
Embryonic Environmental Niche Reprograms Somatic Cells to Express Pluripotency Markers and Participate in Adult Chimaeras

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Outline
2.2. Animals
2.3. Experimental Procedures
2.3.1. Embryo Collection
2.3.2. Mouse Embryonic Fibroblast Isolation
2.3.3. Adult Somatic Cells Isolation
2.3.4. Embryo Manipulation
2.3.5. Embryo Culture Conditions
2.3.6. Embryo Transfer
2.3.7. Obtaining Chimaeric Foetuses and Animals
2.3.8. Immunostaining
2.4. Analyses of Samples
2.4.1. Image Acquisition, Processing and Analysis
2.4.2. Staging of Foetuses
2.4.3. Cytofluorometric Evaluation of Ploidy
2.4.4. Cell Sorting (FACS)
2.4.5. Samples for DNA Analysis and DNA Isolation
2.4.6. DNA Genotyping of the lacZ Product
2.4.7. DNA Genotyping of Microsatellites
2.4.8. Sex Identification in Foetuses
3. Results
3.1. Preimplantation Development of Presumptive Embryonic–Somatic Chimaeras Obtained by MEF Introduction to recipient morulae
3.1.1. MEFs Introduced into 8-16-Cell Recipient Embryo Can Integrate with the Embryo within 2 Days of Culture
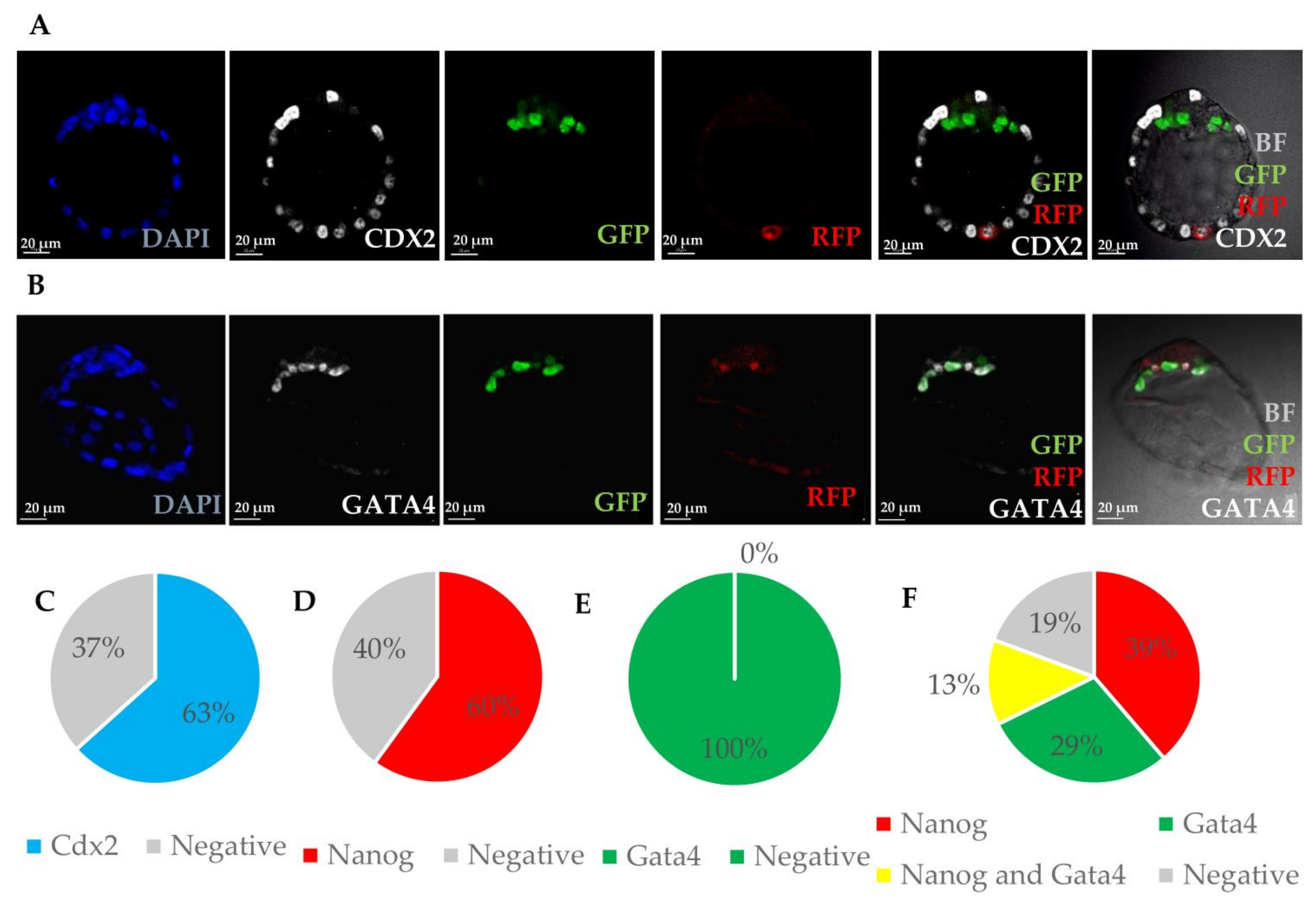
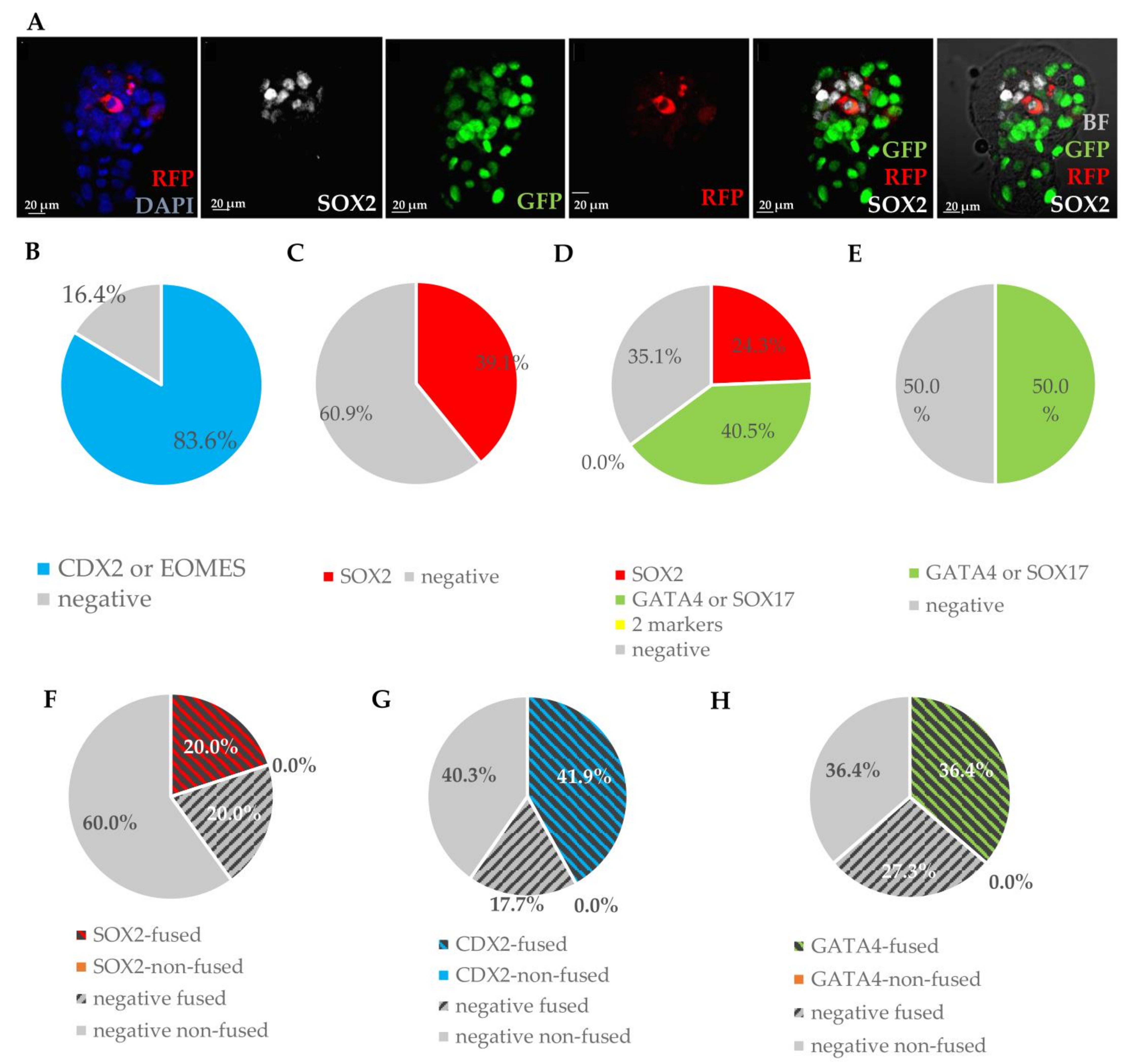
3.1.2. Integrated MEFs Express Markers of Three Blastocyst Lineages
3.1.3. Introduced MEFs Can Undergo Fusion with Blastomeres within 2 Days of Culture
3.2. Postimplantation Development of Presumptive Chimaeras
3.2.1. MEF Progeny Continue Development in Embryonic–Somatic Chimaeras
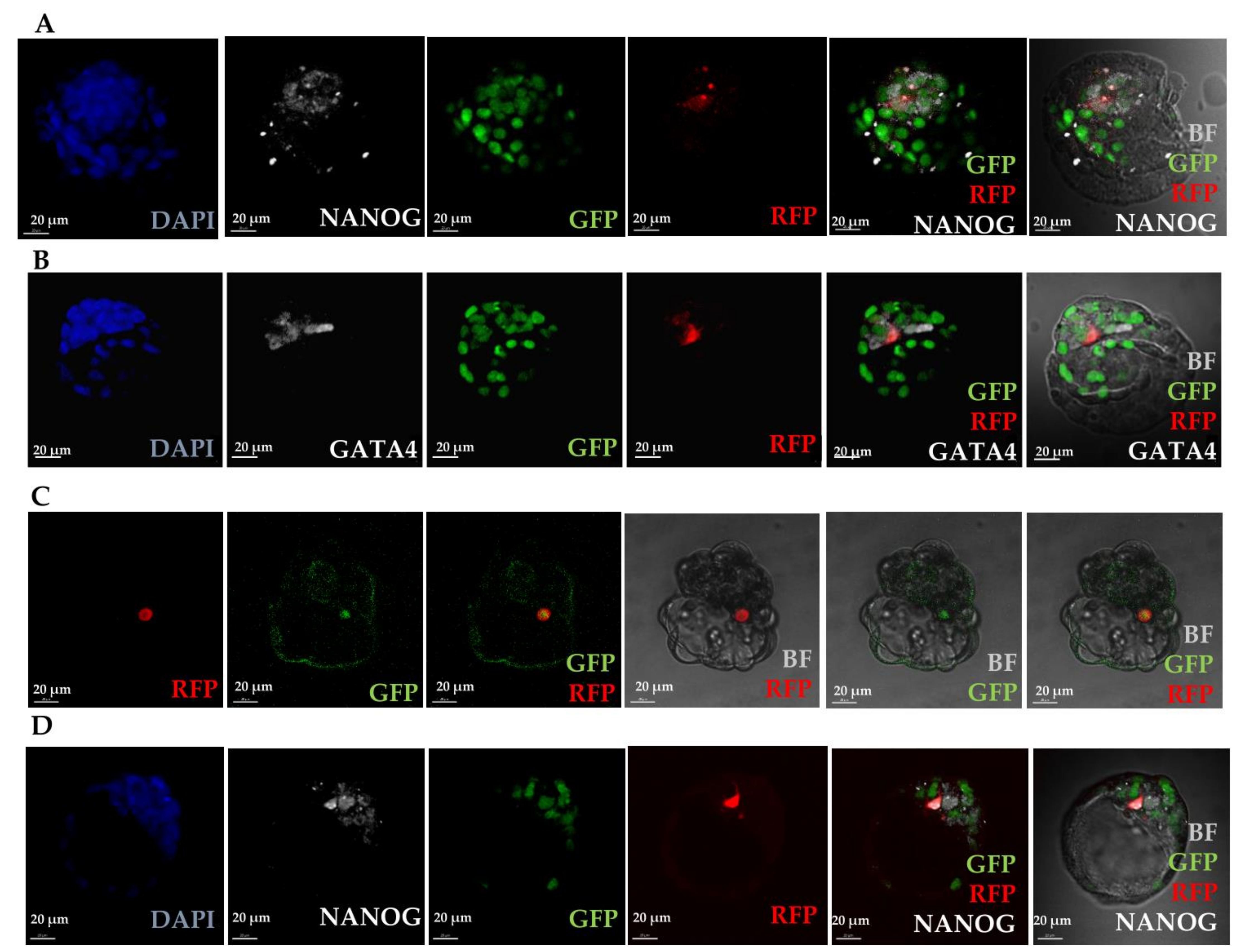
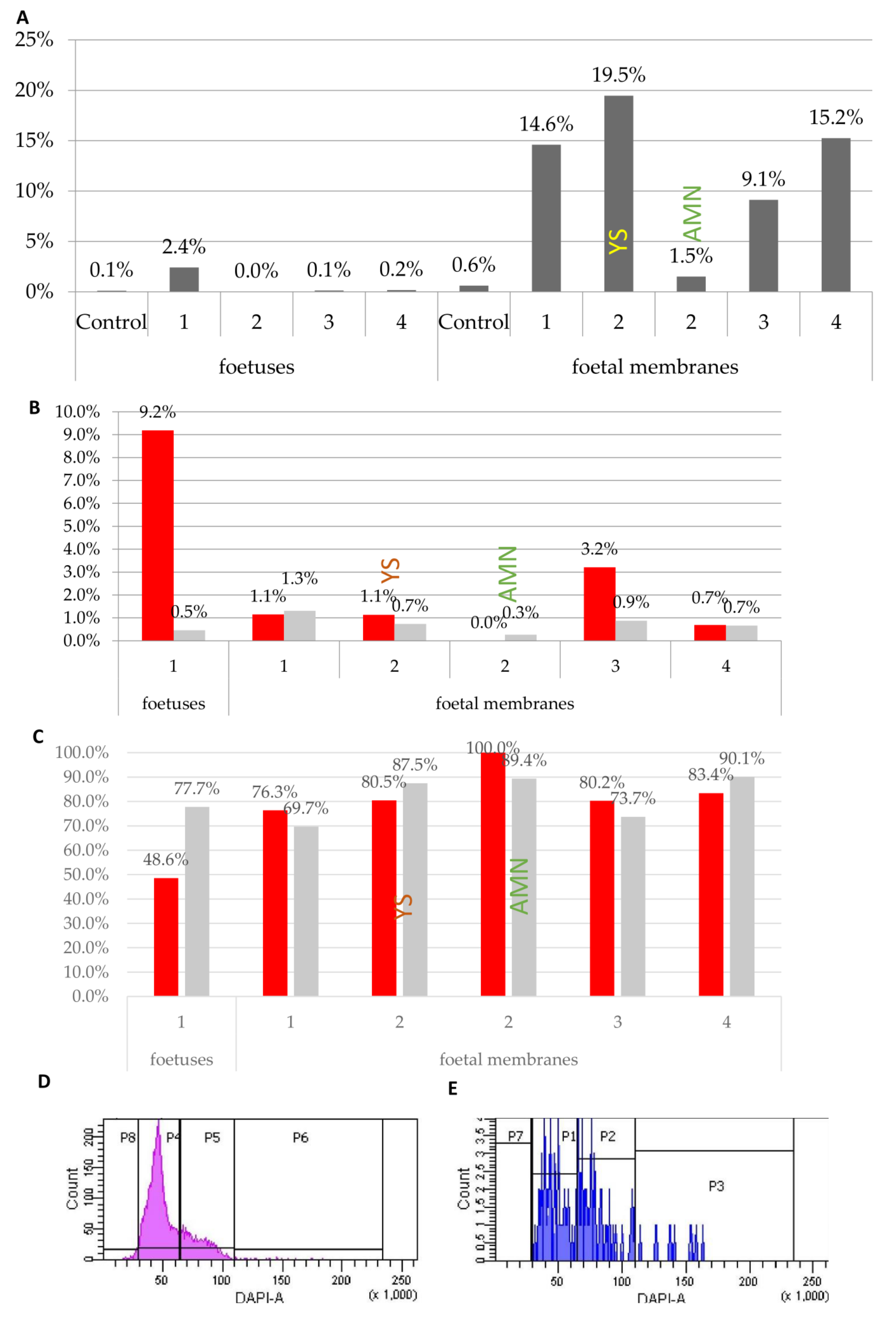
3.2.2. Somatic Cells Introduced into Early Embryos are Present in Chimaeric Foetuses and Foetal Membranes
3.2.3. Donor Markers are Found in both Diploid and Tetraploid Cells
3.2.4. Fluorescently Labelled Progeny of Introduced Cells Are found in Chimaeric Foetuses and Foetal Membranes and Are Frequently Tetraploid
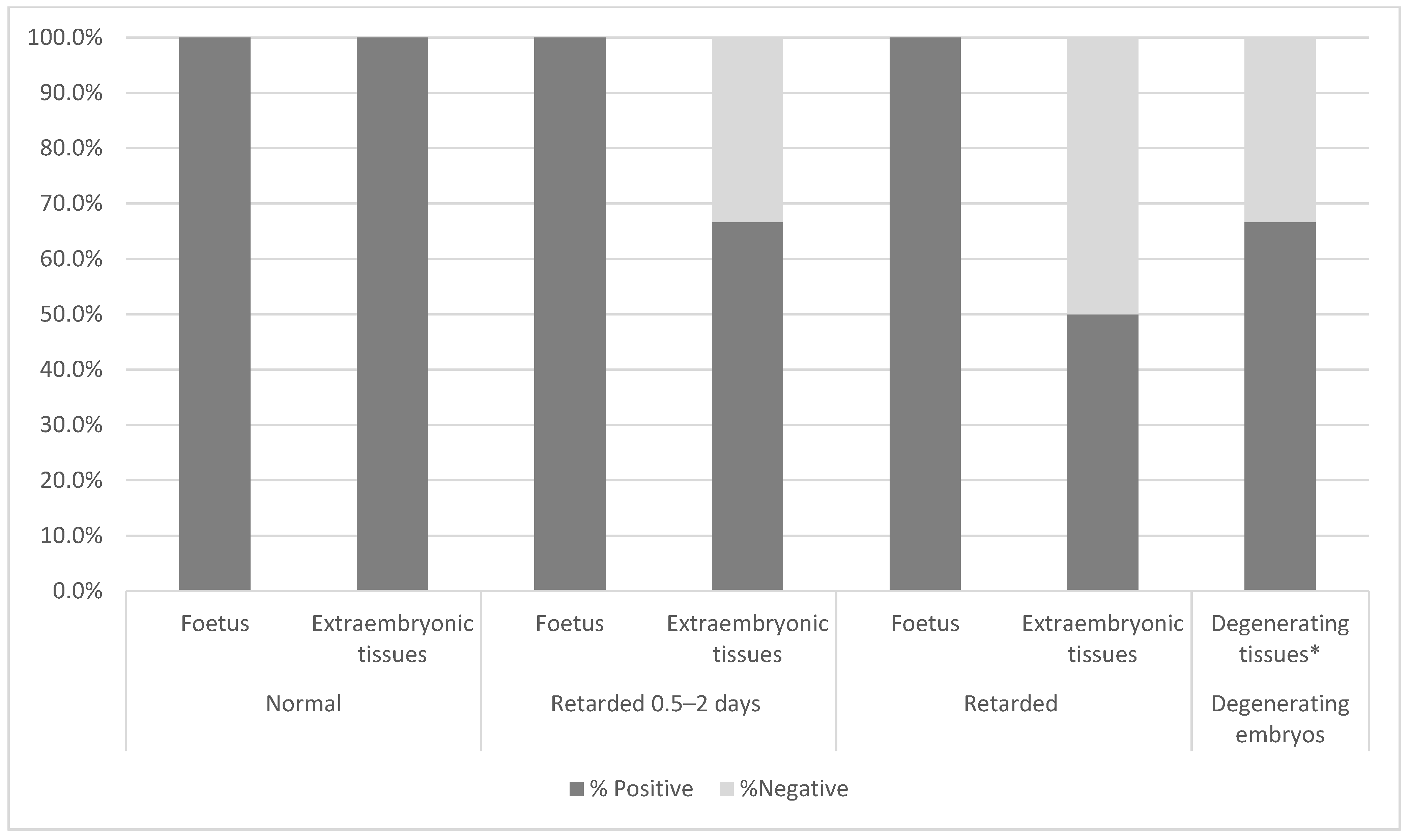
3.2.5. Pregnancy Rate and Postnatal Survival of Chimaeric Animals
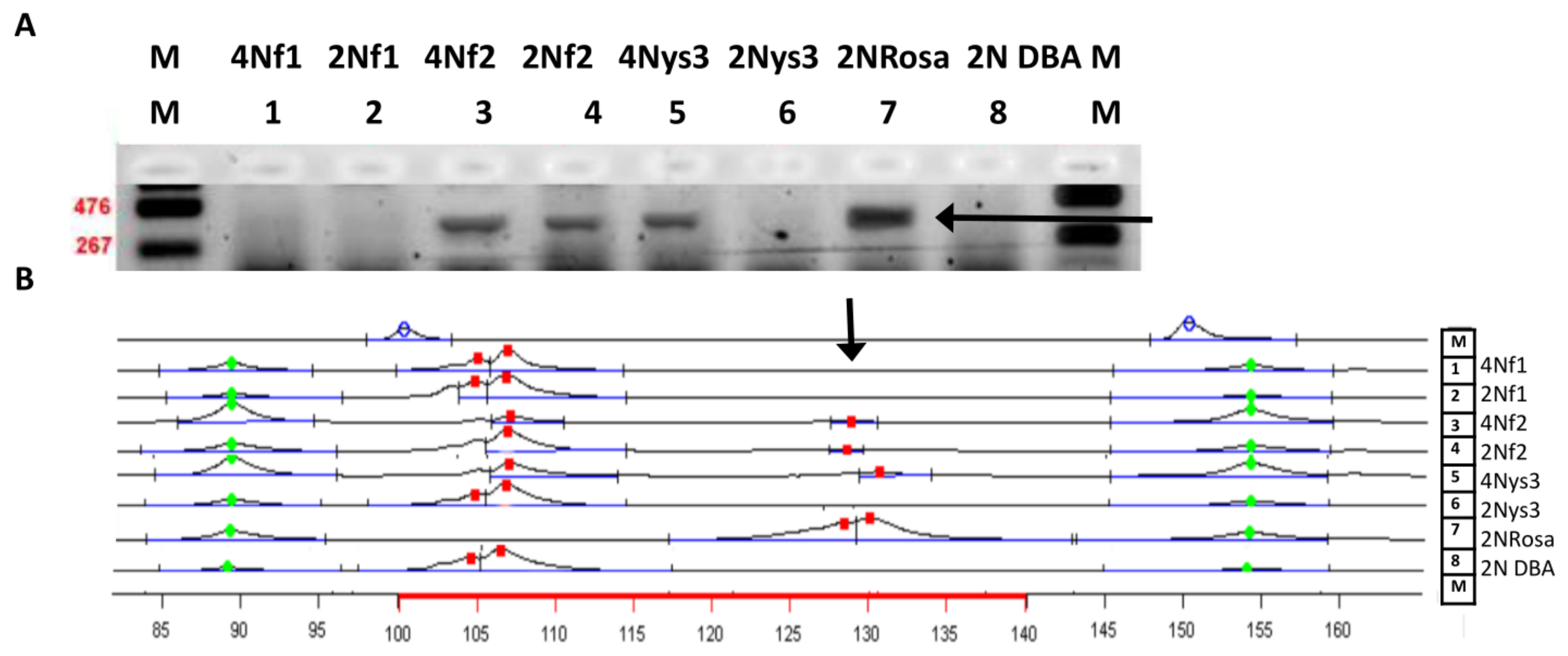
3.2.6. Donor DNA Markers are Present in Adult Tissues
3.2.7. RFP-Positive Progeny of Introduced MEFs Are Present in Adult Tissues
3.2.8. Preimplantation Development of Presumptive Embryonic–Somatic Chimaeras Obtained by Cumulus Cell Introduction
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Waddington, C.H. The Strategy of the Genes; George Allen and Unwin: London, UK, 1957. [Google Scholar]
