
Inflammatory Chemokines in Atherosclerosis

 , ,
, ,
Abstract
:1. Introduction
1.1. CCL2-CCR2
1.2. CCL3
1.3. CCL5-CCR1/CCR5/CCR3
1.4. CCL17
1.5. CCL19, CCL21/CCR7
1.6. CXCL1-CXCR2
1.7. CXCL4
1.8. CXCL8
1.9. CXCL9-CXCL10-CXCL11/CXCR3
1.10. CXCL12-CXCR4/ACKR3
1.11. CXCL16-CXCR6
1.12. CX3CL1-CX3CR1
1.13. MIF-CXCR2/CXCR4/ACKR3
2. Recent Highlights and the Road Ahead
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hughes, C.E.; Nibbs, R.J.B. A guide to chemokines and their receptors. FEBS J. 2018, 285, 2944–2971. [Google Scholar] [CrossRef] [PubMed]
- Raman, D.; Sobolik-Delmaire, T.; Richmond, A. Chemokines in health and disease. Exp. Cell Res. 2011, 317, 575–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lusis, A.J. Atherosclerosis. Nature 2000, 407, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Glass, C.K.; Witztum, J.L. Atherosclerosis. the road ahead. Cell 2001, 104, 503–516. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Martín, L.; Estecha, A.; Samaniego, R.; Sánchez-Ramón, S.; Vega, M.Á.; Sánchez-Mateos, P. The chemokine CXCL12 regulates monocyte-macrophage differentiation and RUNX3 expression. Blood 2011, 117, 88–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zernecke, A.; Shagdarsuren, E.; Weber, C. Chemokines in atherosclerosis: An update. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1897–1908. [Google Scholar] [CrossRef] [Green Version]
- Segers, D.; Lipton, J.A.; Leenen, P.J.M.; Cheng, C.; Tempel, D.; Pasterkamp, G.; Moll, F.L.; de Crom, R.; Krams, R. Atherosclerotic Plaque Stability Is Affected by the Chemokine CXCL10 in Both Mice and Humans. Int. J. Inflam. 2011, 2011, 936109. [Google Scholar] [CrossRef] [Green Version]
- Miller, M.C.; Mayo, K.H. Chemokines from a Structural Perspective. Int. J. Mol. Sci. 2017, 18, 2088. [Google Scholar] [CrossRef] [Green Version]
- Gencer, S.; van der Vorst, E.P.C.; Aslani, M.; Weber, C.; Döring, Y.; Duchene, J. Atypical Chemokine Receptors in Cardiovascular Disease. Thromb. Haemost. 2019, 119, 534–541. [Google Scholar] [CrossRef] [Green Version]
- Deshmane, S.L.; Kremlev, S.; Amini, S.; Sawaya, B.E. Monocyte chemoattractant protein-1 (MCP-1): An overview. J. Interferon Cytokine Res. 2009, 29, 313–326. [Google Scholar] [CrossRef]
- Lu, B.; Rutledge, B.J.; Gu, L.; Fiorillo, J.; Lukacs, N.W.; Kunkel, S.L.; North, R.; Gerard, C.; Rollins, B.J. Abnormalities in monocyte recruitment and cytokine expression in monocyte chemoattractant protein 1-deficient mice. J. Exp. Med. 1998, 187, 601–608. [Google Scholar] [CrossRef] [PubMed]
- Gu, L.; Okada, Y.; Clinton, S.K.; Gerard, C.; Sukhova, G.K.; Libby, P.; Rollins, B.J. Absence of monocyte chemoattractant protein-1 reduces atherosclerosis in low density lipoprotein receptor-deficient mice. Mol. Cell 1998, 2, 275–281. [Google Scholar] [CrossRef]
- Boring, L.; Gosling, J.; Cleary, M.; Charo, I.F. Decreased lesion formation in CCR2-/- mice reveals a role for chemokines in the initiation of atherosclerosis. Nature 1998, 394, 894–897. [Google Scholar] [CrossRef] [PubMed]
- Dawson, T.C.; Kuziel, W.A.; Osahar, T.A.; Maeda, N. Absence of CC chemokine receptor-2 reduces atherosclerosis in apolipoprotein E-deficient mice. Atherosclerosis 1999, 143, 205–211. [Google Scholar] [CrossRef]
- Guo, J.; Van Eck, M.; Twisk, J.; Maeda, N.; Benson, G.M.; Groot, P.H.E.; Van Berkel, T.J.C. Transplantation of monocyte CC-chemokine receptor 2-deficient bone marrow into ApoE3-Leiden mice inhibits atherogenesis. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 447–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winter, C.; Silvestre-Roig, C.; Ortega-Gomez, A.; Lemnitzer, P.; Poelman, H.; Schumski, A.; Winter, J.; Drechsler, M.; de Jong, R.; Immler, R.; et al. Chrono-pharmacological Targeting of the CCL2-CCR2 Axis Ameliorates Atherosclerosis. Cell Metab. 2018, 28, 175–182.e5. [Google Scholar] [CrossRef] [Green Version]
- Schober, A.; Zernecke, A.; Liehn, E.A.; von Hundelshausen, P.; Knarren, S.; Kuziel, W.A.; Weber, C. Crucial role of the CCL2/CCR2 axis in neointimal hyperplasia after arterial injury in hyperlipidemic mice involves early monocyte recruitment and CCL2 presentation on platelets. Circ. Res. 2004, 95, 1125–1133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egashira, K.; Zhao, Q.; Kataoka, C.; Ohtani, K.; Usui, M.; Charo, I.F.; Nishida, K.-I.; Inoue, S.; Katoh, M.; Ichiki, T.; et al. Importance of monocyte chemoattractant protein-1 pathway in neointimal hyperplasia after periarterial injury in mice and monkeys. Circ. Res. 2002, 90, 1167–1172. [Google Scholar] [CrossRef] [Green Version]
- Roque, M.; Kim, W.J.H.; Gazdoin, M.; Malik, A.; Reis, E.D.; Fallon, J.T.; Badimon, J.J.; Charo, I.F.; Taubman, M.B. CCR2 deficiency decreases intimal hyperplasia after arterial injury. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 554–559. [Google Scholar] [CrossRef] [Green Version]
- Zernecke, A.; Bot, I.; Djalali-Talab, Y.; Shagdarsuren, E.; Bidzhekov, K.; Meiler, S.; Krohn, R.; Schober, A.; Sperandio, M.; Soehnlein, O.; et al. Protective role of CXC receptor 4/CXC ligand 12 unveils the importance of neutrophils in atherosclerosis. Circ. Res. 2008, 102, 209–217. [Google Scholar] [CrossRef] [Green Version]
