Using iPSC Models to Understand the Role of Estrogen in Neuron–Glia Interactions in Schizophrenia and Bipolar Disorder

 , , , ,
, , , ,
Abstract
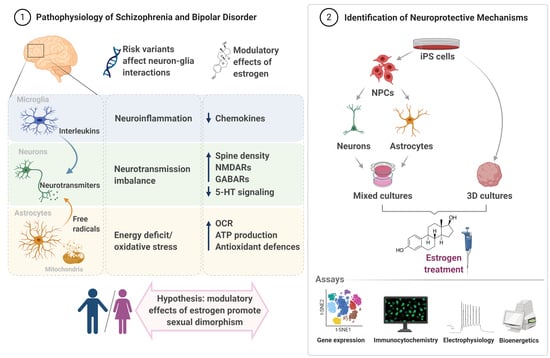
:
1. Introduction
2. Estrogen and Estrogen Receptors in the Brain
3. Genetics of Schizophrenia and Bipolar Disorder
4. Brain Energy Metabolism in Schizophrenia and Bipolar Disorder
4.1. Brain Energy Metabolism Overview
4.2. Brain Energy Metabolism in SCZ and BIP
4.3. Influence of Estrogen on the Mitochondrial Metabolism
5. Neuroinflammation in Schizophrenia and Bipolar Disorder
5.1. Neuroinflammation Overview
5.2. Immune Responses and Inflammatory Glial Functions in SCZ and BIP
5.3. The Effect of Sex Hormones on the Inflammatory Responses of Glial Cells: What Do We Know so Far?
6. Neurotransmission in Schizophrenia and Bipolar Disorder
6.1. Altered Neurotransmission Pathways in SCZ and BIP
6.1.1. The Monoamine Theory
6.1.2. The Glutamatergic and the GABAergic Hypotheses
6.2. The Role of Astrocytes in Neurotransmission
6.3. The Effect of Estrogen on Neurotransmission
7. Transcriptional Effects of Estrogen
8. Induced Pluripotent Stem Cell (iPSC) Models to Study Neuron–Glia Interactions
9. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Whiteford, H.A.; Degenhardt, L.; Rehm, J.; Baxter, A.J.; Ferrari, A.J.; Erskine, H.E.; Charlson, F.J.; Norman, R.E.; Flaxman, A.D.; Johns, N.; et al. Global burden of disease attributable to mental and substance use disorders: Findings from the Global Burden of Disease Study 2010. Lancet 2013, 382, 1575–1586. [Google Scholar] [CrossRef]
- Kessler, R.C.; Angermeyer, M.; Anthony, J.C.; De Graaf, R.; Demyttenaere, K.; Gasquet, I.; De Girolamo, G.; Gluzman, S.; Gureje, O.; Haro, J.M.; et al. Lifetime prevalence and age-of-onset distributions of mental disorders in the World Health Organization’s World Mental Health Survey Initiative. World Psychiatry 2007, 6, 168–176. [Google Scholar] [PubMed]
- Komatsu, H.; Fukuchi, M.; Habata, Y. Potential Utility of Biased GPCR Signaling for Treatment of Psychiatric Disorders. Int. J. Mol. Sci. 2019, 20, 3207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanaway, J.D.; Afshin, A.; Gakidou, E.; Lim, S.S.; Abate, D.; Abate, K.H.; Abbafati, C.; Abbasi, N.; Abbastabar, H.; Abd-Allah, F.; et al. Global, regional, and national comparative risk assessment of 84 behavioural, environmental and occupational, and metabolic risks or clusters of risks for 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1923–1994. [Google Scholar] [CrossRef] [Green Version]
- Vigo, D.; Thornicroft, G.; Atun, R. Estimating the true global burden of mental illness. Lancet Psychiatry 2016, 3, 171–178. [Google Scholar] [CrossRef] [Green Version]
- Freedman, R. Schizophrenia. N. Engl. J. Med. 2003, 349, 1738–1749. [Google Scholar] [CrossRef]
- Grande, I.; Berk, M.; Birmaher, B.; Vieta, E. Bipolar disorder. Lancet 2016, 387, 1561–1572. [Google Scholar] [CrossRef]
- Laursen, T.M. Life expectancy among persons with schizophrenia or bipolar affective disorder. Schizophr. Res. 2011, 131, 101–104. [Google Scholar] [CrossRef]
- Lichtenstein, P.; Yip, B.H.; Björk, C.; Pawitan, Y.; Cannon, T.D.; Sullivan, P.F.; Hultman, C.M. Common genetic determinants of schizophrenia and bipolar disorder in Swedish families: A population-based study. Lancet 2009, 373, 234–239. [Google Scholar] [CrossRef] [Green Version]
- Hill, R.A. Interaction of Sex Steroid Hormones and Brain-Derived Neurotrophic Factor-Tyrosine Kinase B Signalling: Relevance to Schizophrenia and Depression. J. Neuroendocrinol. 2012, 24, 1553–1561. [Google Scholar] [CrossRef]
- Abel, K.M.; Drake, R.; Goldstein, J.M. Sex differences in schizophrenia. Int. Rev. Psychiatry 2010, 22, 417–428. [Google Scholar] [CrossRef] [PubMed]
- Pinares-Garcia, P.; Stratikopoulos, M.; Zagato, A.; Loke, H.; Lee, J. Sex: A Significant Risk Factor for Neurodevelopmental and Neurodegenerative Disorders. Brain Sci. 2018, 8, 154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markham, J.A. Sex steroids and schizophrenia. Rev. Endocr. Metab. Disord. 2012, 13, 187–207. [Google Scholar] [CrossRef] [PubMed]
- Gogos, A.; Ney, L.J.; Seymour, N.; Van Rheenen, T.E.; Felmingham, K.L. Sex differences in schizophrenia, bipolar disorder, and post-traumatic stress disorder: Are gonadal hormones the link? Br. J. Pharmacol. 2019, 176, 4119–4135. [Google Scholar] [CrossRef] [PubMed]
- Owens, S.J.; Purves-Tyson, T.D.; Webster, M.J.; Shannon Weickert, C. Evidence for enhanced androgen action in the prefrontal cortex of people with bipolar disorder but not schizophrenia or major depressive disorder. Psychiatry Res. 2019, 280, 112503. [Google Scholar] [CrossRef]
- Meier, S.M.; Kähler, A.K.; Bergen, S.E.; Sullivan, P.F.; Hultman, C.M.; Mattheisen, M. Chronicity and Sex Affect Genetic Risk Prediction in Schizophrenia. Front. Psychiatry 2020, 11, 313. [Google Scholar] [CrossRef]
- Taylor, C.M.; Pritschet, L.; Yu, S.; Jacobs, E.G. Applying a Women’s Health Lens to the Study of the Aging Brain. Front. Hum. Neurosci. 2019, 13, 224. [Google Scholar] [CrossRef]
- Wise, P.M.; Dubal, D.B.; Wilson, M.E.; Rau, S.W.; Böttner, M. Minireview: Neuroprotective effects of estrogen-new insights into mechanisms of action. Endocrinology 2001, 142, 969–973. [Google Scholar] [CrossRef]
- Lee, J.; Pinares-Garcia, P.; Loke, H.; Ham, S.; Vilain, E.; Harley, V.R. Sex-specific neuroprotection by inhibition of the Y-chromosome gene, SRY, in experimental Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2019, 116, 16577–16582. [Google Scholar] [CrossRef] [Green Version]
- Seeman, M.V. Men and women respond differently to antipsychotic drugs. Neuropharmacology 2020, 163, 107631. [Google Scholar] [CrossRef]
- van der Leeuw, C.; Habets, P.; Gronenschild, E.; Domen, P.; Michielse, S.; van Kroonenburgh, M.; van Os, J.; Marcelis, M. Testing the estrogen hypothesis of schizophrenia: Associations between cumulative estrogen exposure and cerebral structural measures. Schizophr. Res. 2013, 150, 114–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, M.M. Translational Significance of Selective Estrogen Receptor Modulators in Psychiatric Disorders. Int. J. Endocrinol. 2018, 2018, 9516592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulkarni, J.; Butler, S.; Riecher-Rössler, A. Estrogens and SERMS as adjunctive treatments for schizophrenia. Front. Neuroendocrinol. 2019, 53, 100743. [Google Scholar] [CrossRef] [PubMed]
- Riecher-Rössler, A. Sex and gender differences in mental disorders. Lancet Psychiatry 2017, 4, 8–9. [Google Scholar] [CrossRef]
- Acaz-Fonseca, E.; Sanchez-Gonzalez, R.; Azcoitia, I.; Arevalo, M.A.; Garcia-Segura, L.M. Role of astrocytes in the neuroprotective actions of 17β-estradiol and selective estrogen receptor modulators. Mol. Cell. Endocrinol. 2014, 389, 48–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sortino, M.A.; Chisari, M.; Merlo, S.; Vancheri, C.; Caruso, M.; Nicoletti, F.; Canonico, P.L.; Copani, A. Glia Mediates the Neuroprotective Action of Estradiol on β-Amyloid-Induced Neuronal Death. Endocrinology 2004, 145, 5080–5086. [Google Scholar] [CrossRef]
- McGregor, C.; Riordan, A.; Thornton, J. Estrogens and the cognitive symptoms of schizophrenia: Possible neuroprotective mechanisms. Front. Neuroendocrinol. 2017, 47, 19–33. [Google Scholar] [CrossRef]
- Falk, A.; Heine, V.M.; Harwood, A.J.; Sullivan, P.F.; Peitz, M.; Brüstle, O.; Shen, S.; Sun, Y.M.; Glover, J.C.; Posthuma, D.; et al. Modeling psychiatric disorders: From genomic findings to cellular phenotypes. Mol. Psychiatry 2016, 21, 1167. [Google Scholar] [CrossRef] [Green Version]
- Brennand, K.; Savas, J.N.; Kim, Y.; Tran, N.; Simone, A.; Hashimoto-Torii, K.; Beaumont, K.G.; Kim, H.J.; Topol, A.; Ladran, I.; et al. Phenotypic differences in hiPSC NPCs derived from patients with schizophrenia. Mol. Psychiatry 2015, 20, 361–368. [Google Scholar] [CrossRef] [Green Version]
- Akkouh, I.A.; Ueland, T.; Hansson, L.; Inderhaug, E.; Hughes, T.; Steen, N.E.; Aukrust, P.; Andreassen, O.A.; Szabo, A.; Djurovic, S. Decreased IL-1β-induced CCL20 response in human iPSC-astrocytes in schizophrenia: Potential attenuating effects on recruitment of regulatory T cells. Brain Behav. Immun. 2020. [CrossRef]
- Mertens, J.; Wang, Q.-W.; Kim, Y.; Yu, D.X.; Pham, S.; Yang, B.; Zheng, Y.; Diffenderfer, K.E.; Zhang, J.; Soltani, S.; et al. Differential responses to lithium in hyperexcitable neurons from patients with bipolar disorder. Nature 2015, 527, 95–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lancaster, M.A.; Knoblich, J.A. Generation of cerebral organoids from human pluripotent stem cells. Nat. Protoc. 2014, 9, 2329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birey, F.; Andersen, J.; Makinson, C.D.; Islam, S.; Wei, W.; Huber, N.; Fan, H.C.; Metzler, K.R.C.; Panagiotakos, G.; Thom, N.; et al. Assembly of functionally integrated human forebrain spheroids. Nature 2017, 545, 54–59. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Gibbs, R.B. Detection of estradiol in rat brain tissues: Contribution of local versus systemic production. Psychoneuroendocrinology 2019, 102, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Fuentes, N.; Silveyra, P. Estrogen receptor signaling mechanisms. Adv. Protein Chem. Struct. Biol. 2019, 116, 135–170. [Google Scholar] [CrossRef] [PubMed]
- Marino, M.; Galluzzo, P.; Ascenzi, P. Estrogen signaling multiple pathways to impact gene transcription. Curr. Genom. 2006, 7, 497–508. [Google Scholar] [CrossRef] [Green Version]
- Foster, T.C. Role of estrogen receptor alpha and beta expression and signaling on cognitive function during aging. Hippocampus 2012, 22, 656–669. [Google Scholar] [CrossRef] [Green Version]
- Osterlund, M.K.; Keller, E.; Hurd, Y.L. The human forebrain has discrete estrogen receptor alpha messenger RNA expression: High levels in the amygdaloid complex. Neuroscience 2000, 95, 333–342. [Google Scholar] [CrossRef]
- González, M.; Cabrera-Socorro, A.; Pérez-García, C.G.; Fraser, J.D.; López, F.J.; Alonso, R.; Meyer, G. Distribution patterns of estrogen receptor alpha and beta in the human cortex and hippocampus during development and adulthood. J. Comp. Neurol. 2007, 503, 790–802. [Google Scholar] [CrossRef]
- Shughrue, P.J.; Lane, M.V.; Merchenthaler, I. Comparative distribution of estrogen receptor-alpha and -beta mRNA in the rat central nervous system. J. Comp. Neurol. 1997, 388, 507–525. [Google Scholar] [CrossRef]
- Osterlund, M.K.; Hurd, Y.L. Estrogen receptors in the human forebrain and the relation to neuropsychiatric disorders. Prog. Neurobiol. 2001, 64, 251–267. [Google Scholar] [CrossRef]
- Ostlund, H.; Keller, E.; Hurd, Y.L. Estrogen receptor gene expression in relation to neuropsychiatric disorders. Ann. N. Y. Acad. Sci. 2003, 1007, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Mitra, S.W.; Hoskin, E.; Yudkovitz, J.; Pear, L.; Wilkinson, H.A.; Hayashi, S.; Pfaff, D.W.; Ogawa, S.; Rohrer, S.P.; Schaeffer, J.M.; et al. Immunolocalization of estrogen receptor beta in the mouse brain: Comparison with estrogen receptor alpha. Endocrinology 2003, 144, 2055–2067. [Google Scholar] [CrossRef] [PubMed]