- Hochedlinger, K.; Plath, K. Epigenetic reprogramming and induced pluripotency. Development 2009, 136, 509–523. [Google Scholar] [CrossRef] [Green Version]
- Frantz, G.D.; McConnell, S.K. Restriction of Late Cerebral Cortical Progenitors to an Upper-Layer Fate. Neuron 1996, 17, 55–61. [Google Scholar] [CrossRef] [Green Version]
- Morrison, S.J.; Shah, N.M.; Anderson, D.J. Regulatory Mechanisms in Stem Cell Biology. Cell 1997, 88, 287–298. [Google Scholar] [CrossRef] [Green Version]
- Bonifer, C.; Faust, N.; Geiger, H.; Müller, A.M. Developmental Changes in the Differentiation Capacity of Haematopoietic Stem Cells. Immunol. Today 1998, 19, 236–241. [Google Scholar] [CrossRef]
- Campbell, K.H.S.; McWhir, J.; Ritchie, W.A.; Wilmut, I. Sheep Cloned by Nuclear Transfer from a Cultured Cell Line. Nature 1996, 380, 64–66. [Google Scholar] [CrossRef] [PubMed]
- Wilmut, I.; Schnieke, A.E.; McWhir, J.; Kind, A.J.; Campbell, K.H.S. Viable Offspring Derived from Fetal and Adult Mammalian Cells. Nature 1997, 385, 810–813. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Briggs, R.; King, T.J. Transplantation of Living Nuclei from Blastula Cells into Enucleated Frogs′ Eggs. Proc. Natl. Acad. Sci. USA 1952, 38, 455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Briggs, R.; King, T.J. Changes in the Nuclei of Differentiating Gastrula Cells as Revealed by Nuclear Transplantation. Proc. Natl. Acad. Sci. USA 1955, 41, 321–325. [Google Scholar]
- Gurdon, J.B. The Developmental Capacity of Nuclei Taken from Intestinal Epithelium Cells of Feeding Tadpoles. J. Embryol. Exp. Morphol. 1962, 10, 20. [Google Scholar]
- Gurdon, J.B.; Uehlinger, V. “Fertile” Intestine Nuclei. Nature 1966, 210, 1240–1241. [Google Scholar] [CrossRef] [PubMed]
- Cibelli, J.B.; Wilmut, I.; Jaenish, R.; Gurdon, J.B.; Lanza, R. Principles of Cloning, 2nd ed.; Cibelli, J., Wilmut, I.S., Jaenisch, R., Gurdon, J., Lanza, R., West, M., Campbell, K.H., Eds.; Academic Press: Cambridge, MA, USA, 2013. [Google Scholar]
- Tada, M.; Takahama, Y.; Abe, K.; Nakatsuji, N.; Tada, T. Nuclear Reprogramming of Somatic Cells by in Vitro Hybridization with ES Cells. Curr. Biol. 2001, 11, 1553–1558. [Google Scholar] [CrossRef] [Green Version]
- Miller, R.A.; Ruddle, F.H. Pluripotent Teratocarcinoma-Thymus Somatic Cell Hybrids. Cell 1976, 9, 45–55. [Google Scholar] [CrossRef]