- Braunersreuther, V.; Zernecke, A.; Arnaud, C.; Liehn, E.A.; Steffens, S.; Shagdarsuren, E.; Bidzhekov, K.; Burger, F.; Pelli, G.; Luckow, B.; et al. Ccr5 but not Ccr1 deficiency reduces development of diet-induced atherosclerosis in mice. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 373–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyazaki, D.; Nakamura, T.; Toda, M.; Cheung-Chau, K.-W.; Richardson, R.M.; Ono, S.J. Macrophage inflammatory protein-1alpha as a costimulatory signal for mast cell-mediated immediate hypersensitivity reactions. J. Clin. Investig. 2005, 115, 434–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, C. Platelets and chemokines in atherosclerosis: Partners in crime. Circ. Res. 2005, 96, 612–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reichel, C.A.; Rehberg, M.; Lerchenberger, M.; Berberich, N.; Bihari, P.; Khandoga, A.G.; Zahler, S.; Krombach, F. Ccl2 and Ccl3 mediate neutrophil recruitment via induction of protein synthesis and generation of lipid mediators. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 1787–1793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Jager, S.C.A.; Bot, I.; Kraaijeveld, A.O.; Korporaal, S.J.A.; Bot, M.; van Santbrink, P.J.; van Berkel, T.J.C.; Kuiper, J.; Biessen, E.A.L. Leukocyte-specific CCL3 deficiency inhibits atherosclerotic lesion development by affecting neutrophil accumulation. Arterioscler. Thromb. Vasc. Biol. 2013, 33, e75–e83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, L.-X.; Heng, X.-H.; Guo, R.-W.; Si, Y.-K.; Qi, F.; Zhou, X.-B. Atorvastatin inhibits the 5-lipoxygenase pathway and expression of CCL3 to alleviate atherosclerotic lesions in atherosclerotic ApoE knockout mice. J. Cardiovasc. Pharmacol. 2013, 62, 205–211. [Google Scholar] [CrossRef]
- Marques, R.E.; Guabiraba, R.; Russo, R.C.; Teixeira, M.M. Targeting CCL5 in inflammation. Expert Opin. Ther. Targets 2013, 17, 1439–1460. [Google Scholar] [CrossRef]
- von Hundelshausen, P.; Weber, K.S.; Huo, Y.; Proudfoot, A.E.; Nelson, P.J.; Ley, K.; Weber, C. RANTES deposition by platelets triggers monocyte arrest on inflamed and atherosclerotic endothelium. Circulation 2001, 103, 1772–1777. [Google Scholar] [CrossRef]
- Schober, A.; Manka, D.; von Hundelshausen, P.; Huo, Y.; Hanrath, P.; Sarembock, I.J.; Ley, K.; Weber, C. Deposition of platelet RANTES triggering monocyte recruitment requires P-selectin and is involved in neointima formation after arterial injury. Circulation 2002, 106, 1523–1529. [Google Scholar] [CrossRef] [Green Version]
- Veillard, N.R.; Kwak, B.; Pelli, G.; Mulhaupt, F.; James, R.W.; Proudfoot, A.E.I.; Mach, F. Antagonism of RANTES receptors reduces atherosclerotic plaque formation in mice. Circ. Res. 2004, 94, 253–261. [Google Scholar] [CrossRef]
- Fox, J.M.; Kausar, F.; Day, A.; Osborne, M.; Hussain, K.; Mueller, A.; Lin, J.; Tsuchiya, T.; Kanegasaki, S.; Pease, J.E. CXCL4/Platelet Factor 4 is an agonist of CCR1 and drives human monocyte migration. Sci. Rep. 2018, 8, 9466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pitsilos, S.; Hunt, J.; Mohler, E.R.; Prabhakar, A.M.; Poncz, M.; Dawicki, J.; Khalapyan, T.Z.; Wolfe, M.L.; Fairman, R.; Mitchell, M.; et al. Platelet factor 4 localization in carotid atherosclerotic plaques: Correlation with clinical parameters. Thromb. Haemost. 2003, 90, 1112–1120. [Google Scholar] [CrossRef] [PubMed]
- Sachais, B.S.; Turrentine, T.; Dawicki McKenna, J.M.; Rux, A.H.; Rader, D.; Kowalska, M.A. Elimination of platelet factor 4 (PF4) from platelets reduces atherosclerosis in C57Bl/6 and apoE-/- mice. Thromb. Haemost. 2007, 98, 1108–1113. [Google Scholar] [PubMed]
- Huo, Y.; Schober, A.; Forlow, S.B.; Smith, D.F.; Hyman, M.C.; Jung, S.; Littman, D.R.; Weber, C.; Ley, K. Circulating activated platelets exacerbate atherosclerosis in mice deficient in apolipoprotein E. Nat. Med. 2003, 9, 61–67. [Google Scholar] [CrossRef]
- von Hundelshausen, P.; Koenen, R.R.; Sack, M.; Mause, S.F.; Adriaens, W.; Proudfoot, A.E.I.; Hackeng, T.M.; Weber, C. Heterophilic interactions of platelet factor 4 and RANTES promote monocyte arrest on endothelium. Blood 2005, 105, 924–930. [Google Scholar] [CrossRef] [Green Version]
- Vajen, T.; Koenen, R.R.; Werner, I.; Staudt, M.; Projahn, D.; Curaj, A.; Sönmez, T.T.; Simsekyilmaz, S.; Schumacher, D.; Möllmann, J.; et al. Blocking CCL5-CXCL4 heteromerization preserves heart function after myocardial infarction by attenuating leukocyte recruitment and NETosis. Sci. Rep. 2018, 8, 10647. [Google Scholar] [CrossRef]
- Alard, J.-E.; Ortega-Gomez, A.; Wichapong, K.; Bongiovanni, D.; Horckmans, M.; Megens, R.T.A.; Leoni, G.; Ferraro, B.; Rossaint, J.; Paulin, N.; et al. Recruitment of classical monocytes can be inhibited by disturbing heteromers of neutrophil HNP1 and platelet CCL5. Sci. Transl. Med. 2015, 7, 317ra196. [Google Scholar] [CrossRef]
- Soehnlein, O.; Drechsler, M.; Döring, Y.; Lievens, D.; Hartwig, H.; Kemmerich, K.; Ortega-Gómez, A.; Mandl, M.; Vijayan, S.; Projahn, D.; et al. Distinct functions of chemokine receptor axes in the atherogenic mobilization and recruitment of classical monocytes. EMBO Mol. Med. 2013, 5, 471–481. [Google Scholar] [CrossRef]
- Potteaux, S.; Combadière, C.; Esposito, B.; Casanova, S.; Merval, R.; Ardouin, P.; Gao, J.-L.; Murphy, P.M.; Tedgui, A.; Mallat, Z. Chemokine receptor CCR1 disruption in bone marrow cells enhances atherosclerotic lesion development and inflammation in mice. Mol. Med. 2005, 11, 16–20. [Google Scholar] [CrossRef]
- Francisci, D.; Pirro, M.; Schiaroli, E.; Mannarino, M.R.; Cipriani, S.; Bianconi, V.; Alunno, A.; Bagaglia, F.; Bistoni, O.; Falcinelli, E.; et al. Maraviroc Intensification Modulates Atherosclerotic Progression in HIV-Suppressed Patients at High Cardiovascular Risk. A Randomized, Crossover Pilot Study. Open Forum Infect Dis. 2019, 6, ofz112. [Google Scholar] [CrossRef]
- Haley, K.J.; Lilly, C.M.; Yang, J.H.; Feng, Y.; Kennedy, S.P.; Turi, T.G.; Thompson, J.F.; Sukhova, G.H.; Libby, P.; Lee, R.T. Overexpression of eotaxin and the CCR3 receptor in human atherosclerosis: Using genomic technology to identify a potential novel pathway of vascular inflammation. Circulation 2000, 102, 2185–2189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, H.; Li, R.; Choi, S.; Zhou, L.; Pavel, A.; Estrada, Y.D.; Krueger, J.G.; Guttman-Yassky, E. Increased cardiovascular and atherosclerosis markers in blood of older patients with atopic dermatitis. Ann. Allergy Asthma Immunol. 2020, 124, 70–78. [Google Scholar] [CrossRef] [Green Version]
- Brunner, P.M.; Suárez-Fariñas, M.; He, H.; Malik, K.; Wen, H.-C.; Gonzalez, J.; Chan, T.C.-C.; Estrada, Y.; Zheng, X.; Khattri, S.; et al. The atopic dermatitis blood signature is characterized by increases in inflammatory and cardiovascular risk proteins. Sci. Rep. 2017, 7, 8707. [Google Scholar] [CrossRef]
