
Defucosylated Mouse–Dog Chimeric Anti-EGFR Antibody Exerts Antitumor Activities in Mouse Xenograft Models of Canine Tumors

 , , , ,
, , , , 
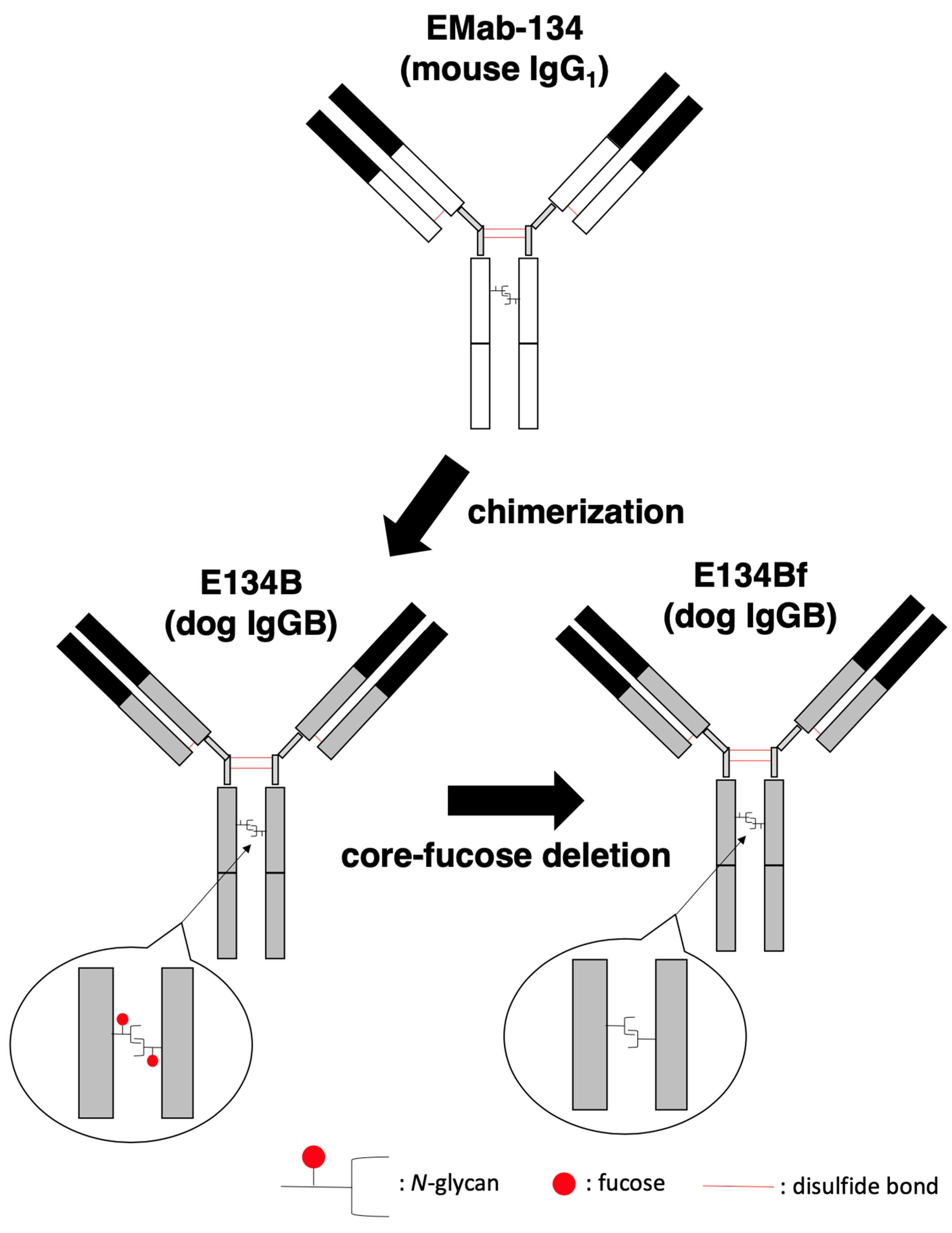{kind=link}
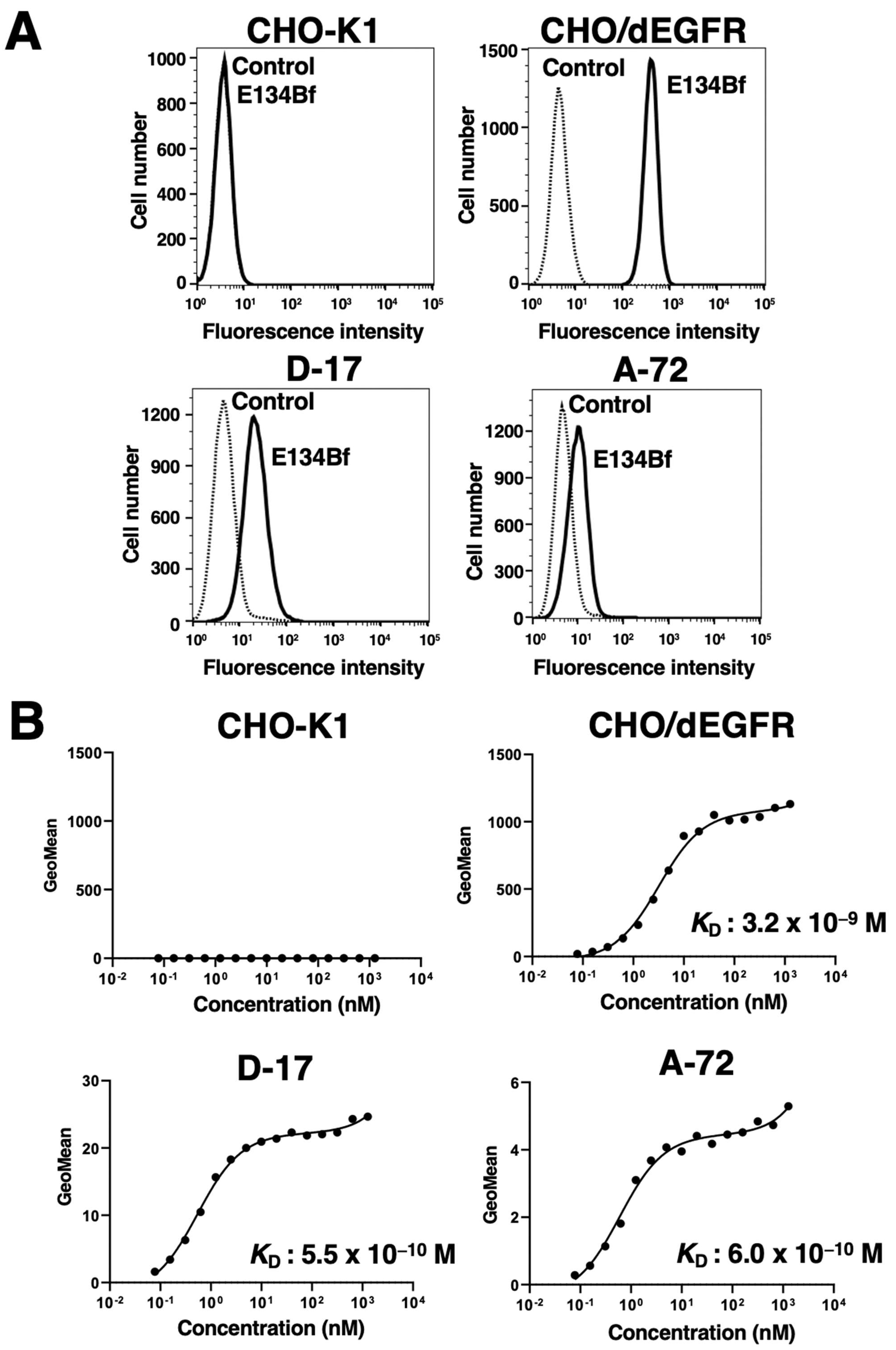{kind=link}
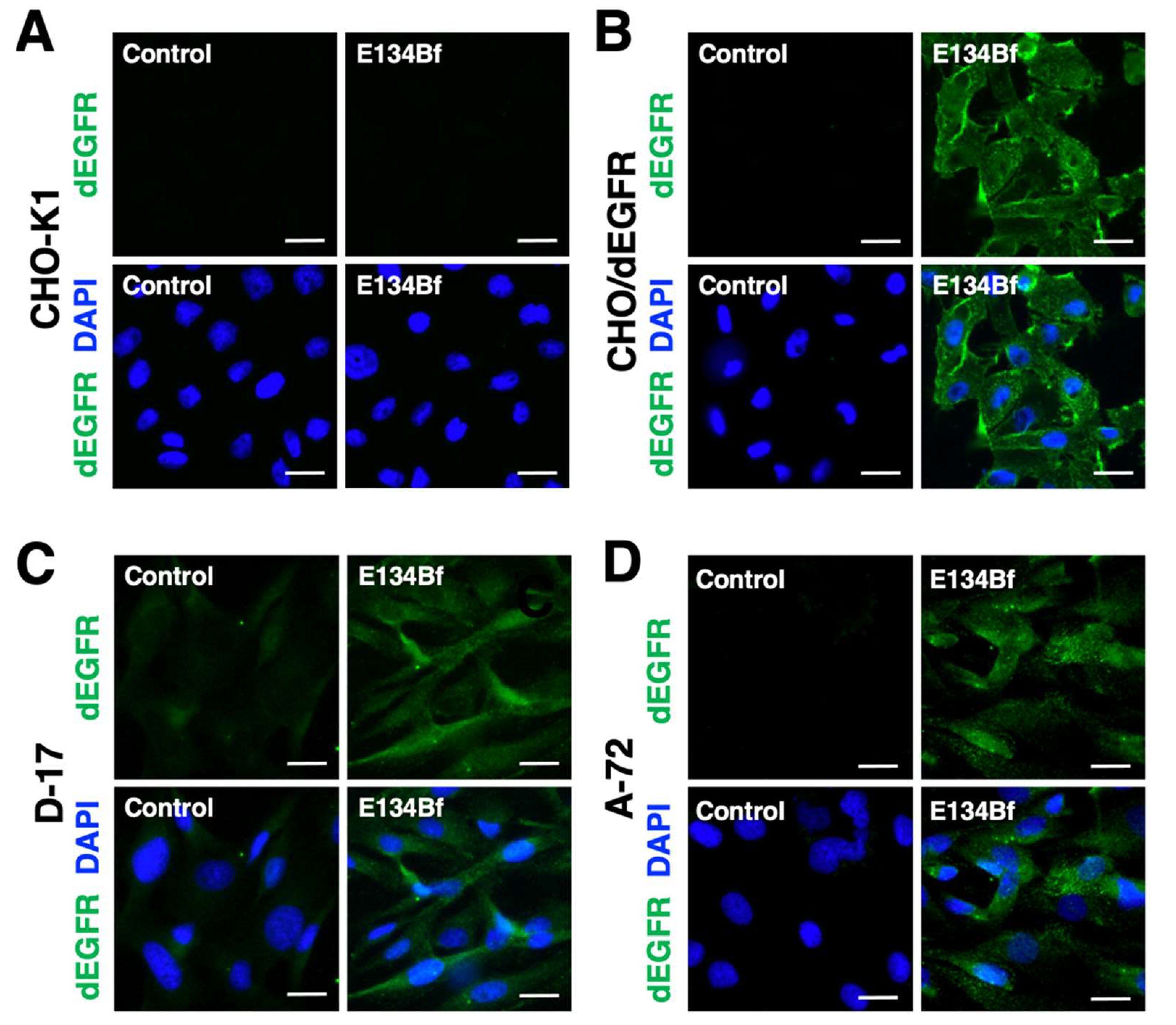{kind=link}
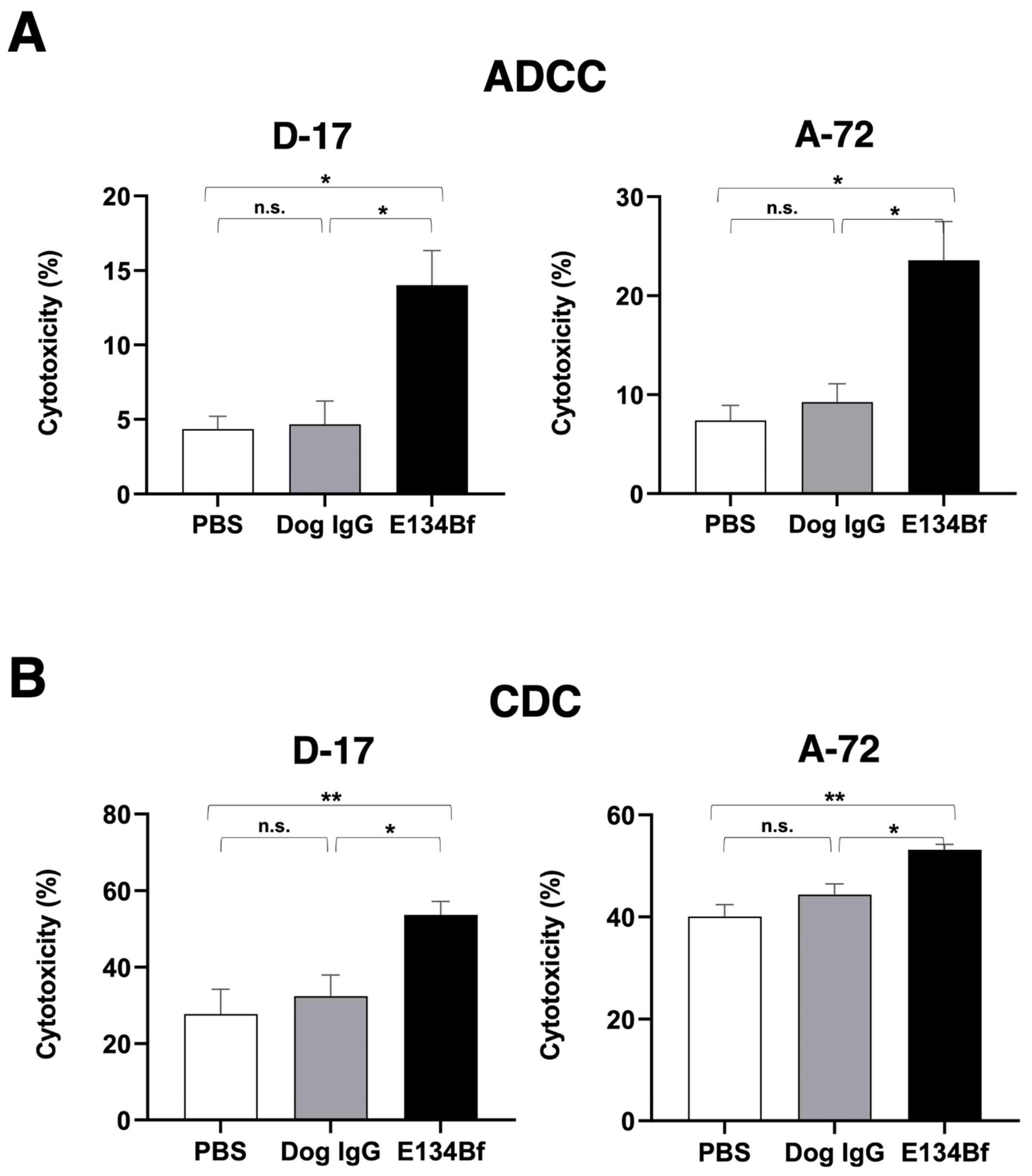{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Animals
2.3. Antibodies
2.4. Flow Cytometry
2.5. Determination of Binding Affinity
2.6. Immunocytochemistry
2.7. ADCC
2.8. CDC
2.9. Influence of E134Bf on EGF-Stimulated Cell Growth
2.10. Antitumor Activity of E134Bf in Xenografts of D-17 and A-72 Cells
2.11. Immunohistochemistry
2.12. Statistical Analyses
3. Results
3.1. Flow Cytometry Analysis against D-17 and A-17 Cells Using E134Bf
3.2. Immunocytochemistry against the D-17 and A-72 Cells Using E134Bf
3.3. E134Bf-Mediated ADCC and CDC in the D-17 and A-72 Cells
3.4. E134Bf Did Not Inhibit the EGF-Stimulated Cell Growth of A-72
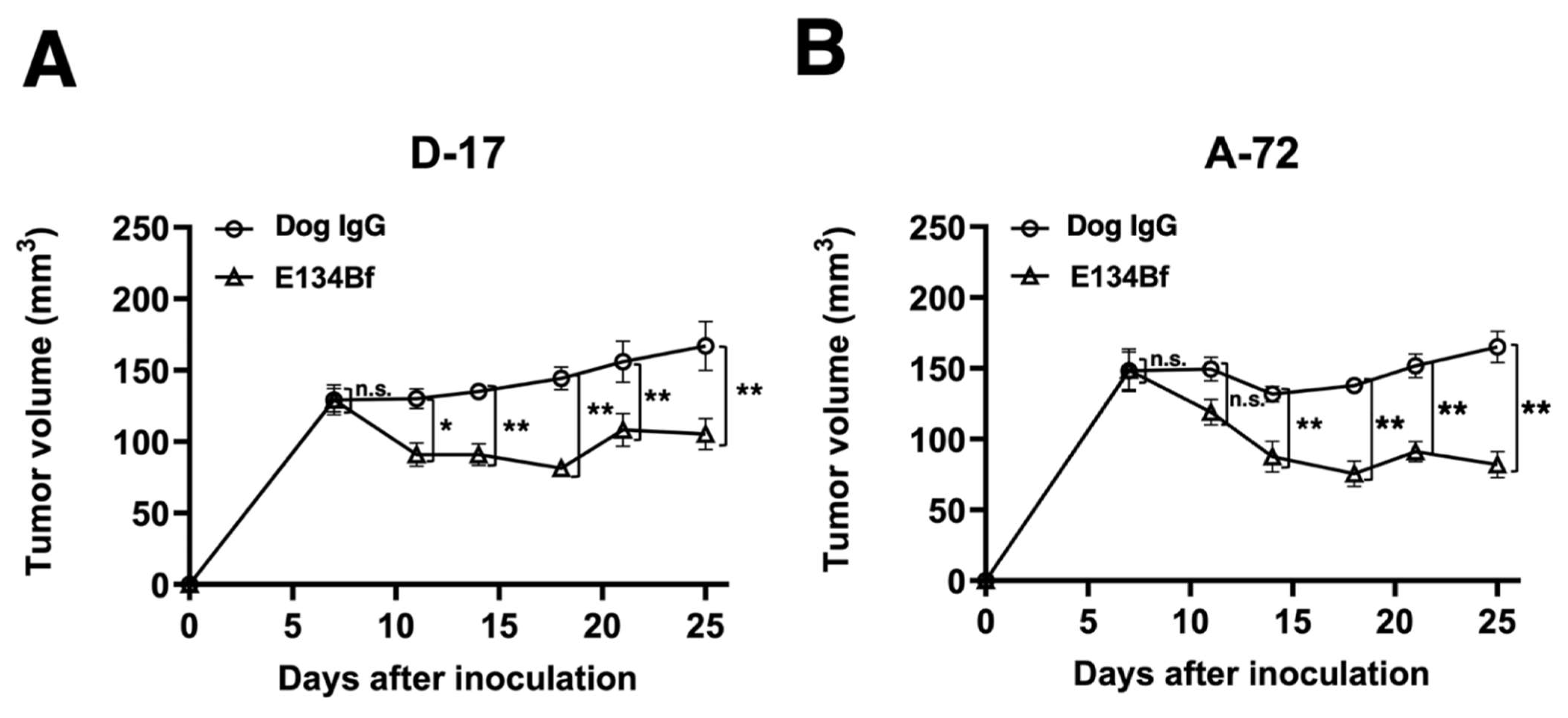
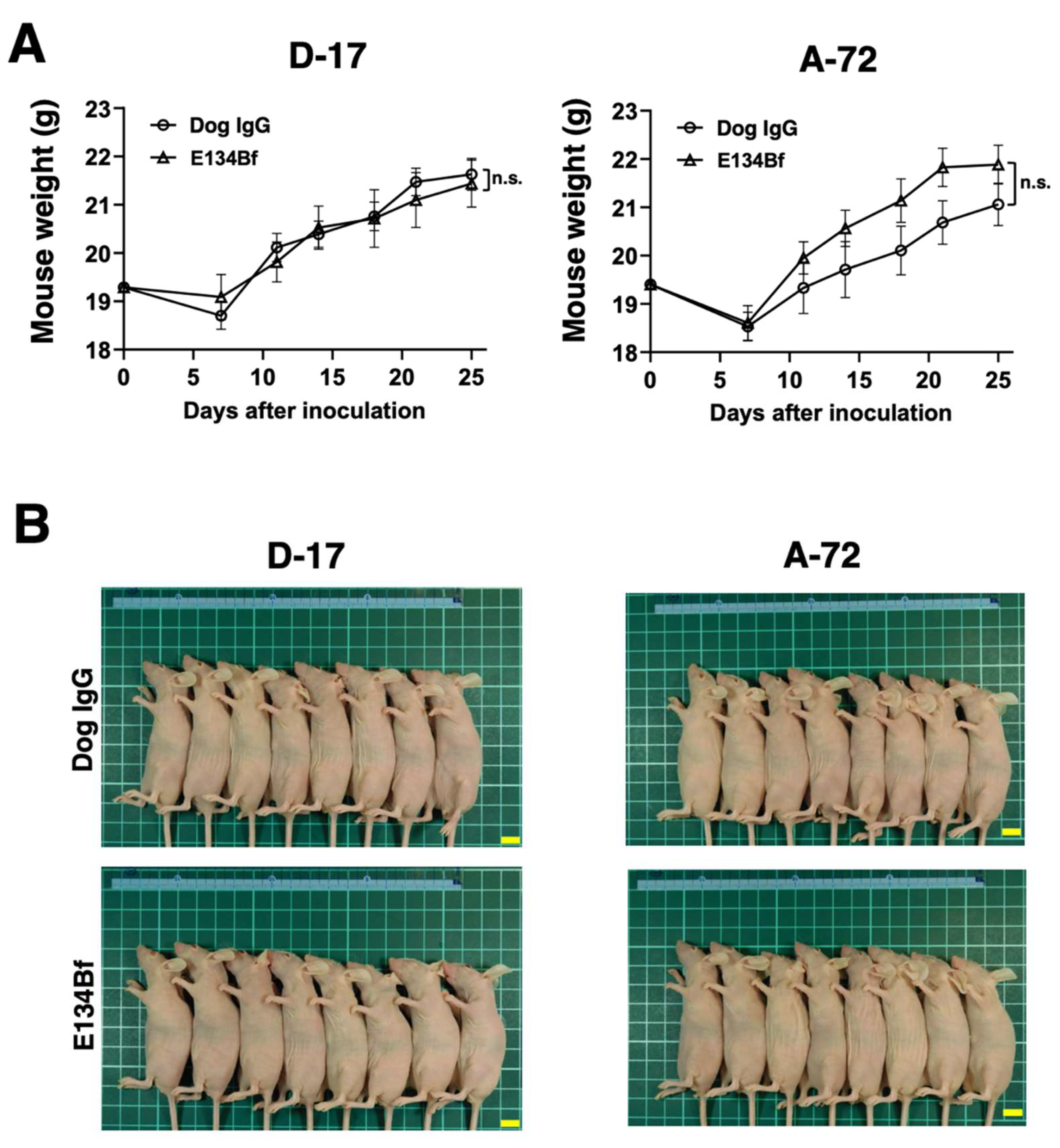
3.5. Antitumor Activities of E134Bf in the Mouse Xenografts of D-17 and A-72 Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Viola, P. The biology of Epidermal Growth Factor Receptor (EGFR) from regulating cell cycle to promoting carcinogenesis: The state of art including treatment options. Ann. Cytol. Pathol. 2020, 5, 048–053. [Google Scholar]
- Lemmon, M.A.; Schlessinger, J.; Ferguson, K.M. The EGFR Family: Not So Prototypical Receptor Tyrosine Kinases. Cold Spring Harb. Perspect. Biol. 2014, 6, a020768. [Google Scholar] [CrossRef] [PubMed]
- Wee, P.; Wang, Z. Epidermal Growth Factor Receptor Cell Proliferation Signaling Pathways. Cancers 2017, 9, 52. [Google Scholar] [CrossRef] [PubMed]
- Bethune, G.; Bethune, D.; Ridgway, N.; Xu, Z. Epidermal growth factor receptor (EGFR) in lung cancer: An overview and update. J. Thorac. Dis. 2010, 2, 48–51. [Google Scholar]
- Gonzalez-Conchas, G.A.; Rodriguez-Romo, L.; Hernandez-Barajas, D.; Gonzalez-Guerrero, J.F.; Rodriguez-Fernandez, I.A.; Verdines-Perez, A.; Templeton, A.J.; Ocana, A.; Seruga, B.; Tannock, I.F.; et al. Epidermal growth factor receptor overexpression and outcomes in early breast cancer: A systematic review and a meta-analysis. Cancer Treat. Rev. 2018, 62, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Spano, J.P.; Fagard, R.; Soria, J.-C.; Rixe, O.; Khayat, D.; Milano, G. Epidermal growth factor receptor signaling in colorectal cancer: Preclinical data and therapeutic perspectives. Ann. Oncol. 2005, 16, 189–194. [Google Scholar] [CrossRef]