- Clarke, D.L. Generalized Potential of Adult Neural Stem Cells. Science 2000, 288, 1660–1663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collas, P.; Taranger, C.K.; Boquest, A.C.; Noer, A.; Dahl, J.A. On the Way to Reprogramming Cells to Pluripotency Using Cell-Free Extracts. Reprod. Biomed. Online 2006, 12, 762–770. [Google Scholar] [CrossRef]
- Maherali, N.; Sridharan, R.; Xie, W.; Utikal, J.; Eminli, S.; Arnold, K.; Stadtfeld, M.; Yachechko, R.; Tchieu, J.; Jaenisch, R.; et al. Directly Reprogrammed Fibroblasts Show Global Epigenetic Remodeling and Widespread Tissue Contribution. Cell Stem Cell 2007, 1, 55–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okita, K.; Nakagawa, M.; Hyenjong, H.; Ichisaka, T.; Yamanaka, S. Generation of Mouse Induced Pluripotent Stem Cells Without Viral Vectors. Science 2008, 322, 949–953. [Google Scholar] [CrossRef] [PubMed]
- Wernig, M.; Meissner, A.; Foreman, R.; Brambrink, T.; Ku, M.; Hochedlinger, K.; Bernstein, B.E.; Jaenisch, R. In Vitro Reprogramming of Fibroblasts into a Pluripotent ES-Cell-like State. Nature 2007, 448, 318–324. [Google Scholar] [CrossRef]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced Pluripotent Stem Cell Lines Derived from Human Somatic Cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef]
- Bradley, A.; Evans, M.; Kaufman, M.H.; Robertson, E. Formation of Germ Line Chimaeras from Embryo-Derived Teratocarcinoma Cell Lines. Nature 1984, 309, 255–256. [Google Scholar] [CrossRef]
- Beddington, R.S. Induction of a Second Neural Axis by the Mouse Node. Development 1994, 120, 613–620. [Google Scholar]
- Nagy, A.; Gocza, E.; Diaz, E.M.; Prideaux, P.; Ivanyi, E.; Markkl’La, M.; Rossant, J. Embryonic Stem Cells Alone Are Able to Support Fetal Development in the Mouse. Development 1990, 110, 815–821. [Google Scholar]
- Modliński, J.A.; Reed, M.A.; Wagner, T.E.; Karasiewicz, J. Embryonic Stem Cells: Developmental Capabilities and Their Possible Use in Mammalian Embryo Cloning. Anim. Reprod. Sci. 1996, 42, 437–446. [Google Scholar] [CrossRef]
- Karasiewicz, J.; Sacharczuk, M.; Was, B.; Guszkiewicz, A.; Korwin-Kossakowski, M.; Gorniewska, M.; Szablisty, E.; Modlinski, J.A. Experimental Embryonic-Somatic Chimaerism in the Sheep Confirmed by Random Amplified Polymorphic DNA Assay. Int. J. Dev. Biol. 2008, 52, 315–322. [Google Scholar] [CrossRef]