- Weber, C.; Meiler, S.; Döring, Y.; Koch, M.; Drechsler, M.; Megens, R.T.A.; Rowinska, Z.; Bidzhekov, K.; Fecher, C.; Ribechini, E.; et al. CCL17-expressing dendritic cells drive atherosclerosis by restraining regulatory T cell homeostasis in mice. J. Clin. Investig. 2011, 121, 2898–2910. [Google Scholar] [CrossRef] [PubMed]
- Andrew, D.P.; Ruffing, N.; Kim, C.H.; Miao, W.; Heath, H.; Li, Y.; Murphy, K.; Campbell, J.J.; Butcher, E.C.; Wu, L. C-C chemokine receptor 4 expression defines a major subset of circulating nonintestinal memory T cells of both Th1 and Th2 potential. J. Immunol. 2001, 166, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Heiseke, A.F.; Faul, A.C.; Lehr, H.-A.; Förster, I.; Schmid, R.M.; Krug, A.B.; Reindl, W. CCL17 promotes intestinal inflammation in mice and counteracts regulatory T cell-mediated protection from colitis. Gastroenterology 2012, 142, 335–345. [Google Scholar] [CrossRef]
- von Hundelshausen, P.; Agten, S.M.; Eckardt, V.; Blanchet, X.; Schmitt, M.M.; Ippel, H.; Neideck, C.; Bidzhekov, K.; Leberzammer, J.; Wichapong, K.; et al. Chemokine interactome mapping enables tailored intervention in acute and chronic inflammation. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef]
- Ye, Y.; Yang, X.; Zhao, X.; Chen, L.; Xie, H.; Zeng, Y.; Shen, Z.; Fan, Z.; Liu, Z.; Zhang, S. Serum chemokine CCL17/thymus activation and regulated chemokine is correlated with coronary artery diseases. Atherosclerosis 2015, 238, 365–369. [Google Scholar] [CrossRef]
- Ye, Y.; Yang, X.; Long, B.; Pang, H.; Zhu, Y.; Zhang, S. Association Between a CCL17 Genetic Variant and Risk of Coronary Artery Disease in a Chinese Han Population. Circ. J. 2017, 82, 224–231. [Google Scholar] [CrossRef] [Green Version]
- Halvorsen, B.; Dahl, T.B.; Smedbakken, L.M.; Singh, A.; Michelsen, A.E.; Skjelland, M.; Krohg-Sørensen, K.; Russell, D.; Höpken, U.E.; Lipp, M.; et al. Increased levels of CCR7 ligands in carotid atherosclerosis: Different effects in macrophages and smooth muscle cells. Cardiovasc. Res. 2014, 102, 148–156. [Google Scholar] [CrossRef] [Green Version]
- Reape, T.J.; Rayner, K.; Manning, C.D.; Gee, A.N.; Barnette, M.S.; Burnand, K.G.; Groot, P.H. Expression and cellular localization of the CC chemokines PARC and ELC in human atherosclerotic plaques. Am. J. Pathol. 1999, 154, 365–374. [Google Scholar] [CrossRef]
- Gunn, M.D.; Tangemann, K.; Tam, C.; Cyster, J.G.; Rosen, S.D.; Williams, L.T. A chemokine expressed in lymphoid high endothelial venules promotes the adhesion and chemotaxis of naive T lymphocytes. Proc. Natl. Acad. Sci. USA 1998, 95, 258–263. [Google Scholar] [CrossRef] [Green Version]
- Luther, S.A.; Tang, H.L.; Hyman, P.L.; Farr, A.G.; Cyster, J.G. Coexpression of the chemokines ELC and SLC by T zone stromal cells and deletion of the ELC gene in the plt/plt mouse. Proc. Natl. Acad. Sci. USA 2000, 97, 12694–12699. [Google Scholar] [CrossRef] [Green Version]
- Laufer, J.M.; Kindinger, I.; Artinger, M.; Pauli, A.; Legler, D.F. CCR7 Is Recruited to the Immunological Synapse, Acts as Co-stimulatory Molecule and Drives LFA-1 Clustering for Efficient T Cell Adhesion Through ZAP70. Front. Immunol. 2018, 9, 3115. [Google Scholar] [CrossRef] [Green Version]
- Burman, A.; Haworth, O.; Hardie, D.L.; Amft, E.N.; Siewert, C.; Jackson, D.G.; Salmon, M.; Buckley, C.D. A chemokine-dependent stromal induction mechanism for aberrant lymphocyte accumulation and compromised lymphatic return in rheumatoid arthritis. J. Immunol. 2005, 174, 1693–1700. [Google Scholar] [CrossRef]
- Marsland, B.J.; Bättig, P.; Bauer, M.; Ruedl, C.; Lässing, U.; Beerli, R.R.; Dietmeier, K.; Ivanova, L.; Pfister, T.; Vogt, L.; et al. CCL19 and CCL21 induce a potent proinflammatory differentiation program in licensed dendritic cells. Immunity 2005, 22, 493–505. [Google Scholar] [CrossRef] [Green Version]
- Benagiano, M.; Azzurri, A.; Ciervo, A.; Amedei, A.; Tamburini, C.; Ferrari, M.; Telford, J.L.; Baldari, C.T.; Romagnani, S.; Cassone, A.; et al. T helper type 1 lymphocytes drive inflammation in human atherosclerotic lesions. Proc. Natl. Acad. Sci. USA 2003, 100, 6658–6663. [Google Scholar] [CrossRef] [Green Version]
- Nickel, T.; Pfeiler, S.; Summo, C.; Kopp, R.; Meimarakis, G.; Sicic, Z.; Lambert, M.; Lackermair, K.; David, R.; Beiras-Fernandez, A.; et al. oxLDL downregulates the dendritic cell homing factors CCR7 and CCL21. Mediators Inflamm. 2012, 2012, 320953. [Google Scholar] [CrossRef]
- Cai, W.; Tao, J.; Zhang, X.; Tian, X.; Liu, T.; Feng, X.; Bai, J.; Yan, C.; Han, Y. Contribution of homeostatic chemokines CCL19 and CCL21 and their receptor CCR7 to coronary artery disease. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1933–1941. [Google Scholar] [CrossRef] [Green Version]
- Wan, W.; Lionakis, M.S.; Liu, Q.; Roffê, E.; Murphy, P.M. Genetic deletion of chemokine receptor Ccr7 exacerbates atherogenesis in ApoE-deficient mice. Cardiovasc. Res. 2013, 97, 580–588. [Google Scholar] [CrossRef] [Green Version]
- Luchtefeld, M.; Grothusen, C.; Gagalick, A.; Jagavelu, K.; Schuett, H.; Tietge, U.J.F.; Pabst, O.; Grote, K.; Drexler, H.; Förster, R.; et al. Chemokine receptor 7 knockout attenuates atherosclerotic plaque development. Circulation 2010, 122, 1621–1628. [Google Scholar] [CrossRef] [Green Version]
- Akhavanpoor, M.; Gleissner, C.A.; Gorbatsch, S.; Doesch, A.O.; Akhavanpoor, H.; Wangler, S.; Jahn, F.; Lasitschka, F.; Katus, H.A.; Erbel, C. CCL19 and CCL21 modulate the inflammatory milieu in atherosclerotic lesions. Drug Des. Dev. Ther. 2014, 8, 2359–2371. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, D.; Andalibi, A.; Chaverri-Almada, L.; Berliner, J.A.; Kirchgessner, T.; Fang, Z.T.; Tekamp-Olson, P.; Lusis, A.J.; Gallegos, C.; Fogelman, A.M. Role of the GRO family of chemokines in monocyte adhesion to MM-LDL-stimulated endothelium. J. Clin. Investig. 1994, 94, 1968–1973. [Google Scholar] [CrossRef] [PubMed]
