
Functional Role of B Cells in Atherosclerosis




Abstract
:1. Introduction
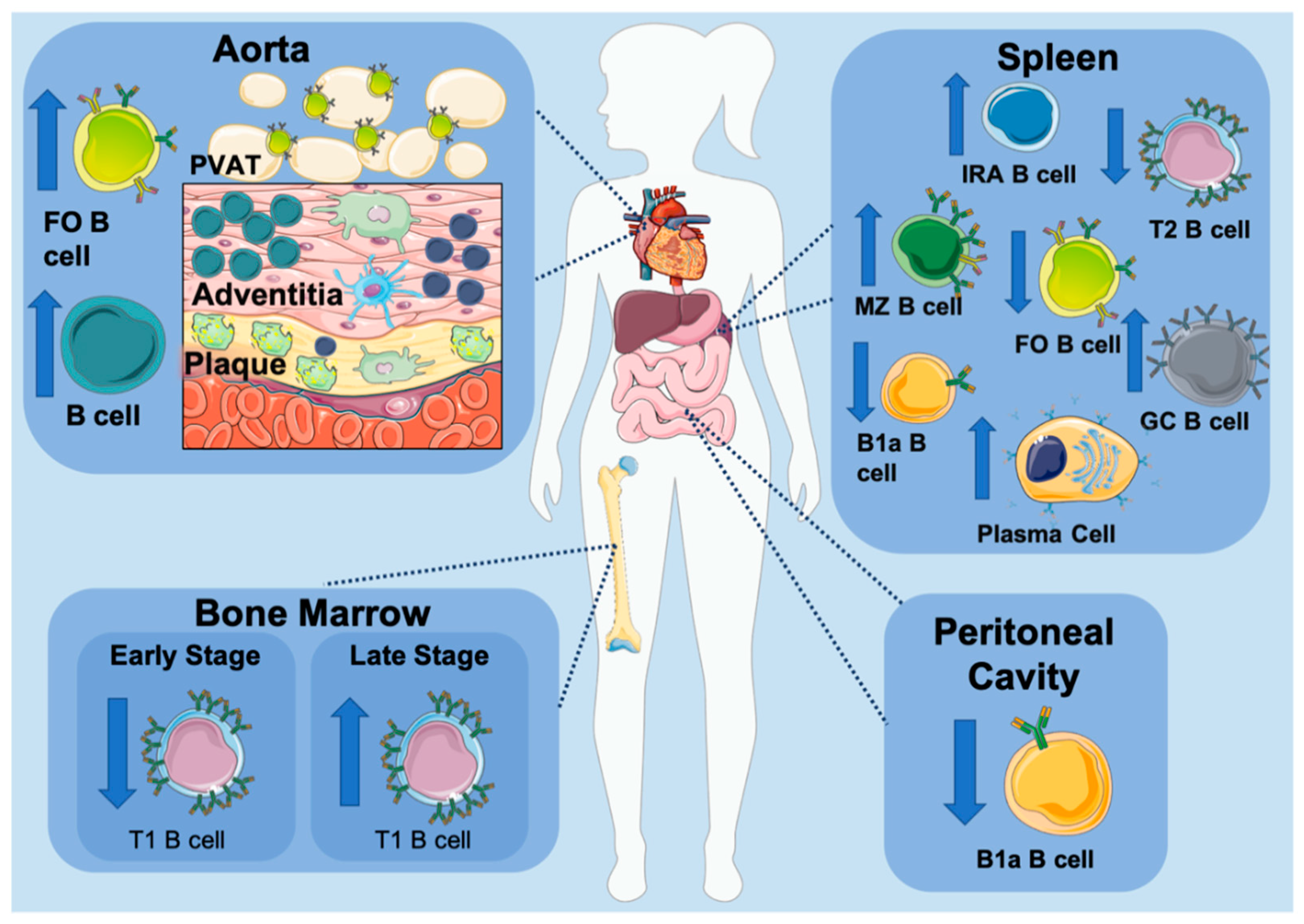
2. B Cells in Atherosclerosis
B Cells within the Aorta
3. BCR Signaling, B Cell Development, and Atherosclerosis
3.1. BCR Signaling
3.2. BCR Signaling in Atherosclerosis
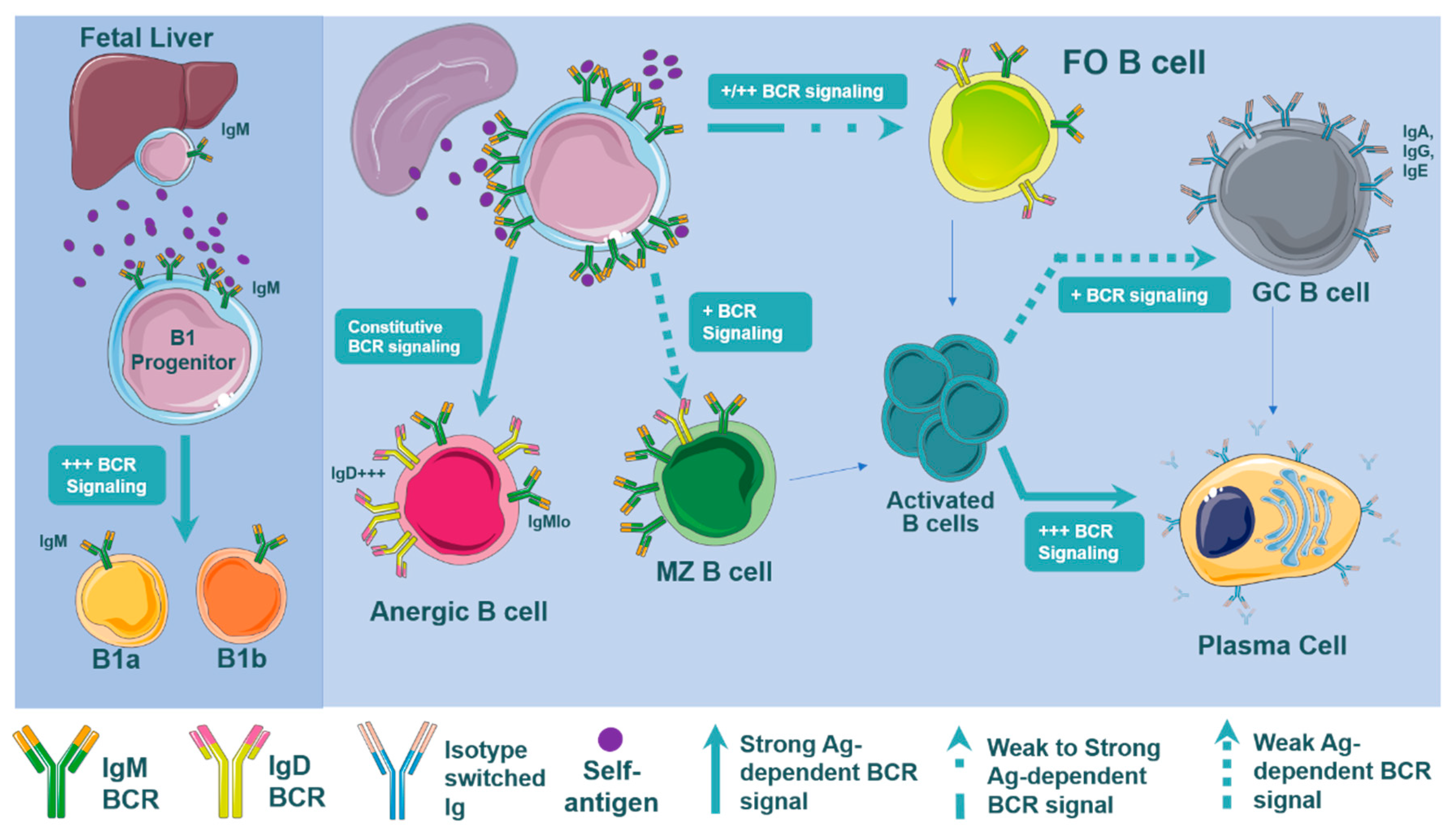
3.3. BCR Signaling and B Cell Development
3.4. B Cell Subset Development and BCR Signaling
3.5. B Cell Subset Development in Atherosclerosis
3.6. Toll Like Receptors, BCR Signaling, and Atherosclerosis
4. B Cell Subsets in Atherosclerosis
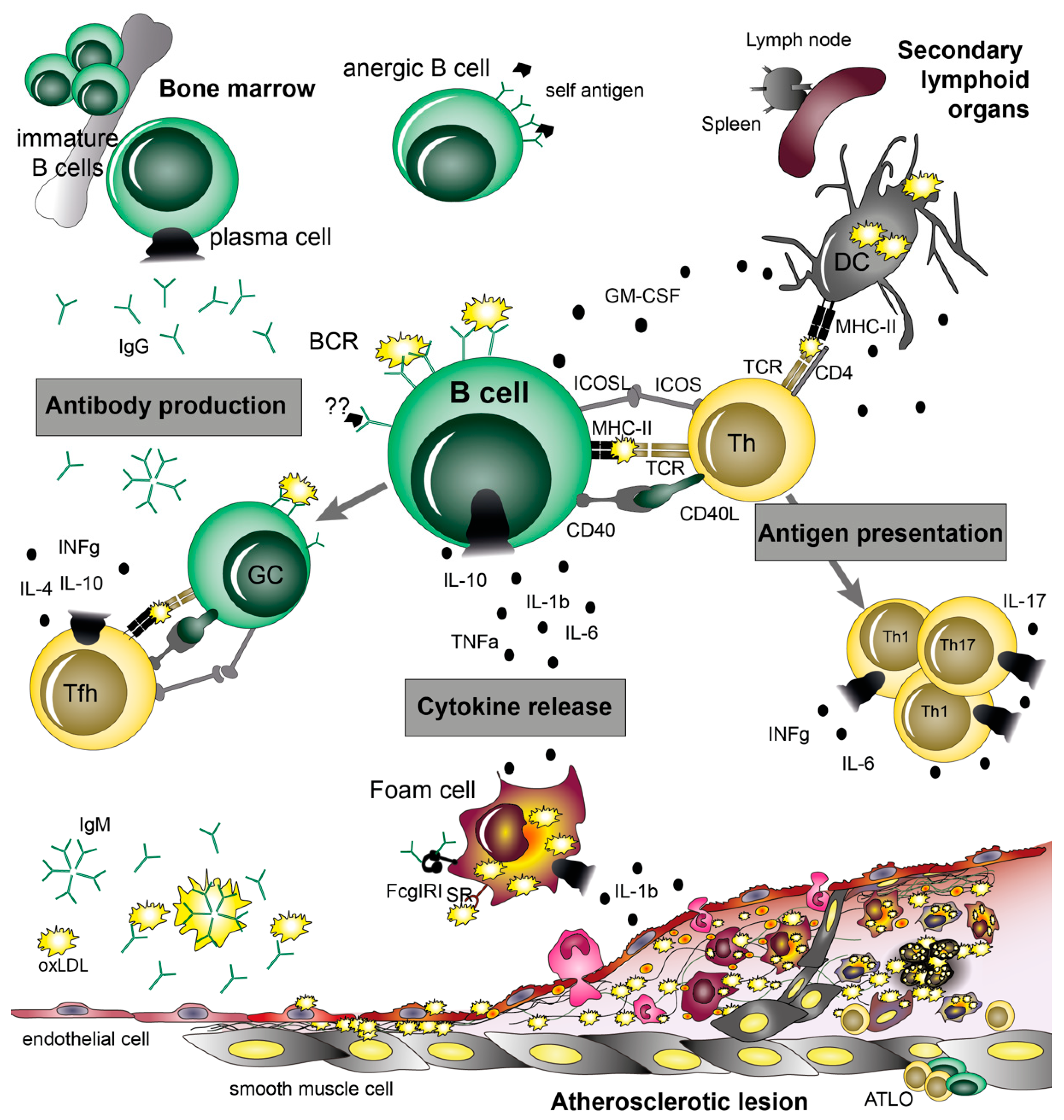
5. B Cell Functions in Atherosclerosis
5.1. Role of Immunoglobulins in Atherogenesis
5.2. IgM
5.3. IgM in Atherosclerosis
5.4. B1a and B1b Cells Produce OSE-Specific IgM
5.5. IgE and Atherosclerosis
5.6. IgG
6. Antigen Presentation and Co-Stimulatory Molecules
7. B Cell Cytokines
8. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Libby, P.; Buring, J.E.; Badimon, L.; Hansson, G.K.; Deanfield, J.; Bittencourt, M.S.; Tokgözoğlu, L.; Lewis, E.F. Atherosclerosis. Nat. Rev. Dis. Primers 2019, 5, 56. [Google Scholar] [CrossRef] [PubMed]
- Mayerl, C.; Lukasser, M.; Sedivy, R.; Niederegger, H.; Seiler, R.; Wick, G. Atherosclerosis research from past to present--on the track of two pathologists with opposing views, Carl von Rokitansky and Rudolf Virchow. Virchows Arch. 2006, 449, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Galkina, E.; Ley, K. Immune and inflammatory mechanisms of atherosclerosis. Annu. Rev. Immunol. 2009, 27, 165–197. [Google Scholar] [CrossRef] [Green Version]
- Galkina, E.; Kadl, A.; Sanders, J.; Varughese, D.; Sarembock, I.J.; Ley, K. Lymphocyte recruitment into the aortic wall before and during development of atherosclerosis is partially L-selectin dependent. J. Exp. Med. 2006, 203, 1273–1282. [Google Scholar] [CrossRef] [Green Version]
- Moore, K.J.; Sheedy, F.J.; Fisher, E.A. Macrophages in atherosclerosis: A dynamic balance. Nat. Rev. Immunol. 2013, 13, 709–721. [Google Scholar] [CrossRef]
- Spann, N.J.; Garmire, L.X.; McDonald, J.G.; Myers, D.S.; Milne, S.B.; Shibata, N.; Reichart, D.; Fox, J.N.; Shaked, I.; Heudobler, D.; et al. Regulated accumulation of desmosterol integrates macrophage lipid metabolism and inflammatory responses. Cell 2012, 151, 138–152. [Google Scholar] [CrossRef] [Green Version]
- Zhu, S.N.; Chen, M.; Jongstra-Bilen, J.; Cybulsky, M.I. GM-CSF regulates intimal cell proliferation in nascent atherosclerotic lesions. J. Exp. Med. 2009, 206, 2141–2149. [Google Scholar] [CrossRef]
- Jongstra-Bilen, J.; Haidari, M.; Zhu, S.N.; Chen, M.; Guha, D.; Cybulsky, M.I. Low-grade chronic inflammation in regions of the normal mouse arterial intima predisposed to atherosclerosis. J. Exp. Med. 2006, 203, 2073–2083. [Google Scholar] [CrossRef]
- Choi, J.H.; Do, Y.; Cheong, C.; Koh, H.; Boscardin, S.B.; Oh, Y.S.; Bozzacco, L.; Trumpfheller, C.; Park, C.G.; Steinman, R.M. Identification of antigen-presenting dendritic cells in mouse aorta and cardiac valves. J. Exp. Med. 2009, 206, 497–505. [Google Scholar] [CrossRef]
- Gil-Pulido, J.; Zernecke, A. Antigen-presenting dendritic cells in atherosclerosis. Eur. J. Pharmacol. 2017, 816, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Saigusa, R.; Winkels, H.; Ley, K. T cell subsets and functions in atherosclerosis. Nat. Rev. Cardiol. 2020, 17, 387–401. [Google Scholar] [CrossRef] [PubMed]
- Allam, G.; Abdel-Moneim, A.; Gaber, A.M. The pleiotropic role of interleukin-17 in atherosclerosis. Biomed. Pharmacother. 2018, 106, 1412–1418. [Google Scholar] [CrossRef] [PubMed]
- Baardman, J.; Lutgens, E. Regulatory T Cell Metabolism in Atherosclerosis. Metabolites 2020, 10, 279. [Google Scholar] [CrossRef]
- Gisterå, A.; Hansson, G.K. The immunology of atherosclerosis. Nat. Rev. Nephrol. 2017, 13, 368–380. [Google Scholar] [CrossRef]
- Hu, D.; Mohanta, S.K.; Yin, C.; Peng, L.; Ma, Z.; Srikakulapu, P.; Grassia, G.; MacRitchie, N.; Dever, G.; Gordon, P.; et al. Artery Tertiary Lymphoid Organs Control Aorta Immunity and Protect against Atherosclerosis via Vascular Smooth Muscle Cell Lymphotoxin β Receptors. Immunity 2015, 42, 1100–1115. [Google Scholar] [CrossRef] [Green Version]
- Zernecke, A.; Winkels, H.; Cochain, C.; Williams, J.W.; Wolf, D.; Soehnlein, O.; Robbins, C.S.; Monaco, C.; Park, I.; McNamara, C.A.; et al. Meta-Analysis of Leukocyte Diversity in Atherosclerotic Mouse Aortas. Circ. Res. 2020, 127, 402–426. [Google Scholar] [CrossRef]
- Sage, A.P.; Tsiantoulas, D.; Binder, C.J.; Mallat, Z. The role of B cells in atherosclerosis. Nat. Rev. Cardiol. 2019, 16, 180–196. [Google Scholar] [CrossRef]
- Liu, X.; Jiang, X.; Liu, R.; Wang, L.; Qian, T.; Zheng, Y.; Deng, Y.; Huang, E.; Xu, F.; Wang, J.Y.; et al. B cells expressing CD11b effectively inhibit CD4+ T-cell responses and ameliorate experimental autoimmune hepatitis in mice. Hepatol. (Baltim. Md.) 2015, 62, 1563–1575. [Google Scholar] [CrossRef] [Green Version]
- Stein, J.V.; Nombela-Arrieta, C. Chemokine control of lymphocyte trafficking: A general overview. Immunology 2005, 116, 1–12. [Google Scholar] [CrossRef]
- Grabner, R.; Lotzer, K.; Dopping, S.; Hildner, M.; Radke, D.; Beer, M.; Spanbroek, R.; Lippert, B.; Reardon, C.A.; Getz, G.S.; et al. Lymphotoxin beta receptor signaling promotes tertiary lymphoid organogenesis in the aorta adventitia of aged ApoE-/- mice. J. Exp. Med. 2009, 206, 233–248. [Google Scholar] [CrossRef] [Green Version]
- Doran, A.C.; Lipinski, M.J.; Oldham, S.N.; Garmey, J.C.; Campbell, K.A.; Skaflen, M.D.; Cutchins, A.; Lee, D.J.; Glover, D.K.; Kelly, K.A.; et al. B-cell aortic homing and atheroprotection depend on Id3. Circ. Res. 2012, 110, e1–e12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dal Porto, J.M.; Gauld, S.B.; Merrell, K.T.; Mills, D.; Pugh-Bernard, A.E.; Cambier, J. B cell antigen receptor signaling 101. Mol. Immunol. 2004, 41, 599–613. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, J.; Burrows, P.D.; Wang, J.Y. B Cell Development and Maturation. Adv. Exp. Med. Biol. 2020, 1254, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Vale, A.M.; Kearney, J.F.; Nobrega, A.; Schroeder, H.W. Development and Function of B Cell Subsets. In Molecular Biology of B Cells; Academic Press: Cambridge, MA, USA, 2015; pp. 99–119. [Google Scholar] [CrossRef]
- Yam-Puc, J.C.; Zhang, L.; Zhang, Y.; Toellner, K.M. Role of B-cell receptors for B-cell development and antigen-induced differentiation. F1000Research 2018, 7, 429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, Y.; Kitabatake, K.; Abe, R.; Tsukimoto, M. Involvement of A2B Receptor in DNA Damage Response and Radiosensitizing Effect of A2B Receptor Antagonists on Mouse B16 Melanoma. Biol. Pharm. Bull. 2020, 43, 516–525. [Google Scholar] [CrossRef] [Green Version]
- Hobeika, E.; Nielsen, P.J.; Medgyesi, D. Signaling mechanisms regulating B-lymphocyte activation and tolerance. J. Mol. Med. 2015, 93, 143–158. [Google Scholar] [CrossRef]
