
Genome-Wide Identification of U-To-C RNA Editing Events for Nuclear Genes in Arabidopsis thaliana




Abstract
:
1. Introduction
2. Materials and Methods
2.1. Plant Growth Conditions and Sample Collection
2.2. RNA Extraction and cDNA Synthesis
2.3. Library Preparation for Transcriptome Sequencing
2.3.1. Clustering and Sequencing
2.3.2. Data Analysis
2.4. Sanger Sequencing
3. Results
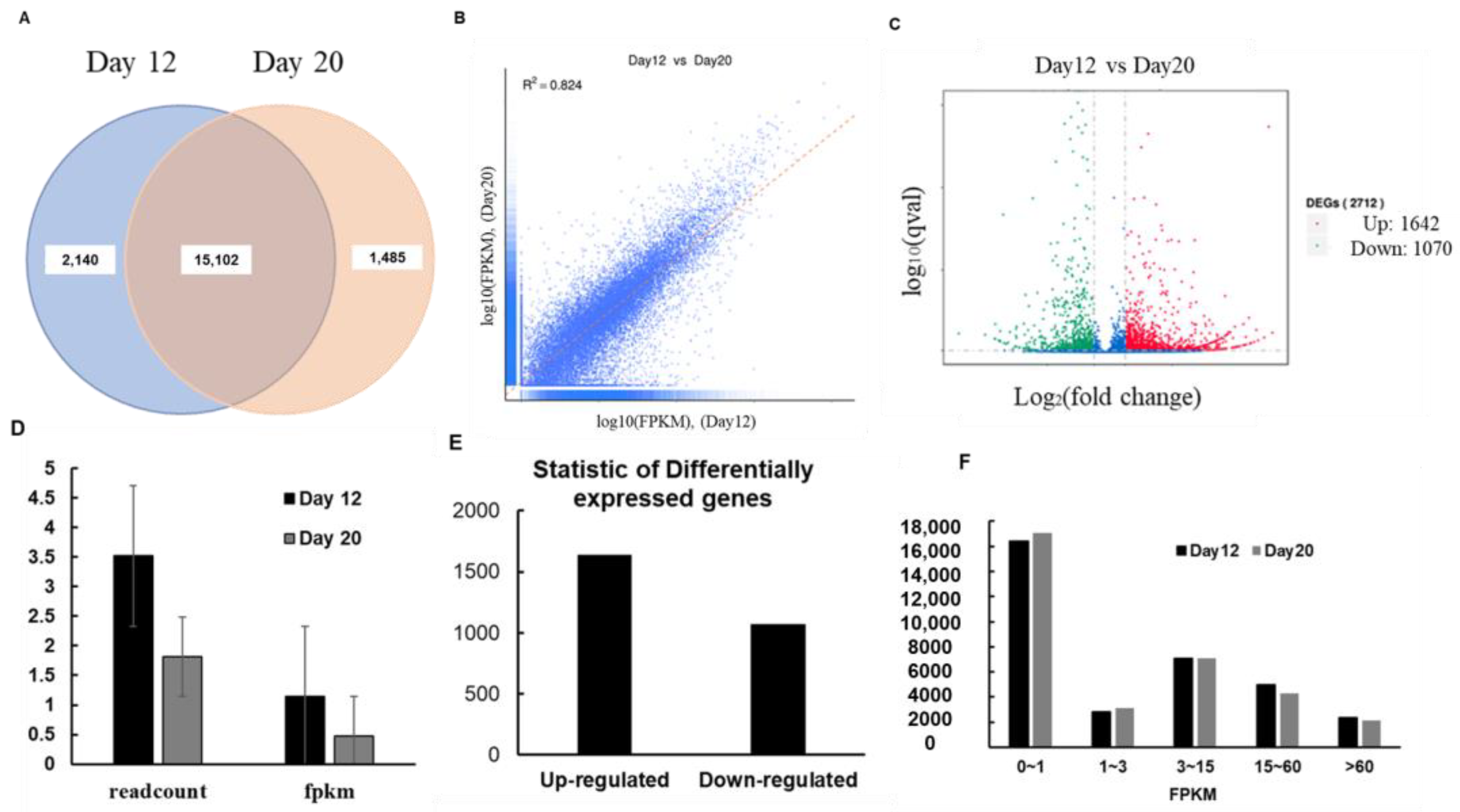
3.1. Identification and Analysis of DEGs by RNA-Seq
3.2. Comparison of Nucleotide Differences between Genomic DNA in Database and RNA-Seq of 12- or 20-D-Old Seedlings
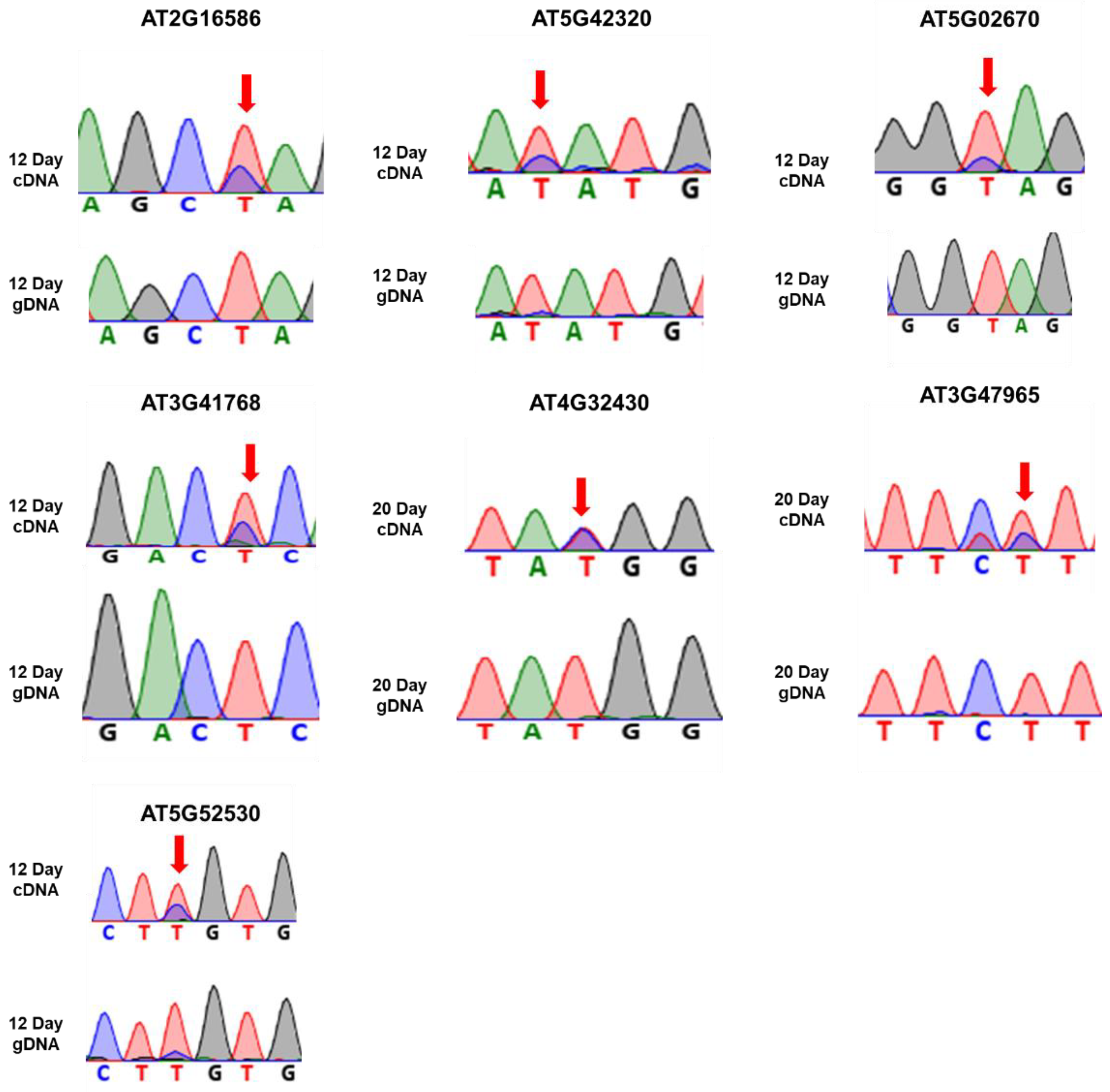
3.3. Identification of Genes Harboring U-To-C RNA Editing Site
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Benne, R. RNA-editing in trypanosome mitochondria. Biochim. Biophys. Acta Gene Struct. Expr. 1989, 1007, 131–139. [Google Scholar] [CrossRef]
- Chen, S.; Habib, G.; Yang, C.; Gu, Z.; Lee, B.; Weng, S.; Silberman, S.R.; Cai, S.; Deslypere, J.; Rosseneu, M. Apolipoprotein B-48 is the product of a messenger RNA with an organ-specific in-frame stop codon. Science 1987, 238, 363–366. [Google Scholar] [CrossRef]
- Walkley, C.R.; Li, J.B. Rewriting the transcriptome: Adenosine-to-inosine RNA editing by ADARs. Genome Biol. 2017, 18, 205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Wang, Q.; He, Y.; Chen, L.; Hao, C.; Jiang, C.; Li, Y.; Dai, Y.; Kang, Z.; Xu, J.R. Genome-wide A-to-I RNA editing in fungi independent of ADAR enzymes. Genome Res. 2016, 26, 499–509. [Google Scholar] [CrossRef] [Green Version]
- Wolf, J.; Gerber, A.P.; Keller, W. tadA, an essential tRNA-specific adenosine deaminase from Escherichia coli. EMBO J. 2002, 21, 3841–3851. [Google Scholar] [CrossRef] [Green Version]
- Bar-Yaacov, D.; Pilpel, Y.; Dahan, O. RNA editing in bacteria: Occurrence, regulation and significance. RNA Biol. 2018, 15, 863–867. [Google Scholar] [CrossRef] [Green Version]
- Thomas, S.M.; Lamb, R.A.; Paterson, R.G. Two mRNAs that differ by two nontemplated nucleotides encode the amino coterminal proteins P and V of the paramyxovirus SV5. Cell 1988, 54, 891–902. [Google Scholar] [CrossRef]