- Geiger, H.; Sick, S.; Bonifer, C.; Müller, A.M. Globin Gene Expression Is Reprogrammed in Chimeras Generated by Injecting Adult Hematopoietic Stem Cells into Mouse Blastocysts. Cell 1998, 93, 1055–1065. [Google Scholar] [CrossRef] [Green Version]
- Piliszek, A.; Modliński, J.A.; Pyśniak, K.; Karasiewicz, J. Foetal Fibroblasts Introduced to Cleaving Mouse Embryos Contribute to Full-Term Development. Reproduction 2007, 133, 207–218. [Google Scholar] [CrossRef] [Green Version]
- Hamilton, T.G.; Klinghoffer, R.A.; Corrin, P.D.; Soriano, P. Evolutionary Divergence of Platelet-Derived Growth Factor Alpha Receptor Signaling Mechanisms. Mol. Cell Biol. 2003, 23, 4013–4025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhee, J.M.; Pirity, M.K.; Lackan, C.S.; Long, J.Z.; Kondoh, G.; Takeda, J.; Hadjantonakis, A.-K. In Vivo Imaging and Differential Localization of Lipid-Modified GFP-Variant Fusions in Embryonic Stem Cells and Mice. Genesis 2006, 44, 202–218. [Google Scholar] [CrossRef] [Green Version]
- Hadjantonakis, A.K.; Papaioannou, V.E. Dynamic in Vivo Imaging and Cell Tracking Using a Histone Fluorescent Protein Fusion in Mice. BMC Biotechnol. 2004, 4, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, J.Z.; Lackan, C.S.; Hadjantonakis, A.K. Genetic and Spectrally Distinct in Vivo Imaging: Embryonic Stem Cells and Mice with Widespread Expression of a Monomeric Red Fluorescent Protein. BMC Biotechnol. 2005, 5, 20. [Google Scholar] [CrossRef] [Green Version]
- Looijenga, L.H.J.; Stoop, H.; de Leeuw, H.P.J.C.; de Gouveia Brazao, C.A.; Gillis, A.J.M.; van Roozendaal, K.E.P.; van Zoelen, E.J.J.; Weber, R.F.A.; Wolffenbuttel, K.P.; van Dekken, H.; et al. POU5F1 (OCT3/4) Identifies Cells with Pluripotent Potential in Human Germ Cell Tumors. Cancer Res. 2003, 63, 2244–2250. [Google Scholar]
- Zheng, X.; Hu, G. Oct4GiP Reporter Assay to Study Genes That Regulate Mouse Embryonic Stem Cell Maintenance and Self-Renewal. J. Vis. Exp. 2012, 3987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vintersten, K.; Monetti, C.; Gertsenstein, M.; Zhang, P.; Laszlo, L.; Biechele, S.; Nagy, A. Mouse in Red: Red Fluorescent Protein Expression in Mouse ES Cells, Embryos, and Adult Animals. Genesis 2004, 40, 241–246. [Google Scholar] [CrossRef] [PubMed]
