The Jekyll and Hyde of Cellular Senescence in Cancer



{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The Hallmarks and Molecular Mechanisms of Cellular Senescence
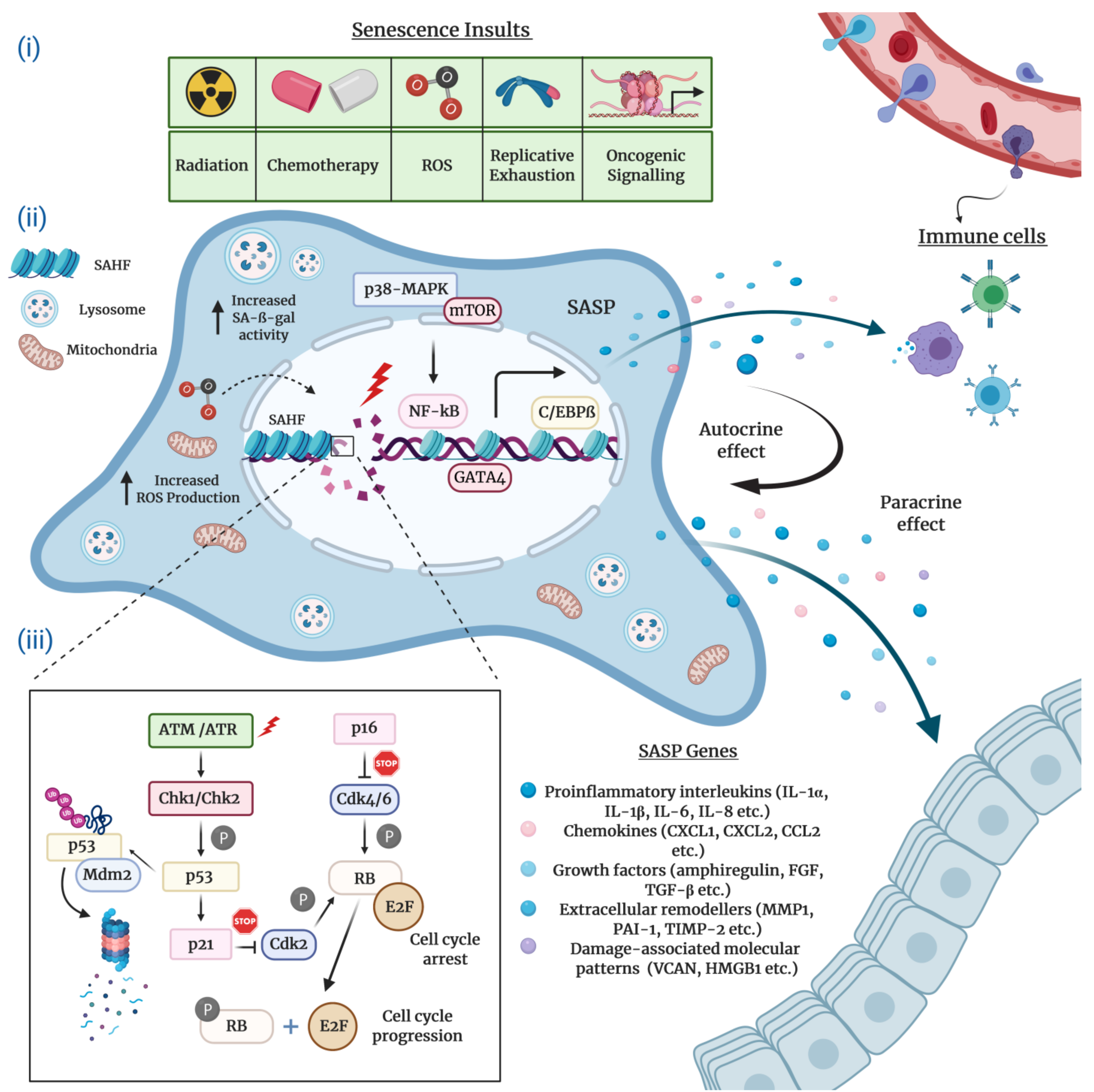
2.1. Morphological and Molecular Biomarkers of Senescent Cells
2.2. The Senescence-Associated Secretory Phenotype (SASP) of Senescent Cells
2.3. Molecular Mechanisms Underlying Cellular Senescence
3. The Significance of Cellular Senescence: From Homeostasis to Cancer
4. The Implications of Therapy-Induced Senescence in Cancer
5. Therapeutic Targeting of Senescent Cells and the SASP in Cancer
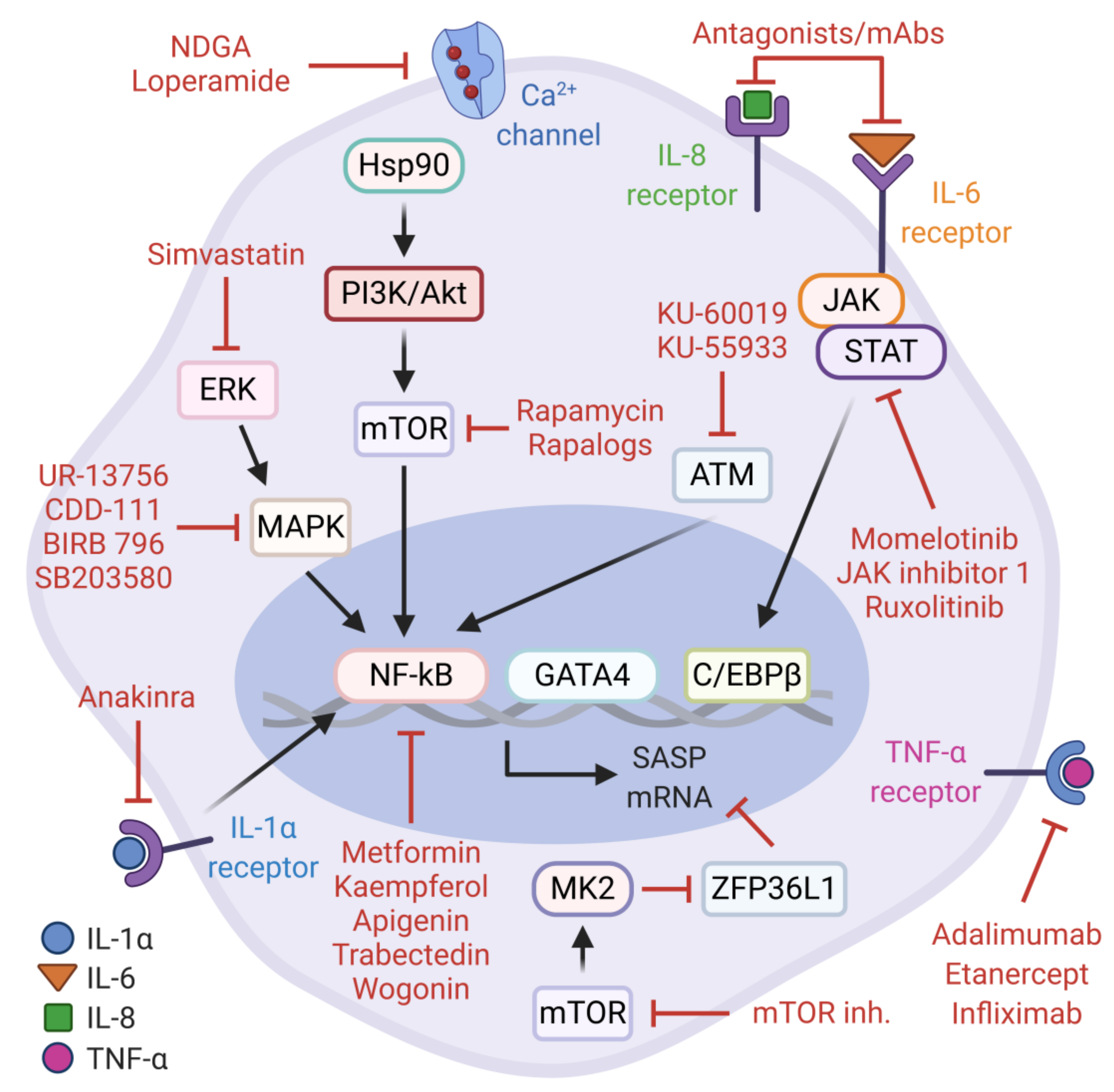
5.1. SASP Activity in Cancer and Anti-SASP Therapies
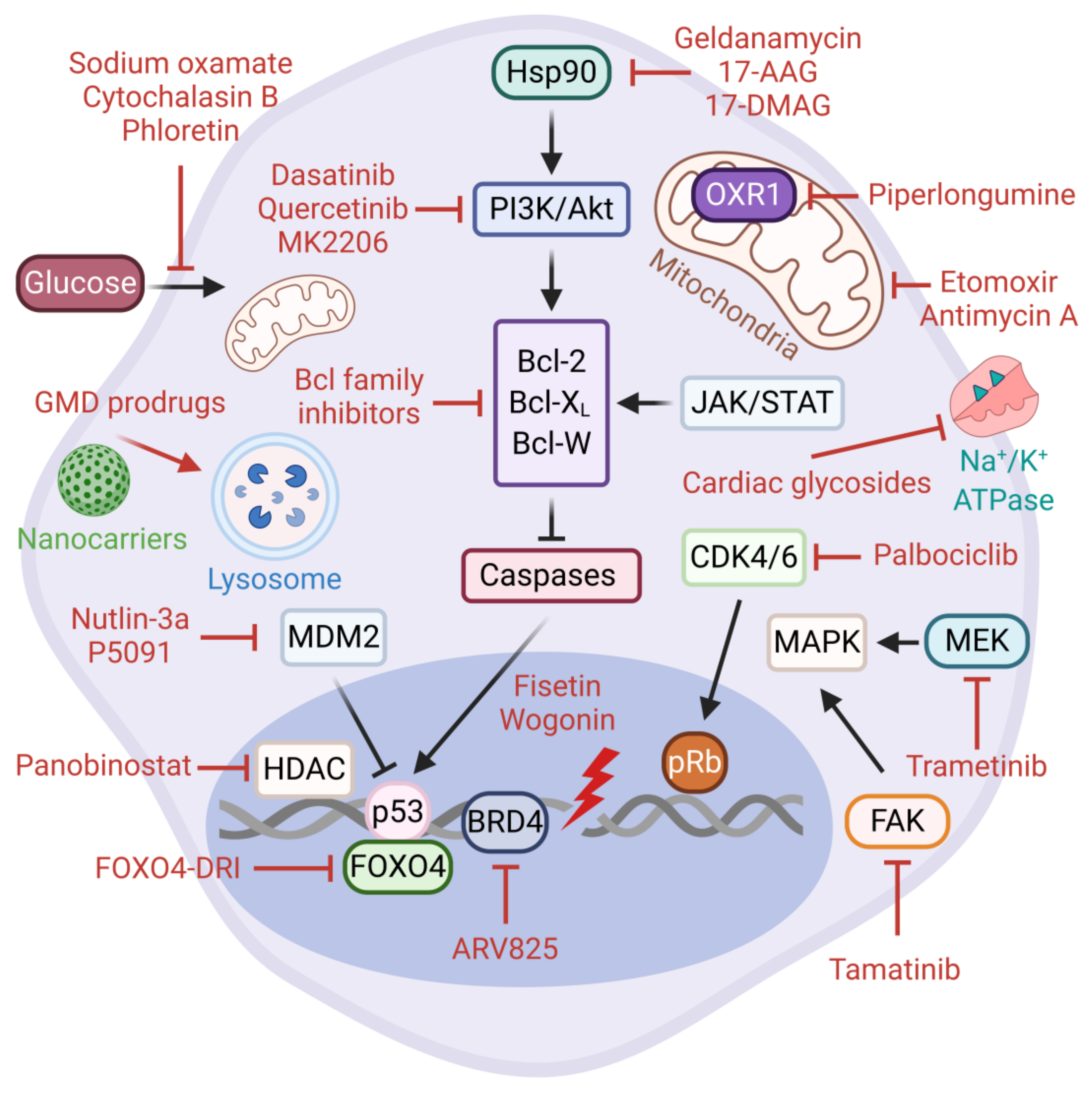
5.2. Senolytic Therapies
5.3. Directed Targeting of Senescent Cells
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hayflick, L.; Moorhead, P.S. The Serial Cultivation of Human Diploid Cell Strains. Exp. Cell Res. 1961, 25, 585–621. [Google Scholar] [CrossRef]
- Hayflick, L. The Limited in Vitro Lifetime of Human Diploid Cell Strains. Exp. Cell Res. 1965, 37, 614–636. [Google Scholar] [CrossRef]
- Campisi, J. Replicative Senescence: An Old Lives’ Tale? Cell 1996, 84, 497–500. [Google Scholar] [CrossRef] [Green Version]
- Shay, J.W.; Wright, W.E. Hayflick, His Limit, and Cellular Ageing. Nat. Rev. Mol. Cell Biol. 2000, 1, 72–76. [Google Scholar] [CrossRef]
- di Fagagna, F.D.; Reaper, P.M.; Clay-Farrace, L.; Fiegler, H.; Carr, P.; von Zglinicki, T.; Saretzki, G.; Carter, N.P.; Jackson, S.P. A DNA Damage Checkpoint Response in Telomere-Initiated Senescence. Nature 2003, 426, 194–198. [Google Scholar] [CrossRef]
- Goldstein, S. Replicative Senescence: The Human Fibroblast Comes of Age. Science 1990, 249, 1129–1133. [Google Scholar] [CrossRef]
- Galluzzi, L.; Yamazaki, T.; Kroemer, G. Linking Cellular Stress Responses to Systemic Homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 731–745. [Google Scholar] [CrossRef]
- Campisi, J.; d’Adda di Fagagna, F. Cellular Senescence: When Bad Things Happen to Good Cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef]
- Van Deursen, J.M. The Role of Senescent Cells in Ageing. Nature 2014, 509, 439–446. [Google Scholar] [CrossRef] [Green Version]
- McHugh, D.; Gil, J. Senescence and Aging: Causes, Consequences, and Therapeutic Avenues. J. Cell Biol. 2018, 217, 65–77. [Google Scholar] [CrossRef]
- Childs, B.G.; Durik, M.; Baker, D.J.; van Deursen, J.M. Cellular Senescence in Aging and Age-Related Disease: From Mechanisms to Therapy. Nat. Med. 2015, 21, 1424–1435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muñoz-Espín, D.; Serrano, M. Cellular Senescence: From Physiology to Pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496. [Google Scholar] [CrossRef] [PubMed]
- Kuilman, T.; Michaloglou, C.; Mooi, W.J.; Peeper, D.S. The Essence of Senescence. Genes Dev. 2010, 24, 2463–2479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narita, M.; Lowe, S.W. Senescence Comes of Age. Nat. Med. 2005, 11, 920–922. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J. Cellular Senescence as a Tumor-Suppressor Mechanism. Trends Cell Biol. 2001, 11, S27–S31. [Google Scholar] [CrossRef] [Green Version]
- Lozano-Torres, B.; Estepa-Fernández, A.; Rovira, M.; Orzáez, M.; Serrano, M.; Martínez-Máñez, R.; Sancenón, F. The Chemistry of Senescence. Nat. Rev. Chem. 2019, 3, 426–441. [Google Scholar] [CrossRef]
- Beauséjour, C.M.; Krtolica, A.; Galimi, F.; Narita, M.; Lowe, S.W.; Yaswen, P.; Campisi, J. Reversal of Human Cellular Senescence: Roles of the P53 and P16 Pathways. EMBO J. 2003, 22, 4212–4222. [Google Scholar] [CrossRef]
- Hoare, M.; Narita, M. Transmitting Senescence to the Cell Neighbourhood. Nat. Cell Biol. 2013, 15, 887–889. [Google Scholar] [CrossRef]
- Mavrogonatou, E.; Pratsinis, H.; Kletsas, D. The Role of Senescence in Cancer Development. Semin. Cancer Biol. 2020, 62, 182–191. [Google Scholar] [CrossRef]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O. A Biomarker That Identifies Senescent Human Cells in Culture and in Aging Skin in Vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef] [Green Version]
- Hernandez-Segura, A.; Nehme, J.; Demaria, M. Hallmarks of Cellular Senescence. Trends Cell Biol. 2018, 28, 436–453. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Chen, W.; Adams, P.D. Molecular Dissection of Formation of Senescence-Associated Heterochromatin Foci. Mol. Cell. Biol. 2007, 27, 2343–2358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sen, P.; Shah, P.P.; Nativio, R.; Berger, S.L. Epigenetic Mechanisms of Longevity and Aging. Cell 2016, 166, 822–839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Victorelli, S.; Passos, J.F. Telomeres and Cell Senescence-Size Matters Not. EBioMedicine 2017, 21, 14–20. [Google Scholar] [CrossRef] [Green Version]
- Kuilman, T.; Peeper, D.S. Senescence-Messaging Secretome: SMS-Ing Cellular Stress. Nat. Rev. Cancer 2009, 9, 81–94. [Google Scholar] [CrossRef]
- Acosta, J.C.; Banito, A.; Wuestefeld, T.; Georgilis, A.; Janich, P.; Morton, J.P.; Athineos, D.; Kang, T.-W.; Lasitschka, F.; Andrulis, M.; et al. A Complex Secretory Program Orchestrated by the Inflammasome Controls Paracrine Senescence. Nat. Cell Biol. 2013, 15, 978–990. [Google Scholar] [CrossRef]
- Tchkonia, T.; Zhu, Y.; van Deursen, J.; Campisi, J.; Kirkland, J.L. Cellular Senescence and the Senescent Secretory Phenotype: Therapeutic Opportunities. J. Clin. Invest. 2013, 123, 966–972. [Google Scholar] [CrossRef] [Green Version]
- Wallis, R.; Mizen, H.; Bishop, C.L. The Bright and Dark Side of Extracellular Vesicles in the Senescence-Associated Secretory Phenotype. Mech. Ageing Dev. 2020, 189, 111263. [Google Scholar] [CrossRef]
- Terlecki-Zaniewicz, L.; Lämmermann, I.; Latreille, J.; Bobbili, M.R.; Pils, V.; Schosserer, M.; Weinmüllner, R.; Dellago, H.; Skalicky, S.; Pum, D.; et al. Small Extracellular Vesicles and Their MiRNA Cargo Are Anti-Apoptotic Members of the Senescence-Associated Secretory Phenotype. Aging 2018, 10, 1103–1132. [Google Scholar] [CrossRef]
- Basisty, N.; Kale, A.; Jeon, O.H.; Kuehnemann, C.; Payne, T.; Rao, C.; Holtz, A.; Shah, S.; Sharma, V.; Ferrucci, L.; et al. A Proteomic Atlas of Senescence-Associated Secretomes for Aging Biomarker Development. PLOS Biol. 2020, 18, e3000599. [Google Scholar] [CrossRef] [Green Version]
- Faget, D.V.; Ren, Q.; Stewart, S.A. Unmasking Senescence: Context-Dependent Effects of SASP in Cancer. Nat. Rev. Cancer 2019, 19, 439–453. [Google Scholar] [CrossRef] [PubMed]
- Freund, A.; Patil, C.K.; Campisi, J. P38MAPK Is a Novel DNA Damage Response-Independent Regulator of the Senescence-Associated Secretory Phenotype. EMBO J. 2011, 30, 1536–1548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birch, J.; Gil, J. Senescence and the SASP: Many Therapeutic Avenues. Genes Dev. 2020, 34, 1565–1576. [Google Scholar] [CrossRef]
