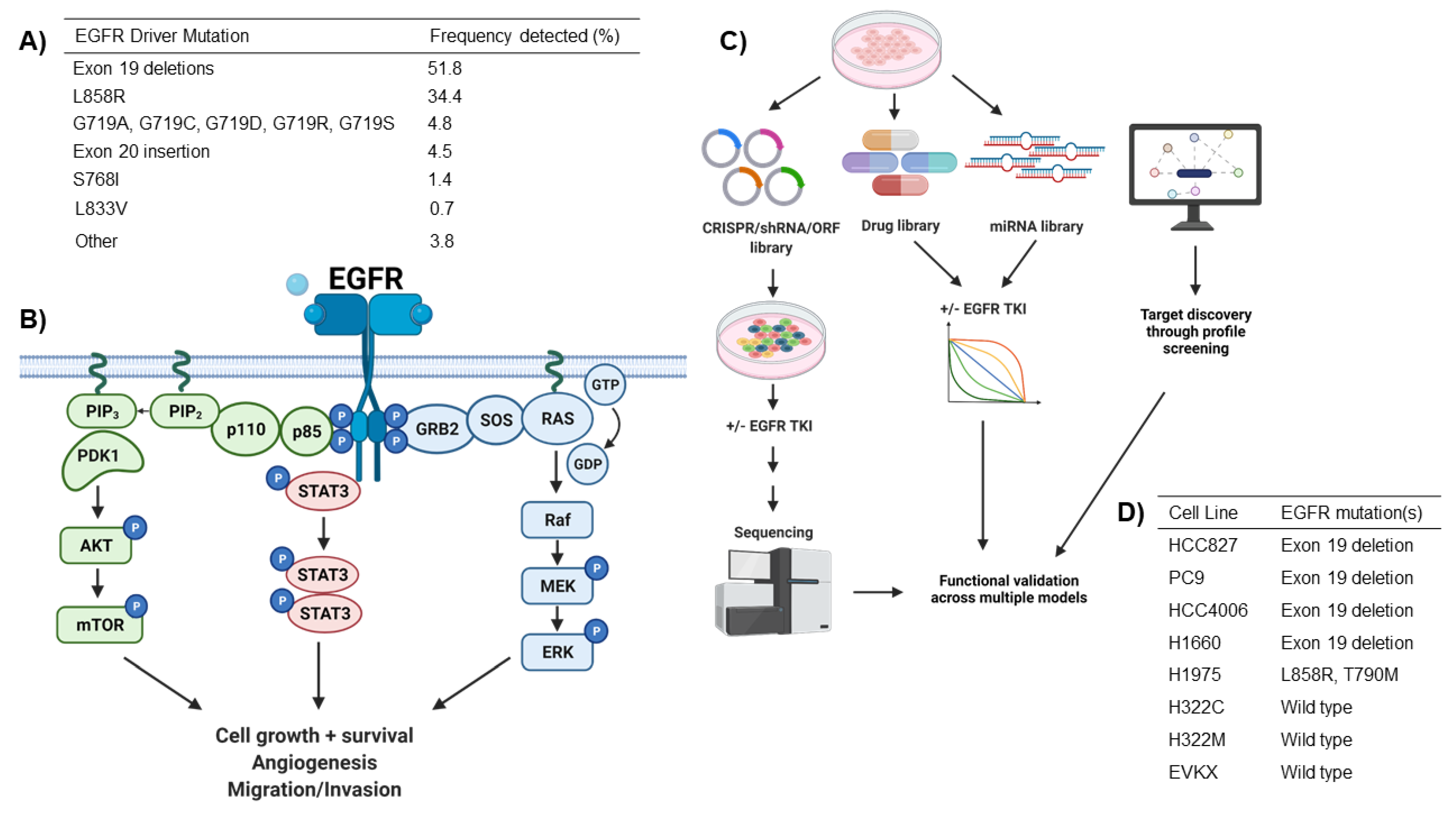
2. EGFR Driver Mutations in NSCLC
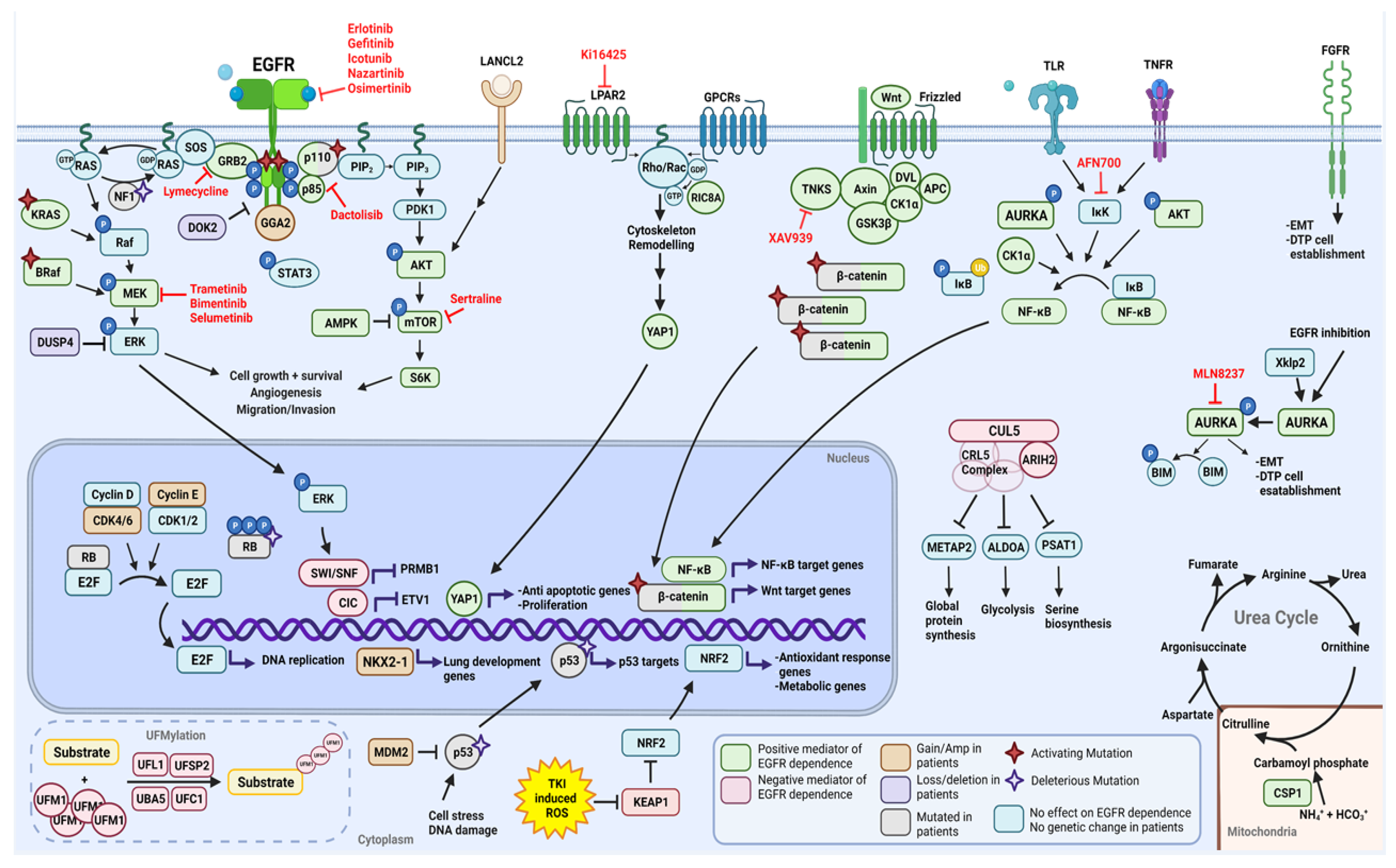
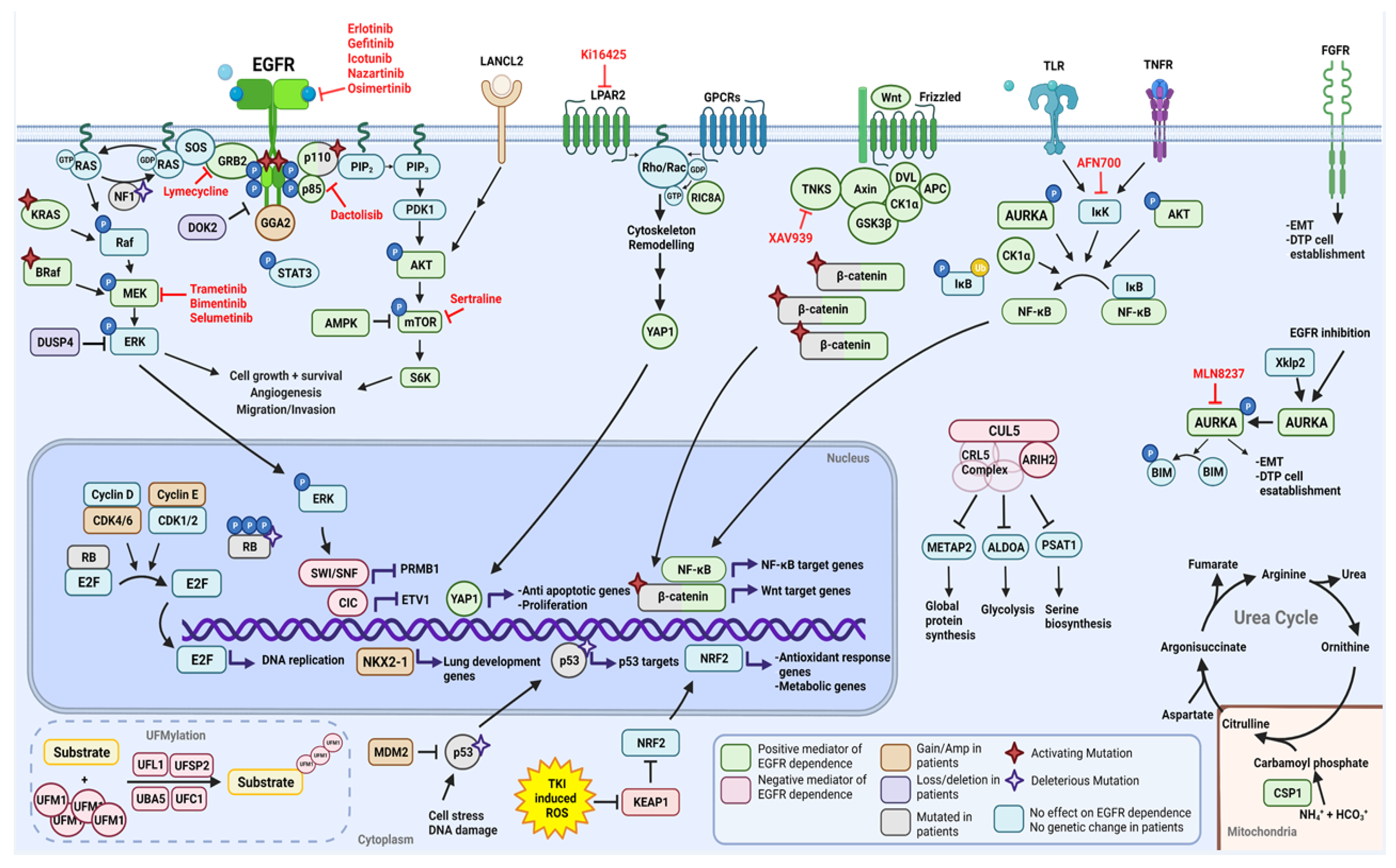
2.1. Functional Modifiers of Mutant EGFR-Induced Lung Tumorigenesis as Targets for Therapeutic Intervention
2.1.1. Positive Moderators of EGFR Dependency
YAP
Wnt/β-Catenin Pathway
NF-κB
Urea Cycle Signaling
FGFR
Aurora Kinase A
Sertraline
Bosutinib
GRB2
Canonical MAPK and PI3K/AKT Signaling Reactivation
2.1.2. Negative Moderators of EGFR Dependency
KEAP1
ARIH2
Ufmylation Pathway
Capicua
SWI/SNF
miRNA
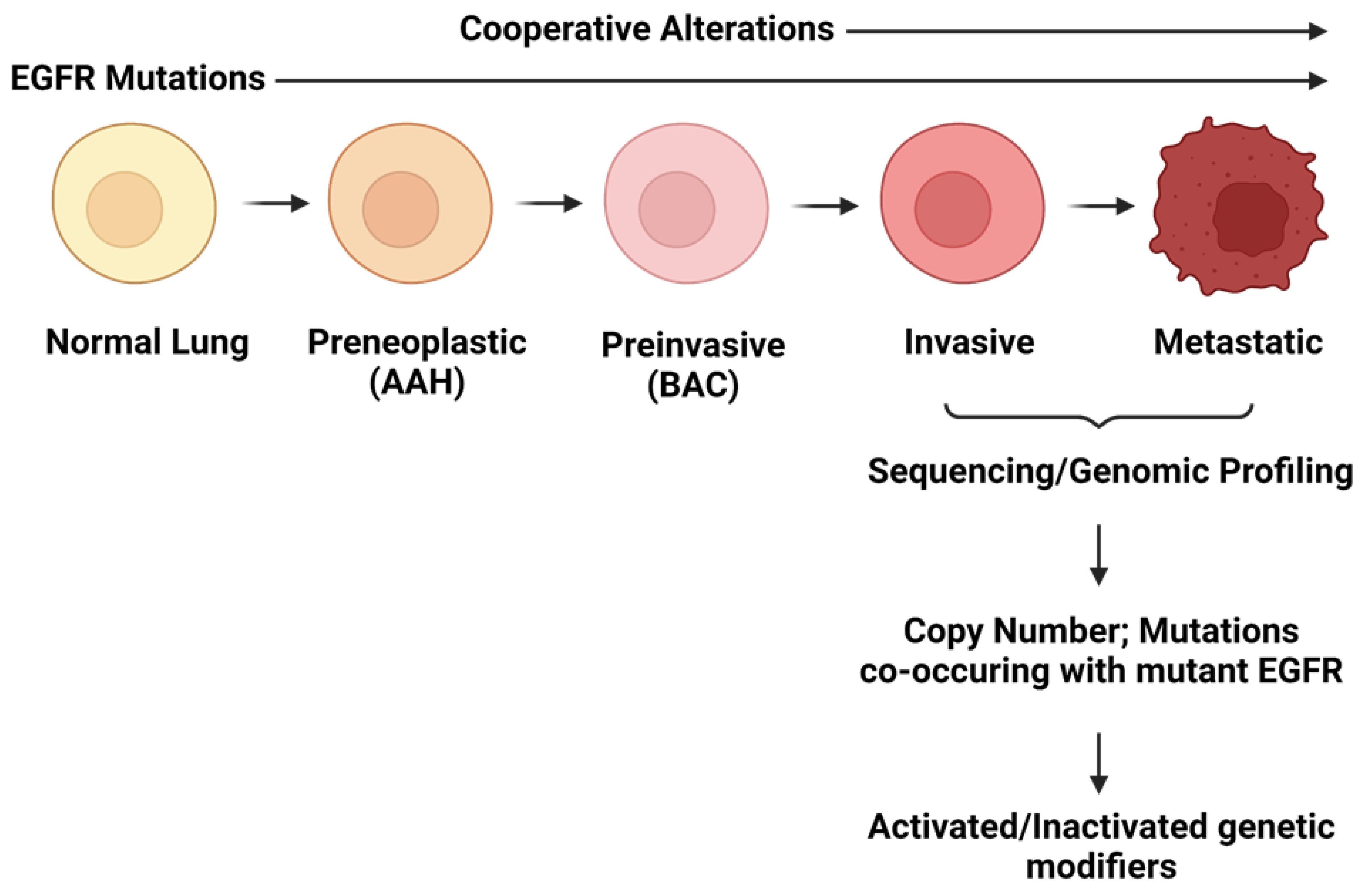
2.2. Cooperating Genomic Alterations in EGFR Mutant NSCLC Cells
2.2.1. Copy Number Alterations
Loss of Chromosome arm 8p Encompassing DUSP4 and DOK2
Amplification of Chromosome Arm 7p: EGFR and LANCL2
Chromosome 16p: GGA2
NKX2-1
MDM2
Cyclins and Cyclin-Dependent Kinases
2.2.2. Mutations
TP53
RB1
PIK3CA
CTNNB1
NF1
3. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wild, C.P.; Weiderpass, E.; Stewart, B.W. (Eds.) World Cancer Report—Cancer Research for Cancer Prevention; International Agency for Research on Cancer: Lyon, France, 2020; ISBN 9789283204237. [Google Scholar]
- Lynch, T.J.; Bell, D.W.; Sordella, R.; Gurubhagavatula, S.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Haserlat, S.M.; Supko, J.G.; Haluska, F.G.; et al. Activating Mutations in the Epidermal Growth Factor Receptor Underlying Responsiveness of Non–Small-Cell Lung Cancer to Gefitinib. N. Engl. J. Med. 2004, 350, 2129–2139. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, F.A.; Rodrigues Pereira, J.; Ciuleanu, T.; Tan, E.H.; Hirsh, V.; Thongprasert, S.; Campos, D.; Maoleekoonpiroj, S.; Smylie, M.; Martins, R.; et al. Erlotinib in Previously Treated Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2005, 353, 123–132. [Google Scholar] [CrossRef] [Green Version]
- Mok, T.S.; Wu, Y.-L.; Thongprasert, S.; Yang, C.-H.; Chu, D.-T.; Saijo, N.; Sunpaweravong, P.; Han, B.; Margono, B.; Ichinose, Y.; et al. Gefitinib or Carboplatin–Paclitaxel in Pulmonary Adenocarcinoma. N. Engl. J. Med. 2009, 361, 947–957. [Google Scholar] [CrossRef]
- Soria, J.-C.; Ohe, Y.; Vansteenkiste, J.; Reungwetwattana, T.; Chewaskulyong, B.; Lee, K.H.; Dechaphunkul, A.; Imamura, F.; Nogami, N.; Kurata, T.; et al. Osimertinib in Untreated EGFR -Mutated Advanced Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 378, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Leonetti, A.; Sharma, S.; Minari, R.; Perego, P.; Giovannetti, E.; Tiseo, M. Resistance Mechanisms to Osimertinib in EGFR-Mutated Non-Small Cell Lung Cancer. Br. J. Cancer 2019, 121, 725–737. [Google Scholar] [CrossRef] [PubMed]
- Sato, M.; Larsen, J.E.; Lee, W.; Sun, H.; Shames, D.S.; Dalvi, M.P.; Ramirez, R.D.; Tang, H.; DiMaio, J.M.; Gao, B.; et al. Human lung epithelial cells progressed to malignancy through specific oncogenic manipulations. Mol. Cancer Res. 2013, 11, 638–650. [Google Scholar] [CrossRef] [Green Version]
- Sato, M.; Vaughan, M.B.; Girard, L.; Peyton, M.; Lee, W.; Shames, D.S.; Ramirez, R.D.; Sunaga, N.; Gazdar, A.F.; Shay, J.W.; et al. Multiple oncogenic changes (K-RASV12, p53 knockdown, mutant EGFRs, p16 bypass, telomerase) are not sufficient to confer a full malignant phenotype on human bronchial epithelial cells. Cancer Res. 2006, 66, 2116–2128. [Google Scholar] [CrossRef] [Green Version]
- Gazdar, A.F.; Minna, J.D. Deregulated EGFR signaling during lung cancer progression: Mutations, amplicons, and autocrine loops. Cancer Prev. Res. 2008, 1, 156–160. [Google Scholar] [CrossRef] [Green Version]
- Shigematsu, H.; Gazdar, A.F. Somatic mutations of epidermal growth factor receptor signaling pathway in lung cancers. Int. J. Cancer 2006, 118, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Metro, G.; Crinò, L. Advances on EGFR mutation for lung cancer. Transl. Lung Cancer Res. 2012, 1, 5–13. [Google Scholar]
- Valley, C.C.; Arndt-Jovin, D.J.; Karedla, N.; Steinkamp, M.P.; Chizhik, A.I.; Hlavacek, W.S.; Wilson, B.S.; Lidke, K.A.; Lidke, D.S. Enhanced dimerization drives ligand-independent activity of mutant epidermal growth factor receptor in lung cancer. Mol. Biol. Cell 2015, 26, 4087–4099. [Google Scholar] [CrossRef] [PubMed]
- Viloria Petit, A.M.; Rak, J.; Hung, M.C.; Rockwell, P.; Goldstein, N.; Fendly, B.; Kerbel, R.S. Neutralizing antibodies against epidermal growth factor and ErbB-2/neu receptor tyrosine kinases down-regulate vascular endothelial growth factor production by tumor cells in vitro and in vivo: Angiogenic implications for signal transduction therapy of so. Am. J. Pathol. 1997, 151, 1523–1530. [Google Scholar]
- Jiang, J.; Greulich, H.; Jänne, P.A.; Sellers, W.R.; Meyerson, M.; Griffin, J.D. Epidermal growth factor-independent transformation of Ba/F3 cells with cancer-derived epidermal growth factor receptor mutants induces gefitinib-sensitive cell cycle progression. Cancer Res. 2005, 65, 8968–8974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sordella, R.; Bell, D.W.; Haber, D.A.; Settleman, J. Gefitinib-sensitizing EGFR mutations in lung cancer activate anti-apoptotic pathways. Science 2004, 305, 1163–1167. [Google Scholar] [CrossRef]
- Herbst, R.S. Review of epidermal growth factor receptor biology. Int. J. Radiat. Oncol. Biol. Phys. 2004, 59, S21–S26. [Google Scholar] [CrossRef]