- Wakayama, T.; Perry, A.C.F.; Zuccotti, M.; Johnson, K.R.; Yanagimachi, R. Full-Term Development of Mice from Enucleated Oocytes Injected with Cumulus Cell Nuclei. Nature 1998, 394, 369–374. [Google Scholar] [CrossRef] [PubMed]
- Stewart, C.L. Production of Chimeras between Embryonic Stem Cells and Embryos. Methods Enzymol. 1993, 225, 823–855. [Google Scholar] [CrossRef] [PubMed]
- Hart, C.Y.T.; Burnett, J.C.; Redfield, M.M. Effects of Avertin versus Xylazine-Ketamine Anesthesia on Cardiac Function in Normal Mice. Am. J. Physiol. Circ. Physiol. 2001, 281, H1938–H1945. [Google Scholar] [CrossRef]
- Kaufman, M.H. The Atlas of Mouse Development; Academic Press Limited: London, UK, 1992. [Google Scholar]
- Beck, F.; Erler, T.; Russell, A.; James, R. Expression of Cdx-2 in the Mouse Embryo and Placenta: Possible Role in Patterning of the Extra-Embryonic Membranes. Dev. Dyn. 1995, 204, 219–227. [Google Scholar] [CrossRef]
- Strumpf, D. Cdx2 Is Required for Correct Cell Fate Specification and Differentiation of Trophectoderm in the Mouse Blastocyst. Development 2005, 132, 2093–2102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurimoto, K.; Yabuta, Y.; Ohinata, Y.; Ono, Y.; Uno, K.D.; Yamada, R.G.; Ueda, H.R.; Saitou, M. An Improved Single-Cell CDNA Amplification Method for Efficient High-Density Oligonucleotide Microarray Analysis. Nucleic Acids Res. 2006, 34, 17. [Google Scholar] [CrossRef] [Green Version]
- Plusa, B.; Piliszek, A.; Frankenberg, S.; Artus, J.; Hadjantonakis, A.-K. Distinct Sequential Cell Behaviours Direct Primitive Endoderm Formation in the Mouse Blastocyst. Development 2008, 135, 3081–3091. [Google Scholar] [CrossRef] [Green Version]
- Chambers, I.; Colby, D.; Robertson, M.; Nichols, J.; Lee, S.; Tweedie, S.; Smith, A. Functional Expression Cloning of Nanog, a Pluripotency Sustaining Factor in Embryonic Stem Cells. Cell 2003, 113, 643–655. [Google Scholar] [CrossRef] [Green Version]
- Mitsui, K.; Tokuzawa, Y.; Itoh, H.; Segawa, K.; Murakami, M.; Takahashi, K.; Maruyama, M.; Maeda, M.; Yamanaka, S. The Homeoprotein Nanog Is Required for Maintenance of Pluripotency in Mouse Epiblast and ES Cells. Cell 2003, 113, 631–642. [Google Scholar] [CrossRef] [Green Version]
- Scott, E.W. Stem Cell Plasticity or Fusion: Two Approaches to Targeted Cell Therapy. Blood Cells Mol. Dis. 2004, 32, 65–67. [Google Scholar] [CrossRef]