- Huo, Y.; Weber, C.; Forlow, S.B.; Sperandio, M.; Thatte, J.; Mack, M.; Jung, S.; Littman, D.R.; Ley, K. The chemokine KC, but not monocyte chemoattractant protein-1, triggers monocyte arrest on early atherosclerotic endothelium. J. Clin. Investig. 2001, 108, 1307–1314. [Google Scholar] [CrossRef]
- Boisvert, W.A.; Rose, D.M.; Johnson, K.A.; Fuentes, M.E.; Lira, S.A.; Curtiss, L.K.; Terkeltaub, R.A. Up-regulated expression of the CXCR2 ligand KC/GRO-alpha in atherosclerotic lesions plays a central role in macrophage accumulation and lesion progression. Am. J. Pathol. 2006, 168, 1385–1395. [Google Scholar] [CrossRef] [Green Version]
- Boisvert, W.A.; Santiago, R.; Curtiss, L.K.; Terkeltaub, R.A. A leukocyte homologue of the IL-8 receptor CXCR-2 mediates the accumulation of macrophages in atherosclerotic lesions of LDL receptor-deficient mice. J. Clin. Investig. 1998, 101, 353–363. [Google Scholar] [CrossRef] [Green Version]
- van Bon, L.; Affandi, A.J.; Broen, J.; Christmann, R.B.; Marijnissen, R.J.; Stawski, L.; Farina, G.A.; Stifano, G.; Mathes, A.L.; Cossu, M.; et al. Proteome-wide analysis and CXCL4 as a biomarker in systemic sclerosis. N. Engl. J. Med. 2014, 370, 433–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gleissner, C.A.; Ley, K. CXCL4 in atherosclerosis: Possible roles in monocyte arrest and macrophage foam cell formation. Thromb. Haemost. 2007, 98, 917–918. [Google Scholar] [CrossRef]
- Scheuerer, B.; Ernst, M.; Dürrbaum-Landmann, I.; Fleischer, J.; Grage-Griebenow, E.; Brandt, E.; Flad, H.D.; Petersen, F. The CXC-chemokine platelet factor 4 promotes monocyte survival and induces monocyte differentiation into macrophages. Blood 2000, 95, 1158–1166. [Google Scholar] [CrossRef]
- Bouhlel, M.A.; Derudas, B.; Rigamonti, E.; Dièvart, R.; Brozek, J.; Haulon, S.; Zawadzki, C.; Jude, B.; Torpier, G.; Marx, N.; et al. PPARgamma activation primes human monocytes into alternative M2 macrophages with anti-inflammatory properties. Cell Metab. 2007, 6, 137–143. [Google Scholar] [CrossRef] [Green Version]
- van der Vorst, E.P.C.; Mandl, M.; Müller, M.; Neideck, C.; Jansen, Y.; Hristov, M.; Gencer, S.; Peters, L.J.F.; Meiler, S.; Feld, M.; et al. Hematopoietic ChemR23 (Chemerin Receptor 23) Fuels Atherosclerosis by Sustaining an M1 Macrophage-Phenotype and Guidance of Plasmacytoid Dendritic Cells to Murine Lesions-Brief Report. Arterioscler. Thromb. Vasc. Biol. 2019, 39, 685–693. [Google Scholar] [CrossRef] [PubMed]
- Gleissner, C.A.; Shaked, I.; Little, K.M.; Ley, K. CXC chemokine ligand 4 induces a unique transcriptome in monocyte-derived macrophages. J. Immunol. 2010, 184, 4810–4818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gleissner, C.A.; Shaked, I.; Erbel, C.; Böckler, D.; Katus, H.A.; Ley, K. CXCL4 downregulates the atheroprotective hemoglobin receptor CD163 in human macrophages. Circ. Res. 2010, 106, 203–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stocker, R.; Perrella, M.A. Heme oxygenase-1: A novel drug target for atherosclerotic diseases? Circulation 2006, 114, 2178–2189. [Google Scholar] [CrossRef] [Green Version]
- Mueller, A.; Meiser, A.; McDonagh, E.M.; Fox, J.M.; Petit, S.J.; Xanthou, G.; Williams, T.J.; Pease, J.E. CXCL4-induced migration of activated T lymphocytes is mediated by the chemokine receptor CXCR3. J. Leukoc. Biol. 2008, 83, 875–882. [Google Scholar] [CrossRef]
- Orekhov, A.N.; Oishi, Y.; Nikiforov, N.G.; Zhelankin, A.V.; Dubrovsky, L.; Sobenin, I.A.; Kel, A.; Stelmashenko, D.; Makeev, V.J.; Foxx, K.; et al. Modified LDL Particles Activate Inflammatory Pathways in Monocyte-derived Macrophages: Transcriptome Analysis. Curr. Pharm. Des. 2018, 24, 3143–3151. [Google Scholar] [CrossRef]
- Simonini, A.; Moscucci, M.; Muller, D.W.; Bates, E.R.; Pagani, F.D.; Burdick, M.D.; Strieter, R.M. IL-8 is an angiogenic factor in human coronary atherectomy tissue. Circulation 2000, 101, 1519–1526. [Google Scholar] [CrossRef]
- Matsuo, Y.; Raimondo, M.; Woodward, T.A.; Wallace, M.B.; Gill, K.R.; Tong, Z.; Burdick, M.D.; Yang, Z.; Strieter, R.M.; Hoffman, R.M.; et al. CXC-chemokine/CXCR2 biological axis promotes angiogenesis in vitro and in vivo in pancreatic cancer. Int. J. Cancer 2009, 125, 1027–1037. [Google Scholar] [CrossRef]
- Lv, G.; Zhu, H.; Li, C.; Wang, J.; Zhao, D.; Li, S.; Ma, L.; Sun, G.; Li, F.; Zhao, Y.; et al. Inhibition of IL-8-mediated endothelial adhesion, VSMCs proliferation and migration by siRNA-TMEM98 suggests TMEM98’s emerging role in atherosclerosis. Oncotarget 2017, 8, 88043–88058. [Google Scholar] [CrossRef] [Green Version]
- Tang, X.-E.; Li, H.; Chen, L.-Y.; Xia, X.-D.; Zhao, Z.-W.; Zheng, X.-L.; Zhao, G.-J.; Tang, C.-K. IL-8 negatively regulates ABCA1 expression and cholesterol efflux via upregulating miR-183 in THP-1 macrophage-derived foam cells. Cytokine 2019, 122, 154385. [Google Scholar] [CrossRef]
- Qin, Y.; Mao, W.; Pan, L.; Sun, Y.; Fan, F.; Zhao, Y.; Cui, Y.; Wei, X.; Kohama, K.; Li, F.; et al. Inhibitory effect of recombinant human CXCL8(3-72)K11R/G31P on atherosclerotic plaques in a mouse model of atherosclerosis. Immunopharmacol. Immunotoxicol. 2019, 41, 446–454. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Town, J.R.; Li, F.; Zhang, X.; Cockcroft, D.W.; Gordon, J.R. ELR-CXC chemokine receptor antagonism targets inflammatory responses at multiple levels. J. Immunol. 2009, 182, 3213–3222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Peng, J.; Sun, W.; Yang, S.; Deng, G.; Li, F.; Cheng, J.-W.; Gordon, J.R. G31P, an antagonist against CXC chemokine receptors 1 and 2, inhibits growth of human prostate cancer cells in nude mice. Tohoku J. Exp. Med. 2012, 228, 147–156. [Google Scholar] [CrossRef] [Green Version]
- Kokje, V.B.C.; Gäbel, G.; Dalman, R.L.; Koole, D.; Northoff, B.H.; Holdt, L.M.; Hamming, J.F.; Lindeman, J.H.N. CXCL8 hyper-signaling in the aortic abdominal aneurysm. Cytokine 2018, 108, 96–104. [Google Scholar] [CrossRef] [PubMed]
- An, Z.; Li, J.; Yu, J.; Wang, X.; Gao, H.; Zhang, W.; Wei, Z.; Zhang, J.; Zhang, Y.; Zhao, J.; et al. Neutrophil extracellular traps induced by IL-8 aggravate atherosclerosis via activation NF-κB signaling in macrophages. Cell Cycle 2019, 18, 2928–2938. [Google Scholar] [CrossRef]