- Huang, C. Germinal Center Reaction. Adv. Exp. Med. Biol. 2020, 1254, 47–53. [Google Scholar] [CrossRef]
- Soh, S.Y.; Faveeuw, C.; Thiam, C.H.; Khoo, L.H.; Yeo, K.P.; Lim, S.Y.; Lim, H.Y.; Ng, J.X.; Angeli, V. NKT Cell Hyporesponsiveness Leads to Unrestrained Accumulation of Marginal Zone B Cells in Hypercholesterolemic Apolipoprotein E-Deficient Mice. J. Immunol. 2016, 197, 3894–3904. [Google Scholar] [CrossRef] [Green Version]
- Grasset, E.K.; Duhlin, A.; Agardh, H.E.; Ovchinnikova, O.; Hägglöf, T.; Forsell, M.N.; Paulsson-Berne, G.; Hansson, G.K.; Ketelhuth, D.F.J.; Karlsson, M.C.I. Sterile inflammation in the spleen during atherosclerosis provides oxidation-specific epitopes that induce a protective B-cell response. Proc. Natl. Acad. Sci. USA 2015, 112, E2030–E2038. [Google Scholar] [CrossRef] [Green Version]
- Caldeira, D.; Alves, D.; Costa, J.; Ferreira, J.J.; Pinto, F.J. Ibrutinib increases the risk of hypertension and atrial fibrillation: Systematic review and meta-analysis. PLoS ONE 2019, 14, e0211228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Myers, D.R.; Zikherman, J.; Roose, J.P. Tonic Signals: Why Do Lymphocytes Bother? Trends Immunol. 2017, 38, 844–857. [Google Scholar] [CrossRef] [PubMed]
- Melchers, F. Checkpoints that control B cell development. J. Clin. Investig. 2015, 125, 2203–2210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noviski, M.; Tan, C.; Huizar, J.; Vykunta, V.; Mueller, J.L.; Zikherman, J. Optimal Development of Mature B Cells Requires Recognition of Endogenous Antigens. J. Immunol. 2019, 203, 418–428. [Google Scholar] [CrossRef] [PubMed]
- Wardemann, H.; Yurasov, S.; Schaefer, A.; Young, J.W.; Meffre, E.; Nussenzweig, M.C. Predominant autoantibody production by early human B cell precursors. Science 2003, 301, 1374–1377. [Google Scholar] [CrossRef] [Green Version]
- Cambier, J.C.; Gauld, S.B.; Merrell, K.T.; Vilen, B.J. B-cell anergy: From transgenic models to naturally occurring anergic B cells? Nat. Rev. Immunol. 2007, 7, 633–643. [Google Scholar] [CrossRef]
- Noviski, M.; Mueller, J.L.; Satterthwaite, A.; Garrett-Sinha, L.A.; Brombacher, F.; Zikherman, J. IgM and IgD B cell receptors differentially respond to endogenous antigens and control B cell fate. eLife 2018, 7. [Google Scholar] [CrossRef]
- Cariappa, A.; Tang, M.; Parng, C.; Nebelitskiy, E.; Carroll, M.; Georgopoulos, K.; Pillai, S. The follicular versus marginal zone B lymphocyte cell fate decision is regulated by Aiolos, Btk, and CD21. Immunity 2001, 14, 603–615. [Google Scholar] [CrossRef] [Green Version]
- Tanigaki, K.; Han, H.; Yamamoto, N.; Tashiro, K.; Ikegawa, M.; Kuroda, K.; Suzuki, A.; Nakano, T.; Honjo, T. Notch-RBP-J signaling is involved in cell fate determination of marginal zone B cells. Nat. Immunol. 2002, 3, 443–450. [Google Scholar] [CrossRef]
- Wong, J.B.; Hewitt, S.L.; Heltemes-Harris, L.M.; Mandal, M.; Johnson, K.; Rajewsky, K.; Koralov, S.B.; Clark, M.R.; Farrar, M.A.; Skok, J.A. B-1a cells acquire their unique characteristics by bypassing the pre-BCR selection stage. Nat. Commun. 2019, 10, 4768. [Google Scholar] [CrossRef] [Green Version]
- Hardy, R.R. B-1 B cell development. J. Immunol. 2006, 177, 2749–2754. [Google Scholar] [CrossRef] [PubMed]
- Baumgarth, N. The double life of a B-1 cell: Self-reactivity selects for protective effector functions. Nat. Rev. Immunol. 2011, 11, 34–46. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.T.; Kläsener, K.; Zürn, C.; Castillo, P.A.; Brust-Mascher, I.; Imai, D.M.; Bevins, C.L.; Reardon, C.; Reth, M.; Baumgarth, N. The IgM receptor FcμR limits tonic BCR signaling by regulating expression of the IgM BCR. Nat. Immunol. 2017, 18, 321–333. [Google Scholar] [CrossRef] [PubMed]
- Pillai, S.; Cariappa, A. The follicular versus marginal zone B lymphocyte cell fate decision. Nat. Rev. Immunol. 2009, 9, 767–777. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, K.; Asano, M.; Shinton, S.A.; Gui, M.; Wen, L.J.; Dashoff, J.; Hardy, R.R. Positive selection of anti-thy-1 autoreactive B-1 cells and natural serum autoantibody production independent from bone marrow B cell development. J. Exp. Med. 2003, 197, 87–99. [Google Scholar] [CrossRef]
- Tsiantoulas, D.; Kiss, M.; Bartolini-Gritti, B.; Bergthaler, A.; Mallat, Z.; Jumaa, H.; Binder, C.J. Secreted IgM deficiency leads to increased BCR signaling that results in abnormal splenic B cell development. Sci. Rep. 2017, 7, 3540. [Google Scholar] [CrossRef]
- Tan, C.; Noviski, M.; Huizar, J.; Zikherman, J. Self-reactivity on a spectrum: A sliding scale of peripheral B cell tolerance. Immunol. Rev. 2019, 292, 37–60. [Google Scholar] [CrossRef]
- Ma, Z.; Choudhury, A.; Kang, S.A.; Monestier, M.; Cohen, P.L.; Eisenberg, R.A. Accelerated atherosclerosis in ApoE deficient lupus mouse models. Clin. Immunol. 2008, 127, 168–175. [Google Scholar] [CrossRef] [Green Version]
- Gruber, S.; Hendrikx, T.; Tsiantoulas, D.; Ozsvar-Kozma, M.; Göderle, L.; Mallat, Z.; Witztum, J.L.; Shiri-Sverdlov, R.; Nitschke, L.; Binder, C.J. Sialic Acid-Binding Immunoglobulin-like Lectin G Promotes Atherosclerosis and Liver Inflammation by Suppressing the Protective Functions of B-1 Cells. Cell Rep. 2016, 14, 2348–2361. [Google Scholar] [CrossRef] [Green Version]
- Jellusova, J.; Nitschke, L. Regulation of B cell functions by the sialic acid-binding receptors siglec-G and CD22. Front. Immunol. 2011, 2, 96. [Google Scholar] [CrossRef] [Green Version]
- Noviski, M.; Zikherman, J. Control of autoreactive B cells by IgM and IgD B cell receptors: Maintaining a fine balance. Curr. Opin. Immunol. 2018, 55, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Waisman, A.; Kraus, M.; Seagal, J.; Ghosh, S.; Melamed, D.; Song, J.; Sasaki, Y.; Classen, S.; Lutz, C.; Brombacher, F.; et al. IgG1 B cell receptor signaling is inhibited by CD22 and promotes the development of B cells whose survival is less dependent on Ig alpha/beta. J. Exp. Med. 2007, 204, 747–758. [Google Scholar] [CrossRef] [PubMed]
- Tsiantoulas, D.; Bot, I.; Ozsvar-Kozma, M.; Goderle, L.; Perkmann, T.; Hartvigsen, K.; Conrad, D.H.; Kuiper, J.; Mallat, Z.; Binder, C.J. Increased Plasma IgE Accelerate Atherosclerosis in Secreted IgM Deficiency. Circ. Res. 2017, 120, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Nus, M.; Basatemur, G.; Galan, M.; Cros-Brunsó, L.; Zhao, T.X.; Masters, L.; Harrison, J.; Figg, N.; Tsiantoulas, D.; Geissmann, F.; et al. NR4A1 Deletion in Marginal Zone B Cells Exacerbates Atherosclerosis in Mice-Brief Report. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 2598–2604. [Google Scholar] [CrossRef]
- Nus, M.; Sage, A.P.; Lu, Y.; Masters, L.; Lam, B.Y.H.; Newland, S.; Weller, S.; Tsiantoulas, D.; Raffort, J.; Marcus, D.; et al. Marginal zone B cells control the response of follicular helper T cells to a high-cholesterol diet. Nat. Med. 2017, 23, 601–610. [Google Scholar] [CrossRef] [Green Version]