- Lopes-Paciencia, S.; Saint-Germain, E.; Rowell, M.-C.; Ruiz, A.F.; Kalegari, P.; Ferbeyre, G. The Senescence-Associated Secretory Phenotype and Its Regulation. Cytokine 2019, 117, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Glück, S.; Guey, B.; Gulen, M.F.; Wolter, K.; Kang, T.-W.; Schmacke, N.A.; Bridgeman, A.; Rehwinkel, J.; Zender, L.; Ablasser, A. Innate Immune Sensing of Cytosolic Chromatin Fragments through CGAS Promotes Senescence. Nat. Cell Biol. 2017, 19, 1061–1070. [Google Scholar] [CrossRef]
- Dou, Z.; Ghosh, K.; Vizioli, M.G.; Zhu, J.; Sen, P.; Wangensteen, K.J.; Simithy, J.; Lan, Y.; Lin, Y.; Zhou, Z.; et al. Cytoplasmic Chromatin Triggers Inflammation in Senescence and Cancer. Nature 2017, 550, 402–406. [Google Scholar] [CrossRef] [Green Version]
- Chien, Y.; Scuoppo, C.; Wang, X.; Fang, X.; Balgley, B.; Bolden, J.E.; Premsrirut, P.; Luo, W.; Chicas, A.; Lee, C.S.; et al. Control of the Senescence-Associated Secretory Phenotype by NF-ΚB Promotes Senescence and Enhances Chemosensitivity. Genes Dev. 2011, 25, 2125–2136. [Google Scholar] [CrossRef] [Green Version]
- Salotti, J.; Johnson, P.F. Regulation of Senescence and the SASP by the Transcription Factor C/EBPβ. Exp. Gerontol. 2019, 128, 110752. [Google Scholar] [CrossRef]
- Acosta, J.C.; O’Loghlen, A.; Banito, A.; Guijarro, M.V.; Augert, A.; Raguz, S.; Fumagalli, M.; Da Costa, M.; Brown, C.; Popov, N.; et al. Chemokine Signaling via the CXCR2 Receptor Reinforces Senescence. Cell 2008, 133, 1006–1018. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Schmitt, C.A. The Dynamic Nature of Senescence in Cancer. Nat. Cell Biol. 2019, 21, 94–101. [Google Scholar] [CrossRef]
- Hoare, M.; Ito, Y.; Kang, T.-W.; Weekes, M.P.; Matheson, N.J.; Patten, D.A.; Shetty, S.; Parry, A.J.; Menon, S.; Salama, R.; et al. NOTCH1 Mediates a Switch between Two Distinct Secretomes during Senescence. Nat. Cell Biol. 2016, 18, 979–992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodier, F.; Coppé, J.-P.; Patil, C.K.; Hoeijmakers, W.A.M.; Muñoz, D.P.; Raza, S.R.; Freund, A.; Campeau, E.; Davalos, A.R.; Campisi, J. Persistent DNA Damage Signalling Triggers Senescence-Associated Inflammatory Cytokine Secretion. Nat. Cell Biol. 2009, 11, 973–979. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.; Xu, Q.; Martin, T.D.; Li, M.Z.; Demaria, M.; Aron, L.; Lu, T.; Yankner, B.A.; Campisi, J.; Elledge, S.J. The DNA Damage Response Induces Inflammation and Senescence by Inhibiting Autophagy of GATA4. Science 2015, 349, aaa5612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaplon, J.; Zheng, L.; Meissl, K.; Chaneton, B.; Selivanov, V.A.; Mackay, G.; van der Burg, S.H.; Verdegaal, E.M.E.; Cascante, M.; Shlomi, T.; et al. A Key Role for Mitochondrial Gatekeeper Pyruvate Dehydrogenase in Oncogene-Induced Senescence. Nature 2013, 498, 109–112. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Espín, D.; Cañamero, M.; Maraver, A.; Gómez-López, G.; Contreras, J.; Murillo-Cuesta, S.; Rodríguez-Baeza, A.; Varela-Nieto, I.; Ruberte, J.; Collado, M.; et al. Programmed Cell Senescence during Mammalian Embryonic Development. Cell 2013, 155, 1104–1118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, T.; Teo, Y.V.; Evans, S.A.; Neretti, N.; Sedivy, J.M. Regulation of Cellular Senescence by Polycomb Chromatin Modifiers through Distinct DNA Damage- and Histone Methylation-Dependent Pathways. Cell Rep. 2018, 22, 3480–3492. [Google Scholar] [CrossRef] [Green Version]
- Wiley, C.D.; Velarde, M.C.; Lecot, P.; Liu, S.; Sarnoski, E.A.; Freund, A.; Shirakawa, K.; Lim, H.W.; Davis, S.S.; Ramanathan, A.; et al. Mitochondrial Dysfunction Induces Senescence with a Distinct Secretory Phenotype. Cell Metab. 2016, 23, 303–314. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Ruiz, P.D.; McKimpson, W.M.; Novikov, L.; Kitsis, R.N.; Gamble, M.J. MacroH2A1 and ATM Play Opposing Roles in Paracrine Senescence and the Senescence-Associated Secretory Phenotype. Mol. Cell 2015, 59, 719–731. [Google Scholar] [CrossRef] [Green Version]
- Contrepois, K.; Coudereau, C.; Benayoun, B.A.; Schuler, N.; Roux, P.-F.; Bischof, O.; Courbeyrette, R.; Carvalho, C.; Thuret, J.-Y.; Ma, Z.; et al. Histone Variant H2A.J Accumulates in Senescent Cells and Promotes Inflammatory Gene Expression. Nat. Commun. 2017, 8, 14995. [Google Scholar] [CrossRef] [Green Version]
- Davalos, A.R.; Kawahara, M.; Malhotra, G.K.; Schaum, N.; Huang, J.; Ved, U.; Beausejour, C.M.; Coppe, J.-P.; Rodier, F.; Campisi, J. P53-Dependent Release of Alarmin HMGB1 Is a Central Mediator of Senescent Phenotypes. J. Cell Biol. 2013, 201, 613–629. [Google Scholar] [CrossRef]
- Pazolli, E.; Alspach, E.; Milczarek, A.; Prior, J.; Piwnica-Worms, D.; Stewart, S.A. Chromatin Remodeling Underlies the Senescence-Associated Secretory Phenotype of Tumor Stromal Fibroblasts That Supports Cancer Progression. Cancer Res. 2012, 72, 2251–2261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tasdemir, N.; Banito, A.; Roe, J.-S.; Alonso-Curbelo, D.; Camiolo, M.; Tschaharganeh, D.F.; Huang, C.-H.; Aksoy, O.; Bolden, J.E.; Chen, C.-C.; et al. BRD4 Connects Enhancer Remodeling to Senescence Immune Surveillance. Cancer Discov. 2016, 6, 612–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capell, B.C.; Drake, A.M.; Zhu, J.; Shah, P.P.; Dou, Z.; Dorsey, J.; Simola, D.F.; Donahue, G.; Sammons, M.; Rai, T.S.; et al. MLL1 Is Essential for the Senescence-Associated Secretory Phenotype. Genes Dev. 2016, 30, 321–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayakawa, T.; Iwai, M.; Aoki, S.; Takimoto, K.; Maruyama, M.; Maruyama, W.; Motoyama, N. SIRT1 Suppresses the Senescence-Associated Secretory Phenotype through Epigenetic Gene Regulation. PLoS ONE 2015, 10, e0116480. [Google Scholar] [CrossRef]
- Alspach, E.; Flanagan, K.C.; Luo, X.; Ruhland, M.K.; Huang, H.; Pazolli, E.; Donlin, M.J.; Marsh, T.; Piwnica-Worms, D.; Monahan, J.; et al. P38MAPK Plays a Crucial Role in Stromal-Mediated Tumorigenesis. Cancer Discov. 2014, 4, 716–729. [Google Scholar] [CrossRef] [Green Version]
- Herranz, N.; Gallage, S.; Mellone, M.; Wuestefeld, T.; Klotz, S.; Hanley, C.J.; Raguz, S.; Acosta, J.C.; Innes, A.J.; Banito, A.; et al. MTOR Regulates MAPKAPK2 Translation to Control the Senescence-Associated Secretory Phenotype. Nat. Cell Biol. 2015, 17, 1205–1217. [Google Scholar] [CrossRef] [Green Version]
- Paez-Ribes, M.; González-Gualda, E.; Doherty, G.J.; Muñoz-Espín, D. Targeting Senescent Cells in Translational Medicine. EMBO Mol. Med. 2019, 11, e10234. [Google Scholar] [CrossRef]
- Herranz, N.; Gil, J. Mechanisms and Functions of Cellular Senescence. J. Clin. Invest. 2018, 128, 1238–1246. [Google Scholar] [CrossRef]