- Xu, H.; Zong, H.; Ma, C.; Ming, S.; Shang, M.; Hailiang, Z.; He, X.; Du, H.; Cao, L. Epidermal growth factor receptor in glioblastoma. Oncol. Lett. 2017, 14, 512–516. [Google Scholar] [CrossRef]
- Wen, Y.H.; Koeppen, H.; Garcia, R.; Chiriboga, L.; Tarlow, B.; Peters, B.; Eigenbrot, C.; Yee, H.; Steiner, G.; Greco, M.A. Epidermal growth factor receptor in osteosarcoma: Expression and mutational analysis. Hum. Pathol. 2007, 38, 1184–1191. [Google Scholar] [CrossRef]
- Kersting, C.; Gebert, C.; Agelopoulos, K.; Schmidt, H.; van Diest, P.J.; Juergens, H.; Winkelmann, W.; Kevric, M.; Gosheger, G.; Brandt, B.; et al. Epidermal Growth Factor Receptor Expression in High-Grade Osteosarcomas Is Associated with a Good Clinical Outcome. Clin. Cancer Res. 2007, 13, 2998–3005. [Google Scholar] [CrossRef] [PubMed]
- Szewczyk, M.; Lechowski, R.; Zabielska, K. What do we know about canine osteosarcoma treatment?—Review. Vet. Res. Commun. 2014, 39, 61–67. [Google Scholar] [CrossRef]
- Simpson, S.; Dunning, M.D.; de Brot, S.; Grau-Roma, L.; Mongan, N.P.; Rutland, C.S. Comparative review of human and canine osteosarcoma: Morphology, epidemiology, prognosis, treatment and genetics. Acta Vet. Scand. 2017, 59, 71. [Google Scholar] [CrossRef]
- Boston, S.E.; Ehrhart, N.P.; Dernell, W.S.; Lafferty, M.; Withrow, S.J. Evaluation of survival time in dogs with stage III osteosarcoma that undergo treatment: 90 cases (1985–2004). J. Am. Vet. Med. Assoc. 2006, 228, 1905–1908. [Google Scholar] [CrossRef]
- Selmic, L.; Burton, J.; Thamm, D.; Withrow, S.; Lana, S. Comparison of Carboplatin and Doxorubicin-Based Chemotherapy Protocols in 470 Dogs after Amputation for Treatment of Appendicular Osteosarcoma. J. Vet. Intern. Med. 2014, 28, 554–563. [Google Scholar] [CrossRef] [PubMed]
- Itai, S.; Yamada, S.; Kaneko, M.K.; Chang, Y.-W.; Harada, H.; Kato, Y. Establishment of EMab-134, a Sensitive and Specific Anti-Epidermal Growth Factor Receptor Monoclonal Antibody for Detecting Squamous Cell Carcinoma Cells of the Oral Cavity. Monoclon. Antibodies Immunodiagn. Immunother. 2017, 36, 272–281. [Google Scholar] [CrossRef]