- de Oliveira, R.T.; Mamoni, R.L.; Souza, J.R.; Fernandes, J.L.; Rios, F.J.; Gidlund, M.; Coelho, O.R.; Blotta, M.H. Differential expression of cytokines, chemokines and chemokine receptors in patients with coronary artery disease. Int. J. Cardiol. 2009, 136, 17–26. [Google Scholar] [CrossRef]
- Qin, S.; Rottman, J.B.; Myers, P.; Kassam, N.; Weinblatt, M.; Loetscher, M.; Koch, A.E.; Moser, B.; Mackay, C.R. The chemokine receptors CXCR3 and CCR5 mark subsets of T cells associated with certain inflammatory reactions. J. Clin. Investig. 1998, 101, 746–754. [Google Scholar] [CrossRef]
- Karin, N.; Wildbaum, G.; Thelen, M. Biased signaling pathways via CXCR3 control the development and function of CD4+ T cell subsets. J. Leukocyte Biol. 2016, 99, 857–862. [Google Scholar] [CrossRef] [Green Version]
- Metzemaekers, M.; Vanheule, V.; Janssens, R.; Struyf, S.; Proost, P. Overview of the Mechanisms that May Contribute to the Non-Redundant Activities of Interferon-Inducible CXC Chemokine Receptor 3 Ligands. Front. Immunol. 2017, 8, 1970. [Google Scholar] [CrossRef]
- Cole, K.E.; Strick, C.A.; Paradis, T.J.; Ogborne, K.T.; Loetscher, M.; Gladue, R.P.; Lin, W.; Boyd, J.G.; Moser, B.; Wood, D.E.; et al. Interferon-inducible T cell alpha chemoattractant (I-TAC): A novel non-ELR CXC chemokine with potent activity on activated T cells through selective high affinity binding to CXCR3. J. Exp. Med. 1998, 187, 2009–2021. [Google Scholar] [CrossRef]
- Colvin, R.A.; Campanella, G.S.; Sun, J.; Luster, A.D. Intracellular domains of CXCR3 that mediate CXCL9, CXCL10, and CXCL11 function. J. Biol. chem. 2004, 279, 30219–30227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mach, F.; Sauty, A.; Iarossi, A.S.; Sukhova, G.K.; Neote, K.; Libby, P.; Luster, A.D. Differential expression of three T lymphocyte-activating CXC chemokines by human atheroma-associated cells. J. Clin. Investig. 1999, 104, 1041–1050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatterjee, M.; Rath, D.; Gawaz, M. Role of chemokine receptors CXCR4 and CXCR7 for platelet function. Biochem. Soc. Trans. 2015, 43, 720–726. [Google Scholar] [CrossRef] [PubMed]
- Veillard, N.R.; Steffens, S.; Pelli, G.; Lu, B.; Kwak, B.R.; Gerard, C.; Charo, I.F.; Mach, F. Differential influence of chemokine receptors CCR2 and CXCR3 in development of atherosclerosis in vivo. Circulation 2005, 112, 870–878. [Google Scholar] [CrossRef] [Green Version]
- van Wanrooij, E.J.; de Jager, S.C.; van Es, T.; de Vos, P.; Birch, H.L.; Owen, D.A.; Watson, R.J.; Biessen, E.A.; Chapman, G.A.; van Berkel, T.J.; et al. CXCR3 antagonist NBI-74330 attenuates atherosclerotic plaque formation in LDL receptor-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 251–257. [Google Scholar] [CrossRef] [Green Version]
- Heller, E.A.; Liu, E.; Tager, A.M.; Yuan, Q.; Lin, A.Y.; Ahluwalia, N.; Jones, K.; Koehn, S.L.; Lok, V.M.; Aikawa, E.; et al. Chemokine CXCL10 promotes atherogenesis by modulating the local balance of effector and regulatory T cells. Circulation 2006, 113, 2301–2312. [Google Scholar] [CrossRef]
- Niki, T.; Soeki, T.; Yamaguchi, K.; Taketani, Y.; Yagi, S.; Iwase, T.; Yamada, H.; Wakatsuki, T.; Shimabukuro, M.; Sata, M. Elevated Concentration of Interferon-Inducible Protein of 10 kD (IP-10) Is Associated With Coronary Atherosclerosis. Int. Heart J. 2015, 56, 269–272. [Google Scholar] [CrossRef] [Green Version]
- Lapidot, T.; Petit, I. Current understanding of stem cell mobilization: The roles of chemokines, proteolytic enzymes, adhesion molecules, cytokines, and stromal cells. Exp. Hematol. 2002, 30, 973–981. [Google Scholar] [CrossRef]
- Tachibana, K.; Hirota, S.; Iizasa, H.; Yoshida, H.; Kawabata, K.; Kataoka, Y.; Kitamura, Y.; Matsushima, K.; Yoshida, N.; Nishikawa, S.; et al. The chemokine receptor CXCR4 is essential for vascularization of the gastrointestinal tract. Nature 1998, 393, 591–594. [Google Scholar] [CrossRef]
- Yellowley, C. CXCL12/CXCR4 signaling and other recruitment and homing pathways in fracture repair. Bonekey Rep. 2013, 2, 300. [Google Scholar] [CrossRef] [Green Version]
- Nagasawa, T. The chemokine CXCL12 and regulation of HSC and B lymphocyte development in the bone marrow niche. Adv. Exp. Med. Biol. 2007, 602, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Gerrits, H.; van Ingen Schenau, D.S.; Bakker, N.E.C.; van Disseldorp, A.J.M.; Strik, A.; Hermens, L.S.; Koenen, T.B.; Krajnc-Franken, M.A.M.; Gossen, J.A. Early postnatal lethality and cardiovascular defects in CXCR7-deficient mice. Genesis 2008, 46, 235–245. [Google Scholar] [CrossRef] [PubMed]
- Burns, J.M.; Summers, B.C.; Wang, Y.; Melikian, A.; Berahovich, R.; Miao, Z.; Penfold, M.E.T.; Sunshine, M.J.; Littman, D.R.; Kuo, C.J.; et al. A novel chemokine receptor for SDF-1 and I-TAC involved in cell survival, cell adhesion, and tumor development. J. Exp. Med. 2006, 203, 2201–2213. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.J.; Banisadr, G.; Bhattacharyya, B.J. CXCR4 signaling in the regulation of stem cell migration and development. J. Neuroimmunol. 2008, 198, 31–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Q.; Jones, D.; Borghesani, P.R.; Segal, R.A.; Nagasawa, T.; Kishimoto, T.; Bronson, R.T.; Springer, T.A. Impaired B-lymphopoiesis, myelopoiesis, and derailed cerebellar neuron migration in CXCR4- and SDF-1-deficient mice. Proc. Natl. Acad. Sci. USA 1998, 95, 9448–9453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samani, N.J.; Erdmann, J.; Hall, A.S.; Hengstenberg, C.; Mangino, M.; Mayer, B.; Dixon, R.J.; Meitinger, T.; Braund, P.; Wichmann, H.E.; et al. Genomewide association analysis of coronary artery disease. N. Engl. J. Med. 2007, 357, 443–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sjaarda, J.; Gerstein, H.; Chong, M.; Yusuf, S.; Meyre, D.; Anand, S.S.; Hess, S.; Paré, G. Blood CSF1 and CXCL12 as Causal Mediators of Coronary Artery Disease. J. Am. Coll. Cardiol. 2018, 72, 300–310. [Google Scholar] [CrossRef]