- Sherr, C.J.; McCormick, F. The RB and P53 Pathways in Cancer. Cancer Cell 2002, 2, 103–112. [Google Scholar] [CrossRef] [Green Version]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef]
- Narita, M.; Nũnez, S.; Heard, E.; Narita, M.; Lin, A.W.; Hearn, S.A.; Spector, D.L.; Hannon, G.J.; Lowe, S.W. Rb-Mediated Heterochromatin Formation and Silencing of E2F Target Genes during Cellular Senescence. Cell 2003, 113, 703–716. [Google Scholar] [CrossRef] [Green Version]
- Chicas, A.; Wang, X.; Zhang, C.; McCurrach, M.; Zhao, Z.; Mert, O.; Dickins, R.A.; Narita, M.; Zhang, M.; Lowe, S.W. Dissecting the Unique Role of the Retinoblastoma Tumor Suppressor during Cellular Senescence. Cancer Cell 2010, 17, 376–387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hara, E.; Smith, R.; Parry, D.; Tahara, H.; Stone, S.; Peters, G. Regulation of P16CDKN2 Expression and Its Implications for Cell Immortalization and Senescence. Mol. Cell. Biol. 1996, 16, 859–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Trotman, L.C.; Shaffer, D.; Lin, H.-K.; Dotan, Z.A.; Niki, M.; Koutcher, J.A.; Scher, H.I.; Ludwig, T.; Gerald, W.; et al. Crucial Role of P53-Dependent Cellular Senescence in Suppression of Pten-Deficient Tumorigenesis. Nature 2005, 436, 725–730. [Google Scholar] [CrossRef] [Green Version]
- Itahana, K.; Dimri, G.; Campisi, J. Regulation of Cellular Senescence by P53: P53 and Cellular Senescence. Eur. J. Biochem. 2001, 268, 2784–2791. [Google Scholar] [CrossRef]
- Rufini, A.; Tucci, P.; Celardo, I.; Melino, G. Senescence and Aging: The Critical Roles of P53. Oncogene 2013, 32, 5129–5143. [Google Scholar] [CrossRef]
- Chan, S.R.W.L.; Blackburn, E.H. Telomeres and Telomerase. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2004, 359, 109–121. [Google Scholar] [CrossRef]
- McClintock, B. The Stability of Broken Ends of Chromosomes in Zea Mays. Genetics 1941, 26, 234–282. [Google Scholar]
- Olovnikov, A.M. A Theory of Marginotomy. The Incomplete Copying of Template Margin in Enzymic Synthesis of Polynucleotides and Biological Significance of the Phenomenon. J. Theor. Biol. 1973, 41, 181–190. [Google Scholar] [CrossRef]
- Watson, J.D. Origin of Concatemeric T7 DNA. Nat. New Biol. 1972, 239, 197–201. [Google Scholar] [CrossRef]
- Blackburn, E.H. Switching and Signaling at the Telomere. Cell 2001, 106, 661–673. [Google Scholar] [CrossRef] [Green Version]
- Wright, W.E.; Piatyszek, M.A.; Rainey, W.E.; Byrd, W.; Shay, J.W. Telomerase Activity in Human Germline and Embryonic Tissues and Cells. Dev. Genet. 1996, 18, 173–179. [Google Scholar] [CrossRef]
- Counter, C.M.; Avilion, A.A.; LeFeuvre, C.E.; Stewart, N.G.; Greider, C.W.; Harley, C.B.; Bacchetti, S. Telomere Shortening Associated with Chromosome Instability Is Arrested in Immortal Cells Which Express Telomerase Activity. EMBO J. 1992, 11, 1921–1929. [Google Scholar] [CrossRef] [PubMed]
- Harley, C.B.; Futcher, A.B.; Greider, C.W. Telomeres Shorten during Ageing of Human Fibroblasts. Nature 1990, 345, 458–460. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.W.; Piatyszek, M.A.; Prowse, K.R.; Harley, C.B.; West, M.D.; Ho, P.L.; Coviello, G.M.; Wright, W.E.; Weinrich, S.L.; Shay, J.W. Specific Association of Human Telomerase Activity with Immortal Cells and Cancer. Science 1994, 266, 2011–2015. [Google Scholar] [CrossRef]
- Bodnar, A.G.; Ouellette, M.; Frolkis, M.; Holt, S.E.; Chiu, C.P.; Morin, G.B.; Harley, C.B.; Shay, J.W.; Lichtsteiner, S.; Wright, W.E. Extension of Life-Span by Introduction of Telomerase into Normal Human Cells. Science 1998, 279, 349–352. [Google Scholar] [CrossRef] [Green Version]
- Vaziri, H.; Benchimol, S. Reconstitution of Telomerase Activity in Normal Human Cells Leads to Elongation of Telomeres and Extended Replicative Life Span. Curr. Biol. CB 1998, 8, 279–282. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Huang, F.W.; Hodis, E.; Xu, M.J.; Kryukov, G.V.; Chin, L.; Garraway, L.A. Highly Recurrent TERT Promoter Mutations in Human Melanoma. Science 2013, 339, 957–959. [Google Scholar] [CrossRef] [Green Version]
- Horn, S.; Figl, A.; Rachakonda, P.S.; Fischer, C.; Sucker, A.; Gast, A.; Kadel, S.; Moll, I.; Nagore, E.; Hemminki, K.; et al. TERT Promoter Mutations in Familial and Sporadic Melanoma. Science 2013, 339, 959–961. [Google Scholar] [CrossRef] [Green Version]
- Borah, S.; Xi, L.; Zaug, A.J.; Powell, N.M.; Dancik, G.M.; Cohen, S.B.; Costello, J.C.; Theodorescu, D.; Cech, T.R. Cancer. TERT Promoter Mutations and Telomerase Reactivation in Urothelial Cancer. Science 2015, 347, 1006–1010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shay, J.W.; Wright, W.E. Role of Telomeres and Telomerase in Cancer. Semin. Cancer Biol. 2011, 21, 349–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, X.; Larsson, C.; Xu, D. Mechanisms Underlying the Activation of TERT Transcription and Telomerase Activity in Human Cancer: Old Actors and New Players. Oncogene 2019, 38, 6172–6183. [Google Scholar] [CrossRef] [Green Version]
- Senturk, S.; Mumcuoglu, M.; Gursoy-Yuzugullu, O.; Cingoz, B.; Akcali, K.C.; Ozturk, M. Transforming Growth Factor-Beta Induces Senescence in Hepatocellular Carcinoma Cells and Inhibits Tumor Growth. Hepatol. Baltim. Md 2010, 52, 966–974. [Google Scholar] [CrossRef] [Green Version]
- Serrano, M.; Lin, A.W.; McCurrach, M.E.; Beach, D.; Lowe, S.W. Oncogenic Ras Provokes Premature Cell Senescence Associated with Accumulation of P53 and P16INK4a. Cell 1997, 88, 593–602. [Google Scholar] [CrossRef] [Green Version]
- Davalli, P.; Mitic, T.; Caporali, A.; Lauriola, A.; D’Arca, D. ROS, Cell Senescence, and Novel Molecular Mechanisms in Aging and Age-Related Diseases. Oxid. Med. Cell. Longev. 2016, 2016, 3565127. [Google Scholar] [CrossRef] [Green Version]
- Di Micco, R.; Fumagalli, M.; Cicalese, A.; Piccinin, S.; Gasparini, P.; Luise, C.; Schurra, C.; Garre’, M.; Nuciforo, P.G.; Bensimon, A.; et al. Oncogene-Induced Senescence Is a DNA Damage Response Triggered by DNA Hyper-Replication. Nature 2006, 444, 638–642. [Google Scholar] [CrossRef]
- Vasileiou, P.V.S.; Evangelou, K.; Vlasis, K.; Fildisis, G.; Panayiotidis, M.I.; Chronopoulos, E.; Passias, P.-G.; Kouloukoussa, M.; Gorgoulis, V.G.; Havaki, S. Mitochondrial Homeostasis and Cellular Senescence. Cells 2019, 8, 686. [Google Scholar] [CrossRef] [Green Version]
- Pluquet, O.; Pourtier, A.; Abbadie, C. The Unfolded Protein Response and Cellular Senescence. A Review in the Theme: Cellular Mechanisms of Endoplasmic Reticulum Stress Signaling in Health and Disease. Am. J. Physiol. Cell Physiol. 2015, 308, C415–C425. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Rebollo, E.; Franzen, J.; Goetzke, R.; Hollmann, J.; Ostrowska, A.; Oliverio, M.; Sieben, T.; Rath, B.; Kornfeld, J.-W.; Wagner, W. Senescence-Associated Metabolomic Phenotype in Primary and IPSC-Derived Mesenchymal Stromal Cells. Stem Cell Rep. 2020, 14, 201–209. [Google Scholar] [CrossRef] [Green Version]