- Hosono, H.; Takei, J.; Ohishi, T.; Sano, M.; Asano, T.; Sayama, Y.; Nakamura, T.; Yanaka, M.; Kawada, M.; Harada, H.; et al. Anti-EGFR monoclonal antibody 134-mG2a exerts antitumor effects in mouse xenograft models of oral squamous cell carcinoma. Int. J. Mol. Med. 2020, 46, 1443–1452. [Google Scholar] [CrossRef]
- Tateyama, N.; Nanamiya, R.; Ohishi, T.; Takei, J.; Nakamura, T.; Yanaka, M.; Hosono, H.; Saito, M.; Asano, T.; Tanaka, T.; et al. Defucosylated Anti-Epidermal Growth Factor Receptor Monoclonal Antibody 134-mG2a-f Exerts Antitumor Activities in Mouse Xenograft Models of Dog Epidermal Growth Factor Receptor-Overexpressed Cells. Monoclon. Antibodies Immunodiagn. Immunother. 2021, 40, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Riggs, J.L.; McAllister, R.M.; Lennette, E.H. Immunofluorescent Studies of RD-114 Virus Replication in Cell Culture. J. Gen. Virol. 1974, 25, 21–29. [Google Scholar] [CrossRef]
- Binn, L.N.; Marchwicki, R.H.; Stephenson, E.H. Establishment of a canine cell line: Derivation, characterization, and viral spectrum. Am. J. Vet. Res. 1980, 41, 855–860. [Google Scholar] [PubMed]
- Tateyama, N.; Asano, T.; Ohishi, T.; Takei, J.; Hosono, H.; Nanamiya, R.; Tanaka, T.; Sano, M.; Saito, M.; Kawada, M.; et al. An Anti-HER2 Monoclonal Antibody H2Mab-41 Exerts Antitumor Activities in Mouse Xenograft Model Using Dog HER2-Overexpressed Cells. Monoclon. Antibodies Immunodiagn. Immunother. 2021, 40, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Asano, T.; Ohishi, T.; Takei, J.; Nakamura, T.; Nanamiya, R.; Hosono, H.; Tanaka, T.; Sano, M.; Harada, H.; Kawada, M.; et al. Anti-HER3 monoclonal antibody exerts antitumor activity in a mouse model of colorectal adenocarcinoma. Oncol. Rep. 2021, 46, 1–10. [Google Scholar] [CrossRef]
- Tanaka, T.; Ohishi, T.; Asano, T.; Takei, J.; Nanamiya, R.; Hosono, H.; Sano, M.; Harada, H.; Kawada, M.; Kaneko, M.K.; et al. An anti-TROP2 monoclonal antibody TrMab-6 exerts antitumor activity in breast cancer mouse xenograft models. Oncol. Rep. 2021, 46, 1–10. [Google Scholar] [CrossRef]
- Hosono, H.; Ohishi, T.; Takei, J.; Asano, T.; Sayama, Y.; Kawada, M.; Kaneko, M.K.; Kato, Y. The anti-epithelial cell adhesion molecule (EpCAM) monoclonal antibody EpMab-16 exerts antitumor activity in a mouse model of colorectal adenocarcinoma. Oncol. Lett. 2020, 20, 383. [Google Scholar] [CrossRef]