- Gaudet, H.M.; Cheng, S.B.; Christensen, E.M.; Filardo, E.J. The G-protein coupled estrogen receptor, GPER: The inside and inside-out story. Mol. Cell. Endocrinol. 2015, 418 Pt 3, 207–219. [Google Scholar] [CrossRef]
- Toran-Allerand, C.D.; Guan, X.; MacLusky, N.J.; Horvath, T.L.; Diano, S.; Singh, M.; Connolly, E.S., Jr.; Nethrapalli, I.S.; Tinnikov, A.A. ER-X: A novel, plasma membrane-associated, putative estrogen receptor that is regulated during development and after ischemic brain injury. J. Neurosci. 2002, 22, 8391–8401. [Google Scholar] [CrossRef]
- Weickert, C.S.; Miranda-Angulo, A.L.; Wong, J.; Perlman, W.R.; Ward, S.E.; Radhakrishna, V.; Straub, R.E.; Weinberger, D.R.; Kleinman, J.E. Variants in the estrogen receptor alpha gene and its mRNA contribute to risk for schizophrenia. Hum. Mol. Genet. 2008, 17, 2293–2309. [Google Scholar] [CrossRef]
- Min, J.A.; Kim, J.J.; Pae, C.U.; Kim, K.H.; Lee, C.U.; Lee, C.; Paik, I.H. Association of estrogen receptor genes and schizophrenia: A preliminary study. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2012, 36, 1–4. [Google Scholar] [CrossRef]
- Perlman, W.R.; Tomaskovic-Crook, E.; Montague, D.M.; Webster, M.J.; Rubinow, D.R.; Kleinman, J.E.; Weickert, C.S. Alteration in estrogen receptor alpha mRNA levels in frontal cortex and hippocampus of patients with major mental illness. Biol. Psychiatry 2005, 58, 812–824. [Google Scholar] [CrossRef]
- Blurton-Jones, M.; Tuszynski, M.H. Estrogen receptor-beta colocalizes extensively with parvalbumin-labeled inhibitory neurons in the cortex, amygdala, basal forebrain, and hippocampal formation of intact and ovariectomized adult rats. J. Comp. Neurol. 2002, 452, 276–287. [Google Scholar] [CrossRef]
- Zhang, Z.J.; Reynolds, G.P. A selective decrease in the relative density of parvalbumin-immunoreactive neurons in the hippocampus in schizophrenia. Schizophr. Res. 2002, 55, 1–10. [Google Scholar] [CrossRef]
- Zhang, X.Y.; Chen, D.C.; Xiu, M.H.; Yang, F.D.; Haile, C.N.; Kosten, T.A.; Kosten, T.R. Gender differences in never-medicated first-episode schizophrenia and medicated chronic schizophrenia patients. J. Clin. Psychiatry 2012, 73, 1025–1033. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, K.; Zsiros, V.; Jiang, Z.; Nakao, K.; Kolata, S.; Zhang, S.; Belforte, J.E. GABAergic interneuron origin of schizophrenia pathophysiology. Neuropharmacology 2012, 62, 1574–1583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sullivan, P.F.; Kendler, K.S.; Neale, M.C. Schizophrenia as a complex trait: Evidence from a meta-analysis of twin studies. Arch. Gen. Psychiatry 2003, 60, 1187–1192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hilker, R.; Helenius, D.; Fagerlund, B.; Skytthe, A.; Christensen, K.; Werge, T.M.; Nordentoft, M.; Glenthøj, B. Heritability of Schizophrenia and Schizophrenia Spectrum Based on the Nationwide Danish Twin Register. Biol. Psychiatry 2018, 83, 492–498. [Google Scholar] [CrossRef] [Green Version]
- Smoller, J.W.; Finn, C.T. Family, twin, and adoption studies of bipolar disorder. Am. J. Med Genet. Part C Semin. Med. Genet. 2003, 123, 48–58. [Google Scholar] [CrossRef]
- Johansson, V.; Kuja-Halkola, R.; Cannon, T.D.; Hultman, C.M.; Hedman, A.M. A population-based heritability estimate of bipolar disorder—In a Swedish twin sample. Psychiatry Res. 2019, 278, 180–187. [Google Scholar] [CrossRef]
- A framework for interpreting genome-wide association studies of psychiatric disorders. Mol. Psychiatry 2009, 14, 10–17. [CrossRef] [Green Version]
- Lam, M.; Chen, C.Y.; Li, Z.; Martin, A.R.; Bryois, J.; Ma, X.; Gaspar, H.; Ikeda, M.; Benyamin, B.; Brown, B.C.; et al. Comparative genetic architectures of schizophrenia in East Asian and European populations. Nat. Genet. 2019, 51, 1670–1678. [Google Scholar] [CrossRef]
- Stahl, E.A.; Breen, G.; Forstner, A.J.; McQuillin, A.; Ripke, S.; Trubetskoy, V.; Mattheisen, M.; Wang, Y.; Coleman, J.R.I.; Gaspar, H.A.; et al. Genome-wide association study identifies 30 Loci Associated with Bipolar Disorder. bioRxiv 2018, 173062. [Google Scholar] [CrossRef]
- Smeland, O.B.; Frei, O.; Dale, A.M.; Andreassen, O.A. The polygenic architecture of schizophrenia—Rethinking pathogenesis and nosology. Nat. Rev. Neurol. 2020, 16, 366–379. [Google Scholar] [CrossRef]
- Schizophrenia Working Group of the Psychiatric Genomics Consortium; Ripke, S.; Neale, B.M.; Corvin, A.; Walters, J.T.R.; Farh, K.-H.; Holmans, P.A.; Lee, P.; Bulik-Sullivan, B.; Collier, D.A.; et al. Biological insights from 108 schizophrenia-associated genetic loci. Nature 2014, 511, 421. [Google Scholar] [CrossRef] [Green Version]
- Ripke, S.; Walters, J.T.; O’Donovan, M.C. Mapping genomic loci prioritises genes and implicates synaptic biology in schizophrenia. MedRxiv 2020. [CrossRef]
- Wray, N.R.; Ripke, S.; Mattheisen, M.; Trzaskowski, M.; Byrne, E.M.; Abdellaoui, A.; Adams, M.J.; Agerbo, E.; Air, T.M.; Andlauer, T.M.F.; et al. Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nat. Genet. 2018, 50, 668–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mäki-Marttunen, T.; Halnes, G.; Devor, A.; Witoelar, A.; Bettella, F.; Djurovic, S.; Wang, Y.; Einevoll, G.T.; Andreassen, O.A.; Dale, A.M. Functional Effects of Schizophrenia-Linked Genetic Variants on Intrinsic Single-Neuron Excitability: A Modeling Study. Biol. Psychiatry Cogn. Neurosci. Neuroimaging 2016, 1, 49–59. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Fan, C.C.; Mäki-Marttunen, T.; Thompson, W.K.; Schork, A.J.; Bettella, F.; Consortium, S.W.G.o.t.P.G.; Djurovic, S.; Dale, A.M.; Andreassen, O.A.; et al. A molecule-based genetic association approach implicates a range of voltage-gated calcium channels associated with schizophrenia. Am. J. Med. Genet. Part B Neuropsychi Genet. 2018, 177, 454–467. [Google Scholar] [CrossRef]
- Smeland, O.B.; Bahrami, S.; Frei, O.; Shadrin, A.; O’Connell, K.; Savage, J.; Watanabe, K.; Krull, F.; Bettella, F.; Steen, N.E.; et al. Genome-wide analysis reveals extensive genetic overlap between schizophrenia, bipolar disorder, and intelligence. Mol. Psychiatry 2019, 1. [Google Scholar] [CrossRef] [Green Version]
- Mullins, N.; Forstner, A.J.; O’Connell, K.S.; Coombes, B.; Coleman, J.R.I.; Qiao, Z.; Als, T.D.; Bigdeli, T.B.; Børte, S.; Bryois, J.; et al. Genome-wide association study of over 40,000 bipolar disorder cases provides novel biological insights. MedRxiv 2020. [CrossRef]
- Battle, A.; Brown, C.D.; Engelhardt, B.E.; Montgomery, S.B. Genetic effects on gene expression across human tissues. Nature 2017, 550, 204–213. [Google Scholar] [CrossRef]
- Fromer, M.; Roussos, P.; Sieberts, S.K.; Johnson, J.S.; Kavanagh, D.H.; Perumal, T.M.; Ruderfer, D.M.; Oh, E.C.; Topol, A.; Shah, H.R.; et al. Gene expression elucidates functional impact of polygenic risk for schizophrenia. Nat. Neurosci. 2016, 19, 1442–1453. [Google Scholar] [CrossRef] [Green Version]
- Wang, D.; Liu, S.; Warrell, J.; Won, H.; Shi, X.; Navarro, F.C.P.; Clarke, D.; Gu, M.; Emani, P.; Yang, Y.T.; et al. Comprehensive functional genomic resource and integrative model for the human brain. Science 2018, 362. [Google Scholar] [CrossRef] [Green Version]
- Gandal, M.J.; Haney, J.R.; Parikshak, N.N.; Leppa, V.; Ramaswami, G.; Hartl, C.; Schork, A.J.; Appadurai, V.; Buil, A.; Werge, T.M.; et al. Shared molecular neuropathology across major psychiatric disorders parallels polygenic overlap. Science 2018, 359, 693–697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuccoli, G.S.; Saia-Cereda, V.M.; Nascimento, J.M.; Martins-de-Souza, D. The Energy Metabolism Dysfunction in Psychiatric Disorders Postmortem Brains: Focus on Proteomic Evidence. Front. Neurosci. 2017, 11. [Google Scholar] [CrossRef] [PubMed]
- Afridi, R.; Kim, J.-H.; Rahman, M.H.; Suk, K. Metabolic Regulation of Glial Phenotypes: Implications in Neuron-Glia Interactions and Neurological Disorders. Front. Cell. Neurosci. 2020, 14, 20. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Kim, J.H.; Song, G.G. Pathway analysis of a genome-wide association study in schizophrenia. Gene 2013, 525, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Smeeth, D.M.; Dima, D.; Jones, L.; Jones, I.; Craddock, N.; Owen, M.J.; Rietschel, M.; Maier, W.; Korszun, A.; Rice, J.P.; et al. Polygenic risk for circulating reproductive hormone levels and their influence on hippocampal volume and depression susceptibility. Psychoneuroendocrinology 2019, 106, 284–292. [Google Scholar] [CrossRef]
- Levey, D.F.; Gelernter, J.; Polimanti, R.; Zhou, H.; Cheng, Z.; Aslan, M.; Quaden, R.; Concato, J.; Radhakrishnan, K.; Bryois, J.; et al. Reproducible Genetic Risk Loci for Anxiety: Results From ∼200,000 Participants in the Million Veteran Program. Am. J. Psychiatry 2020, 177, 223–232. [Google Scholar] [CrossRef]
- Pinsonneault, J.K.; Frater, J.T.; Kompa, B.; Mascarenhas, R.; Wang, D.; Sadee, W. Intronic SNP in ESR1 encoding human estrogen receptor alpha is associated with brain ESR1 mRNA isoform expression and behavioral traits. PLoS ONE 2017, 12, e0179020. [Google Scholar] [CrossRef] [Green Version]
- Bordone, M.P.; Salman, M.M.; Titus, H.E.; Amini, E.; Andersen, J.V.; Chakraborti, B.; Diuba, A.V.; Dubouskaya, T.G.; Ehrke, E.; Espindola de Freitas, A.; et al. The energetic brain—A review from students to students. J. Neurochem. 2019, 151, 139–165. [Google Scholar] [CrossRef]
- de Lores Arnaiz, G.R.; Ordieres, M.G. Brain Na(+), K(+)-ATPase Activity In Aging and Disease. Int. J. Biomed. Sci. IJBS 2014, 10, 85–102. [Google Scholar]
- Stepanova, A.; Konrad, C.; Manfredi, G.; Springett, R.; Ten, V.; Galkin, A. The dependence of brain mitochondria reactive oxygen species production on oxygen level is linear, except when inhibited by antimycin A. J. Neurochem. 2019, 148, 731–745. [Google Scholar] [CrossRef] [Green Version]
- McAvoy, K.; Kawamata, H. Glial mitochondrial function and dysfunction in health and neurodegeneration. Mol. Cell. Neurosci. 2019, 101, 103417. [Google Scholar] [CrossRef] [PubMed]
- Ioannou, M.S.; Jackson, J.; Sheu, S.H.; Chang, C.L.; Weigel, A.V.; Liu, H.; Pasolli, H.A.; Xu, C.S.; Pang, S.; Matthies, D.; et al. Neuron-Astrocyte Metabolic Coupling Protects against Activity-Induced Fatty Acid Toxicity. Cell 2019, 177, 1522–1535. [Google Scholar] [CrossRef] [PubMed]
- Pellerin, L.; Magistretti, P.J. Glutamate uptake into astrocytes stimulates aerobic glycolysis: A mechanism coupling neuronal activity to glucose utilization. Proc. Natl. Acad. Sci. USA 1994, 91, 10625–10629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, M.B.; Jackson, J.G. Astroglial glutamate transporters coordinate excitatory signaling and brain energetics. Neurochem. Int. 2016, 98, 56–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dienel, G.A. Brain lactate metabolism: The discoveries and the controversies. J. Cereb. Blood Flow Metab. 2012, 32, 1107–1138. [Google Scholar] [CrossRef] [Green Version]