- Wyld, L.; Bellantuono, I.; Tchkonia, T.; Morgan, J.; Turner, O.; Foss, F.; George, J.; Danson, S.; Kirkland, J.L. Senescence and Cancer: A Review of Clinical Implications of Senescence and Senotherapies. Cancers 2020, 12, 2134. [Google Scholar] [CrossRef] [PubMed]
- Davaapil, H.; Brockes, J.P.; Yun, M.H. Conserved and Novel Functions of Programmed Cellular Senescence during Vertebrate Development. Dev. Camb. Engl. 2017, 144, 106–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salama, R.; Sadaie, M.; Hoare, M.; Narita, M. Cellular Senescence and Its Effector Programs. Genes Dev. 2014, 28, 99–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burton, D.G.A.; Stolzing, A. Cellular Senescence: Immunosurveillance and Future Immunotherapy. Ageing Res. Rev. 2018, 43, 17–25. [Google Scholar] [CrossRef] [Green Version]
- Finkel, T.; Serrano, M.; Blasco, M.A. The Common Biology of Cancer and Ageing. Nature 2007, 448, 767–774. [Google Scholar] [CrossRef] [Green Version]
- Coppé, J.-P.; Desprez, P.-Y.; Krtolica, A.; Campisi, J. The Senescence-Associated Secretory Phenotype: The Dark Side of Tumor Suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef] [Green Version]
- Sturmlechner, I.; Durik, M.; Sieben, C.J.; Baker, D.J.; van Deursen, J.M. Cellular Senescence in Renal Ageing and Disease. Nat. Rev. Nephrol. 2017, 13, 77–89. [Google Scholar] [CrossRef]
- Lunyak, V.V.; Amaro-Ortiz, A.; Gaur, M. Mesenchymal Stem Cells Secretory Responses: Senescence Messaging Secretome and Immunomodulation Perspective. Front. Genet. 2017, 8, 220. [Google Scholar] [CrossRef]
- Schmitt, C.A.; Lowe, S.W. Apoptosis and Therapy. J. Pathol. 1999, 187, 127–137. [Google Scholar] [CrossRef]
- Hannun, Y.A. Apoptosis and the Dilemma of Cancer Chemotherapy. Blood 1997, 89, 1845–1853. [Google Scholar] [CrossRef]
- Pommier, Y.; Sordet, O.; Antony, S.; Hayward, R.L.; Kohn, K.W. Apoptosis Defects and Chemotherapy Resistance: Molecular Interaction Maps and Networks. Oncogene 2004, 23, 2934–2949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Longley, D.B.; Johnston, P.G. Molecular Mechanisms of Drug Resistance. J. Pathol. 2005, 205, 275–292. [Google Scholar] [CrossRef] [PubMed]
- Housman, G.; Byler, S.; Heerboth, S.; Lapinska, K.; Longacre, M.; Snyder, N.; Sarkar, S. Drug Resistance in Cancer: An Overview. Cancers 2014, 6, 1769–1792. [Google Scholar] [CrossRef] [Green Version]
- Millar, A.W.; Lynch, K.P. Rethinking Clinical Trials for Cytostatic Drugs. Nat. Rev. Cancer 2003, 3, 540–545. [Google Scholar] [CrossRef] [PubMed]
- Nardella, C.; Clohessy, J.G.; Alimonti, A.; Pandolfi, P.P. Pro-Senescence Therapy for Cancer Treatment. Nat. Rev. Cancer 2011, 11, 503–511. [Google Scholar] [CrossRef]
- Lee, M.; Lee, J.-S. Exploiting Tumor Cell Senescence in Anticancer Therapy. BMB Rep. 2014, 47, 51–59. [Google Scholar] [CrossRef]
- Ewald, J.A.; Desotelle, J.A.; Wilding, G.; Jarrard, D.F. Therapy-Induced Senescence in Cancer. J. Natl. Cancer Inst. 2010, 102, 1536–1546. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Wong, S.C.; Pan, J.; Tsao, S.W.; Fung, K.H.; Kwong, D.L.; Sham, J.S.; Nicholls, J.M. Evidence of Cisplatin-Induced Senescent-like Growth Arrest in Nasopharyngeal Carcinoma Cells. Cancer Res. 1998, 58, 5019–5022. [Google Scholar]
- Elmore, L.W.; Rehder, C.W.; Di, X.; McChesney, P.A.; Jackson-Cook, C.K.; Gewirtz, D.A.; Holt, S.E. Adriamycin-Induced Senescence in Breast Tumor Cells Involves Functional P53 and Telomere Dysfunction. J. Biol. Chem. 2002, 277, 35509–35515. [Google Scholar] [CrossRef] [Green Version]
- te Poele, R.H.; Okorokov, A.L.; Jardine, L.; Cummings, J.; Joel, S.P. DNA Damage Is Able to Induce Senescence in Tumor Cells in Vitro and in Vivo. Cancer Res. 2002, 62, 1876–1883. [Google Scholar]
- Gewirtz, D.A.; Holt, S.E.; Elmore, L.W. Accelerated Senescence: An Emerging Role in Tumor Cell Response to Chemotherapy and Radiation. Biochem. Pharmacol. 2008, 76, 947–957. [Google Scholar] [CrossRef] [PubMed]
- Petrova, N.V.; Velichko, A.K.; Razin, S.V.; Kantidze, O.L. Small Molecule Compounds That Induce Cellular Senescence. Aging Cell 2016, 15, 999–1017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malaquin, N.; Olivier, M.-A.; Martinez, A.; Nadeau, S.; Sawchyn, C.; Coppé, J.-P.; Cardin, G.; Mallette, F.A.; Campisi, J.; Rodier, F. Non-Canonical ATM/MRN Activities Temporally Define the Senescence Secretory Program. EMBO Rep. 2020, 21, e50718. [Google Scholar] [CrossRef] [PubMed]
- Bertran-Alamillo, J.; Cattan, V.; Schoumacher, M.; Codony-Servat, J.; Giménez-Capitán, A.; Cantero, F.; Burbridge, M.; Rodríguez, S.; Teixidó, C.; Roman, R.; et al. AURKB as a Target in Non-Small Cell Lung Cancer with Acquired Resistance to Anti-EGFR Therapy. Nat. Commun. 2019, 10, 1812. [Google Scholar] [CrossRef] [PubMed]
- Wagner, V.; Gil, J. Senescence as a Therapeutically Relevant Response to CDK4/6 Inhibitors. Oncogene 2020, 39, 5165–5176. [Google Scholar] [CrossRef]
- Kalathur, M.; Toso, A.; Chen, J.; Revandkar, A.; Danzer-Baltzer, C.; Guccini, I.; Alajati, A.; Sarti, M.; Pinton, S.; Brambilla, L.; et al. A Chemogenomic Screening Identifies CK2 as a Target for Pro-Senescence Therapy in PTEN-Deficient Tumours. Nat. Commun. 2015, 6, 7227. [Google Scholar] [CrossRef]
- Fleury, H.; Malaquin, N.; Tu, V.; Gilbert, S.; Martinez, A.; Olivier, M.-A.; Sauriol, A.; Communal, L.; Leclerc-Desaulniers, K.; Carmona, E.; et al. Exploiting Interconnected Synthetic Lethal Interactions between PARP Inhibition and Cancer Cell Reversible Senescence. Nat. Commun. 2019, 10, 2556. [Google Scholar] [CrossRef] [Green Version]
- Demaria, M.; O’Leary, M.N.; Chang, J.; Shao, L.; Liu, S.; Alimirah, F.; Koenig, K.; Le, C.; Mitin, N.; Deal, A.M.; et al. Cellular Senescence Promotes Adverse Effects of Chemotherapy and Cancer Relapse. Cancer Discov. 2017, 7, 165–176. [Google Scholar] [CrossRef] [Green Version]
- Saleh, T.; Tyutyunyk-Massey, L.; Gewirtz, D.A. Tumor Cell Escape from Therapy-Induced Senescence as a Model of Disease Recurrence after Dormancy. Cancer Res. 2019, 79, 1044–1046. [Google Scholar] [CrossRef] [Green Version]
- Elmore, L.W.; Di, X.; Dumur, C.; Holt, S.E.; Gewirtz, D.A. Evasion of a Single-Step, Chemotherapy-Induced Senescence in Breast Cancer Cells: Implications for Treatment Response. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2005, 11, 2637–2643. [Google Scholar] [CrossRef] [Green Version]
- Chakradeo, S.; Elmore, L.W.; Gewirtz, D.A. Is Senescence Reversible? Curr. Drug Targets 2016, 17, 460–466. [Google Scholar] [CrossRef] [PubMed]