- Kaneko, M.K.; Ohishi, T.; Takei, J.; Sano, M.; Nakamura, T.; Hosono, H.; Yanaka, M.; Asano, T.; Sayama, Y.; Harada, H.; et al. Anti-EpCAM monoclonal antibody exerts antitumor activity against oral squamous cell carcinomas. Oncol. Rep. 2020, 44, 2517–2526. [Google Scholar] [CrossRef]
- Kaneko, M.K.; Ohishi, T.; Kawada, M.; Kato, Y. A cancer-specific anti-podocalyxin monoclonal antibody (60-mG2a-f) exerts antitumor effects in mouse xenograft models of pancreatic carcinoma. Biochem. Biophys. Rep. 2020, 24, 100826. [Google Scholar] [CrossRef]
- Kaneko, M.K.; Ohishi, T.; Nakamura, T.; Inoue, H.; Takei, J.; Sano, M.; Asano, T.; Sayama, Y.; Hosono, H.; Suzuki, H.; et al. Development of Core-Fucose-Deficient Humanized and Chimeric Anti-Human Podoplanin Antibodies. Monoclon. Antibodies Immunodiagn. Immunother. 2020, 39, 167–174. [Google Scholar] [CrossRef]
- Takei, J.; Kaneko, M.K.; Ohishi, T.; Hosono, H.; Nakamura, T.; Yanaka, M.; Sano, M.; Asano, T.; Sayama, Y.; Kawada, M.; et al. A defucosylated anti-CD44 monoclonal antibody 5-mG2a-f exerts antitumor effects in mouse xenograft models of oral squamous cell carcinoma. Oncol. Rep. 2020, 44, 1949–1960. [Google Scholar] [CrossRef] [PubMed]
- Takei, J.; Kaneko, M.K.; Ohishi, T.; Kawada, M.; Harada, H.; Kato, Y. H2Mab-19, an anti-human epidermal growth factor receptor 2 monoclonal antibody exerts antitumor activity in mouse oral cancer xenografts. Exp. Ther. Med. 2020, 20, 846–853. [Google Scholar] [CrossRef] [PubMed]
- Takei, J.; Kaneko, M.K.; Ohishi, T.; Kawada, M.; Harada, H.; Kato, Y. A novel anti-EGFR monoclonal antibody (EMab-17) exerts antitumor activity against oral squamous cell carcinomas via antibody-dependent cellular cytotoxicity and complement-dependent cytotoxicity. Oncol. Lett. 2020, 19, 2809–2816. [Google Scholar] [CrossRef] [PubMed]
- Itai, S.; Ohishi, T.; Kaneko, M.K.; Yamada, S.; Abe, S.; Nakamura, T.; Yanaka, M.; Chang, Y.-W.; Ohba, S.-I.; Nishioka, Y.; et al. Anti-podocalyxin antibody exerts antitumor effects via antibody-dependent cellular cytotoxicity in mouse xenograft models of oral squamous cell carcinoma. Oncotarget 2018, 9, 22480–22497. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ohishi, T.; Kato, Y.; Kaneko, M.K.; Ohba, S.-I.; Inoue, H.; Harakawa, A.; Kawada, M. Anti-Metastatic Activity of an Anti-EGFR Monoclonal Antibody against Metastatic Colorectal Cancer with KRAS p.G13D Mutation. Int. J. Mol. Sci. 2020, 21, 6037. [Google Scholar] [CrossRef]