- Wakeling, A.E.; Guy, S.P.; Woodburn, J.R.; Ashton, S.E.; Curry, B.J.; Barker, A.J.; Gibson, K.H. ZD1839 (Iressa). Cancer Res. 2002, 62, 5749–5754. [Google Scholar]
- Moyer, J.D.; Barbacci, E.G.; Iwata, K.K.; Arnold, L.; Boman, B.; Cunningham, A.; DiOrio, C.; Doty, J.; Morin, M.J.; Moyer, M.P.; et al. Induction of apoptosis and cell cycle arrest by CP-358,774, an inhibitor of epidermal growth factor receptor tyrosine kinase. Cancer Res. 1997, 57, 4838–4848. [Google Scholar]
- Yu, H.A.; Arcila, M.E.; Rekhtman, N.; Sima, C.S.; Zakowski, M.F.; Pao, W.; Kris, M.G.; Miller, V.A.; Ladanyi, M.; Riely, G.J. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin. Cancer Res. 2013, 19, 2240–2247. [Google Scholar] [CrossRef] [Green Version]
- Cross, D.A.E.; Ashton, S.E.; Ghiorghiu, S.; Eberlein, C.; Nebhan, C.A.; Spitzler, P.J.; Orme, J.P.; Finlay, M.R.V.; Ward, R.A.; Mellor, M.J.; et al. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov. 2014, 4, 1046–1061. [Google Scholar] [CrossRef] [Green Version]
- Mok, T.S.; Wu, Y.-L.; Ahn, M.-J.; Garassino, M.C.; Kim, H.R.; Ramalingam, S.S.; Shepherd, F.A.; He, Y.; Akamatsu, H.; Theelen, W.S.M.E.; et al. Osimertinib or Platinum–Pemetrexed in EGFR T790M–Positive Lung Cancer. N. Engl. J. Med. 2016, 376, 629–640. [Google Scholar] [CrossRef] [Green Version]
- La Monica, S.; Fumarola, C.; Cretella, D.; Bonelli, M.; Minari, R.; Cavazzoni, A.; Digiacomo, G.; Galetti, M.; Volta, F.; Mancini, M.; et al. Efficacy of the cdk4/6 dual inhibitor abemaciclib in egfr-mutated nsclc cell lines with different resistance mechanisms to osimertinib. Cancers 2021, 13, 6. [Google Scholar] [CrossRef] [PubMed]
- Ramalingam, S.S.; Cheng, Y.; Zhou, C.; Ohe, Y.; Imamura, F.; Cho, B.C.; Lin, M.-C.; Majem, M.; Shah, R.; Rukazenkov, Y.; et al. Mechanisms of acquired resistance to first-line osimertinib: Preliminary data from the phase III FLAURA study. Ann. Oncol. 2018, 29, viii740. [Google Scholar] [CrossRef]
- Le, X.; Puri, S.; Negrao, M.V.; Nilsson, M.B.; Robichaux, J.; Boyle, T.; Kevin Hicks, J.; Lovinger, K.L.; Roarty, E.; Rinsurongkawong, W.; et al. Landscape of EGFR-dependent and -independent resistance mechanisms to osimertinib and continuation therapy beyond progression in EGFR-mutant NSCLC. Clin. Cancer Res. 2018, 24, 6195–6203. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Yang, N.; Ou, Q.; Xiang, Y.; Jiang, T.; Wu, X.; Bao, H.; Tong, X.; Wang, X.; Shao, Y.W.; et al. Investigating novel resistance mechanisms to third-generation EGFR tyrosine kinase inhibitor osimertinib in non–small cell lung cancer patients. Clin. Cancer Res. 2018, 24, 3097–3107. [Google Scholar] [CrossRef] [Green Version]
- Offin, M.; Somwar, R.; Rekhtman, N.; Benayed, R.; Chang, J.C.; Plodkowski, A.; Lui, A.J.W.; Eng, J.; Rosenblum, M.; Li, B.T.; et al. Acquired ALK and RET Gene Fusions as Mechanisms of Resistance to Osimertinib in EGFR -Mutant Lung Cancers. JCO Precis. Oncol. 2018, 2, 30957057. [Google Scholar] [CrossRef] [PubMed]
- Piotrowska, Z.; Isozaki, H.; Lennerz, J.K.; Gainor, J.F.; Lennes, I.T.; Zhu, V.W.; Marcoux, N.; Banwait, M.K.; Digumarthy, S.R.; Su, W.; et al. Landscape of acquired resistance to osimertinib in EGFR-mutant NSCLC and clinical validation of combined EGFR and RET inhibition with osimertinib and BLU-667 for acquired RET fusion. Cancer Discov. 2018, 8, 1529–1539. [Google Scholar] [CrossRef] [Green Version]
- Zeng, L.; Yang, N.; Zhang, Y. GOPC-ROS1 Rearrangement as an Acquired Resistance Mechanism to Osimertinib and Responding to Crizotinib Combined Treatments in Lung Adenocarcinoma. J. Thorac. Oncol. 2018, 13, e114–e116. [Google Scholar] [CrossRef] [Green Version]
- Kim, T.M.; Song, A.; Kim, D.W.; Kim, S.; Ahn, Y.O.; Keam, B.; Jeon, Y.K.; Lee, S.H.; Chung, D.H.; Heo, D.S. Mechanisms of Acquired Resistance to AZD9291: A Mutation-Selective, Irreversible EGFR Inhibitor. J. Thorac. Oncol. 2015, 10, 1736–1744. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Wang, H.; Li, C.; Wang, Z.; Zhang, P.; Yan, X. Transformation to small-cell carcinoma as an acquired resistance mechanism to AZD9291: A case report. Oncotarget 2017, 8, 18609–18614. [Google Scholar] [CrossRef] [Green Version]
- Minari, R.; Bordi, P.; Del Re, M.; Facchinetti, F.; Mazzoni, F.; Barbieri, F.; Camerini, A.; Comin, C.E.; Gnetti, L.; Azzoni, C.; et al. Primary resistance to osimertinib due to SCLC transformation: Issue of T790M determination on liquid re-biopsy. Lung Cancer 2018, 115, 21–27. [Google Scholar] [CrossRef]
- Jackman, D.; Pao, W.; Riely, G.J.; Engelman, J.A.; Kris, M.G.; Jänne, P.A.; Lynch, T.; Johnson, B.E.; Miller, V.A. Clinical definition of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancer. J. Clin. Oncol. 2010, 28, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Ying, H.Q.; Chen, J.; He, B.S.; Pan, Y.Q.; Wang, F.; Deng, Q.W.; Sun, H.L.; Liu, X.; Wang, S.K. The effect of BIM deletion polymorphism on intrinsic resistance and clinical outcome of cancer patient with kinase inhibitor therapy. Sci. Rep. 2015, 5, 11348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taniguchi, H.; Yamada, T.; Wang, R.; Tanimura, K.; Adachi, Y.; Nishiyama, A.; Tanimoto, A.; Takeuchi, S.; Araujo, L.H.; Boroni, M.; et al. AXL confers intrinsic resistance to osimertinib and advances the emergence of tolerant cells. Nat. Commun. 2019, 10, 259. [Google Scholar] [CrossRef] [PubMed]