- Chamberlain, K.A.; Sheng, Z.H. Mechanisms for the maintenance and regulation of axonal energy supply. J. Neurosci. Res. 2019, 97, 897–913. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.F.; Shao, L.; Sun, X.; Young, L.T. Increased oxidative stress in the anterior cingulate cortex of subjects with bipolar disorder and schizophrenia. Bipolar Disord. 2009, 11, 523–529. [Google Scholar] [CrossRef]
- Manji, H.; Kato, T.; Di Prospero, N.A.; Ness, S.; Beal, M.F.; Krams, M.; Chen, G. Impaired mitochondrial function in psychiatric disorders. Nat. Rev. Neurosci. 2012, 13, 293–307. [Google Scholar] [CrossRef]
- Paulsen, B.; de Moraes, R.; Galina, A.; Souza, M.; dos Santos, C.; Drummond, H.; Nascimento, E.; Silva, J.; Chicaybam, L.; Massuda, R.; et al. Altered oxygen metabolism associated to neurogenesis of induced pluripotent stem cells derived from a schizophrenic patient. Cell Transplant. 2012, 21, 1547–1559. [Google Scholar] [CrossRef] [Green Version]
- Paulsen, B.; Silveira, M.; Galina, A.; Rehen, S. Pluripotent stem cells as a model to study oxygen metabolism in neurogenesis and neurodevelopmental disorders. Arch. Biochem. Biophys. 2013, 534, 3–10. [Google Scholar] [CrossRef]
- Rajasekaran, A.; Venkatasubramanian, G.; Berk, M.; Debnath, M. Mitochondrial dysfunction in schizophrenia: Pathways, mechanisms and implications. Neurosci. Biobehav. Rev. 2015, 48, 10–21. [Google Scholar] [CrossRef] [PubMed]
- Kato, T. Neurobiological basis of bipolar disorder: Mitochondrial dysfunction hypothesis and beyond. Schizophr. Res. 2017, 187, 62–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sigitova, E.; Fišar, Z.; Hroudová, J.; Cikánková, T.; Raboch, J. Biological hypotheses and biomarkers of bipolar disorder. Psychiatry Clin. Neurosci. 2017, 71, 77–103. [Google Scholar] [CrossRef] [PubMed]
- Andreazza, A.C.; Shao, L.; Wang, J.F.; Young, L.T. Mitochondrial complex I activity and oxidative damage to mitochondrial proteins in the prefrontal cortex of patients with bipolar disorder. Arch. Gen. Psychiatry 2010, 67, 360–368. [Google Scholar] [CrossRef] [Green Version]
- Cavelier, L.; Jazin, E.E.; Eriksson, I.; Prince, J.; Båve, U.; Oreland, L.; Gyllensten, U. Decreased cytochrome-c oxidase activity and lack of age-related accumulation of mitochondrial DNA deletions in the brains of schizophrenics. Genomics 1995, 29, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Prince, J.A.; Blennow, K.; Gottfries, C.G.; Karlsson, I.; Oreland, L. Mitochondrial function is differentially altered in the basal ganglia of chronic schizophrenics. Neuropsychopharmacology 1999, 21, 372–379. [Google Scholar] [CrossRef]
- Ben-Shachar, D.; Zuk, R.; Gazawi, H.; Reshef, A.; Sheinkman, A.; Klein, E. Increased mitochondrial complex I activity in platelets of schizophrenic patients. Int. J. Neuropsychopharmacol. 1999, 2, 245–253. [Google Scholar] [CrossRef] [Green Version]
- Ni, P.; Noh, H.; Park, G.H.; Shao, Z.; Guan, Y.; Park, J.M.; Yu, S.; Park, J.S.; Coyle, J.T.; Weinberger, D.R.; et al. iPSC-derived homogeneous populations of developing schizophrenia cortical interneurons have compromised mitochondrial function. Mol. Psychiatry 2019. [CrossRef]
- Adzic, M.; Brkic, Z.; Bulajic, S.; Mitic, M.; Radojcic, M.B. Antidepressant Action on Mitochondrial Dysfunction in Psychiatric Disorders. Drug Dev. Res. 2016, 77, 400–406. [Google Scholar] [CrossRef]
- Martin, S.A.; Souder, D.C.; Miller, K.N.; Clark, J.P.; Sagar, A.K.; Eliceiri, K.W.; Puglielli, L.; Beasley, T.M.; Anderson, R.M. GSK3β Regulates Brain Energy Metabolism. Cell Rep. 2018, 23, 1922–1931. [Google Scholar] [CrossRef]
- Palikaras, K.; Tavernarakis, N. Regulation and roles of mitophagy at synapses. Mech. Ageing Dev. 2020, 187, 111216. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.L.; Miller, A.M.; Woesner, M.E. Autophagy and Schizophrenia: A Closer Look at How Dysregulation of Neuronal Cell Homeostasis Influences the Pathogenesis of Schizophrenia. Einstein J. Biol. Med. 2016, 31, 34–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merenlender-Wagner, A.; Shemer, Z.; Touloumi, O.; Lagoudaki, R.; Giladi, E.; Andrieux, A.; Grigoriadis, N.C.; Gozes, I. New horizons in schizophrenia treatment: Autophagy protection is coupled with behavioral improvements in a mouse model of schizophrenia. Autophagy 2014, 10, 2324–2332. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, H.-G.; Keilhoff, G.; Dobrowolny, H.; Steiner, J. Enhanced mitochondrial autophagy (mitophagy) in oligodendrocytes might play a role in white matter pathology in schizophrenia. Med. Hypotheses 2020, 134, 109443. [Google Scholar] [CrossRef]
- Scaini, G.; Barichello, T.; Fries, G.R.; Kennon, E.A.; Andrews, T.; Nix, B.R.; Zunta-Soares, G.; Valvassori, S.S.; Soares, J.C.; Quevedo, J. TSPO upregulation in bipolar disorder and concomitant downregulation of mitophagic proteins and NLRP3 inflammasome activation. Neuropsychopharmacology 2019, 44, 1291–1299. [Google Scholar] [CrossRef]
- Vucicevic, L.; Misirkic-Marjanovic, M.; Harhaji-Trajkovic, L.; Maric, N.; Trajkovic, V. Mechanisms and therapeutic significance of autophagy modulation by antipsychotic drugs. Cell Stress 2018, 2, 282–291. [Google Scholar] [CrossRef]
- Lisek, M.; Boczek, T.; Zylinska, L. Calcium as a Trojan horse in mental diseases-The role of PMCA and PMCA-interacting proteins in bipolar disorder and schizophrenia. Neurosci. Lett. 2018, 663, 48–54. [Google Scholar] [CrossRef]
- Rushlow, W.J.; Seah, C.; Sutton, L.P.; Bjelica, A.; Rajakumar, N. Antipsychotics affect multiple calcium calmodulin dependent proteins. Neuroscience 2009, 161, 877–886. [Google Scholar] [CrossRef]
- Sczekan, S.R.; Strumwasser, F. Antipsychotic drugs block IP3-dependent Ca2+-release from rat brain microsomes. Biol. Psychiatry 1996, 40, 497–502. [Google Scholar] [CrossRef]
- Akimoto, T.; Kusumi, I.; Suzuki, K.; Koyama, T. Effects of calmodulin and protein kinase C modulators on transient Ca2+ increase and capacitative Ca2+ entry in human platelets: Relevant to pathophysiology of bipolar disorder. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2007, 31, 136–141. [Google Scholar] [CrossRef] [Green Version]
- Rípová, D.; Strunecká, A.; Nemcová, V.; Farská, I. Phospholipids and calcium alterations in platelets of schizophrenic patients. Physiol. Res. 1997, 46, 59–68. [Google Scholar] [PubMed]
- Dedic, N.; Pöhlmann, M.L.; Richter, J.S.; Mehta, D.; Czamara, D.; Metzger, M.W.; Dine, J.; Bedenk, B.T.; Hartmann, J.; Wagner, K.V.; et al. Cross-disorder risk gene CACNA1C differentially modulates susceptibility to psychiatric disorders during development and adulthood. Mol. Psychiatry 2018, 23, 533–543. [Google Scholar] [CrossRef] [PubMed]
- Lidow, M.S. Calcium signaling dysfunction in schizophrenia: A unifying approach. Brain Res. Rev. 2003, 43, 70–84. [Google Scholar] [CrossRef]
- Michels, S.; Wöhr, M.; Schwarting, R.K.W.; Culmsee, C. Psychiatric risk gene Cacna1c determines mitochondrial resilience against oxidative stress in neurons. Cell Death Dis. 2018, 9, 645. [Google Scholar] [CrossRef]
- Irwin, R.W.; Yao, J.; To, J.; Hamilton, R.T.; Cadenas, E.; Brinton, R.D. Selective oestrogen receptor modulators differentially potentiate brain mitochondrial function. J. Neuroendocrinol. 2012, 24, 236–248. [Google Scholar] [CrossRef]
- Flynn, J.M.; Dimitrijevich, S.D.; Younes, M.; Skliris, G.; Murphy, L.C.; Cammarata, P.R. Role of wild-type estrogen receptor-beta in mitochondrial cytoprotection of cultured normal male and female human lens epithelial cells. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E637–E647. [Google Scholar] [CrossRef]
- Van Itallie, C.M.; Dannies, P.S. Estrogen induces accumulation of the mitochondrial ribonucleic acid for subunit II of cytochrome oxidase in pituitary tumor cells. Mol. Endocrinol. 1988, 2, 332–337. [Google Scholar] [CrossRef] [Green Version]
- Bettini, E.; Maggi, A. Estrogen induction of cytochrome c oxidase subunit III in rat hippocampus. J. Neurochem. 1992, 58, 1923–1929. [Google Scholar] [CrossRef]
- Watanabe, T.; Inoue, S.; Hiroi, H.; Orimo, A.; Kawashima, H.; Muramatsu, M. Isolation of estrogen-responsive genes with a CpG island library. Mol. Cell. Biol. 1998, 18, 442–449. [Google Scholar] [CrossRef] [Green Version]
- Araújo, G.W.; Beyer, C.; Arnold, S. Oestrogen influences on mitochondrial gene expression and respiratory chain activity in cortical and mesencephalic astrocytes. J. Neuroendocrinol. 2008, 20, 930–941. [Google Scholar] [CrossRef]
- Mattingly, K.A.; Ivanova, M.M.; Riggs, K.A.; Wickramasinghe, N.S.; Barch, M.J.; Klinge, C.M. Estradiol stimulates transcription of nuclear respiratory factor-1 and increases mitochondrial biogenesis. Mol. Endocrinol. 2008, 22, 609–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnold, S.; Beyer, C. Neuroprotection by estrogen in the brain: The mitochondrial compartment as presumed therapeutic target. J. Neurochem. 2009, 110. [Google Scholar] [CrossRef] [PubMed]
- Arnold, S.; de Araújo, G.W.; Beyer, C. Gender-specific regulation of mitochondrial fusion and fission gene transcription and viability of cortical astrocytes by steroid hormones. J. Mol. Endocrinol. 2008, 41, 289–300. [Google Scholar] [CrossRef] [PubMed]
- Lobatón, C.D.; Vay, L.; Hernández-Sanmiguel, E.; Santodomingo, J.; Moreno, A.; Montero, M.; Alvarez, J. Modulation of mitochondrial Ca2+ uptake by estrogen receptor agonists and antagonists. Br. J. Pharmacol. 2005, 145, 862–871. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.L.; Qin, P.; Liu, Y.; Zhang, L.X.; Guo, H.; Deng, Y.L.; Yizhao, L.; Hou, Y.S.; Wang, L.Y.; Miao, Y.; et al. Alleviation of ischaemia-reperfusion injury by endogenous estrogen involves maintaining Bcl-2 expression via the ERα signalling pathway. Brain Res. 2017, 1661, 15–23. [Google Scholar] [CrossRef]
- Chiueh, C.; Lee, S.; Andoh, T.; Murphy, D. Induction of antioxidative and antiapoptotic thioredoxin supports neuroprotective hypothesis of estrogen. Endocrine 2003, 21, 27–31. [Google Scholar] [CrossRef]
- Schmidt, A.J.; Krieg, J.; Vedder, H. Differential effects of glucocorticoids and gonadal steroids on glutathione levels in neuronal and glial cell systems. J. Neurosci. Res. 2002, 67, 544–550. [Google Scholar] [CrossRef]
- Zhang, Y.; Bhavnani, B.R. Glutamate-induced apoptosis in primary cortical neurons is inhibited by equine estrogens via down-regulation of caspase-3 and prevention of mitochondrial cytochrome c release. BMC Neurosci. 2005, 6, 13. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Duckles, S.P.; Weiss, J.H.; Li, X.; Krause, D.N. 17β-Estradiol prevents cell death and mitochondrial dysfunction by an estrogen receptor-dependent mechanism in astrocytes after oxygen-glucose deprivation/reperfusion. Free Radic. Biol. Med. 2012, 52, 2151–2160. [Google Scholar] [CrossRef] [Green Version]