- Sacquin, A.; Gador, M.; Fazilleau, N. The strength of BCR signaling shapes terminal development of follicular helper T cells in mice. Eur. J. Immunol. 2017, 47, 1295–1304. [Google Scholar] [CrossRef] [Green Version]
- Lechouane, F.; Bonaud, A.; Delpy, L.; Casola, S.; Oruc, Z.; Chemin, G.; Cogné, M.; Sirac, C. B-cell receptor signal strength influences terminal differentiation. Eur. J. Immunol. 2013, 43, 619–628. [Google Scholar] [CrossRef]
- Chan, T.D.; Gatto, D.; Wood, K.; Camidge, T.; Basten, A.; Brink, R. Antigen affinity controls rapid T-dependent antibody production by driving the expansion rather than the differentiation or extrafollicular migration of early plasmablasts. J. Immunol. 2009, 183, 3139–3149. [Google Scholar] [CrossRef] [Green Version]
- Nutt, S.L.; Hodgkin, P.D.; Tarlinton, D.M.; Corcoran, L.M. The generation of antibody-secreting plasma cells. Nat. Rev. Immunol. 2015, 15, 160–171. [Google Scholar] [CrossRef]
- Paus, D.; Phan, T.G.; Chan, T.D.; Gardam, S.; Basten, A.; Brink, R. Antigen recognition strength regulates the choice between extrafollicular plasma cell and germinal center B cell differentiation. J. Exp. Med. 2006, 203, 1081–1091. [Google Scholar] [CrossRef] [Green Version]
- Rubtsov, A.V.; Swanson, C.L.; Troy, S.; Strauch, P.; Pelanda, R.; Torres, R.M. TLR Agonists Promote Marginal Zone B Cell Activation and Facilitate T-Dependent IgM Responses. J. Immunol. 2008, 180, 3882–3888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rawlings, D.J.; Schwartz, M.A.; Jackson, S.W.; Meyer-Bahlburg, A. Integration of B cell responses through Toll-like receptors and antigen receptors. Nat. Rev. Immunol. 2012, 12, 282–294. [Google Scholar] [CrossRef] [PubMed]
- Azulay-Debby, H.; Edry, E.; Melamed, D. CpG DNA stimulates autoreactive immature B cells in the bone marrow. Eur. J. Immunol. 2007, 37, 1463–1475. [Google Scholar] [CrossRef]
- Meyer-Bahlburg, A.; Rawlings, D.J. Differential impact of Toll-like receptor signaling on distinct B cell subpopulations. Front. Biosci. (Landmark Ed.) 2012, 17, 1499–1516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cinamon, G.; Matloubian, M.; Lesneski, M.J.; Xu, Y.; Low, C.; Lu, T.; Proia, R.L.; Cyster, J.G. Sphingosine 1-phosphate receptor 1 promotes B cell localization in the splenic marginal zone. Nat. Immunol. 2004, 5, 713–720. [Google Scholar] [CrossRef]
- Lund, F.E. Cytokine-producing B lymphocytes-key regulators of immunity. Curr. Opin. Immunol. 2008, 20, 332–338. [Google Scholar] [CrossRef] [Green Version]
- Meyer-Bahlburg, A.; Bandaranayake, A.D.; Andrews, S.F.; Rawlings, D.J. Reduced c-myc expression levels limit follicular mature B cell cycling in response to TLR signals. J. Immunol. 2009, 182, 4065–4075. [Google Scholar] [CrossRef] [Green Version]
- Gururajan, M.; Jacob, J.; Pulendran, B. Toll-like receptor expression and responsiveness of distinct murine splenic and mucosal B-cell subsets. PLoS ONE 2007, 2, e863. [Google Scholar] [CrossRef] [Green Version]
- Meyer-Bahlburg, A.; Khim, S.; Rawlings, D.J. B cell intrinsic TLR signals amplify but are not required for humoral immunity. J. Exp. Med. 2007, 204, 3095–3101. [Google Scholar] [CrossRef] [Green Version]
- Cole, J.E.; Georgiou, E.; Monaco, C. The expression and functions of toll-like receptors in atherosclerosis. Mediat. Inflamm 2010, 2010, 393946. [Google Scholar] [CrossRef] [Green Version]
- Edfeldt, K.; Swedenborg, J.; Hansson, G.K.; Yan, Z.Q. Expression of toll-like receptors in human atherosclerotic lesions: A possible pathway for plaque activation. Circulation 2002, 105, 1158–1161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roshan, M.H.; Tambo, A.; Pace, N.P. The Role of TLR2, TLR4, and TLR9 in the Pathogenesis of Atherosclerosis. Int. J. Inflam. 2016, 2016, 1532832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cole, J.E.; Mitra, A.T.; Monaco, C. Treating atherosclerosis: The potential of Toll-like receptors as therapeutic targets. Expert Rev. Cardiovasc. 2010, 8, 1619–1635. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, C.; Papadopoulos, G.; Gudino, C.V.; Weinberg, E.O.; Barth, K.R.; Madrigal, A.G.; Chen, Y.; Ning, H.; LaValley, M.; Gibson, F.C., 3rd; et al. Protective role for TLR4 signaling in atherosclerosis progression as revealed by infection with a common oral pathogen. J. Immunol. 2012, 189, 3681–3688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michelsen, K.S.; Wong, M.H.; Shah, P.K.; Zhang, W.; Yano, J.; Doherty, T.M.; Akira, S.; Rajavashisth, T.B.; Arditi, M. Lack of Toll-like receptor 4 or myeloid differentiation factor 88 reduces atherosclerosis and alters plaque phenotype in mice deficient in apolipoprotein E. Proc. Natl. Acad. Sci. USA 2004, 101, 10679–10684. [Google Scholar] [CrossRef] [Green Version]
- Koulis, C.; Chen, Y.C.; Hausding, C.; Ahrens, I.; Kyaw, T.S.; Tay, C.; Allen, T.; Jandeleit-Dahm, K.; Sweet, M.J.; Akira, S.; et al. Protective role for Toll-like receptor-9 in the development of atherosclerosis in apolipoprotein E-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 516–525. [Google Scholar] [CrossRef] [Green Version]
- Hosseini, H.; Li, Y.; Kanellakis, P.; Tay, C.; Cao, A.; Liu, E.; Peter, K.; Tipping, P.; Toh, B.-H.; Bobik, A.; et al. Toll-Like Receptor (TLR)4 and MyD88 are Essential for Atheroprotection by Peritoneal B1a B Cells. J. Am. Heart Assoc. 2016, 5, e002947. [Google Scholar] [CrossRef]
- Karper, J.C.; de Jager, S.C.; Ewing, M.M.; de Vries, M.R.; Bot, I.; van Santbrink, P.J.; Redeker, A.; Mallat, Z.; Binder, C.J.; Arens, R.; et al. An unexpected intriguing effect of Toll-like receptor regulator RP105 (CD180) on atherosclerosis formation with alterations on B-cell activation. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2810–2817. [Google Scholar] [CrossRef] [Green Version]
- Suthers, A.N.; Sarantopoulos, S. TLR7/TLR9- and B Cell Receptor-Signaling Crosstalk: Promotion of Potentially Dangerous B Cells. Front. Immunol. 2017, 8, 775. [Google Scholar] [CrossRef] [Green Version]
- Baumgarth, N. A Hard(y) Look at B-1 Cell Development and Function. J. Immunol. 2017, 199, 3387–3394. [Google Scholar] [CrossRef]
- Tsiantoulas, D.; Sage, A.P.; Mallat, Z.; Binder, C.J. Targeting B cells in atherosclerosis: Closing the gap from bench to bedside. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 296–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haas, K.M.; Poe, J.C.; Steeber, D.A.; Tedder, T.F. B-1a and B-1b cells exhibit distinct developmental requirements and have unique functional roles in innate and adaptive immunity to S. pneumoniae. Immunity 2005, 23, 7–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, B.; Shukla, S.; Łyszkiewicz, M.; Krey, M.; Viegas, N.; Düber, S.; Weiss, S. Somatic hypermutation in peritoneal B1b cells. Mol. Immunol. 2009, 46, 1613–1619. [Google Scholar] [CrossRef] [PubMed]