- Yano, S.; Yamada, T.; Takeuchi, S.; Tachibana, K.; Minami, Y.; Yatabe, Y.; Mitsudomi, T.; Tanaka, H.; Kimura, T.; Kudoh, S.; et al. Hepatocyte growth factor expression in EGFR mutant lung cancer with intrinsic and acquired resistance to tyrosine kinase inhibitors in a japanese cohort. J. Thorac. Oncol. 2011, 6, 2011–2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herbst, R.S.; Baas, P.; Kim, D.W.; Felip, E.; Pérez-Gracia, J.L.; Han, J.Y.; Molina, J.; Kim, J.H.; Arvis, C.D.; Ahn, M.J.; et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): A randomised controlled trial. Lancet 2016, 387, 1540–1550. [Google Scholar] [CrossRef]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef] [PubMed]
- Rittmeyer, A.; Barlesi, F.; Waterkamp, D.; Park, K.; Ciardiello, F.; von Pawel, J.; Gadgeel, S.M.; Hida, T.; Kowalski, D.M.; Dols, M.C.; et al. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): A phase 3, open-label, multicentre randomised controlled trial. Lancet 2017, 389, 255–265. [Google Scholar] [CrossRef]
- Lee, C.K.; Man, J.; Lord, S.; Links, M.; Gebski, V.; Mok, T.; Yang, J.C.H. Checkpoint Inhibitors in Metastatic EGFR-Mutated Non–Small Cell Lung Cancer—A Meta-Analysis. J. Thorac. Oncol. 2017, 12, 403–407. [Google Scholar] [CrossRef] [Green Version]
- Spigel, D.R.; Schrock, A.B.; Fabrizio, D.; Frampton, G.M.; Sun, J.; He, J.; Gowen, K.; Johnson, M.L.; Bauer, T.M.; Kalemkerian, G.P.; et al. Total mutation burden (TMB) in lung cancer (LC) and relationship with response to PD-1/PD-L1 targeted therapies. J. Clin. Oncol. 2016, 34, 9017. [Google Scholar] [CrossRef]
- Pal, A.S.; Bains, M.; Agredo, A.; Kasinski, A.L. Identification of microRNAs that promote erlotinib resistance in non-small cell lung cancer. Biochem. Pharmacol. 2021, 189, 114154. [Google Scholar] [CrossRef]
- Bolan, P.O.; Zviran, A.; Brenan, L.; Schiffman, J.S.; Dusaj, N.; Goodale, A.; Piccioni, F.; Johannessen, C.M.; Landau, D.A. Genotype-Fitness Maps of EGFR-Mutant Lung Adenocarcinoma Chart the Evolutionary Landscape of Resistance for Combination Therapy Optimization. Cell Syst. 2020, 10, 52–65.e7. [Google Scholar] [CrossRef]
- Chen, Y.; Wu, J.; Yan, H.; Cheng, Y.; Wang, Y.; Yang, Y.; Deng, M.; Che, X.; Hou, K.; Qu, X.; et al. Lymecycline reverses acquired EGFR-TKI resistance in non–small-cell lung cancer by targeting GRB2. Pharmacol. Res. 2020, 159, 105007. [Google Scholar] [CrossRef]
- Zeng, H.; Castillo-Cabrera, J.; Manser, M.; Lu, B.; Yang, Z.; Strande, V.; Begue, D.; Zamponi, R.; Qiu, S.; Sigoillot, F.; et al. Genome-wide Crispr screening reveals genetic modifiers of mutant EGFR dependence in human NSCLC. Elife 2019, 8, e50223. [Google Scholar] [CrossRef]
- Pham-Danis, C.; Gehrke, S.; Danis, E.; Rozhok, A.I.; Daniels, M.W.; Gao, D.; Collins, C.; Di Paola, J.T.; D’Alessandro, A.; DeGregori, J. Urea Cycle Sustains Cellular Energetics upon EGFR Inhibition in EGFR-Mutant NSCLC. Mol. Cancer Res. 2019, 17, 1351–1364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raoof, S.; Mulford, I.J.; Frisco-Cabanos, H.; Nangia, V.; Timonina, D.; Labrot, E.; Hafeez, N.; Bilton, S.J.; Drier, Y.; Ji, F.; et al. Targeting FGFR overcomes EMT-mediated resistance in EGFR mutant non-small cell lung cancer. Oncogene 2019, 38, 6399–6413. [Google Scholar] [CrossRef]
- Shah, K.N.; Bhatt, R.; Rotow, J.; Rohrberg, J.; Olivas, V.; Wang, V.E.; Hemmati, G.; Martins, M.M.; Maynard, A.; Kuhn, J.; et al. Aurora kinase A drives the evolution of resistance to third-generation EGFR inhibitors in lung cancer. Nat. Med. 2019, 25, 111–118. [Google Scholar] [CrossRef]
- Terai, H.; Kitajima, S.; Potter, D.S.; Matsui, Y.; Quiceno, L.G.; Chen, T.; Kim, T.; Rusan, M.; Thai, T.C.; Piccioni, F.; et al. ER stress signaling promotes the survival of cancer ‘persister cells’ tolerant to EGFR tyrosine kinase inhibitors. Cancer Res. 2018, 78, 1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, X.; Lu, W.; Shen, X.; Wang, Q.; Lv, J.; Liu, M.; Cheng, F.; Zhao, Z.; Pang, X. Repurposing sertraline sensitizes non-small cell lung cancer cells to erlotinib by inducing autophagy. JCI Insight 2018, 3, e98921. [Google Scholar] [CrossRef] [PubMed]
- Krall, E.B.; Wang, B.; Munoz, D.M.; Ilic, N.; Raghavan, S.; Niederst, M.J.; Yu, K.; Ruddy, D.A.; Aguirre, A.J.; Kim, J.W.; et al. KEAP1 loss modulates sensitivity to kinase targeted therapy in lung cancer. Elife 2017, 6, e18970. [Google Scholar] [CrossRef] [Green Version]
- Liao, S.; Davoli, T.; Leng, Y.; Li, M.Z.; Xu, Q.; Elledge, S.J. A genetic interaction analysis identifies cancer drivers that modify EGFR dependency. Genes Dev. 2017, 31, 184–196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, H.; Zhang, Z.; Rodriguez-Barrueco, R.R.; Borczuk, A.; Liu, H.; Yu, J.; Silva, J.M.; Cheng, S.K.; Perez-Soler, R.; Halmos, B. Functional genomics screen identifies YAP1 as a key determinant to enhance treatment sensitivity in lung cancer cells. Oncotarget 2016, 7, 28976–28988. [Google Scholar] [CrossRef] [Green Version]