- Tavakolian Ferdousie, V.; Mohammadi, M.; Hassanshahi, G.; Khorramdelazad, H.; Khanamani Falahati-Pour, S.; Mirzaei, M.; Allah Tavakoli, M.; Kamiab, Z.; Ahmadi, Z.; Vazirinejad, R.; et al. Serum CXCL10 and CXCL12 chemokine levels are associated with the severity of coronary artery disease and coronary artery occlusion. Int. J. Cardiol. 2017, 233, 23–28. [Google Scholar] [CrossRef]
- Döring, Y.; van der Vorst, E.P.C.; Duchene, J.; Jansen, Y.; Gencer, S.; Bidzhekov, K.; Atzler, D.; Santovito, D.; Rader, D.J.; Saleheen, D.; et al. CXCL12 Derived From Endothelial Cells Promotes Atherosclerosis to Drive Coronary Artery Disease. Circulation 2019, 139, 1338–1340. [Google Scholar] [CrossRef]
- Yamaguchi, J.-I.; Kusano, K.F.; Masuo, O.; Kawamoto, A.; Silver, M.; Murasawa, S.; Bosch-Marce, M.; Masuda, H.; Losordo, D.W.; Isner, J.M.; et al. Stromal cell-derived factor-1 effects on ex vivo expanded endothelial progenitor cell recruitment for ischemic neovascularization. Circulation 2003, 107, 1322–1328. [Google Scholar] [CrossRef] [Green Version]
- Schober, A.; Knarren, S.; Lietz, M.; Lin, E.A.; Weber, C. Crucial role of stromal cell-derived factor-1alpha in neointima formation after vascular injury in apolipoprotein E-deficient mice. Circulation 2003, 108, 2491–2497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zernecke, A.; Schober, A.; Bot, I.; von Hundelshausen, P.; Liehn, E.A.; Möpps, B.; Mericskay, M.; Gierschik, P.; Biessen, E.A.; Weber, C. SDF-1alpha/CXCR4 axis is instrumental in neointimal hyperplasia and recruitment of smooth muscle progenitor cells. Circ. Res. 2005, 96, 784–791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akhtar, S.; Gremse, F.; Kiessling, F.; Weber, C.; Schober, A. CXCL12 promotes the stabilization of atherosclerotic lesions mediated by smooth muscle progenitor cells in Apoe-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 679–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Döring, Y.; Noels, H.; van der Vorst, E.P.C.; Neideck, C.; Egea, V.; Drechsler, M.; Mandl, M.; Pawig, L.; Jansen, Y.; Schröder, K.; et al. Vascular CXCR4 Limits Atherosclerosis by Maintaining Arterial Integrity: Evidence From Mouse and Human Studies. Circulation 2017, 136, 388–403. [Google Scholar] [CrossRef] [PubMed]
- Gomes, A.L.; Carvalho, T.; Serpa, J.; Torre, C.; Dias, S. Hypercholesterolemia promotes bone marrow cell mobilization by perturbing the SDF-1:CXCR4 axis. Blood 2010, 115, 3886–3894. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Zhu, M.; Penfold, M.E.; Koenen, R.R.; Thiemann, A.; Heyll, K.; Akhtar, S.; Koyadan, S.; Wu, Z.; Gremse, F.; et al. Activation of CXCR7 limits atherosclerosis and improves hyperlipidemia by increasing cholesterol uptake in adipose tissue. Circulation 2014, 129, 1244–1253. [Google Scholar] [CrossRef] [Green Version]
- Matloubian, M.; David, A.; Engel, S.; Ryan, J.E.; Cyster, J.G. A transmembrane CXC chemokine is a ligand for HIV-coreceptor Bonzo. Nat. Immunol. 2000, 1, 298–304. [Google Scholar] [CrossRef]
- Borst, O.; Münzer, P.; Gatidis, S.; Schmidt, E.-M.; Schönberger, T.; Schmid, E.; Towhid, S.T.; Stellos, K.; Seizer, P.; May, A.E.; et al. The inflammatory chemokine CXC motif ligand 16 triggers platelet activation and adhesion via CXC motif receptor 6-dependent phosphatidylinositide 3-kinase/Akt signaling. Circ. Res. 2012, 111, 1297–1307. [Google Scholar] [CrossRef] [Green Version]
- Ludwig, A.; Weber, C. Transmembrane chemokines: Versatile ’special agents’ in vascular inflammation. Thromb. Haemost. 2007, 97, 694–703. [Google Scholar]
- Calabresi, P.A.; Yun, S.H.; Allie, R.; Whartenby, K.A. Chemokine receptor expression on MBP-reactive T cells: CXCR6 is a marker of IFNgamma-producing effector cells. J. Neuroimmunol. 2002, 127, 96–105. [Google Scholar] [CrossRef]
- Galkina, E.; Harry, B.L.; Ludwig, A.; Liehn, E.A.; Sanders, J.M.; Bruce, A.; Weber, C.; Ley, K. CXCR6 promotes atherosclerosis by supporting T-cell homing, interferon-gamma production, and macrophage accumulation in the aortic wall. Circulation 2007, 116, 1801–1811. [Google Scholar] [CrossRef] [Green Version]
- Meyer Dos Santos, S.; Blankenbach, K.; Scholich, K.; Dörr, A.; Monsefi, N.; Keese, M.; Linke, B.; Deckmyn, H.; Nelson, K.; Harder, S. Platelets from flowing blood attach to the inflammatory chemokine CXCL16 expressed in the endothelium of the human vessel wall. Thromb. Haemost. 2015, 114, 297–312. [Google Scholar] [CrossRef]
- Linke, B.; Meyer Dos Santos, S.; Picard-Willems, B.; Keese, M.; Harder, S.; Geisslinger, G.; Scholich, K. CXCL16/CXCR6-mediated adhesion of human peripheral blood mononuclear cells to inflamed endothelium. Cytokine 2019, 122, 154081. [Google Scholar] [CrossRef]
- Minami, M.; Kume, N.; Shimaoka, T.; Kataoka, H.; Hayashida, K.; Akiyama, Y.; Nagata, I.; Ando, K.; Nobuyoshi, M.; Hanyuu, M.; et al. Expression of SR-PSOX, a novel cell-surface scavenger receptor for phosphatidylserine and oxidized LDL in human atherosclerotic lesions. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 1796–1800. [Google Scholar] [CrossRef] [Green Version]
- Shimaoka, T.; Kume, N.; Minami, M.; Hayashida, K.; Kataoka, H.; Kita, T.; Yonehara, S. Molecular cloning of a novel scavenger receptor for oxidized low density lipoprotein, SR-PSOX, on macrophages. J. Biol. Chem. 2000, 275, 40663–40666. [Google Scholar] [CrossRef] [Green Version]
- Ma, K.L.; Wu, Y.; Zhang, Y.; Wang, G.H.; Hu, Z.B.; Ruan, X.Z. Activation of the CXCL16/CXCR6 pathway promotes lipid deposition in fatty livers of apolipoprotein E knockout mice and HepG2 cells. Am. J. Transl. Res. 2018, 10, 1802–1816. [Google Scholar]
- Wågsäter, D.; Olofsson, P.S.; Norgren, L.; Stenberg, B.; Sirsjö, A. The chemokine and scavenger receptor CXCL16/SR-PSOX is expressed in human vascular smooth muscle cells and is induced by interferon gamma. Biochem. Biophys. Res. Commun. 2004, 325, 1187–1193. [Google Scholar] [CrossRef]
- Xiao, Q.; Zhu, X.; Yang, S.; Wang, J.; Yin, R.; Song, J.; Ma, A.; Pan, X. LPS induces CXCL16 expression in HUVECs through the miR-146a-mediated TLR4 pathway. Int. Immunopharmacol. 2019, 69, 143–149. [Google Scholar] [CrossRef]