- Shaw, P.X.; Hörkkö, S.; Chang, M.-K.; Curtiss, L.K.; Palinski, W.; Silverman, G.J.; Witztum, J.L. Natural antibodies with the T15 idiotype may act in atherosclerosis, apoptotic clearance, and protective immunity. J. Clin. Investig. 2000, 105, 1731–1740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masmoudi, H.; Mota-Santos, T.; Huetz, F.; Coutinho, A.; Cazenave, P.A. All T15 Id-positive antibodies (but not the majority of VHT15+ antibodies) are produced by peritoneal CD5+ B lymphocytes. Int. Immunol. 1990, 2, 515–520. [Google Scholar] [CrossRef] [PubMed]
- Kyaw, T.; Tay, C.; Krishnamurthi, S.; Kanellakis, P.; Agrotis, A.; Tipping, P.; Bobik, A.; Toh, B.H. B1a B lymphocytes are atheroprotective by secreting natural IgM that increases IgM deposits and reduces necrotic cores in atherosclerotic lesions. Circ. Res. 2011, 109, 830–840. [Google Scholar] [CrossRef]
- Rosenfeld, S.M.; Perry, H.M.; Gonen, A.; Prohaska, T.A.; Srikakulapu, P.; Grewal, S.; Das, D.; McSkimming, C.; Taylor, A.M.; Tsimikas, S.; et al. B-1b Cells Secrete Atheroprotective IgM and Attenuate Atherosclerosis. Circ. Res. 2015, 117, e28–e39. [Google Scholar] [CrossRef] [Green Version]
- Hardy, R.R.; Hayakawa, K. B cell development pathways. Annu. Rev. Immunol. 2001, 19, 595–621. [Google Scholar] [CrossRef]
- Caligiuri, G.; Nicoletti, A.; Poirier, B.; Hansson, G.K. Protective immunity against atherosclerosis carried by B cells of hypercholesterolemic mice. J. Clin. Investig. 2002, 109, 745–753. [Google Scholar] [CrossRef]
- Rauch, P.J.; Chudnovskiy, A.; Robbins, C.S.; Weber, G.F.; Etzrodt, M.; Hilgendorf, I.; Tiglao, E.; Figueiredo, J.L.; Iwamoto, Y.; Theurl, I.; et al. Innate response activator B cells protect against microbial sepsis. Science 2012, 335, 597–601. [Google Scholar] [CrossRef] [Green Version]
- Hilgendorf, I.; Theurl, I.; Gerhardt, L.M.S.; Robbins, C.S.; Weber, G.F.; Gonen, A.; Iwamoto, Y.; Degousee, N.; Holderried, T.A.W.; Winter, C.; et al. Innate Response Activator B Cells Aggravate Atherosclerosis by Stimulating T Helper-1 Adaptive Immunity. Circulation 2014, 129, 1677–1687. [Google Scholar] [CrossRef] [PubMed]
- Tay, C.; Liu, Y.H.; Hosseini, H.; Kanellakis, P.; Cao, A.; Peter, K.; Tipping, P.; Bobik, A.; Toh, B.H.; Kyaw, T. B-cell-specific depletion of tumour necrosis factor alpha inhibits atherosclerosis development and plaque vulnerability to rupture by reducing cell death and inflammation. Cardiovasc. Res. 2016, 111, 385–397. [Google Scholar] [CrossRef]
- Tsiantoulas, D.; Sage, A.P.; Goderle, L.; Ozsvar-Kozma, M.; Murphy, D.; Porsch, F.; Pasterkamp, G.; Menche, J.; Schneider, P.; Mallat, Z.; et al. B Cell-Activating Factor Neutralization Aggravates Atherosclerosis. Circulation 2018, 138, 2263–2273. [Google Scholar] [CrossRef] [PubMed]
- Ait-Oufella, H.; Herbin, O.; Bouaziz, J.D.; Binder, C.J.; Uyttenhove, C.; Laurans, L.; Taleb, S.; Van Vré, E.; Esposito, B.; Vilar, J.; et al. B cell depletion reduces the development of atherosclerosis in mice. J. Exp. Med. 2010, 207, 1579–1587. [Google Scholar] [CrossRef] [PubMed]
- Kyaw, T.; Tay, C.; Khan, A.; Dumouchel, V.; Cao, A.; To, K.; Kehry, M.; Dunn, R.; Agrotis, A.; Tipping, P.; et al. Conventional B2 B cell depletion ameliorates whereas its adoptive transfer aggravates atherosclerosis. J. Immunol. 2010, 185, 4410–4419. [Google Scholar] [CrossRef]
- Kyaw, T.; Cui, P.; Tay, C.; Kanellakis, P.; Hosseini, H.; Liu, E.; Rolink, A.G.; Tipping, P.; Bobik, A.; Toh, B.H. BAFF receptor mAb treatment ameliorates development and progression of atherosclerosis in hyperlipidemic ApoE(-/-) mice. PLoS ONE 2013, 8, e60430. [Google Scholar] [CrossRef] [Green Version]
- Major, A.S.; Fazio, S.; Linton, M.F. B-lymphocyte deficiency increases atherosclerosis in LDL receptor-null mice. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 1892–1898. [Google Scholar] [CrossRef] [Green Version]
- Sage, A.P.; Tsiantoulas, D.; Baker, L.; Harrison, J.; Masters, L.; Murphy, D.; Loinard, C.; Binder, C.J.; Mallat, Z. BAFF receptor deficiency reduces the development of atherosclerosis in mice--brief report. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1573–1576. [Google Scholar] [CrossRef] [Green Version]
- Tay, C.; Liu, Y.H.; Kanellakis, P.; Kallies, A.; Li, Y.; Cao, A.; Hosseini, H.; Tipping, P.; Toh, B.H.; Bobik, A.; et al. Follicular B Cells Promote Atherosclerosis via T Cell-Mediated Differentiation into Plasma Cells and Secreting Pathogenic Immunoglobulin G. Arterioscler. Thromb. Vasc. Biol. 2018, 38, e71–e84. [Google Scholar] [CrossRef] [Green Version]
- Mauri, C.; Bosma, A. Immune regulatory function of B cells. Annu. Rev. Immunol. 2012, 30, 221–241. [Google Scholar] [CrossRef]
- Casola, S.; Otipoby, K.L.; Alimzhanov, M.; Humme, S.; Uyttersprot, N.; Kutok, J.L.; Carroll, M.C.; Rajewsky, K. B cell receptor signal strength determines B cell fate. Nat. Immunol. 2004, 5, 317–327. [Google Scholar] [CrossRef]
- Baumgarth, N.; Herman, O.C.; Jager, G.C.; Brown, L.E.; Herzenberg, L.A.; Chen, J. B-1 and B-2 cell-derived immunoglobulin M antibodies are nonredundant components of the protective response to influenza virus infection. J. Exp. Med. 2000, 192, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Lam, K.P.; Kühn, R.; Rajewsky, K. In vivo ablation of surface immunoglobulin on mature B cells by inducible gene targeting results in rapid cell death. Cell 1997, 90, 1073–1083. [Google Scholar] [CrossRef] [Green Version]
- Srinivasan, L.; Sasaki, Y.; Calado, D.P.; Zhang, B.; Paik, J.H.; DePinho, R.A.; Kutok, J.L.; Kearney, J.F.; Otipoby, K.L.; Rajewsky, K. PI3 kinase signals BCR-dependent mature B cell survival. Cell 2009, 139, 573–586. [Google Scholar] [CrossRef] [Green Version]
- Ehrenstein, M.R.; Notley, C.A. The importance of natural IgM: Scavenger, protector and regulator. Nat. Rev. Immunol. 2010, 10, 778–786. [Google Scholar] [CrossRef] [PubMed]
- Woof, J.M.; Mestecky, J. Mucosal immunoglobulins. Immunol. Rev. 2005, 206, 64–82. [Google Scholar] [CrossRef]
- Coutinho, A.; Kazatchkine, M.D.; Avrameas, S. Natural autoantibodies. Curr. Opin. Immunol. 1995, 7, 812–818. [Google Scholar] [CrossRef]
- Jones, K.; Savulescu, A.F.; Brombacher, F.; Hadebe, S. Immunoglobulin M in Health and Diseases: How Far Have We Come and What Next? Front. Immunol. 2020, 11, 595535. [Google Scholar] [CrossRef]
- Martin, F.; Oliver, A.M.; Kearney, J.F. Marginal zone and B1 B cells unite in the early response against T-independent blood-borne particulate antigens. Immunity 2001, 14, 617–629. [Google Scholar] [CrossRef] [Green Version]
- Bohannon, C.; Powers, R.; Satyabhama, L.; Cui, A.; Tipton, C.; Michaeli, M.; Skountzou, I.; Mittler, R.S.; Kleinstein, S.H.; Mehr, R.; et al. Long-lived antigen-induced IgM plasma cells demonstrate somatic mutations and contribute to long-term protection. Nat. Commun. 2016, 7, 11826. [Google Scholar] [CrossRef] [Green Version]
- Binder, C.J.; Papac-Milicevic, N.; Witztum, J.L. Innate sensing of oxidation-specific epitopes in health and disease. Nat. Rev. Immunol. 2016, 16, 485–497. [Google Scholar] [CrossRef] [PubMed]