- Burstein, S.R.; Kim, H.J.; Fels, J.A.; Qian, L.; Zhang, S.; Zhou, P.; Starkov, A.A.; Iadecola, C.; Manfredi, G. Estrogen receptor beta modulates permeability transition in brain mitochondria. Biochim. Biophys. Acta Bioenerg. 2018, 1859, 423–433. [Google Scholar] [CrossRef]
- Dantzer, R. Cytokine-induced sickness behaviour: A neuroimmune response to activation of innate immunity. Eur. J. Pharmacol. 2004, 500, 399–411. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.H.; Maletic, V.; Raison, C.L. Inflammation and its discontents: The role of cytokines in the pathophysiology of major depression. Biol. Psychiatry 2009, 65, 732–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khandaker, G.M.; Cousins, L.; Deakin, J.; Lennox, B.R.; Yolken, R.; Jones, P.B. Inflammation and immunity in schizophrenia: Implications for pathophysiology and treatment. Lancet Psychiatry 2015, 2, 258–270. [Google Scholar] [CrossRef] [Green Version]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef] [Green Version]
- Theoharides, T.C.; Tsilioni, I.; Patel, A.B.; Doyle, R. Atopic diseases and inflammation of the brain in the pathogenesis of autism spectrum disorders. Transl. Psychiatry 2016, 6, e844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siniscalco, D.; Schultz, S.; Brigida, A.L.; Antonucci, N. Inflammation and Neuro-Immune Dysregulations in Autism Spectrum Disorders. Pharmaceuticals 2018, 11, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanamsagar, R.; Bilbo, S.D. Sex differences in neurodevelopmental and neurodegenerative disorders: Focus on microglial function and neuroinflammation during development. J. Steroid Biochem. Mol. Biol. 2016, 160, 127–133. [Google Scholar] [CrossRef] [Green Version]
- Kamitaki, N.; Sekar, A.; Handsaker, R.E.; de Rivera, H.; Tooley, K.; Morris, D.L.; Taylor, K.E.; Whelan, C.W.; Tombleson, P.; Loohuis, L.M.O.; et al. Complement genes contribute sex-biased vulnerability in diverse disorders. Nature 2020, 582, 577–581. [Google Scholar] [CrossRef]
- Benedetti, F.; Aggio, V.; Pratesi, M.L.; Greco, G.; Furlan, R. Neuroinflammation in Bipolar Depression. Front. Psychiatry 2020, 11, 71. [Google Scholar] [CrossRef] [Green Version]
- Iwasaki, A.; Medzhitov, R. Control of adaptive immunity by the innate immune system. Nat. Immunol. 2015, 16, 343–353. [Google Scholar] [CrossRef]
- Chaplin, D.D. Overview of the immune response. J. Allergy Clin. Immunol. 2010, 125, S3–S23. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.S.; Derkits, E.J. Prenatal infection and schizophrenia: A review of epidemiologic and translational studies. Am. J. Psychiatry 2010, 167, 261–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, B.J.; Buckley, P.; Seabolt, W.; Mellor, A.; Kirkpatrick, B. Meta-analysis of cytokine alterations in schizophrenia: Clinical status and antipsychotic effects. Biol. Psychiatry 2011, 70, 663–671. [Google Scholar] [CrossRef]
- Khandaker, G.M.; Zimbron, J.; Dalman, C.; Lewis, G.; Jones, P.B. Childhood infection and adult schizophrenia: A meta-analysis of population-based studies. Schizophr. Res. 2012, 139, 161–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khandaker, G.M.; Zimbron, J.; Lewis, G.; Jones, P.B. Prenatal maternal infection, neurodevelopment and adult schizophrenia: A systematic review of population-based studies. Psychol. Med. 2013, 43, 239–257. [Google Scholar] [CrossRef] [Green Version]
- Aas, M.; Dieset, I.; Hope, S.; Hoseth, E.; Mørch, R.; Reponen, E.; Steen, N.E.; Laskemoen, J.F.; Ueland, T.; Aukrust, P.; et al. Childhood maltreatment severity is associated with elevated C-reactive protein and body mass index in adults with schizophrenia and bipolar diagnoses. Brain Behav. Immun. 2017, 65, 342–349. [Google Scholar] [CrossRef] [Green Version]
- Benros, M.E.; Nielsen, P.R.; Nordentoft, M.; Eaton, W.W.; Dalton, S.O.; Mortensen, P.B. Autoimmune diseases and severe infections as risk factors for schizophrenia: A 30-year population-based register study. Am. J. Psychiatry 2011, 168, 1303–1310. [Google Scholar] [CrossRef]
- Tarasov, V.V.; Svistunov, A.A.; Chubarev, V.N.; Sologova, S.S.; Mukhortova, P.; Levushkin, D.; Somasundaram, S.G.; Kirkland, C.E.; Bachurin, S.O.; Aliev, G. Alterations of Astrocytes in the Context of Schizophrenic Dementia. Front. Pharmacol. 2019, 10, 1612. [Google Scholar] [CrossRef] [Green Version]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef] [Green Version]
- Wolf, S.A.; Boddeke, H.W.; Kettenmann, H. Microglia in Physiology and Disease. Annu. Rev. Physiol. 2017, 79, 619–643. [Google Scholar] [CrossRef]
- Prinz, M.; Jung, S.; Priller, J. Microglia Biology: One Century of Evolving Concepts. Cell 2019, 179, 292–311. [Google Scholar] [CrossRef] [PubMed]
- Mrdjen, D.; Pavlovic, A.; Hartmann, F.J.; Schreiner, B.; Utz, S.G.; Leung, B.P.; Lelios, I.; Heppner, F.L.; Kipnis, J.; Merkler, D.; et al. High-Dimensional Single-Cell Mapping of Central Nervous System Immune Cells Reveals Distinct Myeloid Subsets in Health, Aging, and Disease. Immunity 2018, 48, 380–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jordão, M.J.C.; Sankowski, R.; Brendecke, S.M.; Locatelli, G.; Tai, Y.H.; Tay, T.L.; Schramm, E.; Armbruster, S.; Hagemeyer, N.; Groß, O.; et al. Single-cell profiling identifies myeloid cell subsets with distinct fates during neuroinflammation. Science 2019, 363. [Google Scholar] [CrossRef] [PubMed]
- van Berckel, B.N.; Bossong, M.G.; Boellaard, R.; Kloet, R.; Schuitemaker, A.; Caspers, E.; Luurtsema, G.; Windhorst, A.D.; Cahn, W.; Lammertsma, A.A.; et al. Microglia activation in recent-onset schizophrenia: A quantitative (R)-[11C]PK11195 positron emission tomography study. Biol. Psychiatry 2008, 64, 820–822. [Google Scholar] [CrossRef]
- Doorduin, J.; de Vries, E.F.; Willemsen, A.T.; de Groot, J.C.; Dierckx, R.A.; Klein, H.C. Neuroinflammation in schizophrenia-related psychosis: A PET study. J. Nucl. Med. Off. Publ. Soc. Nucl. Med. 2009, 50, 1801–1807. [Google Scholar] [CrossRef] [Green Version]
- Schroder, K.; Sweet, M.J.; Hume, D.A. Signal integration between IFNgamma and TLR signalling pathways in macrophages. Immunobiology 2006, 211, 511–524. [Google Scholar] [CrossRef]
- Perry, V.H.; Nicoll, J.A.; Holmes, C. Microglia in neurodegenerative disease. Nat. Rev. Neurol. 2010, 6, 193–201. [Google Scholar] [CrossRef]
- Haarman, B.C.M.; Riemersma-Van der Lek, R.F.; de Groot, J.C.; Ruhé, H.G.; Klein, H.C.; Zandstra, T.E.; Burger, H.; Schoevers, R.A.; de Vries, E.F.J.; Drexhage, H.A.; et al. Neuroinflammation in bipolar disorder—A [11C]-(R)-PK11195 positron emission tomography study. Brain Behav. Immun. 2014, 40, 219–225. [Google Scholar] [CrossRef]
- Haarman, B.C.; Burger, H.; Doorduin, J.; Renken, R.J.; Sibeijn-Kuiper, A.J.; Marsman, J.B.; de Vries, E.F.; de Groot, J.C.; Drexhage, H.A.; Mendes, R.; et al. Volume, metabolites and neuroinflammation of the hippocampus in bipolar disorder—A combined magnetic resonance imaging and positron emission tomography study. Brain Behav. Immun. 2016, 56, 21–33. [Google Scholar] [CrossRef]
- Manji, H.K.; Moore, G.J.; Rajkowska, G.; Chen, G. Neuroplasticity and cellular resilience in mood disorders. Mol. Psychiatry 2000, 5, 578–593. [Google Scholar] [CrossRef] [Green Version]
- Uranova, N.A.; Vostrikov, V.M.; Orlovskaya, D.D.; Rachmanova, V.I. Oligodendroglial density in the prefrontal cortex in schizophrenia and mood disorders: A study from the Stanley Neuropathology Consortium. Schizophr. Res. 2004, 67, 269–275. [Google Scholar] [CrossRef]
- Konradi, C.; Sillivan, S.E.; Clay, H.B. Mitochondria, oligodendrocytes and inflammation in bipolar disorder: Evidence from transcriptome studies points to intriguing parallels with multiple sclerosis. Neurobiol. Dis. 2012, 45, 37–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheld, M.; Fragoulis, A.; Nyamoya, S.; Zendedel, A.; Denecke, B.; Krauspe, B.; Teske, N.; Kipp, M.; Beyer, C.; Clarner, T. Mitochondrial Impairment in Oligodendroglial Cells Induces Cytokine Expression and Signaling. J. Mol. Neurosci. 2019, 67, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Grosse, L.; Hoogenboezem, T.; Ambrée, O.; Bellingrath, S.; Jörgens, S.; de Wit, H.J.; Wijkhuijs, A.M.; Arolt, V.; Drexhage, H.A. Deficiencies of the T and natural killer cell system in major depressive disorder: T regulatory cell defects are associated with inflammatory monocyte activation. Brain Behav. Immun. 2016, 54, 38–44. [Google Scholar] [CrossRef]
- Drexhage, R.C.; Hoogenboezem, T.H.; Versnel, M.A.; Berghout, A.; Nolen, W.A.; Drexhage, H.A. The activation of monocyte and T cell networks in patients with bipolar disorder. Brain Behav. Immun. 2011, 25, 1206–1213. [Google Scholar] [CrossRef]
- Rosenblat, J.D.; McIntyre, R.S. Bipolar Disorder and Immune Dysfunction: Epidemiological Findings, Proposed Pathophysiology and Clinical Implications. Brain Sci. 2017, 7, 144. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.-Y.; Chiang, J.-H.; Chen, S.-F.; Shen, Y.-C. Systemic autoimmune diseases are associated with an increased risk of bipolar disorder: A nationwide population-based cohort study. J. Affect. Disord. 2018, 227, 31–37. [Google Scholar] [CrossRef]
- Aschner, M. Astrocytes as mediators of immune and inflammatory responses in the CNS. Neurotoxicology 1998, 19, 269–281. [Google Scholar]
- Dong, Y.; Benveniste, E.N. Immune function of astrocytes. Glia 2001, 36, 180–190. [Google Scholar] [CrossRef]
- Kim, R.; Healey, K.L.; Sepulveda-Orengo, M.T.; Reissner, K.J. Astroglial correlates of neuropsychiatric disease: From astrocytopathy to astrogliosis. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2018, 87, 126–146. [Google Scholar] [CrossRef]
- Kindler, J.; Lim, C.K.; Weickert, C.S.; Boerrigter, D.; Galletly, C.; Liu, D.; Jacobs, K.R.; Balzan, R.; Bruggemann, J.; O’Donnell, M.; et al. Dysregulation of kynurenine metabolism is related to proinflammatory cytokines, attention, and prefrontal cortex volume in schizophrenia. Mol. Psychiatry 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singhal, G.; Jaehne, E.J.; Corrigan, F.; Toben, C.; Baune, B.T. Inflammasomes in neuroinflammation and changes in brain function: A focused review. Front. Neurosci. 2014, 8, 315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heneka, M.T.; McManus, R.M.; Latz, E. Inflammasome signalling in brain function and neurodegenerative disease. Nat. Rev. Neurosci. 2018, 19, 610–621. [Google Scholar] [CrossRef]
- Swanson, K.V.; Deng, M.; Ting, J.P. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, A.; Rashidi, E.; Amooeian, V.G. Brain, blood, cerebrospinal fluid, and serum biomarkers in schizophrenia. Psychiatry Res. 2018, 265, 25–38. [Google Scholar] [CrossRef]
- Zhu, F.; Zhang, L.; Liu, F.; Wu, R.; Guo, W.; Ou, J.; Zhang, X.; Zhao, J. Altered Serum Tumor Necrosis Factor and Interleukin-1β in First-Episode Drug-Naive and Chronic Schizophrenia. Front. Neurosci. 2018, 12, 296. [Google Scholar] [CrossRef]
- Mangold, C.A.; Wronowski, B.; Du, M.; Masser, D.R.; Hadad, N.; Bixler, G.V.; Brucklacher, R.M.; Ford, M.M.; Sonntag, W.E.; Freeman, W.M. Sexually divergent induction of microglial-associated neuroinflammation with hippocampal aging. J. Neuroinflamm. 2017, 14, 141. [Google Scholar] [CrossRef]