- Wurmser, A.E.; Nakashima, K.; Summers, R.G.; Toni, N.; D’Amour, K.A.; Lie, D.C.; Gage, F.H. Cell Fusion-Independent Differentiation of Neural Stem Cells to the Endothelial Lineage. Nature 2004, 430, 350–356. [Google Scholar] [CrossRef]
- Saburi, S.; Sato, E.; Azuma, S.; Toyoda, Y.; Tachi, C. Developmental Fate of Single Embryonic Stem Cells Microinjected into 8-Cell-Stage Mouse Embryos. Differentiation 1997, 62, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Żyżyńska-Galeńska, K.; Piliszek, A.; Modliński, J.A. From Blastomeres to Somatic Cells: Reflections on Cell Developmental Potential in Light of Chimaera Studies—A Review. Anim. Sci. Pap. Rep. 2017, 35, 225–240. [Google Scholar]
- Suwińska, A.; Czołowska, R.; Ozdzeński, W.; Tarkowski, A.K. Blastomeres of the Mouse Embryo Lose Totipotency after the Fifth Cleavage Division: Expression of Cdx2 and Oct4 and Developmental Potential of Inner and Outer Blastomeres of 16- and 32-Cell Embryos. Dev. Biol. 2008, 322, 133–144. [Google Scholar] [CrossRef]
- Rossant, J.; Vijh, K.M. Ability of Outside Cells from Preimplantation Mouse Embryos to Form Inner Cell Mass Derivatives. Dev. Biol. 1980, 76, 475–482. [Google Scholar] [CrossRef]
- Grabarek, J.B.; Zyzynska, K.; Saiz, N.; Piliszek, A.; Frankenberg, S.; Nichols, J.; Hadjantonakis, A.-K.; Plusa, B. Differential Plasticity of Epiblast and Primitive Endoderm Precursors within the ICM of the Early Mouse Embryo. Development 2012, 139, 129–139. [Google Scholar] [CrossRef] [Green Version]
- Moustafa, L.A.; Brinster, R.L. The Fate of Transplanted Cells in Mouse Blastocysts in Vitro. J. Exp. Zool. 1972, 181, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Gardner, R.L.; Lyon, M.F.; Evans, E.P.; Burtenshaw, M.D. Clonal Analysis of X-Chromosome Inactivation and the Origin of the Germ Line in the Mouse Embryo. J. Embryol. Exp. Morphol. 1985, 88, 349–363. [Google Scholar] [PubMed]
- Schor, S.L.; Schor, A.M.; Rushton, G.; Smith, L. Adult, Foetal and Transformed Fibroblasts Display Different Migratory Phenotypes on Collagen Gels: Evidence for an Isoformic Transition during Foetal Development. J. Cell Sci. 1985, 73, 221–234. [Google Scholar]
- Tang, Q.-M.; Chen, J.L.; Shen, W.L.; Yin, Z.; Liu, H.H.; Fang, Z.; Heng, B.C.; Ouyang, H.W.; Chen, X. Fetal and Adult Fibroblasts Display Intrinsic Differences in Tendon Tissue Engineering and Regeneration. Sci. Rep. 2015, 4, 5515. [Google Scholar] [CrossRef] [Green Version]