- Miller, Y.I.; Choi, S.H.; Wiesner, P.; Fang, L.; Harkewicz, R.; Hartvigsen, K.; Boullier, A.; Gonen, A.; Diehl, C.J.; Que, X.; et al. Oxidation-specific epitopes are danger-associated molecular patterns recognized by pattern recognition receptors of innate immunity. Circ. Res. 2011, 108, 235–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsiantoulas, D.; Perkmann, T.; Afonyushkin, T.; Mangold, A.; Prohaska, T.A.; Papac-Milicevic, N.; Millischer, V.; Bartel, C.; Hörkkö, S.; Boulanger, C.M.; et al. Circulating microparticles carry oxidation-specific epitopes and are recognized by natural IgM antibodies. J. Lipid Res. 2015, 56, 440–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palinski, W.; Hörkkö, S.; Miller, E.; Steinbrecher, U.P.; Powell, H.C.; Curtiss, L.K.; Witztum, J.L. Cloning of monoclonal autoantibodies to epitopes of oxidized lipoproteins from apolipoprotein E-deficient mice. Demonstration of epitopes of oxidized low density lipoprotein in human plasma. J. Clin. Investig. 1996, 98, 800–814. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.-K.; Bergmark, C.; Laurila, A.; Hörkkö, S.; Han, K.-H.; Friedman, P.; Dennis, E.A.; Witztum, J.L. Monoclonal antibodies against oxidized low-density lipoprotein bind to apoptotic cells and inhibit their phagocytosis by elicited macrophages: Evidence that oxidation-specific epitopes mediate macrophage recognition. Proc. Natl. Acad. Sci. USA 1999, 96, 6353–6358. [Google Scholar] [CrossRef] [Green Version]
- Hörkkö, S.; Bird, D.A.; Miller, E.; Itabe, H.; Leitinger, N.; Subbanagounder, G.; Berliner, J.A.; Friedman, P.; Dennis, E.A.; Curtiss, L.K.; et al. Monoclonal autoantibodies specific for oxidized phospholipids or oxidized phospholipid–protein adducts inhibit macrophage uptake of oxidized low-density lipoproteins. J. Clin. Investig. 1999, 103, 117–128. [Google Scholar] [CrossRef] [Green Version]
- Que, X.; Hung, M.Y.; Yeang, C.; Gonen, A.; Prohaska, T.A.; Sun, X.; Diehl, C.; Määttä, A.; Gaddis, D.E.; Bowden, K.; et al. Oxidized phospholipids are proinflammatory and proatherogenic in hypercholesterolaemic mice. Nature 2018, 558, 301–306. [Google Scholar] [CrossRef]
- Cesena, F.H.; Dimayuga, P.C.; Yano, J.; Zhao, X.; Kirzner, J.; Zhou, J.; Chan, L.F.; Lio, W.M.; Cercek, B.; Shah, P.K.; et al. Immune-modulation by polyclonal IgM treatment reduces atherosclerosis in hypercholesterolemic apoE-/- mice. Atherosclerosis 2012, 220, 59–65. [Google Scholar] [CrossRef] [Green Version]
- Binder, C.J.; Hörkkö, S.; Dewan, A.; Chang, M.K.; Kieu, E.P.; Goodyear, C.S.; Shaw, P.X.; Palinski, W.; Witztum, J.L.; Silverman, G.J. Pneumococcal vaccination decreases atherosclerotic lesion formation: Molecular mimicry between Streptococcus pneumoniae and oxidized LDL. Nat. Med. 2003, 9, 736–743. [Google Scholar] [CrossRef]
- Lewis, M.J.; Malik, T.H.; Ehrenstein, M.R.; Boyle, J.J.; Botto, M.; Haskard, D.O. Immunoglobulin M is required for protection against atherosclerosis in low-density lipoprotein receptor-deficient mice. Circulation 2009, 120, 417–426. [Google Scholar] [CrossRef] [Green Version]
- Centa, M.; Gruber, S.; Nilsson, D.; Polyzos, K.A.; Johansson, D.K.; Hansson, G.K.; Ketelhuth, D.F.; Binder, C.J.; Malin, S. Atherosclerosis Susceptibility in Mice Is Independent of the V1 Immunoglobulin Heavy Chain Gene. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 25–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biswas, T.K.; VanderLaan, P.A.; Que, X.; Gonen, A.; Krishack, P.; Binder, C.J.; Witztum, J.L.; Getz, G.S.; Reardon, C.A. CD1d Selectively Down Regulates the Expression of the Oxidized Phospholipid-Specific E06 IgM Natural Antibody in Ldlr(-/-) Mice. Antibodies (Baselswitzerland) 2020, 9, 30. [Google Scholar] [CrossRef] [PubMed]
- Upadhye, A.; Srikakulapu, P.; Gonen, A.; Hendrikx, S.; Perry, H.M.; Nguyen, A.; McSkimming, C.; Marshall, M.A.; Garmey, J.C.; Taylor, A.M.; et al. Diversification and CXCR4-Dependent Establishment of the Bone Marrow B-1a Cell Pool Governs Atheroprotective IgM Production Linked to Human Coronary Atherosclerosis. Circ. Res. 2019, 125, e55–e70. [Google Scholar] [CrossRef] [PubMed]
- Döring, Y.; Jansen, Y.; Cimen, I.; Aslani, M.; Gencer, S.; Peters, L.J.F.; Duchene, J.; Weber, C.; van der Vorst, E.P.C. B-Cell-Specific CXCR4 Protects Against Atherosclerosis Development and Increases Plasma IgM Levels. Circ. Res. 2020, 126, 787–788. [Google Scholar] [CrossRef] [PubMed]
- Prohaska, T.A.; Que, X.; Diehl, C.J.; Hendrikx, S.; Chang, M.W.; Jepsen, K.; Glass, C.K.; Benner, C.; Witztum, J.L. Massively Parallel Sequencing of Peritoneal and Splenic B Cell Repertoires Highlights Unique Properties of B-1 Cell Antibodies. J. Immunol. (Baltimore, Md.: 1950) 2018, 200, 1702–1717. [Google Scholar] [CrossRef] [Green Version]
- Boes, M.; Esau, C.; Fischer, M.B.; Schmidt, T.; Carroll, M.; Chen, J. Enhanced B-1 cell development, but impaired IgG antibody responses in mice deficient in secreted IgM. J. Immunol. 1998, 160, 4776–4787. [Google Scholar] [PubMed]
- Moon, B.G.; Takaki, S.; Miyake, K.; Takatsu, K. The role of IL-5 for mature B-1 cells in homeostatic proliferation, cell survival, and Ig production. J. Immunol. 2004, 172, 6020–6029. [Google Scholar] [CrossRef] [Green Version]
- Binder, C.J.; Hartvigsen, K.; Chang, M.K.; Miller, M.; Broide, D.; Palinski, W.; Curtiss, L.K.; Corr, M.; Witztum, J.L. IL-5 links adaptive and natural immunity specific for epitopes of oxidized LDL and protects from atherosclerosis. J. Clin. Investig. 2004, 114, 427–437. [Google Scholar] [CrossRef]
- Alugupalli, K.R. A distinct role for B1b lymphocytes in T cell-independent immunity. Curr. Top. Microbiol. Immunol. 2008, 319, 105–130. [Google Scholar] [CrossRef] [Green Version]
- Ishizaka, K.; Ishizaka, T. Identification of gamma-E-antibodies as a carrier of reaginic activity. J. Immunol. 1967, 99, 1187–1198. [Google Scholar]
- Wu, L.C.; Zarrin, A.A. The production and regulation of IgE by the immune system. Nat. Rev. Immunol. 2014, 14, 247–259. [Google Scholar] [CrossRef] [PubMed]
- Sanjuan, M.A.; Sagar, D.; Kolbeck, R. Role of IgE in autoimmunity. J. Allergy Clin. Immunol. 2016, 137, 1651–1661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Cheng, X.; Xiang, M.X.; Alanne-Kinnunen, M.; Wang, J.A.; Chen, H.; He, A.; Sun, X.; Lin, Y.; Tang, T.T.; et al. IgE stimulates human and mouse arterial cell apoptosis and cytokine expression and promotes atherogenesis in Apoe-/- mice. J. Clin. Investig. 2011, 121, 3564–3577. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, J.; Luo, S.; Wang, M.; Huang, Q.; Deng, Z.; de Febbo, C.; Daoui, A.; Liew, P.X.; Sukhova, G.K.; et al. IgE Contributes to Atherosclerosis and Obesity by Affecting Macrophage Polarization, Macrophage Protein Network, and Foam Cell Formation. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 597–610. [Google Scholar] [CrossRef]