- Eggert, T.; Wolter, K.; Ji, J.; Ma, C.; Yevsa, T.; Klotz, S.; Medina-Echeverz, J.; Longerich, T.; Forgues, M.; Reisinger, F.; et al. Distinct Functions of Senescence-Associated Immune Responses in Liver Tumor Surveillance and Tumor Progression. Cancer Cell 2016, 30, 533–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberson, R.S.; Kussick, S.J.; Vallieres, E.; Chen, S.-Y.J.; Wu, D.Y. Escape from Therapy-Induced Accelerated Cellular Senescence in P53-Null Lung Cancer Cells and in Human Lung Cancers. Cancer Res. 2005, 65, 2795–2803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.; Fang, J.; Chen, J. Tumor Cell Senescence Response Produces Aggressive Variants. Cell Death Discov. 2017, 3, 17049. [Google Scholar] [CrossRef] [PubMed]
- Milanovic, M.; Fan, D.N.Y.; Belenki, D.; Däbritz, J.H.M.; Zhao, Z.; Yu, Y.; Dörr, J.R.; Dimitrova, L.; Lenze, D.; Monteiro Barbosa, I.A.; et al. Senescence-Associated Reprogramming Promotes Cancer Stemness. Nature 2018, 553, 96–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milanovic, M.; Yu, Y.; Schmitt, C.A. The Senescence-Stemness Alliance-A Cancer-Hijacked Regeneration Principle. Trends Cell Biol. 2018, 28, 1049–1061. [Google Scholar] [CrossRef]
- Patel, P.L.; Suram, A.; Mirani, N.; Bischof, O.; Herbig, U. Derepression of HTERT Gene Expression Promotes Escape from Oncogene-Induced Cellular Senescence. Proc. Natl. Acad. Sci. USA 2016, 113, E5024–E5033. [Google Scholar] [CrossRef] [Green Version]
- Ozturk, N.; Erdal, E.; Mumcuoglu, M.; Akcali, K.C.; Yalcin, O.; Senturk, S.; Arslan-Ergul, A.; Gur, B.; Yulug, I.; Cetin-Atalay, R.; et al. Reprogramming of Replicative Senescence in Hepatocellular Carcinoma-Derived Cells. Proc. Natl. Acad. Sci. USA 2006, 103, 2178–2183. [Google Scholar] [CrossRef] [Green Version]
- Reyes, J.; Chen, J.-Y.; Stewart-Ornstein, J.; Karhohs, K.W.; Mock, C.S.; Lahav, G. Fluctuations in P53 Signaling Allow Escape from Cell-Cycle Arrest. Mol. Cell 2018, 71, 581–591.e5. [Google Scholar] [CrossRef] [Green Version]
- Sieben, C.J.; Sturmlechner, I.; van de Sluis, B.; van Deursen, J.M. Two-Step Senescence-Focused Cancer Therapies. Trends Cell Biol. 2018, 28, 723–737. [Google Scholar] [CrossRef] [Green Version]
- Childs, B.G.; Gluscevic, M.; Baker, D.J.; Laberge, R.-M.; Marquess, D.; Dananberg, J.; van Deursen, J.M. Senescent Cells: An Emerging Target for Diseases of Ageing. Nat. Rev. Drug Discov. 2017, 16, 718–735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- von Kobbe, C. Targeting Senescent Cells: Approaches, Opportunities, Challenges. Aging 2019, 11, 12844–12861. [Google Scholar] [CrossRef] [PubMed]
- Amaya-Montoya, M.; Pérez-Londoño, A.; Guatibonza-García, V.; Vargas-Villanueva, A.; Mendivil, C.O. Cellular Senescence as a Therapeutic Target for Age-Related Diseases: A Review. Adv. Ther. 2020, 37, 1407–1424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ovadya, Y.; Krizhanovsky, V. Strategies Targeting Cellular Senescence. J. Clin. Invest. 2018, 128, 1247–1254. [Google Scholar] [CrossRef] [PubMed]
- Lujambio, A. To Clear, or Not to Clear (Senescent Cells)? That Is the Question. BioEssays News Rev. Mol. Cell. Dev. Biol. 2016, 38 (Suppl. 1), S56–S64. [Google Scholar] [CrossRef] [PubMed]
- Prata, L.G.P.L.; Ovsyannikova, I.G.; Tchkonia, T.; Kirkland, J.L. Senescent Cell Clearance by the Immune System: Emerging Therapeutic Opportunities. Semin. Immunol. 2018, 40, 101275. [Google Scholar] [CrossRef] [PubMed]
- Saleh, T.; Tyutynuk-Massey, L.; Cudjoe, E.K.; Idowu, M.O.; Landry, J.W.; Gewirtz, D.A. Non-Cell Autonomous Effects of the Senescence-Associated Secretory Phenotype in Cancer Therapy. Front. Oncol. 2018, 8, 164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer Immunoediting: From Immunosurveillance to Tumor Escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef]
- Triana-Martínez, F.; Loza, M.I.; Domínguez, E. Beyond Tumor Suppression: Senescence in Cancer Stemness and Tumor Dormancy. Cells 2020, 9, 346. [Google Scholar] [CrossRef] [Green Version]
- Ritschka, B.; Storer, M.; Mas, A.; Heinzmann, F.; Ortells, M.C.; Morton, J.P.; Sansom, O.J.; Zender, L.; Keyes, W.M. The Senescence-Associated Secretory Phenotype Induces Cellular Plasticity and Tissue Regeneration. Genes Dev. 2017, 31, 172–183. [Google Scholar] [CrossRef] [Green Version]
- Laberge, R.-M.; Zhou, L.; Sarantos, M.R.; Rodier, F.; Freund, A.; de Keizer, P.L.J.; Liu, S.; Demaria, M.; Cong, Y.-S.; Kapahi, P.; et al. Glucocorticoids Suppress Selected Components of the Senescence-Associated Secretory Phenotype. Aging Cell 2012, 11, 569–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fontana, L.; Nehme, J.; Demaria, M. Caloric Restriction and Cellular Senescence. Mech. Ageing Dev. 2018, 176, 19–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grabowska, W.; Sikora, E.; Bielak-Zmijewska, A. Sirtuins, a Promising Target in Slowing down the Ageing Process. Biogerontology 2017, 18, 447–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Magalhães, J.P.; Passos, J.F. Stress, Cell Senescence and Organismal Ageing. Mech. Ageing Dev. 2018, 170, 2–9. [Google Scholar] [CrossRef]
- Kang, C.; Elledge, S.J. How Autophagy Both Activates and Inhibits Cellular Senescence. Autophagy 2016, 12, 898–899. [Google Scholar] [CrossRef]
- Bjedov, I.; Toivonen, J.M.; Kerr, F.; Slack, C.; Jacobson, J.; Foley, A.; Partridge, L. Mechanisms of Life Span Extension by Rapamycin in the Fruit Fly Drosophila Melanogaster. Cell Metab. 2010, 11, 35–46. [Google Scholar] [CrossRef] [Green Version]
- Miller, R.A.; Harrison, D.E.; Astle, C.M.; Fernandez, E.; Flurkey, K.; Han, M.; Javors, M.A.; Li, X.; Nadon, N.L.; Nelson, J.F.; et al. Rapamycin-Mediated Lifespan Increase in Mice Is Dose and Sex Dependent and Metabolically Distinct from Dietary Restriction. Aging Cell 2014, 13, 468–477. [Google Scholar] [CrossRef]
- Harrison, D.E.; Strong, R.; Sharp, Z.D.; Nelson, J.F.; Astle, C.M.; Flurkey, K.; Nadon, N.L.; Wilkinson, J.E.; Frenkel, K.; Carter, C.S.; et al. Rapamycin Fed Late in Life Extends Lifespan in Genetically Heterogeneous Mice. Nature 2009, 460, 392–395. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Yu, Z.; Sunchu, B.; Shoaf, J.; Dang, I.; Zhao, S.; Caples, K.; Bradley, L.; Beaver, L.M.; Ho, E.; et al. Rapamycin Inhibits the Secretory Phenotype of Senescent Cells by a Nrf2-Independent Mechanism. Aging Cell 2017, 16, 564–574. [Google Scholar] [CrossRef]
- Laberge, R.-M.; Sun, Y.; Orjalo, A.V.; Patil, C.K.; Freund, A.; Zhou, L.; Curran, S.C.; Davalos, A.R.; Wilson-Edell, K.A.; Liu, S.; et al. MTOR Regulates the Pro-Tumorigenic Senescence-Associated Secretory Phenotype by Promoting IL1A Translation. Nat. Cell Biol. 2015, 17, 1049–1061. [Google Scholar] [CrossRef]