- Spychala, M.S.; Honarpisheh, P.; McCullough, L.D. Sex differences in neuroinflammation and neuroprotection in ischemic stroke. J. Neurosci. Res. 2017, 95, 462–471. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Li, Z.; Li, J.; Siegel, C.; Yuan, R.; McCullough, L.D. Sex differences in caspase activation after stroke. Stroke 2009, 40, 1842–1848. [Google Scholar] [CrossRef]
- Manwani, B.; Liu, F.; Scranton, V.; Hammond, M.D.; Sansing, L.H.; McCullough, L.D. Differential effects of aging and sex on stroke induced inflammation across the lifespan. Exp. Neurol. 2013, 249, 120–131. [Google Scholar] [CrossRef] [Green Version]
- Klein, S.L.; Marriott, I.; Fish, E.N. Sex-based differences in immune function and responses to vaccination. Trans. R. Soc. Trop. Med. Hyg. 2015, 109, 9–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirza, M.A.; Ritzel, R.; Xu, Y.; McCullough, L.D.; Liu, F. Sexually dimorphic outcomes and inflammatory responses in hypoxic-ischemic encephalopathy. J. Neuroinflamm. 2015, 12, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibanez, C.; Shields, S.A.; El-Etr, M.; Baulieu, E.E.; Schumacher, M.; Franklin, R.J. Systemic progesterone administration results in a partial reversal of the age-associated decline in CNS remyelination following toxin-induced demyelination in male rats. Neuropathol. Appl. Neurobiol. 2004, 30, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Arevalo, M.A.; Diz-Chaves, Y.; Santos-Galindo, M.; Bellini, M.J.; Garcia-Segura, L.M. Selective oestrogen receptor modulators decrease the inflammatory response of glial cells. J. Neuroendocrinol. 2012, 24, 183–190. [Google Scholar] [CrossRef] [Green Version]
- Velez-Perez, A.; Holder, M.K.; Fountain, S.; Blaustein, J.D. Estradiol Increases Microglial Response to Lipopolysaccharide in the Ventromedial Hypothalamus during the Peripubertal Sensitive Period in Female Mice. eNeuro 2020, 7. [Google Scholar] [CrossRef]
- Hall, O.J.; Klein, S.L. Progesterone-based compounds affect immune responses and susceptibility to infections at diverse mucosal sites. Mucosal Immunol. 2017, 10, 1097–1107. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Bian, C.; Luo, Z.; Guille, C.; Ogunrinde, E.; Wu, J.; Zhao, M.; Fitting, S.; Kamen, D.L.; Oates, J.C.; et al. Progesterone decreases gut permeability through upregulating occludin expression in primary human gut tissues and Caco-2 cells. Sci. Rep. 2019, 9, 8367. [Google Scholar] [CrossRef] [Green Version]
- Ghoumari, A.M.; Baulieu, E.E.; Schumacher, M. Progesterone increases oligodendroglial cell proliferation in rat cerebellar slice cultures. Neuroscience 2005, 135, 47–58. [Google Scholar] [CrossRef]
- Zuo, W.; Zhang, W.; Chen, N.H. Sexual dimorphism in cerebral ischemia injury. Eur. J. Pharmacol. 2013, 711, 73–79. [Google Scholar] [CrossRef]
- Demarest, T.G.; McCarthy, M.M. Sex differences in mitochondrial (dys)function: Implications for neuroprotection. J. Bioenerg. Biomembr. 2015, 47, 173–188. [Google Scholar] [CrossRef]
- Lenz, K.M.; Nugent, B.M.; Haliyur, R.; McCarthy, M.M. Microglia are essential to masculinization of brain and behavior. J. Neurosci. 2013, 33, 2761–2772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarz, J.M.; Sholar, P.W.; Bilbo, S.D. Sex differences in microglial colonization of the developing rat brain. J. Neurochem. 2012, 120, 948–963. [Google Scholar] [CrossRef] [PubMed]
- Lenz, K.M.; McCarthy, M.M. A starring role for microglia in brain sex differences. Neurosci. A Rev. J. Bringing Neurobiol. Neurol. Psychiatry 2015, 21, 306–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hui, C.W.; St-Pierre, A.; El Hajj, H.; Remy, Y.; Hébert, S.S.; Luheshi, G.N.; Srivastava, L.K.; Tremblay, M. Prenatal Immune Challenge in Mice Leads to Partly Sex-Dependent Behavioral, Microglial, and Molecular Abnormalities Associated with Schizophrenia. Front. Mol. Neurosci. 2018, 11, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, J.; Badner, J.A.; Hattori, E.; Potash, J.B.; Willour, V.L.; McMahon, F.J.; Gershon, E.S.; Liu, C. Neurotransmission and bipolar disorder: A systematic family-based association study. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. Off. Publ. Int. Soc. Psychiatr. Genet. 2008, 147, 1270–1277. [Google Scholar] [CrossRef] [Green Version]
- Howes, O.; McCutcheon, R.; Stone, J. Glutamate and dopamine in schizophrenia: An update for the 21st century. J. Psychopharmacol. 2015, 29, 97–115. [Google Scholar] [CrossRef] [Green Version]
- Bradford, A. The dopamine and glutamate theories of schizophrenia: A short review. Curr. Anaesth. Crit. Care 2009, 20, 240–241. [Google Scholar] [CrossRef]
- Lewis, D.A.; Moghaddam, B. Cognitive dysfunction in schizophrenia: Convergence of gamma-aminobutyric acid and glutamate alterations. Arch. Neurol. 2006, 63, 1372–1376. [Google Scholar] [CrossRef]
- Hillhouse, T.M.; Porter, J.H. A brief history of the development of antidepressant drugs: From monoamines to glutamate. Exp. Clin. Psychopharmacol. 2015, 23, 1–21. [Google Scholar] [CrossRef]
- Schildkraut, J.J. The catecholamine hypothesis of affective disorders: A review of supporting evidence. Am. J. Psychiatry 1965, 122, 509–522. [Google Scholar] [CrossRef]
- Sedvall, G. Monoamines and schizophrenia. Acta Psychiatr. Scand. 1990, 82, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Stone, J.M.; Morrison, P.D.; Pilowsky, L.S. Glutamate and dopamine dysregulation in schizophrenia—A synthesis and selective review. J. Psychopharmacol. 2007, 21, 440–452. [Google Scholar] [CrossRef] [PubMed]
- Snyder, S.H. Dopamine receptors, neuroleptics, and schizophrenia. Am. J. Psychiatry 1981, 138, 460–464. [Google Scholar] [CrossRef] [PubMed]
- Ashok, A.H.; Marques, T.R.; Jauhar, S.; Nour, M.M.; Goodwin, G.M.; Young, A.H.; Howes, O.D. The dopamine hypothesis of bipolar affective disorder: The state of the art and implications for treatment. Mol. Psychiatry 2017, 22, 666–679. [Google Scholar] [CrossRef]
- Berk, M.; Dodd, S.; Kauer-Sant’anna, M.; Malhi, G.S.; Bourin, M.; Kapczinski, F.; Norman, T. Dopamine dysregulation syndrome: Implications for a dopamine hypothesis of bipolar disorder. Acta Psychiatr. Scand. Suppl. 2007, 116, 41–49. [Google Scholar] [CrossRef]
- Cousins, D.A.; Butts, K.; Young, A.H. The role of dopamine in bipolar disorder. Bipolar Disord. 2009, 11, 787–806. [Google Scholar] [CrossRef]
- Jones, C.A.; Watson, D.J.; Fone, K.C. Animal models of schizophrenia. Br. J. Pharmacol. 2011, 164, 1162–1194. [Google Scholar] [CrossRef]
- Angrist, B.; Sathananthan, G.; Wilk, S.; Gershon, S. Amphetamine psychosis: Behavioral and biochemical aspects. J. Psychiatr. Res. 1974, 11, 13–23. [Google Scholar] [CrossRef]
- Collo, G.; Mucci, A.; Giordano, G.M.; Merlo Pich, E.; Galderisi, S. Negative Symptoms of Schizophrenia and Dopaminergic Transmission: Translational Models and Perspectives Opened by iPSC Techniques. Front. Neurosci. 2020, 14. [Google Scholar] [CrossRef]
- Konopaske, G.T.; Coyle, J.T. Schizophrenia. In Neurobiology of Brain Disorders; Zigmond, M.J., Rowland, L.P., Coyle, J.T., Eds.; Academic Press: San Diego, CA, USA, 2015; Chapter 39; pp. 639–654. [Google Scholar] [CrossRef]
- Manji, H.K.; Quiroz, J.A.; Payne, J.L.; Singh, J.; Lopes, B.P.; Viegas, J.S.; Zarate, C.A. The underlying neurobiology of bipolar disorder. World Psychiatry 2003, 2, 136–146. [Google Scholar]
- Eggers, A.E. A serotonin hypothesis of schizophrenia. Med. Hypotheses 2013, 80, 791–794. [Google Scholar] [CrossRef] [PubMed]
- Gogos, A.; Kwek, P.; Chavez, C.; van den Buuse, M. Estrogen Treatment Blocks 8-Hydroxy-2-dipropylaminotetralin- and Apomorphine-Induced Disruptions of Prepulse Inhibition: Involvement of Dopamine D1 or D2 or Serotonin 5-HT1A 5-HT2A, or 5-HT7 Receptors. J. Pharmacol. Exp. Ther. 2010, 333, 218–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bleich, A.; Brown, S.-L.; Kahn, R.; van Praag, H.M. The Role of Serotonin in Schizophrenia. Schizophr. Bull. 1988, 14, 297–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higgs, B.W.; Elashoff, M.; Richman, S.; Barci, B. An online database for brain disease research. BMC Genom. 2006, 7, 70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lener, M.S.; Niciu, M.J.; Ballard, E.D.; Park, M.; Park, L.T.; Nugent, A.C.; Zarate, C.A., Jr. Glutamate and Gamma-Aminobutyric Acid Systems in the Pathophysiology of Major Depression and Antidepressant Response to Ketamine. Biol. Psychiatry 2017, 81, 886–897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gigante, A.D.; Bond, D.J.; Lafer, B.; Lam, R.W.; Young, L.T.; Yatham, L.N. Brain glutamate levels measured by magnetic resonance spectroscopy in patients with bipolar disorder: A meta-analysis. Bipolar Disord. 2012, 14, 478–487. [Google Scholar] [CrossRef]
- Vöhringer, P.A.; Barroilhet, S.A.; Amerio, A.; Reale, M.L.; Alvear, K.; Vergne, D.; Ghaemi, S.N. Cognitive impairment in bipolar disorder and schizophrenia: A systematic review. Front. Psychiatry 2013, 4, 87. [Google Scholar] [CrossRef] [Green Version]
- Adler, C.M.; Malhotra, A.K.; Elman, I.; Goldberg, T.; Egan, M.; Pickar, D.; Breier, A. Comparison of ketamine-induced thought disorder in healthy volunteers and thought disorder in schizophrenia. Am. J. Psychiatry 1999, 156, 1646–1649. [Google Scholar] [CrossRef]
- Javitt, D.C.; Zukin, S.R. Recent advances in the phencyclidine model of schizophrenia. Am. J. Psychiatry 1991, 148, 1301–1308. [Google Scholar] [CrossRef]
- Moghaddam, B.; Krystal, J.H. Capturing the Angel in “Angel Dust”: Twenty Years of Translational Neuroscience Studies of NMDA Receptor Antagonists in Animals and Humans. Schizophr. Bull. 2012, 38, 942–949. [Google Scholar] [CrossRef] [Green Version]
- Mouri, A.; Noda, Y.; Enomoto, T.; Nabeshima, T. Phencyclidine animal models of schizophrenia: Approaches from abnormality of glutamatergic neurotransmission and neurodevelopment. Neurochem. Int. 2007, 51, 173–184. [Google Scholar] [CrossRef] [PubMed]
- Homayoun, H.; Moghaddam, B. NMDA Receptor Hypofunction Produces Opposite Effects on Prefrontal Cortex Interneurons and Pyramidal Neurons. J. Neurosci. 2007, 27, 11496–11500. [Google Scholar] [CrossRef]
- Olney, J.W.; Farber, N.B. Glutamate Receptor Dysfunction and Schizophrenia. Arch. Gen. Psychiatry 1995, 52, 998–1007. [Google Scholar] [CrossRef] [PubMed]
- Olney, J.W.; Newcomer, J.W.; Farber, N.B. NMDA receptor hypofunction model of schizophrenia. J. Psychiatr. Res. 1999, 33, 523–533. [Google Scholar] [CrossRef]
- Coyle, J.T.; Tsai, G.; Goff, D. Converging evidence of NMDA receptor hypofunction in the pathophysiology of schizophrenia. Ann. N. Y. Acad. Sci. 2003, 1003, 318–327. [Google Scholar] [CrossRef] [PubMed]
- Coyle, J.T.; Basu, A.; Benneyworth, M.; Balu, D.; Konopaske, G. Glutamatergic synaptic dysregulation in schizophrenia: Therapeutic implications. Handb. Exp. Pharmacol. 2012, 267–295. [Google Scholar] [CrossRef] [Green Version]