- Vidarsson, G.; Dekkers, G.; Rispens, T. IgG subclasses and allotypes: From structure to effector functions. Front. Immunol. 2014, 5, 520. [Google Scholar] [CrossRef] [Green Version]
- Ferrante, A.; Beard, L.J.; Feldman, R.G. IgG subclass distribution of antibodies to bacterial and viral antigens. Pediatric Infect. Dis. J. 1990, 9, S16–S24. [Google Scholar] [CrossRef]
- Siber, G.R.; Schur, P.H.; Aisenberg, A.C.; Weitzman, S.A.; Schiffman, G. Correlation between serum IgG-2 concentrations and the antibody response to bacterial polysaccharide antigens. N. Engl. J. Med. 1980, 303, 178–182. [Google Scholar] [CrossRef]
- Ottesen, E.A.; Skvaril, F.; Tripathy, S.P.; Poindexter, R.W.; Hussain, R. Prominence of IgG4 in the IgG antibody response to human filariasis. J. Immunol. 1985, 134, 2707–2712. [Google Scholar]
- Leibundgut, G.; Witztum, J.L.; Tsimikas, S. Oxidation-specific epitopes and immunological responses: Translational biotheranostic implications for atherosclerosis. Curr. Opin. Pharmacol. 2013, 13, 168–179. [Google Scholar] [CrossRef] [Green Version]
- Tsimikas, S.; Brilakis, E.S.; Lennon, R.J.; Miller, E.R.; Witztum, J.L.; McConnell, J.P.; Kornman, K.S.; Berger, P.B. Relationship of IgG and IgM autoantibodies to oxidized low density lipoprotein with coronary artery disease and cardiovascular events. J. Lipid Res. 2007, 48, 425–433. [Google Scholar] [CrossRef] [Green Version]
- Palinski, W.; Miller, E.; Witztum, J.L. Immunization of low density lipoprotein (LDL) receptor-deficient rabbits with homologous malondialdehyde-modified LDL reduces atherogenesis. Proc. Natl. Acad. Sci. USA 1995, 92, 821–825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsimikas, S.; Palinski, W.; Witztum, J.L. Circulating autoantibodies to oxidized LDL correlate with arterial accumulation and depletion of oxidized LDL in LDL receptor-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 95–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansson, G.K.; Nilsson, J. Vaccination against atherosclerosis? Induction of atheroprotective immunity. Semin. Immunopathol. 2009, 31, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Tsimikas, S.; Miyanohara, A.; Hartvigsen, K.; Merki, E.; Shaw, P.X.; Chou, M.Y.; Pattison, J.; Torzewski, M.; Sollors, J.; Friedmann, T.; et al. Human oxidation-specific antibodies reduce foam cell formation and atherosclerosis progression. J. Am. Coll. Cardiol. 2011, 58, 1715–1727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kyaw, T.; Tay, C.; Hosseini, H.; Kanellakis, P.; Gadowski, T.; MacKay, F.; Tipping, P.; Bobik, A.; Toh, B.H. Depletion of B2 but not B1a B cells in BAFF receptor-deficient ApoE mice attenuates atherosclerosis by potently ameliorating arterial inflammation. PLoS ONE 2012, 7, e29371. [Google Scholar] [CrossRef] [PubMed]
- Klimov, A.N.; Denisenko, A.D.; Popov, A.V.; Nagornev, V.A.; Pleskov, V.M.; Vinogradov, A.G.; Denisenko, T.V.; Magracheva, E.; Kheifes, G.M.; Kuznetzov, A.S. Lipoprotein-antibody immune complexes. Their catabolism and role in foam cell formation. Atherosclerosis 1985, 58, 1–15. [Google Scholar] [CrossRef]
- Schiopu, A.; Bengtsson, J.; Söderberg, I.; Janciauskiene, S.; Lindgren, S.; Ares, M.P.; Shah, P.K.; Carlsson, R.; Nilsson, J.; Fredrikson, G.N. Recombinant human antibodies against aldehyde-modified apolipoprotein B-100 peptide sequences inhibit atherosclerosis. Circulation 2004, 110, 2047–2052. [Google Scholar] [CrossRef] [Green Version]
- Fredrikson, G.N.; Söderberg, I.; Lindholm, M.; Dimayuga, P.; Chyu, K.Y.; Shah, P.K.; Nilsson, J. Inhibition of atherosclerosis in apoE-null mice by immunization with apoB-100 peptide sequences. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 879–884. [Google Scholar] [CrossRef] [Green Version]
- Hernández-Vargas, P.; Ortiz-Muñoz, G.; López-Franco, O.; Suzuki, Y.; Gallego-Delgado, J.; Sanjuán, G.; Lázaro, A.; López-Parra, V.; Ortega, L.; Egido, J.; et al. Fcgamma receptor deficiency confers protection against atherosclerosis in apolipoprotein E knockout mice. Circ. Res. 2006, 99, 1188–1196. [Google Scholar] [CrossRef]
- Clement, M.; Guedj, K.; Andreata, F.; Morvan, M.; Bey, L.; Khallou-Laschet, J.; Gaston, A.T.; Delbosc, S.; Alsac, J.M.; Bruneval, P.; et al. Control of the T follicular helper-germinal center B-cell axis by CD8⁺ regulatory T cells limits atherosclerosis and tertiary lymphoid organ development. Circulation 2015, 131, 560–570. [Google Scholar] [CrossRef] [Green Version]
- Gaddis, D.E.; Padgett, L.E.; Wu, R.; McSkimming, C.; Romines, V.; Taylor, A.M.; McNamara, C.A.; Kronenberg, M.; Crotty, S.; Thomas, M.J.; et al. Apolipoprotein AI prevents regulatory to follicular helper T cell switching during atherosclerosis. Nat. Commun. 2018, 9, 1095. [Google Scholar] [CrossRef] [PubMed]
- Sage, A.P.; Nus, M.; Bagchi Chakraborty, J.; Tsiantoulas, D.; Newland, S.A.; Finigan, A.J.; Masters, L.; Binder, C.J.; Mallat, Z. X-Box Binding Protein-1 Dependent Plasma Cell Responses Limit the Development of Atherosclerosis. Circ. Res. 2017, 121, 270–281. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.C.; Dougan, S.K.; McGehee, A.M.; Love, J.C.; Ploegh, H.L. XBP-1 regulates signal transduction, transcription factors and bone marrow colonization in B cells. EMBO J. 2009, 28, 1624–1636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reimold, A.M.; Iwakoshi, N.N.; Manis, J.; Vallabhajosyula, P.; Szomolanyi-Tsuda, E.; Gravallese, E.M.; Friend, D.; Grusby, M.J.; Alt, F.; Glimcher, L.H. Plasma cell differentiation requires the transcription factor XBP-1. Nature 2001, 412, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Tellier, J.; Shi, W.; Minnich, M.; Liao, Y.; Crawford, S.; Smyth, G.K.; Kallies, A.; Busslinger, M.; Nutt, S.L. Blimp-1 controls plasma cell function through the regulation of immunoglobulin secretion and the unfolded protein response. Nat. Immunol. 2016, 17, 323–330. [Google Scholar] [CrossRef] [Green Version]
- Centa, M.; Jin, H.; Hofste, L.; Hellberg, S.; Busch, A.; Baumgartner, R.; Verzaal, N.J.; Lind Enoksson, S.; Perisic Matic, L.; Boddul, S.V.; et al. Germinal Center-Derived Antibodies Promote Atherosclerosis Plaque Size and Stability. Circulation 2019, 139, 2466–2482. [Google Scholar] [CrossRef]
- Lorenzo, C.; Delgado, P.; Busse, C.E.; Sanz-Bravo, A.; Martos-Folgado, I.; Bonzon-Kulichenko, E.; Ferrarini, A.; Gonzalez-Valdes, I.B.; Mur, S.M.; Roldán-Montero, R.; et al. ALDH4A1 is an atherosclerosis auto-antigen targeted by protective antibodies. Nature 2020. [Google Scholar] [CrossRef]
- Yuseff, M.I.; Pierobon, P.; Reversat, A.; Lennon-Dumenil, A.M. How B cells capture, process and present antigens: A crucial role for cell polarity. Nat. Rev. Immunol. 2013, 13, 475–486. [Google Scholar] [CrossRef]
- Wigren, M.; Rattik, S.; Yao Mattisson, I.; Tomas, L.; Gronberg, C.; Soderberg, I.; Alm, R.; Sundius, L.; Ljungcrantz, I.; Bjorkbacka, H.; et al. Lack of Ability to Present Antigens on Major Histocompatibility Complex Class II Molecules Aggravates Atherosclerosis in ApoE(-/-) Mice. Circulation 2019, 139, 2554–2566. [Google Scholar] [CrossRef]