- Broker, B.J.; Chakrabarti, R.; Blynman, T.; Roesler, J.; Wang, M.B.; Srivatsan, E.S. Comparison of Growth Factor Expression in Fetal and Adult Fibroblasts: A Preliminary Report. Arch. Otolaryngol. Head Neck Surg. 1999, 125, 676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masaki, H.; Kato-Itoh, M.; Takahashi, Y.; Umino, A.; Sato, H.; Ito, K.; Yanagida, A.; Nishimura, T.; Yamaguchi, T.; Hirabayashi, M.; et al. Inhibition of Apoptosis Overcomes Stage-Related Compatibility Barriers to Chimera Formation in Mouse Embryos. Cell Stem Cell 2016, 19, 587–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ton-That, H.; Kaestner, K.H.; Shields, J.M.; Mahatanankoon, C.S.; Yang, V.W. Expression of the Gut-Enriched Krüppel-like Factor Gene during Development and Intestinal Tumorigenesis. FEBS Lett. 1997, 419, 239–243. [Google Scholar] [CrossRef] [Green Version]
- Wei, Z.; Yang, Y.; Zhang, P.; Andrianakos, R.; Hasegawa, K.; Lyu, J.; Chen, X.; Bai, G.; Liu, C.; Pera, M.; et al. Klf4 Directly Interacts with Oct4 and Sox2 to Promote Reprogramming. Stem Cells 2009, 27, 2969–2978. [Google Scholar] [CrossRef] [PubMed]
- Hagos, E.G.; Ghaleb, A.M.; Dalton, W.B.; Bialkowska, A.B.; Yang, V.W. Mouse Embryonic Fibroblasts Null for the Krüppel-like Factor 4 Gene Are Genetically Unstable. Oncogene 2009, 28, 1197–1205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Desponts, C.; Do, J.T.; Hahm, H.S.; Schöler, H.R.; Ding, S. Induction of Pluripotent Stem Cells from Mouse Embryonic Fibroblasts by Oct4 and Klf4 with Small-Molecule Compounds. Cell Stem Cell 2008, 3, 568–574. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.B.; Sebastiano, V.; Wu, G.; Araúzo-Bravo, M.; Sasse, P.; Gentile, L.; Ko, K.; Ruau, D.; Ehrich, M.; van den Boom, D.; et al. Oct4-Induced Pluripotency in Adult Neural Stem Cells. Cell 2009, 136, 411–419. [Google Scholar] [CrossRef]
- Ho, P.J.; Yen, M.L.; Lin, J.-D.; Chen, L.-S.; Hu, H.-I.; Yeh, C.-K.; Peng, C.-Y.; Lin, C.-Y.; Yet, S.-F.; Yen, B.L. Endogenous KLF4 Expression in Human Fetal Endothelial Cells Allows for Reprogramming to Pluripotency With Just OCT3/4 and SOX2—Brief Report. Arter. Thromb. Vasc. Biol. 2010, 30, 1905–1907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ter Huurne, M.; Chappell, J.; Dalton, S.; Stunnenberg, H.G. Distinct Cell-Cycle Control in Two Different States of Mouse Pluripotency. Cell Stem Cell 2017, 21, 449–455. [Google Scholar] [CrossRef] [Green Version]