- Alimbetov, D.; Davis, T.; Brook, A.J.C.; Cox, L.S.; Faragher, R.G.A.; Nurgozhin, T.; Zhumadilov, Z.; Kipling, D. Suppression of the Senescence-Associated Secretory Phenotype (SASP) in Human Fibroblasts Using Small Molecule Inhibitors of P38 MAP Kinase and MK2. Biogerontology 2016, 17, 305–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, H.T.; Park, J.T.; Choi, K.; Kim, Y.; Choi, H.J.C.; Jung, C.W.; Lee, Y.-S.; Park, S.C. Chemical Screening Identifies ATM as a Target for Alleviating Senescence. Nat. Chem. Biol. 2017, 13, 616–623. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Zhang, L.; Lu, A.; Han, Y.; Colangelo, D.; Bukata, C.; Scibetta, A.; Yousefzadeh, M.J.; Li, X.; Gurkar, A.U.; et al. ATM Is a Key Driver of NF-ΚB-Dependent DNA-Damage-Induced Senescence, Stem Cell Dysfunction and Aging. Aging 2020, 12, 4688–4710. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Fu, D.; Xu, Q.; Cong, X.; Wu, C.; Zhong, X.; Ma, Y.; Lv, Z.; Chen, F.; Han, L.; et al. The Senescence-Associated Secretory Phenotype Is Potentiated by Feedforward Regulatory Mechanisms Involving Zscan4 and TAK1. Nat. Commun. 2018, 9, 1723. [Google Scholar] [CrossRef]
- Camorani, S.; Cerchia, L.; Fedele, M.; Erba, E.; D’Incalci, M.; Crescenzi, E. Trabectedin Modulates the Senescence-Associated Secretory Phenotype and Promotes Cell Death in Senescent Tumor Cells by Targeting NF-ΚB. Oncotarget 2018, 9, 19929–19944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasznicki, J.; Sliwinska, A.; Drzewoski, J. Metformin in Cancer Prevention and Therapy. Ann. Transl. Med. 2014, 2, 57. [Google Scholar] [CrossRef]
- Saraei, P.; Asadi, I.; Kakar, M.A.; Moradi-Kor, N. The Beneficial Effects of Metformin on Cancer Prevention and Therapy: A Comprehensive Review of Recent Advances. Cancer Manag. Res. 2019, 11, 3295–3313. [Google Scholar] [CrossRef] [Green Version]
- Bailey, C.J.; Turner, R.C. Metformin. N. Engl. J. Med. 1996, 334, 574–579. [Google Scholar] [CrossRef]
- Kulkarni, A.S.; Gubbi, S.; Barzilai, N. Benefits of Metformin in Attenuating the Hallmarks of Aging. Cell Metab. 2020, 32, 15–30. [Google Scholar] [CrossRef]
- Hu, Q.; Peng, J.; Jiang, L.; Li, W.; Su, Q.; Zhang, J.; Li, H.; Song, M.; Cheng, B.; Xia, J.; et al. Metformin as a Senostatic Drug Enhances the Anticancer Efficacy of CDK4/6 Inhibitor in Head and Neck Squamous Cell Carcinoma. Cell Death Dis. 2020, 11, 925. [Google Scholar] [CrossRef]
- Soto-Gamez, A.; Demaria, M. Therapeutic Interventions for Aging: The Case of Cellular Senescence. Drug Discov. Today 2017, 22, 786–795. [Google Scholar] [CrossRef] [PubMed]
- Moiseeva, O.; Deschênes-Simard, X.; St-Germain, E.; Igelmann, S.; Huot, G.; Cadar, A.E.; Bourdeau, V.; Pollak, M.N.; Ferbeyre, G. Metformin Inhibits the Senescence-Associated Secretory Phenotype by Interfering with IKK/NF-ΚB Activation. Aging Cell 2013, 12, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Bian, Y.; Wei, J.; Zhao, C.; Li, G. Natural Polyphenols Targeting Senescence: A Novel Prevention and Therapy Strategy for Cancer. Int. J. Mol. Sci. 2020, 21, 684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velarde, M.C.; Demaria, M. Targeting Senescent Cells: Possible Implications for Delaying Skin Aging: A Mini-Review. Gerontology 2016, 62, 513–518. [Google Scholar] [CrossRef] [Green Version]
- Perrott, K.M.; Wiley, C.D.; Desprez, P.-Y.; Campisi, J. Apigenin Suppresses the Senescence-Associated Secretory Phenotype and Paracrine Effects on Breast Cancer Cells. GeroScience 2017, 39, 161–173. [Google Scholar] [CrossRef] [Green Version]
- Lim, H.; Park, H.; Kim, H.P. Effects of Flavonoids on Senescence-Associated Secretory Phenotype Formation from Bleomycin-Induced Senescence in BJ Fibroblasts. Biochem. Pharmacol. 2015, 96, 337–348. [Google Scholar] [CrossRef]
- Chen, G.Y.; Nuñez, G. Sterile Inflammation: Sensing and Reacting to Damage. Nat. Rev. Immunol. 2010, 10, 826–837. [Google Scholar] [CrossRef] [Green Version]
- Xu, M.; Tchkonia, T.; Ding, H.; Ogrodnik, M.; Lubbers, E.R.; Pirtskhalava, T.; White, T.A.; Johnson, K.O.; Stout, M.B.; Mezera, V.; et al. JAK Inhibition Alleviates the Cellular Senescence-Associated Secretory Phenotype and Frailty in Old Age. Proc. Natl. Acad. Sci. USA 2015, 112, E6301–E6310. [Google Scholar] [CrossRef] [Green Version]
- Griveau, A.; Wiel, C.; Ziegler, D.V.; Bergo, M.O.; Bernard, D. The JAK1/2 Inhibitor Ruxolitinib Delays Premature Aging Phenotypes. Aging Cell 2020, 19, e13122. [Google Scholar] [CrossRef] [Green Version]
- Rikitake, Y.; Liao, J.K. Rho GTPases, Statins, and Nitric Oxide. Circ. Res. 2005, 97, 1232–1235. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Uppal, H.; Demaria, M.; Desprez, P.-Y.; Campisi, J.; Kapahi, P. Simvastatin Suppresses Breast Cancer Cell Proliferation Induced by Senescent Cells. Sci. Rep. 2015, 5, 17895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuhrmann-Stroissnigg, H.; Ling, Y.Y.; Zhao, J.; McGowan, S.J.; Zhu, Y.; Brooks, R.W.; Grassi, D.; Gregg, S.Q.; Stripay, J.L.; Dorronsoro, A.; et al. Identification of HSP90 Inhibitors as a Novel Class of Senolytics. Nat. Commun. 2017, 8, 422. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Kawamoto, S.; Ohtani, N.; Hara, E. Impact of Senescence-Associated Secretory Phenotype and Its Potential as a Therapeutic Target for Senescence-Associated Diseases. Cancer Sci. 2017, 108, 563–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lau, L.; Porciuncula, A.; Yu, A.; Iwakura, Y.; David, G. Uncoupling the Senescence-Associated Secretory Phenotype from Cell Cycle Exit via Interleukin-1 Inactivation Unveils Its Protumorigenic Role. Mol. Cell. Biol. 2019, 39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prattichizzo, F.; De Nigris, V.; La Sala, L.; Procopio, A.D.; Olivieri, F.; Ceriello, A. “Inflammaging” as a Druggable Target: A Senescence-Associated Secretory Phenotype-Centered View of Type 2 Diabetes. Oxid. Med. Cell. Longev. 2016, 2016, 1810327. [Google Scholar] [CrossRef]
- Prattichizzo, F.; Giuliani, A.; Recchioni, R.; Bonafè, M.; Marcheselli, F.; De Carolis, S.; Campanati, A.; Giuliodori, K.; Rippo, M.R.; Brugè, F.; et al. Anti-TNF-α Treatment Modulates SASP and SASP-Related MicroRNAs in Endothelial Cells and in Circulating Angiogenic Cells. Oncotarget 2016, 7, 11945–11958. [Google Scholar] [CrossRef] [Green Version]
- Kirkland, J.L.; Tchkonia, T. Cellular Senescence: A Translational Perspective. EBioMedicine 2017, 21, 21–28. [Google Scholar] [CrossRef] [Green Version]
- Myrianthopoulos, V.; Evangelou, K.; Vasileiou, P.V.S.; Cooks, T.; Vassilakopoulos, T.P.; Pangalis, G.A.; Kouloukoussa, M.; Kittas, C.; Georgakilas, A.G.; Gorgoulis, V.G. Senescence and Senotherapeutics: A New Field in Cancer Therapy. Pharmacol. Ther. 2019, 193, 31–49. [Google Scholar] [CrossRef]
- Kirkland, J.L.; Tchkonia, T.; Zhu, Y.; Niedernhofer, L.J.; Robbins, P.D. The Clinical Potential of Senolytic Drugs. J. Am. Geriatr. Soc. 2017, 65, 2297–2301. [Google Scholar] [CrossRef]
- Hickson, L.J.; Langhi Prata, L.G.P.; Bobart, S.A.; Evans, T.K.; Giorgadze, N.; Hashmi, S.K.; Herrmann, S.M.; Jensen, M.D.; Jia, Q.; Jordan, K.L.; et al. Senolytics Decrease Senescent Cells in Humans: Preliminary Report from a Clinical Trial of Dasatinib plus Quercetin in Individuals with Diabetic Kidney Disease. EBioMedicine 2019, 47, 446–456. [Google Scholar] [CrossRef] [Green Version]