- Adams, V.; Evans, K.M.; Sampson, J.; Wood, J.L.N. Methods and mortality results of a health survey of purebred dogs in the UK. J. Small Anim. Pract. 2010, 51, 512–524. [Google Scholar] [CrossRef] [PubMed]
- Lewis, T.W.; Wiles, B.M.; Llewellyn-Zaidi, A.M.; Evans, K.M.; O’Neill, D.G. Longevity and mortality in Kennel Club registered dog breeds in the UK in 2014. Canine Genet. Epidemiol. 2018, 5, 10. [Google Scholar] [CrossRef]
- Phillips, B.; Powers, B.E.; Dernell, W.S.; Straw, R.C.; Khanna, C.; Hogge, G.S.; Vail, D.M. Use of Single-Agent Carboplatin as Adjuvant or Neoadjuvant Therapy in Conjunction With Amputation for Appendicular Osteosarcoma in Dogs. J. Am. Anim. Hosp. Assoc. 2009, 45, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Withrow, S.J.; Thrall, D.E.; Straw, R.C.; Powers, B.E.; Wrigley, R.H.; LaRue, S.M.; Page, R.L.; Richardson, D.C.; Bissonette, K.W.; Betts, C.W.; et al. Intra-arterial cisplatin with or without radiation in limb-sparing for canine osteosarcoma. Cancer 1993, 71, 2484–2490. [Google Scholar] [CrossRef]
- Berg, J.; Weinstein, M.J.; Springfield, D.S.; Rand, W.M. Results of surgery and doxorubicin chemotherapy in dogs with osteosarcoma. J. Am. Vet. Med. Assoc. 1995, 206, 1555–1560. [Google Scholar]
- Saam, D.E.; Liptak, J.; Stalker, M.J.; Chun, R. Predictors of outcome in dogs treated with adjuvant carboplatin for appendicular osteosarcoma: 65 cases (1996–2006). J. Am. Vet. Med. Assoc. 2011, 238, 195–206. [Google Scholar] [CrossRef]
- Berg, J. Canine osteosarcoma: Amputation and chemotherapy. Vet. Clin. N. Am. Small Anim. Pract. 1996, 26, 111–121. [Google Scholar] [CrossRef]
- Martano, M.; Iussich, S.; Morello, E.; Buracco, P. Canine oral fibrosarcoma: Changes in prognosis over the last 30 years? Vet. J. 2018, 241, 1–7. [Google Scholar] [CrossRef]
- Milovancev, M.; Helfand, S.C.; Marley, K.; Goodall, C.P.; Löhr, C.V.; Bracha, S. Antiproliferative effects of masitinib and imatinib against canine oral fibrosarcoma in vitro. BMC Vet. Res. 2016, 12, 85. [Google Scholar] [CrossRef]
- Beckman, R.A.; von Roemeling, R.; Scott, A.M. Monoclonal antibody dose determination and biodistribution into solid tumors. Ther. Deliv. 2011, 2, 333–344. [Google Scholar] [CrossRef]