- Behrens, M.M.; Sejnowski, T.J. Does schizophrenia arise from oxidative dysregulation of parvalbumin-interneurons in the developing cortex? Neuropharmacology 2009, 57, 193–200. [Google Scholar] [CrossRef] [Green Version]
- Akbarian, S.; Huang, H.-S. Molecular and cellular mechanisms of altered GAD1/GAD67 expression in schizophrenia and related disorders. Brain Res. Rev. 2006, 52, 293–304. [Google Scholar] [CrossRef]
- Lewis, D.A.; Hashimoto, T.; Volk, D.W. Cortical inhibitory neurons and schizophrenia. Nat. Rev. Neurosci. 2005, 6, 312–324. [Google Scholar] [CrossRef]
- Gonzalez-Burgos, G.; Hashimoto, T.; Lewis, D.A. Alterations of Cortical GABA Neurons and Network Oscillations in Schizophrenia. Curr. Psychiatry Rep. 2010, 12, 335–344. [Google Scholar] [CrossRef]
- Chang, C.Y.; Chen, Y.W.; Wang, T.W.; Lai, W.S. Akting up in the GABA hypothesis of schizophrenia: Akt1 deficiency modulates GABAergic functions and hippocampus-dependent functions. Sci. Rep. 2016, 6, 33095. [Google Scholar] [CrossRef] [PubMed]
- Behrens, C.J.; Van Den Boom, L.P.; Heinemann, U. Effects of the GABAA receptor antagonists bicuculline and gabazine on stimulus-induced sharp wave-ripple complexes in adult rat hippocampus in vitro. Eur. J. Neurosci. 2007, 25, 2170–2181. [Google Scholar] [CrossRef] [PubMed]
- Delini-Stula, A.; Berdah-Tordjman, D. Benzodiazepines and GABA hypothesis of schizophrenia. J. Psychopharmacol. 1995, 9, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Chiapponi, C.; Piras, F.; Piras, F.; Caltagirone, C.; Spalletta, G. GABA System in Schizophrenia and Mood Disorders: A Mini Review on Third-Generation Imaging Studies. Front. Psychiatry 2016, 7. [Google Scholar] [CrossRef] [Green Version]
- Shorter, E. The history of lithium therapy. Bipolar Disord. 2009, 11, 4–9. [Google Scholar] [CrossRef] [Green Version]
- Malhi, G.S.; Tanious, M.; Das, P.; Coulston, C.M.; Berk, M. Potential Mechanisms of Action of Lithium in Bipolar Disorder. CNS Drugs 2013, 27, 135–153. [Google Scholar] [CrossRef]
- Gottesfeld, Z.; Ebstein, B.S.; Samuel, D. Effect of Lithium on Concentrations of Glutamate and GABA Levels in Amygdala and Hypothalamus of Rat. Nat. New Biol. 1971, 234, 124–125. [Google Scholar] [CrossRef]
- Berridge, M.J. Calcium signalling and psychiatric disease: Bipolar disorder and schizophrenia. Cell Tissue Res. 2014, 357, 477–492. [Google Scholar] [CrossRef]
- Mei, Y.-Y.; Wu, D.C.; Zhou, N. Astrocytic Regulation of Glutamate Transmission in Schizophrenia. Front. Psychiatry 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Eid, T.; Ghosh, A.; Wang, Y.; Beckström, H.; Zaveri, H.P.; Lee, T.S.; Lai, J.C.; Malthankar-Phatak, G.H.; de Lanerolle, N.C. Recurrent seizures and brain pathology after inhibition of glutamine synthetase in the hippocampus in rats. Brain 2008, 131, 2061–2070. [Google Scholar] [CrossRef] [Green Version]
- Iglesias, J.; Morales, L.; Barreto, G.E. Metabolic and Inflammatory Adaptation of Reactive Astrocytes: Role of PPARs. Mol. Neurobiol. 2017, 54, 2518–2538. [Google Scholar] [CrossRef]
- Nedic Erjavec, G.; Konjevod, M.; Nikolac Perkovic, M.; Svob Strac, D.; Tudor, L.; Barbas, C.; Grune, T.; Zarkovic, N.; Pivac, N. Short overview on metabolomic approach and redox changes in psychiatric disorders. Redox Biol. 2018, 14, 178–186. [Google Scholar] [CrossRef] [PubMed]
- Erli, F.; Palmos, A.B.; Raval, P.; Mukherjee, J.; Sellers, K.J.; Gatford, N.J.F.; Moss, S.J.; Brandon, N.J.; Penzes, P.; Srivastava, D.P. Estradiol reverses excitatory synapse loss in a cellular model of neuropsychiatric disorders. Transl. Psychiatry 2020, 10, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barth, C.; Villringer, A.; Sacher, J. Sex hormones affect neurotransmitters and shape the adult female brain during hormonal transition periods. Front. Neurosci. 2015, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fink, G.; Sumner, B.E.; Rosie, R.; Grace, O.; Quinn, J.P. Estrogen control of central neurotransmission: Effect on mood, mental state, and memory. Cell. Mol. Neurobiol. 1996, 16, 325–344. [Google Scholar] [CrossRef] [PubMed]
- Colciago, A.; Bonalume, V.; Melfi, V.; Magnaghi, V. Genomic and Non-genomic Action of Neurosteroids in the Peripheral Nervous System. Front. Neurosci. 2020, 14, 796. [Google Scholar] [CrossRef]
- McEwen, B.S.; Alves, S.E. Estrogen Actions in the Central Nervous System. Endocr. Rev. 1999, 20, 279–307. [Google Scholar] [CrossRef]
- Hiroi, R.; Neumaier, J.F. Estrogen decreases 5-HT1B autoreceptor mRNA in selective subregion of rat dorsal raphe nucleus: Inverse association between gene expression and anxiety behavior in the open field. Neuroscience 2009, 158, 456–464. [Google Scholar] [CrossRef] [Green Version]
- Gundlah, C.; Lu, N.Z.; Bethea, C.L. Ovarian steroid regulation of monoamine oxidase-A and B mRNAs in the macaque dorsal raphe and hypothalamic nuclei. Psychopharmacology 2002, 160, 271–282. [Google Scholar] [CrossRef]
- Smith, S.S.; Waterhouse, B.D.; Chapin, J.K.; Woodward, D.J. Progesterone alters GABA and glutamate responsiveness: A possible mechanism for its anxiolytic action. Brain Res. 1987, 400, 353–359. [Google Scholar] [CrossRef]
- Woolley, C.S.; Weiland, N.G.; McEwen, B.S.; Schwartzkroin, P.A. Estradiol Increases the Sensitivity of Hippocampal CA1 Pyramidal Cells to NMDA Receptor-Mediated Synaptic Input: Correlation with Dendritic Spine Density. J. Neurosci. 1997, 17, 1848–1859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiland, N.G. Estradiol selectively regulates agonist binding sites on the N-methyl-D-aspartate receptor complex in the CA1 region of the hippocampus. Endocrinology 1992, 131, 662–668. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.; Moss, R. Long-term and short-term electrophysiological effects of estrogen on the synaptic properties of hippocampal CA1 neurons. J. Neurosci. 1992, 12, 3217–3225. [Google Scholar] [CrossRef]
- Kim, M.T.; Soussou, W.; Gholmieh, G.; Ahuja, A.; Tanguay, A.; Berger, T.W.; Brinton, R.D. 17β-Estradiol potentiates field excitatory postsynaptic potentials within each subfield of the hippocampus with greatest potentiation of the associational/commissural afferents of CA3. Neuroscience 2006, 141, 391–406. [Google Scholar] [CrossRef]
- Babayan, A.H.; Kramár, E.A. Rapid Effects of Oestrogen on Synaptic Plasticity: Interactions with Actin and Its Signalling Proteins. J. Neuroendocrinol. 2013, 25, 1163–1172. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Kantorovich, S.; Babayan, A.H.; Hou, B.; Gall, C.M.; Lynch, G. Estrogen’s Effects on Excitatory Synaptic Transmission Entail Integrin and TrkB Transactivation and Depend Upon β1-integrin function. Neuropsychopharmacology 2016, 41, 2723–2732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, D.D.; Cole, N.B.; Greenberger, V.; Segal, M. Estradiol Increases Dendritic Spine Density by Reducing GABA Neurotransmission in Hippocampal Neurons. J. Neurosci. 1998, 18, 2550–2559. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Day, M.; Muñiz, L.C.; Bitran, D.; Arias, R.; Revilla-Sanchez, R.; Grauer, S.; Zhang, G.; Kelley, C.; Pulito, V.; et al. Activation of estrogen receptor-β regulates hippocampal synaptic plasticity and improves memory. Nat. Neurosci. 2008, 11, 334–343. [Google Scholar] [CrossRef]
- Kramár, E.A.; Chen, L.Y.; Brandon, N.J.; Rex, C.S.; Liu, F.; Gall, C.M.; Lynch, G. Cytoskeletal Changes Underlie Estrogen’s Acute Effects on Synaptic Transmission and Plasticity. J. Neurosci. 2009, 29, 12982–12993. [Google Scholar] [CrossRef] [Green Version]
- Hao, J.; Rapp, P.R.; Leffler, A.E.; Leffler, S.R.; Janssen, W.G.M.; Lou, W.; McKay, H.; Roberts, J.A.; Wearne, S.L.; Hof, P.R.; et al. Estrogen Alters Spine Number and Morphology in Prefrontal Cortex of Aged Female Rhesus Monkeys. J. Neurosci. 2006, 26, 2571–2578. [Google Scholar] [CrossRef] [Green Version]
- Heldring, N.; Pike, A.; Andersson, S.; Matthews, J.; Cheng, G.; Hartman, J.; Tujague, M.; Ström, A.; Treuter, E.; Warner, M.; et al. Estrogen Receptors: How Do They Signal and What Are Their Targets. Physiol. Rev. 2007, 87, 905–931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ross, N.R.; Porter, L.L. Effects of dopamine and estrogen upon cortical neurons that express parvalbumin in vitro. Dev. Brain Res. 2002, 137, 23–34. [Google Scholar] [CrossRef]
- Welboren, W.J.; Stunnenberg, H.G.; Sweep, F.C.; Span, P.N. Identifying estrogen receptor target genes. Mol. Oncol. 2007, 1, 138–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charpentier, A.H.; Bednarek, A.K.; Daniel, R.L.; Hawkins, K.A.; Laflin, K.J.; Gaddis, S.; MacLeod, M.C.; Aldaz, C.M. Effects of estrogen on global gene expression: Identification of novel targets of estrogen action. Cancer Res. 2000, 60, 5977–5983. [Google Scholar] [PubMed]
- Inoue, A.; Yoshida, N.; Omoto, Y.; Oguchi, S.; Yamori, T.; Kiyama, R.; Hayashi, S. Development of cDNA microarray for expression profiling of estrogen-responsive genes. J. Mol. Endocrinol. 2002, 29, 175–192. [Google Scholar] [CrossRef] [Green Version]
- Seth, P.; Krop, I.; Porter, D.; Polyak, K. Novel estrogen and tamoxifen induced genes identified by SAGE (Serial Analysis of Gene Expression). Oncogene 2002, 21, 836–843. [Google Scholar] [CrossRef] [Green Version]
- Cunliffe, H.E.; Ringnér, M.; Bilke, S.; Walker, R.L.; Cheung, J.M.; Chen, Y.; Meltzer, P.S. The gene expression response of breast cancer to growth regulators: Patterns and correlation with tumor expression profiles. Cancer Res. 2003, 63, 7158–7166. [Google Scholar]
- Frasor, J.; Danes, J.M.; Komm, B.; Chang, K.C.; Lyttle, C.R.; Katzenellenbogen, B.S. Profiling of estrogen up- and down-regulated gene expression in human breast cancer cells: Insights into gene networks and pathways underlying estrogenic control of proliferation and cell phenotype. Endocrinology 2003, 144, 4562–4574. [Google Scholar] [CrossRef]
- Wang, J.; Cheng, C.M.; Zhou, J.; Smith, A.; Weickert, C.S.; Perlman, W.R.; Becker, K.G.; Powell, D.; Bondy, C.A. Estradiol alters transcription factor gene expression in primate prefrontal cortex. J. Neurosci. Res. 2004, 76, 306–314. [Google Scholar] [CrossRef]
- Sarvari, M.; Hrabovszky, E.; Kallo, I.; Solymosi, N.; Toth, K.; Liko, I.; Szeles, J.; Maho, S.; Molnar, B.; Liposits, Z. Estrogens regulate neuroinflammatory genes via estrogen receptors alpha and beta in the frontal cortex of middle-aged female rats. J. Neuroinflamm. 2011, 8, 82. [Google Scholar] [CrossRef] [Green Version]
- Block, M.L.; Zecca, L.; Hong, J.S. Microglia-mediated neurotoxicity: Uncovering the molecular mechanisms. Nat. Rev. Neurosci. 2007, 8, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Gogos, A.; Sbisa, A.M.; Sun, J.; Gibbons, A.; Udawela, M.; Dean, B. A Role for Estrogen in Schizophrenia: Clinical and Preclinical Findings. Int. J. Endocrinol. 2015, 2015, 615356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bortolato, B.; Miskowiak, K.W.; Köhler, C.A.; Vieta, E.; Carvalho, A.F. Cognitive dysfunction in bipolar disorder and schizophrenia: A systematic review of meta-analyses. Neuropsychiatr. Dis. Treat. 2015, 11, 3111–3125. [Google Scholar] [CrossRef] [Green Version]