- Farr, J.N.; Xu, M.; Weivoda, M.M.; Monroe, D.G.; Fraser, D.G.; Onken, J.L.; Negley, B.A.; Sfeir, J.G.; Ogrodnik, M.B.; Hachfeld, C.M.; et al. Targeting Cellular Senescence Prevents Age-Related Bone Loss in Mice. Nat. Med. 2017, 23, 1072–1079. [Google Scholar] [CrossRef] [PubMed]
- Ogrodnik, M.; Miwa, S.; Tchkonia, T.; Tiniakos, D.; Wilson, C.L.; Lahat, A.; Day, C.P.; Burt, A.; Palmer, A.; Anstee, Q.M.; et al. Cellular Senescence Drives Age-Dependent Hepatic Steatosis. Nat. Commun. 2017, 8, 15691. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Pirtskhalava, T.; Farr, J.N.; Weigand, B.M.; Palmer, A.K.; Weivoda, M.M.; Inman, C.L.; Ogrodnik, M.B.; Hachfeld, C.M.; Fraser, D.G.; et al. Senolytics Improve Physical Function and Increase Lifespan in Old Age. Nat. Med. 2018, 24, 1246–1256. [Google Scholar] [CrossRef]
- Schafer, M.J.; White, T.A.; Iijima, K.; Haak, A.J.; Ligresti, G.; Atkinson, E.J.; Oberg, A.L.; Birch, J.; Salmonowicz, H.; Zhu, Y.; et al. Cellular Senescence Mediates Fibrotic Pulmonary Disease. Nat. Commun. 2017, 8, 14532. [Google Scholar] [CrossRef]
- Justice, J.N.; Nambiar, A.M.; Tchkonia, T.; LeBrasseur, N.K.; Pascual, R.; Hashmi, S.K.; Prata, L.; Masternak, M.M.; Kritchevsky, S.B.; Musi, N.; et al. Senolytics in Idiopathic Pulmonary Fibrosis: Results from a First-in-Human, Open-Label, Pilot Study. EBioMedicine 2019, 40, 554–563. [Google Scholar] [CrossRef] [Green Version]
- Chang, J.; Wang, Y.; Shao, L.; Laberge, R.-M.; Demaria, M.; Campisi, J.; Janakiraman, K.; Sharpless, N.E.; Ding, S.; Feng, W.; et al. Clearance of Senescent Cells by ABT263 Rejuvenates Aged Hematopoietic Stem Cells in Mice. Nat. Med. 2016, 22, 78–83. [Google Scholar] [CrossRef] [Green Version]
- Shahbandi, A.; Rao, S.G.; Anderson, A.Y.; Frey, W.D.; Olayiwola, J.O.; Ungerleider, N.A.; Jackson, J.G. BH3 Mimetics Selectively Eliminate Chemotherapy-Induced Senescent Cells and Improve Response in TP53 Wild-Type Breast Cancer. Cell Death Differ. 2020, 27, 3097–3116. [Google Scholar] [CrossRef]
- Zhu, Y.; Tchkonia, T.; Fuhrmann-Stroissnigg, H.; Dai, H.M.; Ling, Y.Y.; Stout, M.B.; Pirtskhalava, T.; Giorgadze, N.; Johnson, K.O.; Giles, C.B.; et al. Identification of a Novel Senolytic Agent, Navitoclax, Targeting the Bcl-2 Family of Anti-Apoptotic Factors. Aging Cell 2016, 15, 428–435. [Google Scholar] [CrossRef]
- Ritschka, B.; Knauer-Meyer, T.; Gonçalves, D.S.; Mas, A.; Plassat, J.-L.; Durik, M.; Jacobs, H.; Pedone, E.; Di Vicino, U.; Cosma, M.P.; et al. The Senotherapeutic Drug ABT-737 Disrupts Aberrant P21 Expression to Restore Liver Regeneration in Adult Mice. Genes Dev. 2020, 34, 489–494. [Google Scholar] [CrossRef]
- Yosef, R.; Pilpel, N.; Tokarsky-Amiel, R.; Biran, A.; Ovadya, Y.; Cohen, S.; Vadai, E.; Dassa, L.; Shahar, E.; Condiotti, R.; et al. Directed Elimination of Senescent Cells by Inhibition of BCL-W and BCL-XL. Nat. Commun. 2016, 7, 11190. [Google Scholar] [CrossRef]
- He, Y.; Zhang, X.; Chang, J.; Kim, H.-N.; Zhang, P.; Wang, Y.; Khan, S.; Liu, X.; Zhang, X.; Lv, D.; et al. Using Proteolysis-Targeting Chimera Technology to Reduce Navitoclax Platelet Toxicity and Improve Its Senolytic Activity. Nat. Commun. 2020, 11, 1996. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Doornebal, E.J.; Pirtskhalava, T.; Giorgadze, N.; Wentworth, M.; Fuhrmann-Stroissnigg, H.; Niedernhofer, L.J.; Robbins, P.D.; Tchkonia, T.; Kirkland, J.L. New Agents That Target Senescent Cells: The Flavone, Fisetin, and the BCL-XL Inhibitors, A1331852 and A1155463. Aging 2017, 9, 955–963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, P.-M.; Tseng, H.-H.; Peng, C.-W.; Chen, W.-S.; Chiu, S.-J. Dietary Flavonoid Fisetin Targets Caspase-3-Deficient Human Breast Cancer MCF-7 Cells by Induction of Caspase-7-Associated Apoptosis and Inhibition of Autophagy. Int. J. Oncol. 2012, 40, 469–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Youns, M.; Abdel Halim Hegazy, W. The Natural Flavonoid Fisetin Inhibits Cellular Proliferation of Hepatic, Colorectal, and Pancreatic Cancer Cells through Modulation of Multiple Signaling Pathways. PLoS ONE 2017, 12, e0169335. [Google Scholar] [CrossRef]
- Chen, Y.-C.; Shen, S.-C.; Lee, W.-R.; Lin, H.-Y.; Ko, C.-H.; Shih, C.-M.; Yang, L.-L. Wogonin and Fisetin Induction of Apoptosis through Activation of Caspase 3 Cascade and Alternative Expression of P21 Protein in Hepatocellular Carcinoma Cells SK-HEP-1. Arch. Toxicol. 2002, 76, 351–359. [Google Scholar] [CrossRef]
- Cho, H.-J.; Yang, E.J.; Park, J.T.; Kim, J.-R.; Kim, E.-C.; Jung, K.-J.; Park, S.C.; Lee, Y.-S. Identification of SYK Inhibitor, R406 as a Novel Senolytic Agent. Aging 2020, 12, 8221–8240. [Google Scholar] [CrossRef]
- Fuhrmann-Stroissnigg, H.; Niedernhofer, L.J.; Robbins, P.D. Hsp90 Inhibitors as Senolytic Drugs to Extend Healthy Aging. Cell Cycle Georget. Tex 2018, 17, 1048–1055. [Google Scholar] [CrossRef] [Green Version]
- Verzella, D.; Pescatore, A.; Capece, D.; Vecchiotti, D.; Ursini, M.V.; Franzoso, G.; Alesse, E.; Zazzeroni, F. Life, Death, and Autophagy in Cancer: NF-ΚB Turns up Everywhere. Cell Death Dis. 2020, 11, 210. [Google Scholar] [CrossRef] [Green Version]
- Pungsrinont, T.; Sutter, M.F.; Ertingshausen, M.C.C.M.; Lakshmana, G.; Kokal, M.; Khan, A.S.; Baniahmad, A. Senolytic Compounds Control a Distinct Fate of Androgen Receptor Agonist- and Antagonist-Induced Cellular Senescent LNCaP Prostate Cancer Cells. Cell Biosci. 2020, 10, 59. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Shi, Z.; Lin, J.; Zhao, S.; Hao, M.; Xu, J.; Li, Y.; Zhao, Q.; Tao, L.; Diao, A. Piperlongumine-Induced Nuclear Translocation of the FOXO3A Transcription Factor Triggers BIM-Mediated Apoptosis in Cancer Cells. Biochem. Pharmacol. 2019, 163, 101–110. [Google Scholar] [CrossRef]
- Liu, X.; Wang, Y.; Zhang, X.; Gao, Z.; Zhang, S.; Shi, P.; Zhang, X.; Song, L.; Hendrickson, H.; Zhou, D.; et al. Senolytic Activity of Piperlongumine Analogues: Synthesis and Biological Evaluation. Bioorg. Med. Chem. 2018, 26, 3925–3938. [Google Scholar] [CrossRef] [PubMed]
- Makhov, P.; Golovine, K.; Teper, E.; Kutikov, A.; Mehrazin, R.; Corcoran, A.; Tulin, A.; Uzzo, R.G.; Kolenko, V.M. Piperlongumine Promotes Autophagy via Inhibition of Akt/MTOR Signalling and Mediates Cancer Cell Death. Br. J. Cancer 2014, 110, 899–907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Zhang, S.; Liu, X.; Wang, Y.; Chang, J.; Zhang, X.; Mackintosh, S.G.; Tackett, A.J.; He, Y.; Lv, D.; et al. Oxidation Resistance 1 Is a Novel Senolytic Target. Aging Cell 2018, 17, e12780. [Google Scholar] [CrossRef]