- Cattaneo, R.; Kaelin, K.; Baczko, K.; Billeter, M.A. Measles virus editing provides an additional cysteine-rich protein. Cell 1989, 56, 759–764. [Google Scholar] [CrossRef]
- Covello, P.S.; Gray, M.W. RNA editing in plant mitochondria. Nature 1989, 341, 662–666. [Google Scholar] [CrossRef]
- Gualberto, J.M.; Lamattina, L.; Bonnard, G.; Weil, J.H.; Grienenberger, J.M. RNA editing in wheat mitochondria results in the conservation of protein sequences. Nature 1989, 341, 660–662. [Google Scholar] [CrossRef]
- Hiesel, R.; Wissinger, B.; Schuster, W.; Brennicke, A. RNA editing in plant mitochondria. Science 1989, 246, 1632–1634. [Google Scholar] [CrossRef] [PubMed]
- Barkan, A.; Small, I. Pentatricopeptide repeat proteins in plants. Annu. Rev. Plant Biol. 2014, 65, 415–442. [Google Scholar] [CrossRef]
- Lurin, C.; Andrés, C.; Aubourg, S.; Bellaoui, M.; Bitton, F.; Bruyère, C.; Caboche, M.; Debast, C.; Gualberto, J.; Hoffmann, B.; et al. Small Genome-wide analysis of Arabidopsis pentatricopeptide repeat proteins reveals their essential role in organelle biogenesis. Plant Cell 2004, 16, 2089–2103. [Google Scholar] [CrossRef] [Green Version]
- Chateigner-Boutin, A.L.; Hanson, M.R. Developmental co-variation of RNA editing extent of plastid editing sites exhibiting similar cis-elements. Nucleic Acids Res. 2003, 31, 2586–2594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schallenberg-Rüdinger, M.; Kindgren, P.; Zehrmann, A.; Small, I.; Knoop, V. A DYW-protein knockout in Physcomitrella affects two closely spaced mitochondrial editing sites and causes a severe developmental phenotype. Plant J. 2013, 76, 420–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leu, K.C.; Hsieh, M.H.; Wang, H.J.; Hsieh, H.L.; Jauh, G.Y. Distinct role of Arabidopsis mitochondrial P-type pentatricopeptide repeat protein-modulating editing protein, PPME, in nad1 RNA editing. RNA Biol. 2016, 13, 593–604. [Google Scholar] [CrossRef] [Green Version]
- Sun, T.; Bentolila, S.; Hanson, M.R. The unexpected diversity of plant organelle RNA editosomes. Trends Plant Sci. 2016, 21, 962–973. [Google Scholar] [CrossRef] [Green Version]