- Sabattini, S.; Mancini, F.R.; Marconato, L.; Bacci, B.; Rossi, F.; Vignoli, M.; Bettini, G. EGFR overexpression in canine primary lung cancer: Pathogenetic implications and impact on survival. Vet. Comp. Oncol. 2012, 12, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Singer, J.; Fazekas, J.; Wang, W.; Weichselbaumer, M.; Matz, M.; Mader, A.; Steinfellner, W.; Meitz, S.; Mechtcheriakova, D.; Sobanov, Y.; et al. Generation of a Canine Anti-EGFR (ErbB-1) Antibody for Passive Immunotherapy in Dog Cancer Patients. Mol. Cancer Ther. 2014, 13, 1777–1790. [Google Scholar] [CrossRef] [PubMed]
- Veloso, E.S.; Gonçalves, I.N.N.; Arantes, J.A.; De Abreu, R.V.S.; Cassali, G.D.; Ferreira, E. Quantification of EGFR family in canine mammary ductal carcinomas in situ: Implications on the histological graduation. Vet. Res. Commun. 2019, 43, 123–129. [Google Scholar] [CrossRef]
- Millanta, F.; Silvestri, G.; Vaselli, C.; Citi, S.; Pisani, G.; Lorenzi, D.; Poli, A. The role of vascular endothelial growth factor and its receptor Flk-1/KDR in promoting tumour angiogenesis in feline and canine mammary carcinomas: A preliminary study of autocrine and paracrine loops. Res. Vet. Sci. 2006, 81, 350–357. [Google Scholar] [CrossRef] [PubMed]
- Jubala, C.M.; Wojcieszyn, J.W.; Valli, V.E.O.; Getzy, D.M.; Fosmire, S.P.; Coffey, D.; Bellgrau, D.; Modiano, J.F. CD20 Expression in Normal Canine B Cells and in Canine non-Hodgkin Lymphoma. Vet. Pathol. 2005, 42, 468–476. [Google Scholar] [CrossRef]
- Mizuno, T.; Kato, Y.; Kaneko, M.K.; Sakai, Y.; Shiga, T.; Kato, M.; Tsukui, T.; Takemoto, H.; Tokimasa, A.; Baba, K.; et al. Generation of a canine anti-canine CD20 antibody for canine lymphoma treatment. Sci. Rep. 2020, 10, 11476. [Google Scholar] [CrossRef]
- Carrasco-Ramirez, P.; Greening, D.W.; Andres, G.; Gopal, S.K.; Martin-Villar, E.; Renart, J.; Simpson, R.J.; Quintanilla, M. Podoplanin is a component of extracellular vesicles that reprograms cell-derived exosomal proteins and modulates lymphatic vessel formation. Oncotarget 2016, 7, 16070–16089. [Google Scholar] [CrossRef]
- Kamoto, S.; Shinada, M.; Kato, D.; Yoshimoto, S.; Ikeda, N.; Tsuboi, M.; Yoshitake, R.; Eto, S.; Hashimoto, Y.; Takahashi, Y.; et al. Phase I/II Clinical Trial of the Anti-Podoplanin Monoclonal Antibody Therapy in Dogs with Malignant Melanoma. Cells 2020, 9, 2529. [Google Scholar] [CrossRef]