- Ma, A.-J.; Zhu, X.-Y.; Yang, S.-N.; Pan, X.-D.; Wang, T.; Wang, Y.; Xiao, X.; Liu, S.-H. Associations of CXCL16, miR-146a and miR-146b in atherosclerotic apolipoprotein E-knockout mice. Mol. Med. Rep. 2018, 18, 2995–3002. [Google Scholar] [CrossRef]
- Yin, R.; Ma, A.; Pan, X.; Yang, S. Biomarkers of cerebral microembolic signals. Clinica chimica acta; international journal of clinical chemistry 2017, 475, 164–168. [Google Scholar] [CrossRef]
- Ma, A.; Pan, X.; Xing, Y.; Wu, M.; Wang, Y.; Ma, C. Elevation of serum CXCL16 level correlates well with atherosclerotic ischemic stroke. Arch. Med. Sci. 2014, 10, 47–52. [Google Scholar] [CrossRef] [Green Version]
- Hu, Z.B.; Chen, Y.; Gong, Y.X.; Gao, M.; Zhang, Y.; Wang, G.H.; Tang, R.N.; Liu, H.; Liu, B.C.; Ma, K.L. Activation of the CXCL16/CXCR6 Pathway by Inflammation Contributes to Atherosclerosis in Patients with End-stage Renal Disease. Int. J. Med. Sci. 2016, 13, 858–867. [Google Scholar] [CrossRef] [Green Version]
- Verge, G.M.; Milligan, E.D.; Maier, S.F.; Watkins, L.R.; Naeve, G.S.; Foster, A.C. Fractalkine (CX3CL1) and fractalkine receptor (CX3CR1) distribution in spinal cord and dorsal root ganglia under basal and neuropathic pain conditions. Eur. J. Neurosci. 2004, 20, 1150–1160. [Google Scholar] [CrossRef]
- Lindia, J.A.; McGowan, E.; Jochnowitz, N.; Abbadie, C. Induction of CX3CL1 expression in astrocytes and CX3CR1 in microglia in the spinal cord of a rat model of neuropathic pain. J. Pain 2005, 6, 434–438. [Google Scholar] [CrossRef]
- Landsman, L.; Bar-On, L.; Zernecke, A.; Kim, K.-W.; Krauthgamer, R.; Shagdarsuren, E.; Lira, S.A.; Weissman, I.L.; Weber, C.; Jung, S. CX3CR1 is required for monocyte homeostasis and atherogenesis by promoting cell survival. Blood 2009, 113, 963–972. [Google Scholar] [CrossRef] [Green Version]
- Stolla, M.; Pelisek, J.; von Brühl, M.-L.; Schäfer, A.; Barocke, V.; Heider, P.; Lorenz, M.; Tirniceriu, A.; Steinhart, A.; Bauersachs, J.; et al. Fractalkine is expressed in early and advanced atherosclerotic lesions and supports monocyte recruitment via CX3CR1. PLoS ONE 2012, 7, e43572. [Google Scholar] [CrossRef] [Green Version]
- Teupser, D.; Pavlides, S.; Tan, M.; Gutierrez-Ramos, J.-C.; Kolbeck, R.; Breslow, J.L. Major reduction of atherosclerosis in fractalkine (CX3CL1)-deficient mice is at the brachiocephalic artery, not the aortic root. Proc. Natl. Acad. Sci. USA 2004, 101, 17795–17800. [Google Scholar] [CrossRef] [Green Version]
- Ancuta, P.; Rao, R.; Moses, A.; Mehle, A.; Shaw, S.K.; Luscinskas, F.W.; Gabuzda, D. Fractalkine preferentially mediates arrest and migration of CD16+ monocytes. J. Exp. Med. 2003, 197, 1701–1707. [Google Scholar] [CrossRef] [Green Version]
- Roy-Chowdhury, E.; Brauns, N.; Helmke, A.; Nordlohne, J.; Bräsen, J.H.; Schmitz, J.; Volkmann, J.; Fleig, S.V.; Kusche-Vihrog, K.; Haller, H.; et al. Human CD16+ monocytes promote a pro-atherosclerotic endothelial cell phenotype via CX3CR1-CX3CL1 interaction. Cardiovasc. Res. 2020. [Google Scholar] [CrossRef]
- Panigrahi, S.; Chen, B.; Fang, M.; Potashnikova, D.; Komissarov, A.A.; Lebedeva, A.; Michaelson, G.M.; Wyrick, J.M.; Morris, S.R.; Sieg, S.F.; et al. CX3CL1 and IL-15 Promote CD8 T cell chemoattraction in HIV and in atherosclerosis. PLoS Pathog. 2020, 16, e1008885. [Google Scholar] [CrossRef]
- Riopel, M.; Vassallo, M.; Ehinger, E.; Pattison, J.; Bowden, K.; Winkels, H.; Wilson, M.; de Jong, R.; Patel, S.; Balakrishna, D.; et al. CX3CL1-Fc treatment prevents atherosclerosis in Ldlr KO mice. Mol. Metab. 2019, 20, 89–101. [Google Scholar] [CrossRef]
- Rowinska, Z.; Koeppel, T.A.; Sanati, M.; Schelzig, H.; Jankowski, J.; Weber, C.; Zernecke, A.; Liehn, E.A. Role of the CX3C chemokine receptor CX3CR1 in the pathogenesis of atherosclerosis after aortic transplantation. PLoS ONE 2017, 12, e0170644. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.J.; Wang, Y.M.; Lee, V.W.S.; Zhang, G.Y.; Medbury, H.; Williams, H.; Wang, Y.; Tan, T.K.; Harris, D.C.H.; Alexander, S.I.; et al. DEC205-DC targeted DNA vaccine against CX3CR1 protects against atherogenesis in mice. PLoS ONE 2018, 13, e0195657. [Google Scholar] [CrossRef]
- Sinitski, D.; Kontos, C.; Krammer, C.; Asare, Y.; Kapurniotu, A.; Bernhagen, J. Macrophage Migration Inhibitory Factor (MIF)-Based Therapeutic Concepts in Atherosclerosis and Inflammation. Thromb. Haemost. 2019, 119, 553–566. [Google Scholar] [CrossRef] [Green Version]
- Burger-Kentischer, A.; Goebel, H.; Seiler, R.; Fraedrich, G.; Schaefer, H.E.; Dimmeler, S.; Kleemann, R.; Bernhagen, J.; Ihling, C. Expression of macrophage migration inhibitory factor in different stages of human atherosclerosis. Circulation 2002, 105, 1561–1566. [Google Scholar] [CrossRef] [Green Version]
- Burger-Kentischer, A.; Göbel, H.; Kleemann, R.; Zernecke, A.; Bucala, R.; Leng, L.; Finkelmeier, D.; Geiger, G.; Schaefer, H.E.; Schober, A.; et al. Reduction of the aortic inflammatory response in spontaneous atherosclerosis by blockade of macrophage migration inhibitory factor (MIF). Atherosclerosis 2006, 184, 28–38. [Google Scholar] [CrossRef]
- Pan, J.-H.; Sukhova, G.K.; Yang, J.-T.; Wang, B.; Xie, T.; Fu, H.; Zhang, Y.; Satoskar, A.R.; David, J.R.; Metz, C.N.; et al. Macrophage migration inhibitory factor deficiency impairs atherosclerosis in low-density lipoprotein receptor-deficient mice. Circulation 2004, 109, 3149–3153. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Sakuma, M.; Zago, A.C.; Zhang, X.; Shi, C.; Leng, L.; Mizue, Y.; Bucala, R.; Simon, D. Evidence for a role of macrophage migration inhibitory factor in vascular disease. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 709–714. [Google Scholar] [CrossRef] [Green Version]
- Schober, A.; Bernhagen, J.; Thiele, M.; Zeiffer, U.; Knarren, S.; Roller, M.; Bucala, R.; Weber, C. Stabilization of atherosclerotic plaques by blockade of macrophage migration inhibitory factor after vascular injury in apolipoprotein E-deficient mice. Circulation 2004, 109, 380–385. [Google Scholar] [CrossRef] [Green Version]