- Aenlle, K.K.; Kumar, A.; Cui, L.; Jackson, T.C.; Foster, T.C. Estrogen effects on cognition and hippocampal transcription in middle-aged mice. Neurobiol. Aging 2009, 30, 932–945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pechenino, A.S.; Frick, K.M. The effects of acute 17beta-estradiol treatment on gene expression in the young female mouse hippocampus. Neurobiol. Learn. Mem. 2009, 91, 315–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Csöregh, L.; Andersson, E.; Fried, G. Transcriptional analysis of estrogen effects in human embryonic neurons and glial cells. Neuroendocrinology 2009, 89, 171–186. [Google Scholar] [CrossRef]
- Solum, D.T.; Handa, R.J. Estrogen regulates the development of brain-derived neurotrophic factor mRNA and protein in the rat hippocampus. J. Neurosci. 2002, 22, 2650–2659. [Google Scholar] [CrossRef]
- Allen, A.L.; McCarson, K.E. Estrogen increases nociception-evoked brain-derived neurotrophic factor gene expression in the female rat. Neuroendocrinology 2005, 81, 193–199. [Google Scholar] [CrossRef]
- Zhou, W.; Cunningham, K.A.; Thomas, M.L. Estrogen regulation of gene expression in the brain: A possible mechanism altering the response to psychostimulants in female rats. Brain Res. Mol. Brain Res. 2002, 100, 75–83. [Google Scholar] [CrossRef]
- Singer, C.A.; Rogers, K.L.; Strickland, T.M.; Dorsa, D.M. Estrogen protects primary cortical neurons from glutamate toxicity. Neurosci. Lett. 1996, 212, 13–16. [Google Scholar] [CrossRef]
- Garcia-Segura, L.M.; Cardona-Gomez, P.; Naftolin, F.; Chowen, J.A. Estradiol upregulates Bcl-2 expression in adult brain neurons. Neuroreport 1998, 9, 593–597. [Google Scholar] [CrossRef] [PubMed]
- Behl, C.; Skutella, T.; Lezoualc’H, F.; Post, A.; Widmann, M.; Newton, C.J.; Holsboer, F. Neuroprotection against Oxidative Stress by Estrogens: Structure-Activity Relationship. Mol. Pharmacol. 1997, 51, 535–541. [Google Scholar] [CrossRef] [PubMed]
- Cyr, M.; Landry, M.; Di Paolo, T. Modulation by Estrogen-Receptor Directed Drugs of 5-Hydroxytryptamine-2A Receptors in Rat Brain. Neuropsychopharmacology 2000, 23, 69–78. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, J.; Cardarelli, R.A.; Cantaut-Belarif, Y.; Deeb, T.Z.; Srivastava, D.P.; Tyagarajan, S.K.; Pangalos, M.N.; Triller, A.; Maguire, J.; Brandon, N.J.; et al. Estradiol modulates the efficacy of synaptic inhibition by decreasing the dwell time of GABAA receptors at inhibitory synapses. Proc. Natl. Acad. Sci. USA 2017, 114, 11763–11768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shum, C.; Macedo, S.C.; Warre-Cornish, K.; Cocks, G.; Price, J.; Srivastava, D.P. Utilizing induced pluripotent stem cells (iPSCs) to understand the actions of estrogens in human neurons. Horm. Behav. 2015, 74, 228–242. [Google Scholar] [CrossRef] [Green Version]
- Nishimura, K.; Doi, D.; Samata, B.; Murayama, S.; Tahara, T.; Onoe, H.; Takahashi, J. Estradiol Facilitates Functional Integration of iPSC-Derived Dopaminergic Neurons into Striatal Neuronal Circuits via Activation of Integrin α5β1. Stem Cell Rep. 2016, 6, 511–524. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [Green Version]
- Gore, A.; Li, Z.; Fung, H.L.; Young, J.E.; Agarwal, S.; Antosiewicz-Bourget, J.; Canto, I.; Giorgetti, A.; Israel, M.A.; Kiskinis, E.; et al. Somatic coding mutations in human induced pluripotent stem cells. Nature 2011, 471, 63–67. [Google Scholar] [CrossRef]
- López-Serrano, C.; Torres-Espín, A.; Hernández, J.; Alvarez-Palomo, A.B.; Requena, J.; Gasull, X.; Edel, M.J.; Navarro, X. Effects of the Post-Spinal Cord Injury Microenvironment on the Differentiation Capacity of Human Neural Stem Cells Derived from Induced Pluripotent Stem Cells. Cell Transplant. 2016, 25, 1833–1852. [Google Scholar] [CrossRef] [Green Version]
- Requena, J.; Alvarez-Palomo, A.B.; Codina-Pascual, M.; Delgado-Morales, R.; Moran, S.; Esteller, M.; Sal, M.; Juan, M.; Boronat Barado, A.; Consiglio, A.; et al. Global Proteomic and Methylome Analysis in Human Induced Pluripotent Stem Cells Reveals Overexpression of a Human TLR3 Affecting Proper Innate Immune Response Signaling. Stem Cells 2019, 37, 476–488. [Google Scholar] [CrossRef] [Green Version]
- Fujimori, K.; Ishikawa, M.; Otomo, A.; Atsuta, N.; Nakamura, R.; Akiyama, T.; Hadano, S.; Aoki, M.; Saya, H.; Sobue, G.; et al. Modeling sporadic ALS in iPSC-derived motor neurons identifies a potential therapeutic agent. Nat. Med. 2018, 24, 1579–1589. [Google Scholar] [CrossRef] [PubMed]
- Birger, A.; Ben-Dor, I.; Ottolenghi, M.; Turetsky, T.; Gil, Y.; Sweetat, S.; Perez, L.; Belzer, V.; Casden, N.; Steiner, D.; et al. Human iPSC-derived astrocytes from ALS patients with mutated C9ORF72 show increased oxidative stress and neurotoxicity. EBioMedicine 2019, 50, 274–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, I.K.; Yang, B.; Forest, C.; Qian, L.; Chan, A.W.S. Amelioration of Huntington’s disease phenotype in astrocytes derived from iPSC-derived neural progenitor cells of Huntington’s disease monkeys. PLoS ONE 2019, 14, e0214156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Leeuw, S.; Tackenberg, C. Alzheimer’s in a dish—Induced pluripotent stem cell-based disease modeling. Transl. Neurodegener. 2019, 8, 21. [Google Scholar] [CrossRef]
- Penney, J.; Ralvenius, W.T.; Tsai, L.H. Modeling Alzheimer’s disease with iPSC-derived brain cells. Mol. Psychiatry 2020, 25, 148–167. [Google Scholar] [CrossRef] [Green Version]
- Brennand, K.; Simone, A.; Jou, J.; Gelboin-Burkhart, C.; Tran, N.; Sangar, S.; Li, Y.; Mu, Y.; Chen, G.; Yu, D.; et al. Modelling schizophrenia using human induced pluripotent stem cells. Nature 2011, 473, 221–225. [Google Scholar] [CrossRef]
- Ishii, T.; Ishikawa, M.; Fujimori, K.; Maeda, T.; Kushima, I.; Arioka, Y.; Mori, D.; Nakatake, Y.; Yamagata, B.; Nio, S.; et al. In Vitro Modeling of the Bipolar Disorder and Schizophrenia Using Patient-Derived Induced Pluripotent Stem Cells with Copy Number Variations of PCDH15 and RELN. eNeuro 2019, 6. [Google Scholar] [CrossRef] [Green Version]
- Nakazawa, T.; Kikuchi, M.; Ishikawa, M.; Yamamori, H.; Nagayasu, K.; Matsumoto, T.; Fujimoto, M.; Yasuda, Y.; Fujiwara, M.; Okada, S.; et al. Differential gene expression profiles in neurons generated from lymphoblastoid B-cell line-derived iPS cells from monozygotic twin cases with treatment-resistant schizophrenia and discordant responses to clozapine. Schizophr. Res. 2017, 181, 75–82. [Google Scholar] [CrossRef] [Green Version]
- Goudriaan, A.; Camargo, N.; Carney, K.E.; Oliet, S.H.; Smit, A.B.; Verheijen, M.H. Novel cell separation method for molecular analysis of neuron-astrocyte co-cultures. Front. Cell Neurosci. 2014, 8, 12. [Google Scholar] [CrossRef] [Green Version]
- Nadadhur, A.G.; Emperador Melero, J.; Meijer, M.; Schut, D.; Jacobs, G.; Li, K.W.; Hjorth, J.J.J.; Meredith, R.M.; Toonen, R.F.; Van Kesteren, R.E.; et al. Multi-level characterization of balanced inhibitory-excitatory cortical neuron network derived from human pluripotent stem cells. PLoS ONE 2017, 12, e0178533. [Google Scholar] [CrossRef] [Green Version]
- Terrasso, A.P.; Silva, A.C.; Filipe, A.; Pedroso, P.; Ferreira, A.L.; Alves, P.M.; Brito, C. Human neuron-astrocyte 3D co-culture-based assay for evaluation of neuroprotective compounds. J. Pharmacol. Toxicol. Methods 2017, 83, 72–79. [Google Scholar] [CrossRef] [PubMed]
- van Deijk, A.F.; Broersen, L.M.; Verkuyl, J.M.; Smit, A.B.; Verheijen, M.H.G. High Content Analysis of Hippocampal Neuron-Astrocyte Co-cultures Shows a Positive Effect of Fortasyn Connect on Neuronal Survival and Postsynaptic Maturation. Front. Neurosci. 2017, 11, 440. [Google Scholar] [CrossRef] [PubMed]
- Aebersold, M.J.; Thompson-Steckel, G.; Joutang, A.; Schneider, M.; Burchert, C.; Forró, C.; Weydert, S.; Han, H.; Vörös, J. Simple and Inexpensive Paper-Based Astrocyte Co-culture to Improve Survival of Low-Density Neuronal Networks. Front. Neurosci. 2018, 12, 94. [Google Scholar] [CrossRef] [PubMed]
- Sim, F.J.; Windrem, M.S.; Goldman, S.A. Fate determination of adult human glial progenitor cells. Neuron Glia Biol. 2009, 5, 45–55. [Google Scholar] [CrossRef] [Green Version]
- Miller, J.A.; Horvath, S.; Geschwind, D.H. Divergence of human and mouse brain transcriptome highlights Alzheimer disease pathways. Proc. Natl. Acad. Sci. USA 2010, 107, 12698–12703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Sloan, S.A.; Clarke, L.E.; Caneda, C.; Plaza, C.A.; Blumenthal, P.D.; Vogel, H.; Steinberg, G.K.; Edwards, M.S.; Li, G.; et al. Purification and Characterization of Progenitor and Mature Human Astrocytes Reveals Transcriptional and Functional Differences with Mouse. Neuron 2016, 89, 37–53. [Google Scholar] [CrossRef] [Green Version]
- Horrobin, D.F. Schizophrenia: The illness that made us human. Med. Hypotheses 1998, 50, 269–288. [Google Scholar] [CrossRef]
- Oberheim, N.A.; Wang, X.; Goldman, S.; Nedergaard, M. Astrocytic complexity distinguishes the human brain. Trends Neurosci. 2006, 29, 547–553. [Google Scholar] [CrossRef]
- Oberheim, N.A.; Takano, T.; Han, X.; He, W.; Lin, J.H.; Wang, F.; Xu, Q.; Wyatt, J.D.; Pilcher, W.; Ojemann, J.G.; et al. Uniquely hominid features of adult human astrocytes. J. Neurosci. 2009, 29, 3276–3287. [Google Scholar] [CrossRef]
- Windrem, M.S.; Osipovitch, M.; Liu, Z.; Bates, J.; Chandler-Militello, D.; Zou, L.; Munir, J.; Schanz, S.; McCoy, K.; Miller, R.H.; et al. Human iPSC Glial Mouse Chimeras Reveal Glial Contributions to Schizophrenia. Cell Stem Cell 2017, 21, 195–208. [Google Scholar] [CrossRef]
- Hedegaard, A.; Monzón-Sandoval, J.; Newey, S.E.; Whiteley, E.S.; Webber, C.; Akerman, C.J. Pro-maturational Effects of Human iPSC-Derived Cortical Astrocytes upon iPSC-Derived Cortical Neurons. Stem Cell Rep. 2020, 15, 38–51. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, M.A.; Renner, M.; Martin, C.A.; Wenzel, D.; Bicknell, L.S.; Hurles, M.E.; Homfray, T.; Penninger, J.M.; Jackson, A.P.; Knoblich, J.A. Cerebral organoids model human brain development and microcephaly. Nature 2013, 501, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Qian, X.; Nguyen, H.N.; Song, M.M.; Hadiono, C.; Ogden, S.C.; Hammack, C.; Yao, B.; Hamersky, G.R.; Jacob, F.; Zhong, C.; et al. Brain-Region-Specific Organoids Using Mini-bioreactors for Modeling ZIKV Exposure. Cell 2016, 165, 1238–1254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renner, M.; Lancaster, M.A.; Bian, S.; Choi, H.; Ku, T.; Peer, A.; Chung, K.; Knoblich, J.A. Self-organized developmental patterning and differentiation in cerebral organoids. EMBO J. 2017, 36, 1316–1329. [Google Scholar] [CrossRef] [PubMed]
- Gerakis, Y.; Hetz, C. Brain organoids: A next step for humanized Alzheimer’s disease models? Mol. Psychiatry 2019, 24, 474–478. [Google Scholar] [CrossRef] [Green Version]