- Wang, Y.; Chang, J.; Liu, X.; Zhang, X.; Zhang, S.; Zhang, X.; Zhou, D.; Zheng, G. Discovery of Piperlongumine as a Potential Novel Lead for the Development of Senolytic Agents. Aging 2016, 8, 2915–2926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samaraweera, L.; Adomako, A.; Rodriguez-Gabin, A.; McDaid, H.M. A Novel Indication for Panobinostat as a Senolytic Drug in NSCLC and HNSCC. Sci. Rep. 2017, 7, 1900. [Google Scholar] [CrossRef]
- He, Y.; Li, W.; Lv, D.; Zhang, X.; Zhang, X.; Ortiz, Y.T.; Budamagunta, V.; Campisi, J.; Zheng, G.; Zhou, D. Inhibition of USP7 Activity Selectively Eliminates Senescent Cells in Part via Restoration of P53 Activity. Aging Cell 2020, 19, e13117. [Google Scholar] [CrossRef]
- Vilgelm, A.E.; Pawlikowski, J.S.; Liu, Y.; Hawkins, O.E.; Davis, T.A.; Smith, J.; Weller, K.P.; Horton, L.W.; McClain, C.M.; Ayers, G.D.; et al. Mdm2 and Aurora Kinase a Inhibitors Synergize to Block Melanoma Growth by Driving Apoptosis and Immune Clearance of Tumor Cells. Cancer Res. 2015, 75, 181–193. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Tindall, D.J. Dynamic FoxO Transcription Factors. J. Cell Sci. 2007, 120, 2479–2487. [Google Scholar] [CrossRef] [Green Version]
- Greer, E.L.; Brunet, A. FOXO Transcription Factors at the Interface between Longevity and Tumor Suppression. Oncogene 2005, 24, 7410–7425. [Google Scholar] [CrossRef] [Green Version]
- Bourgeois, B.; Madl, T. Regulation of Cellular Senescence via the FOXO4-P53 Axis. FEBS Lett. 2018, 592, 2083–2097. [Google Scholar] [CrossRef] [Green Version]
- Baar, M.P.; Brandt, R.M.C.; Putavet, D.A.; Klein, J.D.D.; Derks, K.W.J.; Bourgeois, B.R.M.; Stryeck, S.; Rijksen, Y.; van Willigenburg, H.; Feijtel, D.A.; et al. Targeted Apoptosis of Senescent Cells Restores Tissue Homeostasis in Response to Chemotoxicity and Aging. Cell 2017, 169, 132–147.e16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dörr, J.R.; Yu, Y.; Milanovic, M.; Beuster, G.; Zasada, C.; Däbritz, J.H.M.; Lisec, J.; Lenze, D.; Gerhardt, A.; Schleicher, K.; et al. Synthetic Lethal Metabolic Targeting of Cellular Senescence in Cancer Therapy. Nature 2013, 501, 421–425. [Google Scholar] [CrossRef] [PubMed]
- Wakita, M.; Takahashi, A.; Sano, O.; Loo, T.M.; Imai, Y.; Narukawa, M.; Iwata, H.; Matsudaira, T.; Kawamoto, S.; Ohtani, N.; et al. A BET Family Protein Degrader Provokes Senolysis by Targeting NHEJ and Autophagy in Senescent Cells. Nat. Commun. 2020, 11, 1935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dostanic-Larson, I.; Van Huysse, J.W.; Lorenz, J.N.; Lingrel, J.B. The Highly Conserved Cardiac Glycoside Binding Site of Na,K-ATPase Plays a Role in Blood Pressure Regulation. Proc. Natl. Acad. Sci. USA 2005, 102, 15845–15850. [Google Scholar] [CrossRef] [Green Version]
- Seema Patel Plant-Derived Cardiac Glycosides: Role in Heart Ailments and Cancer Management. Biomed. Pharmacother. Biomed. Pharmacother. 2016, 84, 1036–1041. [CrossRef]
- Triana-Martínez, F.; Picallos-Rabina, P.; Da Silva-Álvarez, S.; Pietrocola, F.; Llanos, S.; Rodilla, V.; Soprano, E.; Pedrosa, P.; Ferreirós, A.; Barradas, M.; et al. Identification and Characterization of Cardiac Glycosides as Senolytic Compounds. Nat. Commun. 2019, 10, 4731. [Google Scholar] [CrossRef]
- Guerrero, A.; Herranz, N.; Sun, B.; Wagner, V.; Gallage, S.; Guiho, R.; Wolter, K.; Pombo, J.; Irvine, E.E.; Innes, A.J.; et al. Cardiac Glycosides Are Broad-Spectrum Senolytics. Nat. Metab. 2019, 1, 1074–1088. [Google Scholar] [CrossRef]
- Muñoz-Espín, D.; Rovira, M.; Galiana, I.; Giménez, C.; Lozano-Torres, B.; Paez-Ribes, M.; Llanos, S.; Chaib, S.; Muñoz-Martín, M.; Ucero, A.C.; et al. A Versatile Drug Delivery System Targeting Senescent Cells. EMBO Mol. Med. 2018, 10. [Google Scholar] [CrossRef]
- Galiana, I.; Lozano-Torres, B.; Sancho, M.; Alfonso, M.; Bernardos, A.; Bisbal, V.; Serrano, M.; Martínez-Máñez, R.; Orzáez, M. Preclinical Antitumor Efficacy of Senescence-Inducing Chemotherapy Combined with a NanoSenolytic. J. Control. Release Off. J. Control. Release Soc. 2020, 323, 624–634. [Google Scholar] [CrossRef]
- González-Gualda, E.; Pàez-Ribes, M.; Lozano-Torres, B.; Macias, D.; Wilson, J.R.; González-López, C.; Ou, H.-L.; Mirón-Barroso, S.; Zhang, Z.; Lérida-Viso, A.; et al. Galacto-Conjugation of Navitoclax as an Efficient Strategy to Increase Senolytic Specificity and Reduce Platelet Toxicity. Aging Cell 2020, 19, e13142. [Google Scholar] [CrossRef]
- Ekpenyong-Akiba, A.E.; Canfarotta, F.; Abd, H.B.; Poblocka, M.; Casulleras, M.; Castilla-Vallmanya, L.; Kocsis-Fodor, G.; Kelly, M.E.; Janus, J.; Althubiti, M.; et al. Detecting and Targeting Senescent Cells Using Molecularly Imprinted Nanoparticles. Nanoscale Horiz. 2019, 4, 757–768. [Google Scholar] [CrossRef]
- Ke, S.; Lai, Y.; Zhou, T.; Li, L.; Wang, Y.; Ren, L.; Ye, S. Molybdenum Disulfide Nanoparticles Resist Oxidative Stress-Mediated Impairment of Autophagic Flux and Mitigate Endothelial Cell Senescence and Angiogenic Dysfunctions. ACS Biomater. Sci. Eng. 2018, 4, 663–674. [Google Scholar] [CrossRef] [PubMed]
- Thapa, R.K.; Nguyen, H.T.; Jeong, J.-H.; Kim, J.R.; Choi, H.-G.; Yong, C.S.; Kim, J.O. Progressive Slowdown/Prevention of Cellular Senescence by CD9-Targeted Delivery of Rapamycin Using Lactose-Wrapped Calcium Carbonate Nanoparticles. Sci. Rep. 2017, 7, 43299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lozano-Torres, B.; Blandez, J.F.; Sancenón, F.; Martínez-Máñez, R. Novel Probes and Carriers to Target Senescent Cells. In Senolytics in Disease, Ageing and Longevity; Muñoz-Espin, D., Demaria, M., Eds.; Healthy Ageing and Longevity; Springer International Publishing: Cham, Switzerland, 2020; Volume 11, pp. 163–180. ISBN 978-3-030-44902-5. [Google Scholar]
- Guerrero, A.; Guiho, R.; Herranz, N.; Uren, A.; Withers, D.J.; Martínez-Barbera, J.P.; Tietze, L.F.; Gil, J. Galactose-Modified Duocarmycin Prodrugs as Senolytics. Aging Cell 2020, 19, e13133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, Y.; Zhou, H.; Zhu, Y.; Sun, Q.; Ji, Y.; Xue, A.; Wang, Y.; Chen, W.; Yu, X.; Wang, L.; et al. Elimination of Senescent Cells by β-Galactosidase-Targeted Prodrug Attenuates Inflammation and Restores Physical Function in Aged Mice. Cell Res. 2020, 30, 574–589. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Demirci, D.; Dayanc, B.; Mazi, F.A.; Senturk, S. The Jekyll and Hyde of Cellular Senescence in Cancer. Cells 2021, 10, 208. https://doi.org/10.3390/cells10020208
Demirci D, Dayanc B, Mazi FA, Senturk S. The Jekyll and Hyde of Cellular Senescence in Cancer. Cells. 2021; 10(2):208. https://doi.org/10.3390/cells10020208
Chicago/Turabian StyleDemirci, Dilara, Bengisu Dayanc, Fatma Aybuke Mazi, and Serif Senturk. 2021. "The Jekyll and Hyde of Cellular Senescence in Cancer" Cells 10, no. 2: 208. https://doi.org/10.3390/cells10020208
APA StyleDemirci, D., Dayanc, B., Mazi, F. A., & Senturk, S. (2021). The Jekyll and Hyde of Cellular Senescence in Cancer. Cells, 10(2), 208. https://doi.org/10.3390/cells10020208