- Bayer-Császár, E.; Haag, S.; Jörg, A.; Glass, F.; Härtel, B.; Obata, T.; Meyer, E.H.; Brennicke, A.; Takenaka, M. The conserved domain in MORF proteins has distinct affinities to the PPR and E elements in PPR RNA editing factors. Biochim. Biophys. Acta Gene Regul. Mech. 2017, 1860, 813–828. [Google Scholar] [CrossRef]
- Schallenberg-Rüdinger, M.; Knoop, V. Coevolution of organelle RNA editing and nuclear specificity factors in early land plants. In Genomes and Evolution of Charophytes, Bryophytes and Ferns. Advances in Botanical Research; Rensing, S.A., Ed.; Elsevier Academic Press: Amsterdam, The Netherlands, 2016; Volume 78. [Google Scholar]
- Gerke, P.; Szövényi, P.; Neubauer, A.; Lenz, H.; Gutmann, B.; McDowell, R.; Small, I.; Schallenberg-Rüdinger, M.; Knoop, V. Towards a Plant Model for Enigmatic U-to-C RNA Editing: The Organelle Genomes, Transcriptomes, Editomes and Candidate RNA Editing Factors in the Hornwort Anthoceros Agrestis. New Phytologist 2020, 225, 1974–1992. [Google Scholar] [CrossRef] [Green Version]
- Oldenkott, B.; Yang, Y.; Lesch, E.; Knoop, V.; Schallenberg-Rüdinger, M. Plant-type pentatricopeptide repeat proteins with a DYW domain drive C-to-U RNA editing in Escherichia coli. Commun. Biol. 2019, 2, 85. [Google Scholar] [CrossRef]
- Gutmann, B.; Royan, S.; Schallenberg-Rüdinger, M.; Lenz, H.; Castleden, I.R.; McDowell, R.; Vacher, M.A.; Tonti-Filippini, J.; Bond, C.S.; Knoop, V.; et al. Small, the expansion and diversification of pentatricopeptide repeat RNA-editing factors in plants. Mol. Plant 2019. [Google Scholar] [CrossRef]
- Qulsum, U.; Azad, T.A.; Tsukahara, T. Analysis of tissue-specific RNA editing events of genes involved in RNA editing in Arabidopsis thaliana. J. Plant Biol. 2019, 62, 351–358. [Google Scholar] [CrossRef]
- Qulsum, U.; Tsukahara, T. Tissue-specific alternative splicing of pentatricopeptide repeat (PPR) family genes in Arabidopsis thaliana. Biosci. Trends 2019, 12, 569–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramaswami, G.; Zhang, R.; Piskol, R.; Keegan, L.P.; Deng, P.; O’Connell, M.A.; Li, J.B. Identifying RNA editing sites using RNA sequencing data alone. Nat. Methods 2013, 10, 128–132. [Google Scholar] [CrossRef] [PubMed]
- Yang, E.; van Nimwegen, E.; Zavolan, M.; Rajewsky, N.; Schroeder, M.; Magnasco, M.; Darnell, J.E., Jr. Decay rates of human mRNAs: Correlation with functional characteristics and sequence attributes. Genome Res. 2003, 13, 1863–1872. [Google Scholar]