- Velasco, S.; Kedaigle, A.J.; Simmons, S.K.; Nash, A.; Rocha, M.; Quadrato, G.; Paulsen, B.; Nguyen, L.; Adiconis, X.; Regev, A.; et al. Individual brain organoids reproducibly form cell diversity of the human cerebral cortex. Nature 2019, 570, 523–527. [Google Scholar] [CrossRef]
- Yoon, S.J.; Elahi, L.S.; Pașca, A.M.; Marton, R.M.; Gordon, A.; Revah, O.; Miura, Y.; Walczak, E.M.; Holdgate, G.M.; Fan, H.C.; et al. Reliability of human cortical organoid generation. Nat. Methods 2019, 16, 75–78. [Google Scholar] [CrossRef]
- Smits, L.M.; Reinhardt, L.; Reinhardt, P.; Glatza, M.; Monzel, A.S.; Stanslowsky, N.; Rosato-Siri, M.D.; Zanon, A.; Antony, P.M.; Bellmann, J.; et al. Modeling Parkinson’s disease in midbrain-like organoids. NPJ Parkinson’s Dis. 2019, 5, 5. [Google Scholar] [CrossRef] [Green Version]
- Muguruma, K. Self-Organized Cerebellar Tissue from Human Pluripotent Stem Cells and Disease Modeling with Patient-Derived iPSCs. Cerebellum 2018, 17, 37–41. [Google Scholar] [CrossRef]
- Silva, T.P.; Fernandes, T.G.; Nogueira, D.E.S.; Rodrigues, C.A.V.; Bekman, E.P.; Hashimura, Y.; Jung, S.; Lee, B.; Carmo-Fonseca, M.; Cabral, J.M.S. Scalable Generation of Mature Cerebellar Organoids from Human Pluripotent Stem Cells and Characterization by Immunostaining. J. Vis. Exp. 2020. [CrossRef]
- Paşca, A.M.; Sloan, S.A.; Clarke, L.E.; Tian, Y.; Makinson, C.D.; Huber, N.; Kim, C.H.; Park, J.Y.; O’Rourke, N.A.; Nguyen, K.D.; et al. Functional cortical neurons and astrocytes from human pluripotent stem cells in 3D culture. Nat. Methods 2015, 12, 671–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sloan, S.A.; Darmanis, S.; Huber, N.; Khan, T.A.; Birey, F.; Caneda, C.; Reimer, R.; Quake, S.R.; Barres, B.A.; Paşca, S.P. Human Astrocyte Maturation Captured in 3D Cerebral Cortical Spheroids Derived from Pluripotent Stem Cells. Neuron 2017, 95, 779–790. [Google Scholar] [CrossRef] [PubMed]
- Mansour, A.A.; Gonçalves, J.T.; Bloyd, C.W.; Li, H.; Fernandes, S.; Quang, D.; Johnston, S.; Parylak, S.L.; Jin, X.; Gage, F.H. An in vivo model of functional and vascularized human brain organoids. Nat. Biotechnol. 2018, 36, 432–441. [Google Scholar] [CrossRef] [PubMed]
- Ormel, P.R.; Vieira de Sá, R.; van Bodegraven, E.J.; Karst, H.; Harschnitz, O.; Sneeboer, M.A.M.; Johansen, L.E.; van Dijk, R.E.; Scheefhals, N.; Berdenis van Berlekom, A.; et al. Microglia innately develop within cerebral organoids. Nat. Commun. 2018, 9, 4167. [Google Scholar] [CrossRef]
- Trujillo, C.A.; Gao, R.; Negraes, P.D.; Gu, J.; Buchanan, J.; Preissl, S.; Wang, A.; Wu, W.; Haddad, G.G.; Chaim, I.A.; et al. Complex Oscillatory Waves Emerging from Cortical Organoids Model Early Human Brain Network Development. Cell Stem Cell 2019, 25, 558–569. [Google Scholar] [CrossRef]
- Adhya, D.; Annuario, E.; Lancaster, M.A.; Price, J.; Baron-Cohen, S.; Srivastava, D.P. Understanding the role of steroids in typical and atypical brain development: Advantages of using a “brain in a dish” approach. J. Neuroendocrinol. 2018, 30. [Google Scholar] [CrossRef] [Green Version]
- de Boer, J.; Prikken, M.; Lei, W.U.; Begemann, M.; Sommer, I. The effect of raloxifene augmentation in men and women with a schizophrenia spectrum disorder: A systematic review and meta-analysis. NPJ Schizophr. 2018, 4, 1. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.M.; Zheng, W.; Li, X.H.; Cai, D.B.; Yang, X.H.; Ungvari, G.S.; Ng, C.H.; Wang, X.P.; Kulkarni, J.; Grigg, J.; et al. Adjunctive raloxifene for postmenopausal women with schizophrenia: A meta-analysis of randomized, double-blind, placebo-controlled trials. Schizophr. Res. 2018, 197, 288–293. [Google Scholar] [CrossRef]
- Palacios, J.; Yildiz, A.; Young, A.H.; Taylor, M.J. Tamoxifen for bipolar disorder: Systematic review and meta-analysis. J. Psychopharmacol. 2019, 33, 177–184. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Experimental Model | Preparation | E2 Effects | Receptor Involved | Reference |
|---|---|---|---|---|
| Ovariectomized mice | Whole brain mitochondria | ↑ activity of complex IV (COX); ↑ expression of nuDNA subunit COX IV, mtDNA subunit COX I, and of the antioxidant enzymes MnSOD, PrdxV, and glutathione peroxidase; ↓ lipoperoxidation; ↑ maximal mitochondrial capacity | ERα or Erβ | Irwin et al., 2012 [115] |
| Human lens cells | Cell culture | ↓ oxidative stress; ↓ mitochondrial membrane permeability | ERβ1 | Flynn et al., 2008 [116] |
| Human pituitary tumor cells | Cell culture | ↑ expression of the COX II subunit | N.I. | Van Itallie et al., 1988 [117] |
| Rats | Hippocampal tissue | ↑ expression of the COX III subunit; ↑ activity of COX | N.I. | Bettini et al., 1992 [118] |
| Several human cell lines | Cell culture | ↑ expression of the COX VII subunit | N.I. | Watanabe et al., 1998 [119] |
| Mouse cortical and mesencephalic astrocytes | Cell culture | ↑ expression of catalytic subunits of the ETC complexes I, III, IV and V; ↑ activity of ATP synthase; ↑ mtDNA/nuDNA ratio | N.I. | Araújo et al., 2008 [120] |
| MCF-7 and H1793 cells | Cell culture | ↑ expression of NRF-1; ↑ mitochondrial biogenesis | N.I. | Mattingly et al., 2008 [121] |
| Mouse astrocytes | Cell culture | ↑ transcription of mitochondrial fusion/fission proteins | N.I. | Arnold et al., 2008 [123] |
| HeLa cells | Cell culture | ↑ MCU-mediated mitochondrial Ca2+ uptake | N.I. | Lobatón et al., 2005 [124] |
| Ischemia-perfusion in Female rats | Brain tissue | ↑ expression of anti-apoptotic proteins (e.g., Bcl-2) | Erα | Zhang et al., 2017 [125] |
| SH-SY5Y cells | Cell culture | ↑ expression of the antioxidant proteins thioredoxin and MnSOD | N.I. | Chiueh et al., 2003 [126] |
| HT22 cells, mouse hippocampal and neocortical, and C6 cells | Cell culture | ↑ expression of glutathione | N.I. | Schmidt et al., 2002 [127] |
| Rat cortical neurons submitted to glutamatergic excitotoxicity | Cell culture | ↓ apoptosis; prevention of cytochrome c release; ↓ expression of caspase-3 | N.I. | Zhang et al., 2005 [128] |
| Pubertal period female mice | Ventromedial hypothalamus | ↑ microglial response to LPS | N.I. | Velez-Perez et al., 2020 [185] |
| Rat cortical neurons transfected with mutated DISC1 | Cell culture | ↑ spine density and synaptic proteins; ↓ DISC1 aggregates | N.I. | Erli et al., 2020 [244] |
| Rat cortical neurons submitted to glutamatergic excitotoxicity | Cell culture | ↓ release of lactate dehydrogenase | Classical estrogen receptors | Singer et al., 1996 [281] |
| Ovariectomized rats | Hypothalamic arcuate nucleus | ↑ number of Bcl-2-immunoreactive neurons | N.I. | Garcia-Segura et al., 1998 [282] |
| Rat hippocampal neurons submitted to oxidative stress inducers | Cell and organotypic culture | Prevention of peroxide accumulation | Independent on any ER activation | Behl et al., 1997 [283] |
| Ovariectomized rats | Several brain regions | Prevention of serotonin-receptor loss due to ovariectomy | N.I. | Cyr et al., 2000 [284] |
| Ovariectomized rats | CA1-region hippocampal neurons | ↑ excitability, independent of NMDA receptors | N.I. | Wong et al., 1992 [254] |
| Male adult rats | Hippocampal slices | ↑ synaptic responses by a mechanism dependent on integrin activation and signaling | Erβ | Wang et al., 2016 [257] |
| Rats and mice | Cell culture and brain slices | ↓ amplitude of inhibitory synaptic currents; destabilization of GABAARs and gephyrin at inhibitory synapses | N.I. | Mukherjee et al., 2017 [285] |
| Rats | Frontal cortex organotypic slice cultures | ↑ cortical expression of parvalbumin in both deep and superficial layers | N.I. | Ross and Porter, 2002 [263] |
| Ovariectomized monkeys | Dorsolateral prefrontal cortex | Regulated 40 genes, including ↑ expression of C-FOS and ↓ expression of E2F1 and TFIIB mRNA and protein | N.I. | Wang et al., 2004 [270] |
| Ovariectomized rats | Frontal cortex | Regulated 16 genes including ↓ expression of complement C3 and C4b, Ccl2, Tgfb1, macrophage expressed gene Mpeg1, RT1-Aw2, Cx3cr1, Fcgr2b, Cd11b, Tlr4 and Tlr9, defensin Np4, RatNP-3b, IgG-2a, Il6 and the ER gene Esr1 | ERα and Erβ | Sárvári et al., 2011 [271] |
| Ovariectomized mice | Hippocampus | Regulated 187 genes, mostly of them involved in transcription, cell signaling, cell growth, and lipid and protein metabolism | N.I. | Aenlle et al., 2009 [275] |
| Female mice | Dorsal hippocampus | Alteration in the expression of 204 genes, of which 23 are involved with learning/memory; changes in the content of the proteins Hsp70, Igfbp2, Actn4, Tubb2a, and Snap25 | N.I. | Pechenino and Frick, 2009 [276] |
| Human fetus | Neuron and glial cell cultures | Altered expression of 199 genes, many implicated in cell differentiation, cell cycle regulation, signal transduction, apoptosis, and ion channels and transporters | N.I. | Csöregh et al., 2009 [277] |
| Gonadectomized male rats | Hippocampus | ↑ levels of BDNF mRNA and protein | Erα | Solum and Handa, 2002 [278] |
| Intact and ovariectomized female rats | Hippocampus, cortex and spinal cord | ↑ expression of the BDNF gene | N.I. | Allen and McCarson, 2005 [279] |
| Ovariectomized rats | Amygdala, hypothalamus, nucleus accumbens, midbrain, and ventral tegmental area | ↑ levels of dopamine and serotonin receptors mRNA; ↓ levels of ERα and ERβ mRNA | N.I. | Zhou et al., 2002 [280] |
| Human cells | iPSC-derived forebrain neurons | ↑ number of dendritic branches | N.I. | Shum et al., 2015 [286] |
| Human cells in a rat model of Parkinson’s disease | iPSC-derived dopaminergic neurons | Activation of integrin α5β1 in the rat striatum; ↑ integration of grafted neurons into host striatum | N.I. | Nishimura et al., 2016 [287] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reis de Assis, D.; Szabo, A.; Requena Osete, J.; Puppo, F.; O’Connell, K.S.; A. Akkouh, I.; Hughes, T.; Frei, E.; A. Andreassen, O.; Djurovic, S. Using iPSC Models to Understand the Role of Estrogen in Neuron–Glia Interactions in Schizophrenia and Bipolar Disorder. Cells 2021, 10, 209. https://doi.org/10.3390/cells10020209
Reis de Assis D, Szabo A, Requena Osete J, Puppo F, O’Connell KS, A. Akkouh I, Hughes T, Frei E, A. Andreassen O, Djurovic S. Using iPSC Models to Understand the Role of Estrogen in Neuron–Glia Interactions in Schizophrenia and Bipolar Disorder. Cells. 2021; 10(2):209. https://doi.org/10.3390/cells10020209
Chicago/Turabian StyleReis de Assis, Denis, Attila Szabo, Jordi Requena Osete, Francesca Puppo, Kevin S. O’Connell, Ibrahim A. Akkouh, Timothy Hughes, Evgeniia Frei, Ole A. Andreassen, and Srdjan Djurovic. 2021. "Using iPSC Models to Understand the Role of Estrogen in Neuron–Glia Interactions in Schizophrenia and Bipolar Disorder" Cells 10, no. 2: 209. https://doi.org/10.3390/cells10020209
APA StyleReis de Assis, D., Szabo, A., Requena Osete, J., Puppo, F., O’Connell, K. S., A. Akkouh, I., Hughes, T., Frei, E., A. Andreassen, O., & Djurovic, S. (2021). Using iPSC Models to Understand the Role of Estrogen in Neuron–Glia Interactions in Schizophrenia and Bipolar Disorder. Cells, 10(2), 209. https://doi.org/10.3390/cells10020209