- Tay, C.; Kanellakis, P.; Hosseini, H.; Cao, A.; Toh, B.H.; Bobik, A.; Kyaw, T. B Cell and CD4 T Cell Interactions Promote Development of Atherosclerosis. Front. Immunol. 2019, 10, 3046. [Google Scholar] [CrossRef]
- Williams, J.W.; Elvington, A.; Kessler, S.; Wohltmann, M.; Wu, G.F.; Randolph, G.J. B Cell-Mediated Antigen Presentation through MHC Class II Is Dispensable for Atherosclerosis Progression. ImmunoHorizons 2019, 3, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Maehr, R.; Kraus, M.; Ploegh, H.L. Mice deficient in invariant-chain and MHC class II exhibit a normal mature B2 cell compartment. Eur. J. Immunol. 2004, 34, 2230–2236. [Google Scholar] [CrossRef] [PubMed]
- Ronchese, F.; Hausmann, B. B lymphocytes in vivo fail to prime naive T cells but can stimulate antigen-experienced T lymphocytes. J. Exp. Med. 1993, 177, 679–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reichardt, P.; Dornbach, B.; Rong, S.; Beissert, S.; Gueler, F.; Loser, K.; Gunzer, M. Naive B cells generate regulatory T cells in the presence of a mature immunologic synapse. Blood 2007, 110, 1519–1529. [Google Scholar] [CrossRef]
- Archambault, A.S.; Carrero, J.A.; Barnett, L.G.; McGee, N.G.; Sim, J.; Wright, J.O.; Raabe, T.; Chen, P.; Ding, H.; Allenspach, E.J.; et al. Cutting edge: Conditional MHC class II expression reveals a limited role for B cell antigen presentation in primary and secondary CD4 T cell responses. J. Immunol. 2013, 191, 545–550. [Google Scholar] [CrossRef] [Green Version]
- Epstein, M.M.; Di Rosa, F.; Jankovic, D.; Sher, A.; Matzinger, P. Successful T cell priming in B cell-deficient mice. J. Exp. Med. 1995, 182, 915–922. [Google Scholar] [CrossRef]
- Freigang, S.; Horkko, S.; Miller, E.; Witztum, J.L.; Palinski, W. Immunization of LDL receptor-deficient mice with homologous malondialdehyde-modified and native LDL reduces progression of atherosclerosis by mechanisms other than induction of high titers of antibodies to oxidative neoepitopes. Arterioscler. Thromb. Vasc. Biol. 1998, 18, 1972–1982. [Google Scholar] [CrossRef] [Green Version]
- Wigren, M.; Kolbus, D.; Duner, P.; Ljungcrantz, I.; Soderberg, I.; Bjorkbacka, H.; Fredrikson, G.N.; Nilsson, J. Evidence for a role of regulatory T cells in mediating the atheroprotective effect of apolipoprotein B peptide vaccine. J. Intern. Med. 2011, 269, 546–556. [Google Scholar] [CrossRef]
- Tse, K.; Gonen, A.; Sidney, J.; Ouyang, H.; Witztum, J.L.; Sette, A.; Tse, H.; Ley, K. Atheroprotective Vaccination with MHC-II Restricted Peptides from ApoB-100. Front. Immunol. 2013, 4, 493. [Google Scholar] [CrossRef]
- Herbin, O.; Ait-Oufella, H.; Yu, W.; Fredrikson, G.N.; Aubier, B.; Perez, N.; Barateau, V.; Nilsson, J.; Tedgui, A.; Mallat, Z. Regulatory T-cell response to apolipoprotein B100-derived peptides reduces the development and progression of atherosclerosis in mice. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 605–612. [Google Scholar] [CrossRef] [Green Version]
- Kimura, T.; Kobiyama, K.; Winkels, H.; Tse, K.; Miller, J.; Vassallo, M.; Wolf, D.; Ryden, C.; Orecchioni, M.; Dileepan, T.; et al. Regulatory CD4(+) T Cells Recognize Major Histocompatibility Complex Class II Molecule-Restricted Peptide Epitopes of Apolipoprotein B. Circulation 2018, 138, 1130–1143. [Google Scholar] [CrossRef] [PubMed]
- Albany, C.J.; Trevelin, S.C.; Giganti, G.; Lombardi, G.; Scotta, C. Getting to the Heart of the Matter: The Role of Regulatory T-Cells (Tregs) in Cardiovascular Disease (CVD) and Atherosclerosis. Front. Immunol. 2019, 10, 2795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, D.M.; Thornton, A.M.; DiPaolo, R.J.; Shevach, E.M. Activated CD4+CD25+ T cells selectively kill B lymphocytes. Blood 2006, 107, 3925–3932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, A.; Liu, Y.; Chen, W.; Wang, J.; Xue, Y.; Huang, F.; Rong, L.; Lin, J.; Liu, D.; Yan, M.; et al. TGF-beta-Induced Regulatory T Cells Directly Suppress B Cell Responses through a Noncytotoxic Mechanism. J. Immunol. 2016, 196, 3631–3641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weingartner, E.; Golding, A. Direct control of B cells by Tregs: An opportunity for long-term modulation of the humoral response. Cell Immunol. 2017, 318, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Seo, S.J.; Fields, M.L.; Buckler, J.L.; Reed, A.J.; Mandik-Nayak, L.; Nish, S.A.; Noelle, R.J.; Turka, L.A.; Finkelman, F.D.; Caton, A.J.; et al. The impact of T helper and T regulatory cells on the regulation of anti-double-stranded DNA B cells. Immunity 2002, 16, 535–546. [Google Scholar] [CrossRef] [Green Version]
- Ludwig-Portugall, I.; Hamilton-Williams, E.E.; Gottschalk, C.; Kurts, C. Cutting edge: CD25+ regulatory T cells prevent expansion and induce apoptosis of B cells specific for tissue autoantigens. J. Immunol. 2008, 181, 4447–4451. [Google Scholar] [CrossRef] [Green Version]
- Glatman Zaretsky, A.; Konradt, C.; Depis, F.; Wing, J.B.; Goenka, R.; Atria, D.G.; Silver, J.S.; Cho, S.; Wolf, A.I.; Quinn, W.J.; et al. T Regulatory Cells Support Plasma Cell Populations in the Bone Marrow. Cell Rep. 2017, 18, 1906–1916. [Google Scholar] [CrossRef]
- Linterman, M.A.; Pierson, W.; Lee, S.K.; Kallies, A.; Kawamoto, S.; Rayner, T.F.; Srivastava, M.; Divekar, D.P.; Beaton, L.; Hogan, J.J.; et al. Foxp3+ follicular regulatory T cells control the germinal center response. Nat. Med. 2011, 17, 975–982. [Google Scholar] [CrossRef] [Green Version]
- Chung, Y.; Tanaka, S.; Chu, F.; Nurieva, R.I.; Martinez, G.J.; Rawal, S.; Wang, Y.H.; Lim, H.; Reynolds, J.M.; Zhou, X.H.; et al. Follicular regulatory T cells expressing Foxp3 and Bcl-6 suppress germinal center reactions. Nat. Med. 2011, 17, 983–988. [Google Scholar] [CrossRef]
- Chien, C.H.; Chiang, B.L. Recent advances in regulatory T cells induced by B cells. Cell Mol. Immunol. 2018, 15, 539–541. [Google Scholar] [CrossRef] [PubMed]
- Wolf, D.; Gerhardt, T.; Winkels, H.; Michel, N.A.; Pramod, A.B.; Ghosheh, Y.; Brunel, S.; Buscher, K.; Miller, J.; McArdle, S.; et al. Pathogenic Autoimmunity in Atherosclerosis Evolves From Initially Protective Apolipoprotein B(100)-Reactive CD4(+) T-Regulatory Cells. Circulation 2020, 142, 1279–1293. [Google Scholar] [CrossRef] [PubMed]
- Butcher, M.J.; Filipowicz, A.R.; Waseem, T.C.; McGary, C.M.; Crow, K.J.; Magilnick, N.; Boldin, M.; Lundberg, P.S.; Galkina, E.V. Atherosclerosis-Driven Treg Plasticity Results in Formation of a Dysfunctional Subset of Plastic IFNγ+ Th1/Tregs. Circ. Res. 2016, 119, 1190–1203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawabe, T.; Naka, T.; Yoshida, K.; Tanaka, T.; Fujiwara, H.; Suematsu, S.; Yoshida, N.; Kishimoto, T.; Kikutani, H. The immune responses in CD40-deficient mice: Impaired immunoglobulin class switching and germinal center formation. Immunity 1994, 1, 167–178. [Google Scholar] [CrossRef]