- Lantermann, A.B.; Chen, D.; McCutcheon, K.; Hoffman, G.; Frias, E.; Ruddy, D.; Rakiec, D.; Korn, J.; McAllister, G.; Stegmeier, F.; et al. Inhibition of Casein Kinase 1 Alpha Prevents Acquired Drug Resistance to Erlotinib in EGFR-Mutant Non–Small Cell Lung Cancer. Cancer Res. 2015, 75, 4937–4948. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Vasu, V.T.; Mishra, R.; Singleton, K.R.; Yoo, M.; Leach, S.M.; Farias-Hesson, E.; Mason, R.J.; Kang, J.; Ramamoorthy, P.; et al. Bioinformatics-driven discovery of rational combination for overcoming EGFR-mutant lung cancer resistance to EGFR therapy. Bioinformatics 2014, 30, 2393–2398. [Google Scholar] [CrossRef]
- Sharifnia, T.; Rusu, V.; Piccioni, F.; Bagul, M.; Imielinski, M.; Cherniack, A.D.; Pedamallu, C.S.; Wong, B.; Wilson, F.H.; Garraway, L.A.; et al. Genetic modifiers of EGFR dependence in non-small cell lung cancer. Proc. Natl. Acad. Sci. USA 2014, 111, 18661–18666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casás-Selves, M.; Kim, J.; Zhang, Z.; Helfrich, B.A.; Gao, D.; Porter, C.C.; Scarborough, H.A.; Bunn, P.A.; Chan, D.C.; Tan, A.C.; et al. Tankyrase and the canonical Wnt pathway protect lung cancer cells from EGFR inhibition. Cancer Res. 2012, 72, 4154–4164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bivona, T.G.; Hieronymus, H.; Parker, J.; Chang, K.; Taron, M.; Rosell, R.; Moonsamy, P.; Dahlman, K.; Miller, V.A.; Costa, C.; et al. FAS and NF-κB signalling modulate dependence of lung cancers on mutant EGFR. Nature 2011, 471, 523–526. [Google Scholar] [CrossRef] [Green Version]
- Chan, P.; Thomas, C.J.; Sprang, S.R.; Tall, G.G. Molecular chaperoning function of Ric-8 is to fold nascent heterotrimeric G protein α subunits. Proc. Natl. Acad. Sci. USA 2013, 110, 3794–3799. [Google Scholar] [CrossRef] [Green Version]
- Kant, R.; Zeng, B.; Thomas, C.J.; Bothner, B.; Sprang, S.R. Ric-8A, a G protein chaperone with nucleotide exchange activity induces long-range secondary structure changes in Gα. Elife 2016, 5, e19238. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Teng, L. YAP/TAZ for cancer therapy: Opportunities and challenges (Review). Int. J. Oncol. 2015, 46, 1444–1452. [Google Scholar] [CrossRef] [Green Version]
- Lin, L.; Sabnis, A.J.; Chan, E.; Olivas, V.; Cade, L.; Pazarentzos, E.; Asthana, S.; Neel, D.; Yan, J.J.; Lu, X.; et al. The Hippo effector YAP promotes resistance to RAF- and MEK-targeted cancer therapies. Nat. Genet. 2015, 47, 250–256. [Google Scholar] [CrossRef]
- Yu, F.X.; Zhao, B.; Panupinthu, N.; Jewell, J.L.; Lian, I.; Wang, L.H.; Zhao, J.; Yuan, H.; Tumaneng, K.; Li, H.; et al. Regulation of the Hippo-YAP pathway by G-protein-coupled receptor signaling. Cell 2012, 150, 780–791. [Google Scholar] [CrossRef] [Green Version]
- Song, S.; Honjo, S.; Jin, J.; Chang, S.S.; Scott, A.W.; Chen, Q.; Kalhor, N.; Correa, A.M.; Hofstetter, W.L.; Albarracin, C.T.; et al. The hippo coactivator YAP1 mediates EGFR overexpression and confers chemoresistance in esophageal cancer. Clin. Cancer Res. 2015, 21, 2580–2590. [Google Scholar] [CrossRef] [Green Version]
- Urtasun, R.; Latasa, M.U.; Demartis, M.I.; Balzani, S.; Goñi, S.; Garcia-Irigoyen, O.; Elizalde, M.; Azcona, M.; Pascale, R.M.; Feo, F.; et al. Connective tissue growth factor autocriny in human hepatocellular carcinoma: Oncogenic role and regulation by epidermal growth factor receptor/yes-associated protein-mediated activation. Hepatology 2011, 54, 2149–2158. [Google Scholar] [CrossRef]
- Sun, P.L.; Kim, J.E.; Yoo, S.B.; Kim, H.; Jin, Y.; Jheon, S.; Kim, K.; Lee, C.T.; Chung, J.H. Cytoplasmic YAP Expression is Associated with Prolonged Survival in Patients with Lung Adenocarcinomas and Epidermal Growth Factor Receptor Tyrosine Kinase Inhibitor Treatment. Ann. Surg. Oncol. 2014, 21, 610–618. [Google Scholar] [CrossRef]
- Pai, S.G.; Carneiro, B.A.; Mota, J.M.; Costa, R.; Leite, C.A.; Barroso-Sousa, R.; Kaplan, J.B.; Chae, Y.K.; Giles, F.J. Wnt/beta-catenin pathway: Modulating anticancer immune response. J. Hematol. Oncol. 2017, 10, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clevers, H.; Nusse, R. Wnt/β-catenin signaling and disease. Cell 2012, 149, 1192–1205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasmi, R.R.; Sakthivel, K.M.; Guruvayoorappan, C. NF-κB inhibitors in treatment and prevention of lung cancer. Biomed. Pharmacother. 2020, 130, 110569. [Google Scholar] [CrossRef]
- Perkins, N.D. Integrating cell-signalling pathways with NF-κB and IKK function. Nat. Rev. Mol. Cell Biol. 2007, 8, 49–62. [Google Scholar] [CrossRef]
- Amit, S.; Hatzubai, A.; Birman, Y.; Andersen, J.S.; Ben-Shushan, E.; Mann, M.; Ben-Neriah, Y.; Alkalay, I. Axin-mediated CKI phosphorylation of β-catenin at Ser 45: A molecular switch for the Wnt pathway. Genes Dev. 2002, 16, 1066–1076. [Google Scholar] [CrossRef] [Green Version]
- Gao, Z.H.; Seeling, J.M.; Hill, V.; Yochum, A.; Virshup, D.M. Casein kinase I phosphorylates and destabilizes the β-catenin degradation complex. Proc. Natl. Acad. Sci. USA 2002, 99, 1182–1187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knippschild, U.; Wolff, S.; Giamas, G.; Brockschmidt, C.; Wittau, M.; Würl, P.U.; Eismann, T.; Stöter, M. The Role of the Casein Kinase 1 (CK1) Family in Different Signaling Pathways Linked to Cancer Development. Oncol. Res. Treat. 2005, 28, 508–514. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elstrom, R.L.; Bauer, D.E.; Buzzai, M.; Karnauskas, R.; Harris, M.H.; Plas, D.R.; Zhuang, H.; Cinalli, R.M.; Alavi, A.; Rudin, C.M.; et al. Akt stimulates aerobic glycolysis in cancer cells. Cancer Res. 2004, 64, 3892–3899. [Google Scholar] [CrossRef] [Green Version]