- Maekawa, N.; Konnai, S.; Okagawa, T.; Nishimori, A.; Ikebuchi, R.; Izumi, Y.; Takagi, S.; Kagawa, Y.; Nakajima, C.; Suzuki, Y.; et al. Immunohistochemical Analysis of PD-L1 Expression in Canine Malignant Cancers and PD-1 Expression on Lymphocytes in Canine Oral Melanoma. PLoS ONE 2016, 11, e0157176. [Google Scholar] [CrossRef]
- Singer, J.; Weichselbaumer, M.; Stockner, T.; Mechtcheriakova, D.; Sobanov, Y.; Bajna, E.; Wrba, F.; Horvat, R.; Thalhammer, J.G.; Willmann, M.; et al. Comparative oncology: ErbB-1 and ErbB-2 homologues in canine cancer are susceptible to cetuximab and trastuzumab targeting. Mol. Immunol. 2012, 50, 200–209. [Google Scholar] [CrossRef]
- Wang, S.; Zhong, G.; Wang, X.-X.; Yu, F.; Weng, D.; Lin, J. Prognostic significance of the expression of HER family members in primary osteosarcoma. Oncol. Lett. 2018, 16, 2185–2194. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Wang, S.-L.; Chen, H.; Shen, R.-K.; Li, X.-D.; Huang, Q.-S.; Wu, C.-Y.; Weng, D.-F.; Lin, J.-H. Clinicopathological and prognostic values of ErbB receptor family amplification in primary osteosarcoma. Scand. J. Clin. Lab. Investig. 2019, 79, 601–612. [Google Scholar] [CrossRef] [PubMed]
- Gentschev, I.; Patil, S.S.; Adelfinger, M.; Weibel, S.; Geissinger, U.; Frentzen, A.; Chen, N.G.; Yu, Y.A.; Zhang, Q.; Ogilvie, G.; et al. Characterization and evaluation of a new oncolytic Vaccinia Virus strain LIVP6.1.1 for canine cancer therapy. Bioengineered 2013, 4, 84–89. [Google Scholar] [CrossRef]
- Park, H.; Park, S.; Bazer, F.W.; Lim, W.; Song, G. Myricetin treatment induces apoptosis in canine osteosarcoma cells by inducing DNA fragmentation, disrupting redox homeostasis, and mediating loss of mitochondrial membrane potential. J. Cell. Physiol. 2018, 233, 7457–7466. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Thamm, D.H.; Misra, V. The effect of Zhangfei/CREBZF on cell growth, differentiation, apoptosis, migration, and the unfolded protein response in several canine osteosarcoma cell lines. BMC Vet. Res. 2015, 11, 22. [Google Scholar] [CrossRef] [PubMed]
- Massimini, M.; Palmieri, C.; De Maria, R.; Romanucci, M.; Malatesta, D.; DE Martinis, M.; Maniscalco, L.; Ciccarelli, A.; Ginaldi, L.; Buracco, P.; et al. 17-AAG and Apoptosis, Autophagy, and Mitophagy in Canine Osteosarcoma Cell Lines. Vet. Pathol. 2016, 54, 405–412. [Google Scholar] [CrossRef]