- Mantani, P.T.; Ljungcrantz, I.; Andersson, L.; Alm, R.; Hedblad, B.; Bjorkbacka, H.; Nilsson, J.; Fredrikson, G.N. Circulating CD40+ and CD86+ B cell subsets demonstrate opposing associations with risk of stroke. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 211–218. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.S.; Kageyama, R.; Eto, D.; Escobar, T.C.; Johnston, R.J.; Monticelli, L.; Lao, C.; Crotty, S. ICOS receptor instructs T follicular helper cell versus effector cell differentiation via induction of the transcriptional repressor Bcl6. Immunity 2011, 34, 932–946. [Google Scholar] [CrossRef] [Green Version]
- Gotsman, I.; Grabie, N.; Gupta, R.; Dacosta, R.; MacConmara, M.; Lederer, J.; Sukhova, G.; Witztum, J.L.; Sharpe, A.H.; Lichtman, A.H. Impaired regulatory T-cell response and enhanced atherosclerosis in the absence of inducible costimulatory molecule. Circulation 2006, 114, 2047–2055. [Google Scholar] [CrossRef]
- Hamel, K.M.; Cao, Y.; Olalekan, S.A.; Finnegan, A. B cell-specific expression of inducible costimulator ligand is necessary for the induction of arthritis in mice. Arthritis Rheumatol. 2014, 66, 60–67. [Google Scholar] [CrossRef]
- Buono, C.; Pang, H.; Uchida, Y.; Libby, P.; Sharpe, A.H.; Lichtman, A.H. B7-1/B7-2 costimulation regulates plaque antigen-specific T-cell responses and atherogenesis in low-density lipoprotein receptor-deficient mice. Circulation 2004, 109, 2009–2015. [Google Scholar] [CrossRef]
- Ait-Oufella, H.; Salomon, B.L.; Potteaux, S.; Robertson, A.K.; Gourdy, P.; Zoll, J.; Merval, R.; Esposito, B.; Cohen, J.L.; Fisson, S.; et al. Natural regulatory T cells control the development of atherosclerosis in mice. Nat. Med. 2006, 12, 178–180. [Google Scholar] [CrossRef]
- Meiler, S.; Smeets, E.; Winkels, H.; Shami, A.; Pascutti, M.F.; Nolte, M.A.; Beckers, L.; Weber, C.; Gerdes, N.; Lutgens, E. Constitutive GITR Activation Reduces Atherosclerosis by Promoting Regulatory CD4+ T-Cell Responses-Brief Report. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1748–1752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamze, M.; Desmetz, C.; Berthe, M.L.; Roger, P.; Boulle, N.; Brancherau, P.; Picard, E.; Guzman, C.; Tolza, C.; Guglielmi, P. Characterization of resident B cells of vascular walls in human atherosclerotic patients. J. Immunol. 2013, 191, 3006–3016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, K.H.; Chen, L.C.; Hsu, K.; Chang, C.C.; Chang, C.Y.; Kao, C.W.; Chang, Y.F.; Chang, M.C.; Chen, C.G. BAFF-driven NLRP3 inflammasome activation in B cells. Cell Death Dis. 2020, 11, 820. [Google Scholar] [CrossRef] [PubMed]
- Potteaux, S.; Esposito, B.; van Oostrom, O.; Brun, V.; Ardouin, P.; Groux, H.; Tedgui, A.; Mallat, Z. Leukocyte-derived interleukin 10 is required for protection against atherosclerosis in low-density lipoprotein receptor knockout mice. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 1474–1478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rincon-Arevalo, H.; Quintero, J.C.; Fortich, F.; Rojas, M.; Vasquez, G.; Castano, D.; Yassin, L.M. Low frequency of IL-10(+) B cells in patients with atherosclerosis is related with inflammatory condition. Heliyon 2020, 6, e03441. [Google Scholar] [CrossRef]
- Rincon-Arevalo, H.; Villa-Pulgarin, J.; Tabares, J.; Rojas, M.; Vasquez, G.; Ramirez-Pineda, J.R.; Castano, D.; Yassin, L.M. Interleukin-10 production and T cell-suppressive capacity in B cell subsets from atherosclerotic apoE (-/-) mice. Immunol. Res. 2017, 65, 995–1008. [Google Scholar] [CrossRef]
- Strom, A.C.; Cross, A.J.; Cole, J.E.; Blair, P.A.; Leib, C.; Goddard, M.E.; Rosser, E.C.; Park, I.; Hultgardh Nilsson, A.; Nilsson, J.; et al. B regulatory cells are increased in hypercholesterolaemic mice and protect from lesion development via IL-10. Thromb. Haemost. 2015, 114, 835–847. [Google Scholar] [CrossRef]
- Fillatreau, S.; Sweenie, C.H.; McGeachy, M.J.; Gray, D.; Anderton, S.M. B cells regulate autoimmunity by provision of IL-10. Nat. Immunol. 2002, 3, 944–950. [Google Scholar] [CrossRef]
- Wu, L.; Parekh, V.V.; Hsiao, J.; Kitamura, D.; Van Kaer, L. Spleen supports a pool of innate-like B cells in white adipose tissue that protects against obesity-associated insulin resistance. Proc. Natl. Acad. Sci. USA 2014, 111, E4638–E4647. [Google Scholar] [CrossRef] [Green Version]
- Sage, A.P.; Nus, M.; Baker, L.L.; Finigan, A.J.; Masters, L.M.; Mallat, Z. Regulatory B cell-specific interleukin-10 is dispensable for atherosclerosis development in mice. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1770–1773. [Google Scholar] [CrossRef] [Green Version]
- Ponnuswamy, P.; Joffre, J.; Herbin, O.; Esposito, B.; Laurans, L.; Binder, C.J.; Tedder, T.F.; Zeboudj, L.; Loyer, X.; Giraud, A.; et al. Angiotensin II synergizes with BAFF to promote atheroprotective regulatory B cells. Sci. Rep. 2017, 7, 4111. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| B Cell Subset | Location | Surface Markers | Functions | Experimental Models |
|---|---|---|---|---|
| B1a | Serosal cavities Spleen-dependent | B220low/mid IgMhi IgDlo CD93- CD43+ CD23- CD5+ [89] | Secrete natural IgM [42,81] T cell-independent | Splenectomy with B1a or B cell transfer [87,90] |
| B1b | Serosal cavities Spleen-dependent | B220low/mid IgMhi IgDlo CD43+ CD23- CD5- [89] | Secrete natural IgM, T cell-dependent and independent responses Isotype switch to IgA [42,81] | Rag−/−Apoe−/− with B1b cell transfer [88] |
| Innate Response Activator B cells | Spleen Differentiated from TLR activated B1a b cells | B220+ IgMhi CD23lo CD21lo CD138hi CD43hi VLA4hi [91] | Produce GM-CSF [91] | Csf2−/−μMT mice (mixed chimera on Ldlr−/−) [92] |
| Marginal Zone B cells | Marginal zone of secondary lymphoid tissues | B220+ IgMhi IgDlo CD21hi CD43- CD23- CD1hi [45] | Shuttle antigens to follicles Migrate to T cell zones Produce mainly IgM plasma cells Isotype switch to IgA or IgG3 T cell-independent | CD79cre/+Rbpjkflox/flox (mixed chimera on Ldlr−/−) [56] |
| Follicular B cells | Recirculating Follicles of secondary lymphoid tissues | B220+ IgMlo IgDhi CD43- CD23+ CD21mid [45] | T cell-dependent GC formation Isotype switched Igs | μMT with B2 B cell transfer [22,93] Anti-BAFF Abs [94] Anti-CD20 and anti-BAFFR [95,96,97] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, S.D.; Mussbacher, M.; Galkina, E.V. Functional Role of B Cells in Atherosclerosis. Cells 2021, 10, 270. https://doi.org/10.3390/cells10020270
Ma SD, Mussbacher M, Galkina EV. Functional Role of B Cells in Atherosclerosis. Cells. 2021; 10(2):270. https://doi.org/10.3390/cells10020270
Chicago/Turabian StyleMa, Shelby D., Marion Mussbacher, and Elena V. Galkina. 2021. "Functional Role of B Cells in Atherosclerosis" Cells 10, no. 2: 270. https://doi.org/10.3390/cells10020270
APA StyleMa, S. D., Mussbacher, M., & Galkina, E. V. (2021). Functional Role of B Cells in Atherosclerosis. Cells, 10(2), 270. https://doi.org/10.3390/cells10020270