- Babic, I.; Anderson, E.S.; Tanaka, K.; Guo, D.; Masui, K.; Li, B.; Zhu, S.; Gu, Y.; Villa, G.R.; Akhavan, D.; et al. EGFR mutation-induced alternative splicing of max contributes to growth of glycolytic tumors in brain cancer. Cell Metab. 2013, 17, 1000–1008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Cheng, K.; Walton, Z.; Wang, Y.; Ebi, H.; Shimamura, T.; Liu, Y.; Tupper, T.; Ouyang, J.; Li, J.; et al. A murine lung cancer co-clinical trial identifies genetic modifiers of therapeutic response. Nature 2012, 483, 613–617. [Google Scholar] [CrossRef]
- Sharma, S.V.; Lee, D.Y.; Li, B.; Quinlan, M.P.; Takahashi, F.; Maheswaran, S.; McDermott, U.; Azizian, N.; Zou, L.; Fischbach, M.A.; et al. A Chromatin-Mediated Reversible Drug-Tolerant State in Cancer Cell Subpopulations. Cell 2010, 141, 69–80. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.; Su, N.; Yang, J.; Tan, Q.; Huang, S.; Jin, M.; Ni, Z.; Zhang, B.; Zhang, D.; Luo, F.; et al. FGF/FGFR signaling in health and disease. Signal Transduct. Target. Ther. 2020, 5, 181. [Google Scholar] [CrossRef] [PubMed]
- Azuma, K.; Kawahara, A.; Sonoda, K.; Nakashima, K.; Tashiro, K.; Watari, K.; Izumi, H.; Kage, M.; Kuwano, M.; Ono, M.; et al. FGFR1 activation is an escape mechanism in human lung cancer cells resistant to afatinib, a pan-EGFR family kinase inhibitor. Oncotarget 2014, 5, 5908–5919. [Google Scholar] [CrossRef] [Green Version]
- Terai, H.; Soejima, K.; Yasuda, H.; Nakayama, S.; Hamamoto, J.; Arai, D.; Ishioka, K.; Ohgino, K.; Ikemura, S.; Sato, T.; et al. Activation of the FGF2-FGFR1 autocrine pathway: A novel mechanism of acquired resistance to gefitinib in NSCLC. Mol. Cancer Res. 2013, 11, 759–767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ware, K.E.; Hinz, T.K.; Kleczko, E.; Singleton, K.R.; Marek, L.A.; Helfrich, B.A.; Cummings, C.T.; Graham, D.K.; Astling, D.; Tan, A.C.; et al. A mechanism of resistance to gefitinib mediated by cellular reprogramming and the acquisition of an FGF2-FGFR1 autocrine growth loop. Oncogenesis 2013, 2, e39. [Google Scholar] [CrossRef] [Green Version]
- Fu, J.; Bian, M.; Jiang, Q.; Zhang, C. Roles of aurora kinases in mitosis and tumorigenesis. Mol. Cancer Res. 2007, 5, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Blakely, C.M.; Gubens, M.A.; Allen, G.M.; Shah, S.; Jereza, M.; Bacaltos, B.; Bandyopadhyay, S. Phase I study of the aurora kinase A inhibitor alisertib in combination with osimertinib in EGFR-mutant lung cancer. J. Clin. Oncol. 2021, 39, 9074. [Google Scholar] [CrossRef]
- Knox, C.; Law, V.; Jewison, T.; Liu, P.; Ly, S.; Frolkis, A.; Pon, A.; Banco, K.; Mak, C.; Neveu, V.; et al. DrugBank 3.0: A comprehensive resource for “Omics” research on drugs. Nucleic Acids Res. 2011, 39 (Suppl. 1), D1035–D1041. [Google Scholar] [CrossRef] [Green Version]
- Hernandez-Boussard, T.; Whirl-Carrillo, M.; Hebert, J.M.; Gong, L.; Owen, R.; Gong, M.; Gor, W.; Liu, F.; Truong, C.; Whaley, R.; et al. The pharmacogenetics and pharmacogenomics knowledge base: Accentuating the knowledge. Nucleic Acids Res. 2008, 36 (Suppl. 1), D913–D918. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, F.; Shi, Z.; Qin, C.; Tao, L.; Liu, X.; Xu, F.; Zhang, L.; Song, Y.; Liu, X.; Zhang, J.; et al. Therapeutic target database update 2012: A resource for facilitating target-oriented drug discovery. Nucleic Acids Res. 2012, 40, D1128–D1136. [Google Scholar] [CrossRef] [PubMed]
- Hamosh, A.; Scott, A.F.; Amberger, J.S.; Bocchini, C.A.; McKusick, V.A. Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res. 2005, 33 (Suppl. 1), D514–D517. [Google Scholar] [CrossRef]
- Yu, W.; Gwinn, M.; Clyne, M.; Yesupriya, A.; Khoury, M.J. A navigator for human genome epidemiology. Nat. Genet. 2008, 40, 124–125. [Google Scholar] [CrossRef]
- Davis, A.P.; King, B.L.; Mockus, S.; Murphy, C.G.; Saraceni-Richards, C.; Rosenstein, M.; Wiegers, T.; Mattingly, C.J. The comparative toxicogenomics database: Update 2011. Nucleic Acids Res. 2011, 39 (Suppl. 1), D1067–D1072. [Google Scholar] [CrossRef] [Green Version]
- Lewis, R.J.; Angier, M.K.; Williamson, K.S.; Johnson, R.D. Analysis of sertraline in postmortem fluids and tissues in 11 aviation accident victims. J. Anal. Toxicol. 2013, 37, 208–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamb, J.; Crawford, E.D.; Peck, D.; Modell, J.W.; Blat, I.C.; Wrobel, M.J.; Lerner, J.; Brunet, J.P.; Subramanian, A.; Ross, K.N.; et al. The connectivity map: Using gene-expression signatures to connect small molecules, genes, and disease. Science 2006, 313, 1929–1935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakatani, K.; Yamaoka, T.; Ohba, M.; Fujita, K.I.; Arata, S.; Kusumoto, S.; Taki-Takemoto, I.; Kamei, D.; Iwai, S.; Tsurutani, J.; et al. KRAS and EGFR amplifications mediate resistance to rociletinib and osimertinib in acquired afatinib-resistant NSCLC harboring exon 19 deletion/T790M in EGFR. Mol. Cancer Ther. 2019, 18, 112–126. [Google Scholar] [CrossRef] [Green Version]
- Ooft, M.L.; Braunius, W.W.; Heus, P.; Stegeman, I.; Van Diest, P.J.; Grolman, W.; Zuur, C.I.; Willems, S.M. Prognostic significance of the EGFR pathway in nasopharyngeal carcinoma: A systematic review and meta-analysis. Biomark. Med. 2015, 9, 997–1010. [Google Scholar] [CrossRef]