- Aksoy, I.; Rognard, C.; Moulin, A.; Marcy, G.; Masfaraud, E.; Wianny, F.; Cortay, V.; Bellemin-Ménard, A.; Doerflinger, N.; Dirheimer, M.; et al. Apoptosis, G1 Phase Stall, and Premature Differentiation Account for Low Chimeric Competence of Human and Rhesus Monkey Naive Pluripotent Stem Cells. Stem Cell Rep. 2021, 16, 56–74. [Google Scholar] [CrossRef] [PubMed]
- Håkelien, A.-M.; Collas, P. Novel Approaches to Transdifferentiation. Cloning Stem Cells 2002, 4, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Taranger, C.K.; Noer, A.; Sørensen, A.L.; Håkelien, A.-M.; Boquest, A.C.; Collas, P. Induction of Dedifferentiation, Genomewide Transcriptional Programming, and Epigenetic Reprogramming by Extracts of Carcinoma and Embryonic Stem Cells. Mol. Biol. Cell 2005, 16, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collas, P.; Taranger, C.K. Epigenetic Reprogramming of Nuclei Using Cell Extracts. Stem Cell Rev. 2006, 2, 309–317. [Google Scholar] [CrossRef]
- Eakin, G.S.; Hadjantonakis, A.-K.; Papaioannou, V.E.; Behringer, R.R. Developmental Potential and Behavior of Tetraploid Cells in the Mouse Embryo. Dev. Biol. 2005, 288, 150–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Żyżyńska-Galeńska, K.; Bernat, A.; Piliszek, A.; Karasiewicz, J.; Szablisty, E.; Sacharczuk, M.; Brewińska-Olchowik, M.; Bochenek, M.; Grabarek, J.; Modliński, J.A. Embryonic Environmental Niche Reprograms Somatic Cells to Express Pluripotency Markers and Participate in Adult Chimaeras. Cells 2021, 10, 490. https://doi.org/10.3390/cells10030490
Żyżyńska-Galeńska K, Bernat A, Piliszek A, Karasiewicz J, Szablisty E, Sacharczuk M, Brewińska-Olchowik M, Bochenek M, Grabarek J, Modliński JA. Embryonic Environmental Niche Reprograms Somatic Cells to Express Pluripotency Markers and Participate in Adult Chimaeras. Cells. 2021; 10(3):490. https://doi.org/10.3390/cells10030490
Chicago/Turabian StyleŻyżyńska-Galeńska, Krystyna, Agnieszka Bernat, Anna Piliszek, Jolanta Karasiewicz, Ewa Szablisty, Mariusz Sacharczuk, Marta Brewińska-Olchowik, Michał Bochenek, Joanna Grabarek, and Jacek Andrzej Modliński. 2021. "Embryonic Environmental Niche Reprograms Somatic Cells to Express Pluripotency Markers and Participate in Adult Chimaeras" Cells 10, no. 3: 490. https://doi.org/10.3390/cells10030490