- Gebhard, C.; Miller, I.; Hummel, K.; Ondrovics, M.N.N.; Schlosser, S.; Walter, I. Comparative proteome analysis of monolayer and spheroid culture of canine osteosarcoma cells. J. Proteom. 2018, 177, 124–136. [Google Scholar] [CrossRef]
- Mantovani, F.B.; Morrison, J.A.; Mutsaers, A.J. Effects of epidermal growth factor receptor kinase inhibition on radiation response in canine osteosarcoma cells. BMC Vet. Res. 2016, 12, 82. [Google Scholar] [CrossRef]
- Selvarajah, G.T.; Verheije, M.H.; Kik, M.; Slob, A.; Rottier, P.J.; Mol, J.A.; Kirpensteijn, J. Expression of epidermal growth factor receptor in canine osteosarcoma: Association with clinicopathological parameters and prognosis. Vet. J. 2012, 193, 412–419. [Google Scholar] [CrossRef] [PubMed]
- Kato, Y.; Ito, Y.; Ohishi, T.; Kawada, M.; Nakamura, T.; Sayama, Y.; Sano, M.; Asano, T.; Yanaka, M.; Okamoto, S.; et al. Antibody–Drug Conjugates Using Mouse–Canine Chimeric Anti-Dog Podoplanin Antibody Exerts Antitumor Activity in a Mouse Xenograft Model. Monoclon. Antibodies Immunodiagn. Immunother. 2020, 39, 37–44. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, G.; Ohishi, T.; Kaneko, M.K.; Takei, J.; Mizuno, T.; Kawada, M.; Saito, M.; Suzuki, H.; Kato, Y. Defucosylated Mouse–Dog Chimeric Anti-EGFR Antibody Exerts Antitumor Activities in Mouse Xenograft Models of Canine Tumors. Cells 2021, 10, 3599. https://doi.org/10.3390/cells10123599
Li G, Ohishi T, Kaneko MK, Takei J, Mizuno T, Kawada M, Saito M, Suzuki H, Kato Y. Defucosylated Mouse–Dog Chimeric Anti-EGFR Antibody Exerts Antitumor Activities in Mouse Xenograft Models of Canine Tumors. Cells. 2021; 10(12):3599. https://doi.org/10.3390/cells10123599
Chicago/Turabian StyleLi, Guanjie, Tomokazu Ohishi, Mika K. Kaneko, Junko Takei, Takuya Mizuno, Manabu Kawada, Masaki Saito, Hiroyuki Suzuki, and Yukinari Kato. 2021. "Defucosylated Mouse–Dog Chimeric Anti-EGFR Antibody Exerts Antitumor Activities in Mouse Xenograft Models of Canine Tumors" Cells 10, no. 12: 3599. https://doi.org/10.3390/cells10123599
APA StyleLi, G., Ohishi, T., Kaneko, M. K., Takei, J., Mizuno, T., Kawada, M., Saito, M., Suzuki, H., & Kato, Y. (2021). Defucosylated Mouse–Dog Chimeric Anti-EGFR Antibody Exerts Antitumor Activities in Mouse Xenograft Models of Canine Tumors. Cells, 10(12), 3599. https://doi.org/10.3390/cells10123599