- Kobayashi, A.; Kang, M.-I.; Okawa, H.; Ohtsuji, M.; Zenke, Y.; Chiba, T.; Igarashi, K.; Yamamoto, M. Oxidative Stress Sensor Keap1 Functions as an Adaptor for Cul3-Based E3 Ligase To Regulate Proteasomal Degradation of Nrf2. Mol. Cell. Biol. 2004, 24, 7130–7139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Xiong, X.; Sun, Y. Cullin-RING Ligase 5: Functional characterization and its role in human cancers. Semin. Cancer Biol. 2020, 67, 61–79. [Google Scholar] [CrossRef] [PubMed]
- Gerakis, Y.; Quintero, M.; Li, H.; Hetz, C. The UFMylation System in Proteostasis and Beyond. Trends Cell Biol. 2019, 29, 974–986. [Google Scholar] [CrossRef]
- Cubillos-Ruiz, J.R.; Bettigole, S.E.; Glimcher, L.H. Tumorigenic and Immunosuppressive Effects of Endoplasmic Reticulum Stress in Cancer. Cell 2017, 168, 692–706. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Liang, S.Q.; Xu, D.; Yang, Z.; Marti, T.M.; Gao, Y.; Kocher, G.J.; Zhao, H.; Schmid, R.A.; Peng, R.W. HSP90/AXL/eIF4E-regulated unfolded protein response as an acquired vulnerability in drug-resistant KRAS-mutant lung cancer. Oncogenesis 2019, 8, 45. [Google Scholar] [CrossRef] [Green Version]
- Inoue, Y.; Nikolic, A.; Farnsworth, D.; Shi, R.; Johnson, F.D.; Liu, A.; Ladanyi, M.; Somwar, R.; Gallo, M.; Lockwood, W.W. Extracellular signal-regulated kinase mediates chromatin rewiring and lineage transformation in lung cancer. Elife 2021, 10, e66524. [Google Scholar] [CrossRef] [PubMed]
- Xia, W.; Nagase, S.; Montia, A.G.; Kalachikov, S.M.; Keniry, M.; Su, T.; Memeo, L.; Hibshoosh, H.; Parsons, R. BAF180 is a critical regulator of p21 induction and a tumor suppressor mutated in breast cancer. Cancer Res. 2008, 68, 1667–1674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Politi, K.; Zakowski, M.F.; Fan, P.D.; Schonfeld, E.A.; Pao, W.; Varmus, H.E. Lung adenocarcinomas induced in mice by mutant EGF receptors found in human lung cancers respond to a tyrosine kinase inhibitor or to down-regulation of the receptors. Genes Dev. 2006, 20, 1496–1510. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.A.; Suzawa, K.; Jordan, E.; Zehir, A.; Ni, A.; Kim, R.; Kris, M.G.; Hellmann, M.D.; Li, B.T.; Somwar, R.; et al. Concurrent alterations in EGFR-mutant lung cancers associated with resistance to EGFR kinase inhibitors and characterization of MTOR as a mediator of resistance. Clin. Cancer Res. 2018, 24, 3108–3118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.; Xu, Y.; Zhao, J.; Zhong, W.; Zhang, L.; Bi, Y.; Wang, M. Concurrent Driver Gene Mutations as Negative Predictive Factors in Epidermal Growth Factor Receptor-Positive Non-Small Cell Lung Cancer. EBioMedicine 2019, 42, 304–310. [Google Scholar] [CrossRef] [Green Version]
- Zheng, C.; Li, X.; Ren, Y.; Yin, Z.; Zhou, B. Coexisting EGFR and TP53 Mutations in Lung Adenocarcinoma Patients Are Associated With COMP and ITGB8 Upregulation and Poor Prognosis. Front. Mol. Biosci. 2020, 7, 30. [Google Scholar] [CrossRef] [Green Version]
- Blakely, C.M.; Watkins, T.B.K.; Wu, W.; Gini, B.; Chabon, J.J.; McCoach, C.E.; McGranahan, N.; Wilson, G.A.; Birkbak, N.J.; Olivas, V.R.; et al. Evolution and clinical impact of co-occurring genetic alterations in advanced-stage EGFR-mutant lung cancers. Nat. Genet. 2017, 49, 1693–1704. [Google Scholar] [CrossRef]
- Eng, J.; Woo, K.M.; Sima, C.S.; Plodkowski, A.; Hellmann, M.D.; Chaft, J.E.; Kris, M.G.; Arcila, M.E.; Ladanyi, M.; Drilon, A. Impact of Concurrent PIK3CA Mutations on Response to EGFR Tyrosine Kinase Inhibition in EGFR-Mutant Lung Cancers and on Prognosis in Oncogene-Driven Lung Adenocarcinomas. J. Thorac. Oncol. 2015, 10, 1713–1719. [Google Scholar] [CrossRef] [Green Version]
- Thomas de Montpréville, V.; Lacroix, L.; Rouleau, E.; Mamodaly, M.; Leclerc, J.; Tutuianu, L.; Planchard, D.; Boulate, D.; Mercier, O.; Besse, B.; et al. Non-small cell lung carcinomas with CTNNB1 (beta-catenin) mutations: A clinicopathological study of 26 cases. Ann. Diagn. Pathol. 2020, 46, 151522. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Li, W.; Shao, J.; Zhao, J.; Chen, C. Analysis of the Clinicopathologic Characteristics of Lung Adenocarcinoma With CTNNB1 Mutation. Front. Genet. 2020, 10, 1367. [Google Scholar] [CrossRef] [PubMed]
- O’Farrell, H.; Harbourne, B.; Kurlawala, Z.; Inoue, Y.; Nagelberg, A.L.; Martinez, V.D.; Lu, D.; Oh, M.H.; Coe, B.P.; Thu, K.L.; et al. Integrative Genomic Analyses Identifies GGA2 as a Cooperative Driver of EGFR-Mediated Lung Tumorigenesis. J. Thorac. Oncol. 2019, 14, 656–671. [Google Scholar] [CrossRef] [Green Version]
- Chitale, D.; Gong, Y.; Taylor, B.S.; Broderick, S.; Brennan, C.; Somwar, R.; Golas, B.; Wang, L.; Motoi, N.; Szoke, J.; et al. An integrated genomic analysis of lung cancer reveals loss of DUSP4 in EGFR-mutant tumors. Oncogene 2009, 28, 2773–2783. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Zhang, J.; Berger, A.H.; Diolombi, M.S.; Ng, C.; Fung, J.; Bronson, R.T.; Castillo-Martin, M.; Thin, T.H.; Cordon-Cardo, C.; et al. Compound haploinsufficiency of Dok2 and Dusp4 promotes lung tumorigenesis. J. Clin. Investig. 2019, 129, 215–222. [Google Scholar] [CrossRef] [Green Version]
- Soh, J.; Okumura, N.; Lockwood, W.W.; Yamamoto, H.; Shigematsu, H.; Zhang, W.; Chari, R.; Shames, D.S.; Tang, X.; MacAulay, C.; et al. Oncogene mutations, copy number gains and mutant allele specific imbalance (MASI) frequently occur together in tumor cells. PLoS ONE 2009, 4, e7464. [Google Scholar] [CrossRef]