- Bernhagen, J.; Krohn, R.; Lue, H.; Gregory, J.L.; Zernecke, A.; Koenen, R.R.; Dewor, M.; Georgiev, I.; Schober, A.; Leng, L.; et al. MIF is a noncognate ligand of CXC chemokine receptors in inflammatory and atherogenic cell recruitment. Nat. Med. 2007, 13, 587–596. [Google Scholar] [CrossRef]
- Alampour-Rajabi, S.; El Bounkari, O.; Rot, A.; Müller-Newen, G.; Bachelerie, F.; Gawaz, M.; Weber, C.; Schober, A.; Bernhagen, J. MIF interacts with CXCR7 to promote receptor internalization, ERK1/2 and ZAP-70 signaling, and lymphocyte chemotaxis. FASEB J. 2015, 29, 4497–4511. [Google Scholar] [CrossRef] [Green Version]
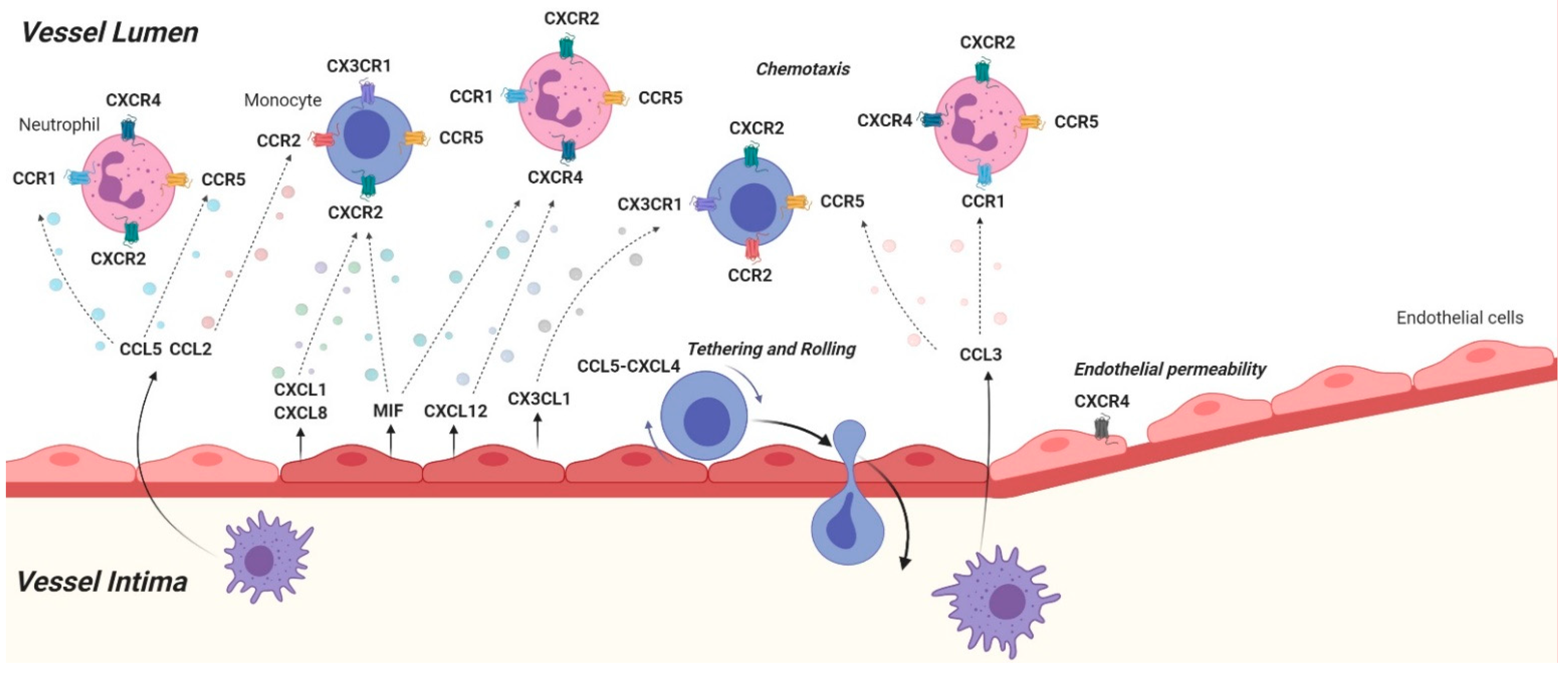


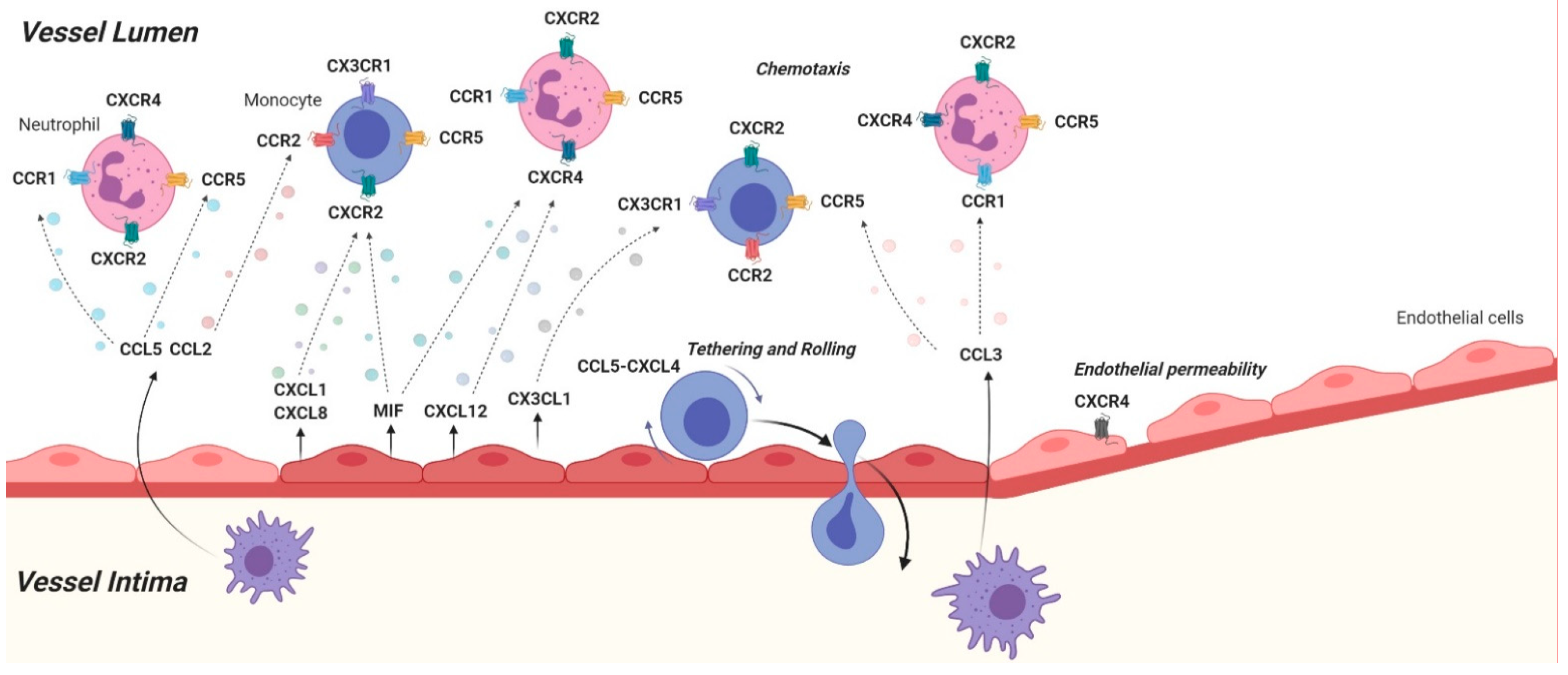{kind=link}
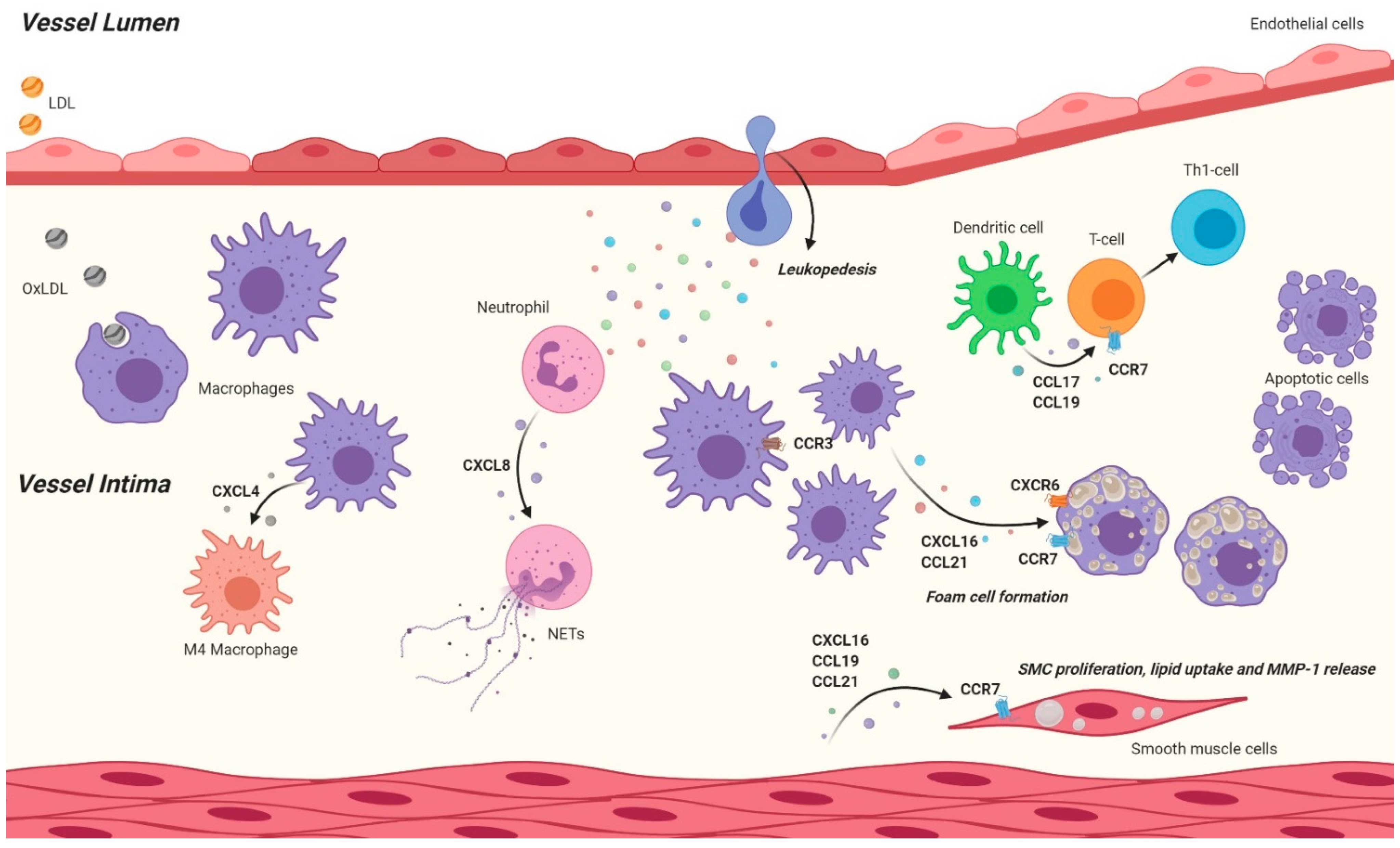{kind=link}
| Chemokine axis | (Mouse) Model | Treatment | Outcome | Ref. |
|---|---|---|---|---|
| CCL2-CCR2 | C57/Bl6 | Thioglycollate (i.p.) injection | Reduced monocyte recruitment | [11] |
| CCL2−/− Ldlr−/− | 12 weeks WD | Smaller lesions, reduced recruitment | [12] | |
| CCL2−/− Apoe−/− & CCL2−/− bone marrow into Apoe3 Leiden mice | 5–13 weeks WD | Smaller lesions, reduced recruitment | [13,14,15] | |
| Apoe−/− | 4 weeks WD, CCR2 Antagonist RS102895 | Smaller lesions, less macrophages | [16] | |
| CCR2−/− Apoe−/− | WD, Cuff placement | Reduced neointimal formation | [17,18,19] | |
| CCL3 | Ldlr-/ transplanted with CCL3−/− BM | 12 weeks WD | Reduced lesion size | [25] |
| Apoe−/− | Atorvastatin, 16 weeks WD | Atorvastatin blocks CCL3 expression and thereby lesion formation | [26] | |
| CCL5-CCR1/CCR3/ CCR5 | Apoe−/− | Wire injury, inhibition of CCL5 | Reduced neointima formation | [29] |
| Apoe−/− | 14 weeks WD, Met-Rantes (CCL5 inhibitor) | Smaller lesions and reduced lesional number of macrophages and T cells | [30] | |
| Apoe−/− | CCL5-CXCL4 heterodimer blocking with MKEY | Reduced scar formation in MI | [36] | |
| CCR5−/− Apoe−/− | WD | Reduced lesion size | [21] | |
| CCR1−/− Apoe−/− | WD | Increased lesion size | [21] | |
| CCR1−/− Apoe−/− | 4 weeks WD | Reduced lesion size | [38] | |
| CCL17 | CCL17e/e Apoe−/− | 4 and 12 weeks WD | Reduced lesion size, increased number of Treg | [44] |
| Apoe−/− | WD, CCL5-CCL17 heterodimer inhibition | Smaller lesions | [47] | |
| CCL19, CCL21/CCR7 | ApoE−/−CCR7−/− | 8 weeks WD | Increased lesion size and T cell number | [60] |
| Ldlr−/−CCR7−/− | 12 weeks WD, adoptive transfer of wild type T cells | Increased lesion size | [61] | |
| CCL19−/− CCL21−/−BM into Ldlr−/− | WD | Increased plaque stability, no change in lesion size | [62] | |
| CXCL1-CXCR2 | Cxcl1−/− Ldlr−/− | 16 weeks WD | Reduced lesion size, less macrophages | [65] |
| Cxcl1−/− BM into Ldlr−/− | 16 weeks WD | No effect | [65] | |
| Cxcr2−/− BM into Ldlr−/− | 16 weeks WD | Reduced lesion size, less macrophages | [66] | |
| Apoe−/− | 8 weeks WD versus chow diet | Increased levels of CXCL1 in the serum, more monocytes in the circulation | [38] | |
| Apoe−/− | CXCL1 neutralization with antibody injections, 4 weeks WD | Reduced lesion size, decreased monocyte mobilization | [38] | |
| CXCL4 | PF4−/− Apoe−/− | WD | Reduced lesion size | [33] |
| CXCL8 | Apoe−/− | Injection of human CXCL8 analog, WD 12 weeks | Reduced lipid levels, reduced lesions? | [81] |
| CXCL12-CXCR4/ACKR3 | Nude mice | Injectionof CXCL12 into hindlimb of mice | Induces ischemic neovascularization | [110] |
| Apoe−/− | Vascular injury | SMC-derived CXCL12 mediates neointima formation | [111] | |
| Apoe−/− lacking CXCL12 specifically in ECs | WD 12 weeks | Reduced lesion formation | [109] | |
| Ldlr−/− with CXCR4−/− BM, or Apoe−/− with CXCR4 blocking | WD, AMD3465 | Enhanced lesion formation | [20] | |
| Apoe−/− | WD and repetitive CXCL12-injections | More stable plaque phenotype in partial ligation, no size differences | [113] | |
| Apoe−/− lacking CXCR4 in ECs or SMCs specifically | WD 12 weeks | Enhanced lesion formation, higher permeability | [114] | |
| C57/Bl6 | 4 weeks high cholesterol diet | Higher CXCL12 levels | [115] | |
| ACKR3−/− Apoe−/− | Wire induced vascular injury, WD | Enhanced peripheral cholesterol levels, increased neointima | [116] | |
| CXCL16-CXCR6 | CXCR6GFP/GFP ApoE−/− | WD | Reduced lesion size, less T cells and macrophages | [121] |
| Apoe−/− | WD versus Chow diet | Upregulation of CXCL16 under WD | [125] | |
| CX3CL1-CX3CR1 | Apoe−/− with CX3CR1−/− BM | 12 weeks WD | Induces monocyte apoptosis, reduces lesion size | [135] |
| CX3CL1−/− Apoe−/− CX3CL1−/− Ldlr−/− | ~12 weeks WD | Reduced lesion size and monocyte recruitment | [136] | |
| Ldlr−/− | Injection of a long lasting FcCX3CL1 version, agonist, WD | Reduced lesion size and monocyte recruitment | [141] | |
| Apoe−/− | Transplantation of aortic segments from Cx3cr−/−ApoE−/− into ApoE−/− | Plaque regression | [142] | |
| Apoe−/− | DNA vaccine which induces antibodies against CX3CR1, chow diet | Reduced lesion size, less macrophage accumulation | [143] | |
| MIF-CXCR2/CXCR4/ACKR3 | Apoe−/− | MIF antibody blocking | Reduced macrophage load in lesions | [147] |
| MIF−/− Ldlr−/− | WD | Reduced lesion size | [148] | |
| Apoe−/− (vascular injury) | MIF antibody blocking | Reduced inflammation and intimal thickening | [149,150] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gencer, S.; Evans, B.R.; van der Vorst, E.P.C.; Döring, Y.; Weber, C. Inflammatory Chemokines in Atherosclerosis. Cells 2021, 10, 226. https://doi.org/10.3390/cells10020226
Gencer S, Evans BR, van der Vorst EPC, Döring Y, Weber C. Inflammatory Chemokines in Atherosclerosis. Cells. 2021; 10(2):226. https://doi.org/10.3390/cells10020226
Chicago/Turabian StyleGencer, Selin, Bryce R. Evans, Emiel P.C. van der Vorst, Yvonne Döring, and Christian Weber. 2021. "Inflammatory Chemokines in Atherosclerosis" Cells 10, no. 2: 226. https://doi.org/10.3390/cells10020226
APA StyleGencer, S., Evans, B. R., van der Vorst, E. P. C., Döring, Y., & Weber, C. (2021). Inflammatory Chemokines in Atherosclerosis. Cells, 10(2), 226. https://doi.org/10.3390/cells10020226