- Berger, A.H.; Chen, M.; Morotti, A.; Janas, J.A.; Niki, M.; Bronson, R.T.; Taylor, B.S.; Ladanyi, M.; Van Aelst, L.; Politi, K.; et al. DOK2 inhibits EGFR-mutated lung adenocarcinoma. PLoS ONE 2013, 8, e79526. [Google Scholar]
- Lou, Y.; Xu, J.; Zhang, Y.; Zhang, W.; Zhang, X.; Gu, P.; Zhong, H.; Wang, H.; Lu, J.; Han, B. Akt kinase LANCL2 functions as a key driver in EGFR-mutant lung adenocarcinoma tumorigenesis. Cell Death Dis. 2021, 12, 170. [Google Scholar] [CrossRef]
- Shanzhi, W.; Yiping, H.; Ling, H.; Jianming, Z.; Qiang, L. The relationship between TTF-1 expression and EGFR mutations in lung adenocarcinomas. PLoS ONE 2014, 9, e95479. [Google Scholar]
- Clarke, N.; Biscocho, J.; Kwei, K.A.; Davidson, J.M.; Sridhar, S.; Gong, X.; Pollack, J.R. Integrative genomics implicates EGFR as a downstream mediator in nkx2-1 amplified non-small cell lung cancer. PLoS ONE 2015, 10, e0142061. [Google Scholar]
- Sun, D.; Zhu, Y.; Zhu, J.; Tao, J.; Wei, X.; Wo, Y.; Hou, H. Primary resistance to first-generation EGFR-TKIs induced by MDM2 amplification in NSCLC. Mol. Med. 2020, 26, 66. [Google Scholar] [CrossRef] [PubMed]
- Skoulidis, F.; Heymach, J.V. Co-occurring genomic alterations in non-small-cell lung cancer biology and therapy. Nat. Rev. Cancer 2019, 19, 495–509. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.M.; Lockwood, W.W.; Buys, T.P.H.; Chari, R.; Coe, B.P.; Lam, S.; Lam, W.L. Integrative genomic and gene expression analysis of chromosome 7 identified novel oncogene loci in non-small cell lung cancer. Genome 2008, 51, 1032–1039. [Google Scholar] [CrossRef] [PubMed]
- Hou, H.; Sun, D.; Zhang, X. The role of MDM2 amplification and overexpression in therapeutic resistance of malignant tumors. Cancer Cell Int. 2019, 19, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Ekholm, S.V.; Reed, S.I. Regulation of G1 cyclin-dependent kinases in the mammalian cell cycle. Curr. Opin. Cell Biol. 2000, 12, 676–684. [Google Scholar] [CrossRef]
- Gini, B.; Thomas, N.; Blakely, C.M. Impact of concurrent genomic alterations in epidermal growth factor receptor (EGFR)-mutated lung cancer. J. Thorac. Dis. 2020, 12, 2883–2895. [Google Scholar] [CrossRef] [PubMed]
- Offin, M.; Chan, J.M.; Tenet, M.; Rizvi, H.A.; Shen, R.; Riely, G.J.; Rekhtman, N.; Daneshbod, Y.; Quintanal-Villalonga, A.; Penson, A.; et al. Concurrent RB1 and TP53 Alterations Define a Subset of EGFR-Mutant Lung Cancers at risk for Histologic Transformation and Inferior Clinical Outcomes. J. Thorac. Oncol. 2019, 14, 1784–1793. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Vogt, P.K. Class I PI3K in oncogenic cellular transformation. Oncogene 2008, 27, 5486–5496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakayama, S.; Sng, N.; Carretero, J.; Welner, R.; Hayashi, Y.; Yamamoto, M.; Tan, A.J.; Yamaguchi, N.; Yasuda, H.; Li, D.; et al. β-catenin contributes to lung tumor development induced by EGFR mutations. Cancer Res. 2014, 74, 5891–5902. [Google Scholar] [CrossRef] [Green Version]
- Ratner, N.; Miller, S.J. A RASopathy gene commonly mutated in cancer: The neurofibromatosis type 1 tumour suppressor. Nat. Rev. Cancer 2015, 15, 290–301. [Google Scholar] [CrossRef]
- de Bruin, E.C.; Cowell, C.; Warne, P.H.; Jiang, M.; Saunders, R.E.; Melnick, M.A.; Gettinger, S.; Walther, Z.; Wurtz, A.; Heynen, G.J.; et al. Reduced NF1 expression confers resistance to EGFR inhibition in lung cancer. Cancer Discov. 2014, 4, 606–619. [Google Scholar] [CrossRef] [Green Version]
- Weinstein, I.B. Cancer: Addiction to oncogenes—The Achilles heal of cancer. Science 2002, 297, 63–64. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, I.B.; Joe, A. Oncogene addiction. Cancer Res. 2008, 68, 3077–3080. [Google Scholar] [CrossRef] [Green Version]
- Sequist, L.V.; Han, J.Y.; Ahn, M.J.; Cho, B.C.; Yu, H.; Kim, S.W.; Yang, J.C.H.; Lee, J.S.; Su, W.C.; Kowalski, D.; et al. Osimertinib plus savolitinib in patients with EGFR mutation-positive, MET-amplified, non-small-cell lung cancer after progression on EGFR tyrosine kinase inhibitors: Interim results from a multicentre, open-label, phase 1b study. Lancet Oncol. 2020, 21, 373–386. [Google Scholar] [CrossRef]
- Wang, D.; Tai, P.W.L.; Gao, G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat. Rev. Drug Discov. 2019, 18, 358–378. [Google Scholar] [CrossRef]
- Ohta, T.; Iijima, K.; Miyamoto, M.; Nakahara, I.; Tanaka, H.; Ohtsuji, M.; Suzuki, T.; Kobayashi, A.; Yokota, J.; Sakiyama, T.; et al. Loss of Keap1 function activates Nrf2 and provides advantages for lung cancer cell growth. Cancer Res. 2008, 68, 1303–1309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rojo de la Vega, M.; Chapman, E.; Zhang, D.D. NRF2 and the Hallmarks of Cancer. Cancer Cell 2018, 34, 21–43. [Google Scholar] [CrossRef]
- Johnson, F.D.; Ferrarone, J.; Liu, A.; Brandstädter, C.; Munuganti, R.; Farnsworth, D.; Lu, D.; Luu, J.; Sihota, T.; Jansen, S.; et al. A novel small molecule that induces cytotoxicity in lung cancer cells inhibits disulfide reductases GSR and TXNRD1. bioRxiv 2021. [Google Scholar]
- Romero, R.; Sayin, V.I.; Davidson, S.M.; Bauer, M.R.; Singh, S.X.; Leboeuf, S.E.; Karakousi, T.R.; Ellis, D.C.; Bhutkar, A.; Sánchez-Rivera, F.J.; et al. Keap1 loss promotes Kras-driven lung cancer and results in dependence on glutaminolysis. Nat. Med. 2017, 23, 1362–1368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nie, W.; Gan, L.; Wang, X.; Gu, K.; Qian, F.F.; Hu, M.J.; Zhang, D.; Chen, S.Q.; Lu, J.; Cao, S.H.; et al. Atezolizumab prolongs overall survival over docetaxel in advanced non-small-cell lung cancer patients harboring STK11 or KEAP1 mutation. Oncoimmunology 2021, 10, 1865670. [Google Scholar] [CrossRef]
- Hata, A.N.; Niederst, M.J.; Archibald, H.L.; Gomez-Caraballo, M.; Siddiqui, F.M.; Mulvey, H.E.; Maruvka, Y.E.; Ji, F.; Bhang, H.E.C.; Radhakrishna, V.K.; et al. Tumor cells can follow distinct evolutionary paths to become resistant to epidermal growth factor receptor inhibition. Nat. Med. 2016, 22, 262–269. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Hits | Screening Methodology | Model | Drug Used | Year | Reference |
|---|---|---|---|---|---|
| miR-5693, miR-3618, and m+B5:G22iR-432-5p | 2019 mature microRNAs | EKVX, H322M | Erlotinib | 2021 | [41] |
| MAPK and AKT pathway | 17,255 ORFs, covering 12,728 genes and 35 mutant oncogenes | PC9 | Erlotinib, Osimertinib, Trametinib | 2020 | [42] |
| GRB2 | CMap analysis | PC9, HCC827 | Icotunib | 2020 | [43] |
| RIC8A, LPAR2, YAP1, ARIH2, KEAP1 | Whole genome CRISPR Cas9 screen (18,360 genes) | HCC827 | Erlotinib | 2019 | [44] |
| CPS1 | shRNAs targeting rate-limiting metabolic enzymes | PC9, HCC4006, H1650, H322C, PC9, PC9-EGFR T790M | Erlotinib | 2019 | [45] |
| FGFR | Whole genome CRISPR Cas9 screen (20,000 genes) | Patient Derived Cells | Nazartinib | 2019 | [46] |
| Aurora kinase A | 94-compound cancer-focused drug library | H1975 | Rociletinib | 2019 | [47] |
| Ufmylation pathway | Whole genome CRISPR Cas9 screen (18,454 genes) | PC9 | Erlotinib, THZ1 | 2018 | [48] |
| Sertraline | Comprehensive drug-gene interactions profile | H522, A549, H1975, PC9 | Erlotinib | 2018 | [49] |
| KEAP1 | CRISPR-Cas9 KO targeted screens | HCC827 | Erlotinib | 2017 | [50] |
| CIC, SWI/SNF | CRISPR and shRNA library (10 gRNAs or shRNAs per gene) targeting 500 potential tumor suppressors | PC9 | Gefitinib | 2017 | [51] |
| YAP1 | shRNA screen (~60,000 individual shRNAs) | PC9 | Cisplatin | 2016 | [52] |
| CK1α | shRNA screen against about 350 potentially cancer-relevant genes (~6500 shRNAs) | HCC827, HCC4006 and PC9 | Erlotinib | 2015 | [53] |
| Bosutinib | 3700 shRNAs targeting ~600 kinases | H1650 | Gefitinib | 2014 | [54] |
| CRKL, SRC, RAF1, FRK, BLK, and HCK | 589 ORFs encoding kinases and kinase related proteins | PC9 | Erlotinib | 2014 | [55] |
| Wnt/β-catenin pathway, tankyrase | SBI shRNA library | H322C, HCC4006 | Gefitinib | 2013 | [56] |
| NF-κB | shRNA screen targeting >2000 cancer-relevant genes | H1650 | Erlotinib | 2011 | [57] |
| Gene Name | Symbol | Chromosome | Alteration Type | Frequency | Pathway | Reference |
|---|---|---|---|---|---|---|
| Tumour protein p53 | TP53 | 17p | Deleterious mutation | 51–60% | P53 | [102,103,104] |
| RB transcriptional corepressor 1 | RB1 | 13q | Deleterious mutation | 10–12% | RB/E2F (G1/S) | [102,103,105] |
| Neurofibromatosis type 1 | NF1 | 17q | Deleterious mutation | 9.40% | Ras | [105] |
| Phosphatidylinositol-4,5-biphosphate 3-kinase catalytic subunit alpha | PIK3CA | 3q | Activating mutation | 12% | PI3K-AKT | [102,106] |
| Beta catenin 1 | CTNNB1 | 3p | Activating mutation | 9% | WNT/β-catenin | [102,105,107,108] |
| Golgi associated, gamma adaptin ear containing, ARF binding protein 2 | GGA2 | 16p | Gain/Amp | 59% | EGFR | [109] |
| Dual specificity phosphatase 4 | DUSP4 | 8p | Loss/Deletion | 49% * | MAPK/ERK | [110,111] |
| Epidermal growth factor receptor | EGFR | 7p | Gain/Amp | 59% | EGFR | [112] |
| Docking protein 2 | DOK2 | 8p | Loss/Deletion | 48–63% | MAPK/ERK | [111,113] |
| LanC like 2 | LANCL2 | 7p | Gain/Amp | 37% ** | Akt phosphorylation | [114] |
| NK2 Homeobox 1 | NKX2-1 | 14q | Gain/Amp | 15% | Regulates P53 transcription | [102,103,115,116] |
| Mouse Double Minute 2 | MDM2 | 12q | Gain/Amp | 12% | MDM2-p53 | [102,117] |
| Cyclin dependent kinase 4 | CDK4 | 12q | Gain/Amp | 10% | CDK4/6 (G1/S) | [102,105,118] |
| Cyclin dependent kinase 6 | CDK6 | 7q | Gain/Amp | 7% | G1/S | [105,118] |
| Cyclin E1 | CCNE1 | 19q | Gain/Amp | 6.90% | G1/S | [105] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Farnsworth, D.A.; Chen, Y.T.; de Rappard Yuswack, G.; Lockwood, W.W. Emerging Molecular Dependencies of Mutant EGFR-Driven Non-Small Cell Lung Cancer. Cells 2021, 10, 3553. https://doi.org/10.3390/cells10123553
Farnsworth DA, Chen YT, de Rappard Yuswack G, Lockwood WW. Emerging Molecular Dependencies of Mutant EGFR-Driven Non-Small Cell Lung Cancer. Cells. 2021; 10(12):3553. https://doi.org/10.3390/cells10123553
Chicago/Turabian StyleFarnsworth, Dylan A., Yankuan T. Chen, Georgia de Rappard Yuswack, and William W. Lockwood. 2021. "Emerging Molecular Dependencies of Mutant EGFR-Driven Non-Small Cell Lung Cancer" Cells 10, no. 12: 3553. https://doi.org/10.3390/cells10123553
APA StyleFarnsworth, D. A., Chen, Y. T., de Rappard Yuswack, G., & Lockwood, W. W. (2021). Emerging Molecular Dependencies of Mutant EGFR-Driven Non-Small Cell Lung Cancer. Cells, 10(12), 3553. https://doi.org/10.3390/cells10123553