- Drescher, A.; Hupfer, H.; Nickel, C.; Albertazzi, F.; Hohmann, U.; Herrmann, R. Maier R C-to-U conversion in the intercistronic ndhI/ndhG RNA of plastids from monocot plants: Conventional editing in an unconventional small reading frame? Mol. Genet. Genom. 2002, 267, 262–269. [Google Scholar] [CrossRef]
- Farré, J.; Aknin, C. Alejandro araya BC RNA editing in mitochondrial for splicing Trans -Introns is required for splicing. PLoS ONE 2012, 7, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Wu, B.; Chen, H.; Shao, J.; Zhang, H.; Wu, K.; Liu, C. Identification of symmetrical RNA editing events in the mitochondria of Salvia miltiorrhiza by strand-specific RNA sequencing. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef]
- Meng, Y.; Chen, D.; Jin, Y.; Mao, C.; Wu, P.; Chen, M. RNA editing of nuclear transcripts in Arabidopsis thaliana. BMC Genom. 2010, 11, 1–7. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S.No. | Position | Reads | Gene ID | Description | |
|---|---|---|---|---|---|
| 12 Days | 20 Days | ||||
| 1 | 3412532 | 56 | 0 | AT2G07715 | Ribosomal Proteins L2, RNA binding domain |
| 2 | 8544440 | 34 | 0 | AT4G14940 | Amine oxidase |
| 3 | 26898977 | 2 | 0 | AT5G67411 | GRAS family transcription factor |
| 4 | 8297931 | 4 | 0 | AT1G23380 | KNOTTED1-like homeobox gene 6 |
| 5 | 14657330 | 14 | 0 | AT4G29950 | Ypt/Rab-GAP domain of gyp1p superfamily protein |
| 6 | 3392826 | 107 | 16 | AT2G07709 | - |
| 7 | 362386 | 175 | 44 | ATMG01390 | - |
| 8 | 7191444 | 105 | 197 | AT2G16586 | Unknown |
| 9 | 3061212 | 498 | 2 | AT4G06477 | - |
| 10 | 9226791 | 28 | 69 | AT4G16330 | 2-oxoglutarate (2OG) and Fe(II)-dependent oxygenase superfamily protein |
| 11 | 5816271 | 12 | 6 | AT3G17050 | - |
| 12 | 9255546 | 268 | 99 | AT4G16380 | Heavy metal transport/detoxification superfamily protein |
| 13 | 14198871 | 647 | 240 | AT3G41768 | - |
| 14 | 16918673 | 55 | 46 | AT5G42320 | Zn-dependent exopeptidases superfamily protein |
| 15 | 17708862 | 0 | 21 | AT3G47965 | Unknown |
| 16 | 24989428 | 27 | 0 | AT5G62220 | glycosyltransferase 18 |
| 17 | 21320395 | 0 | 12 | AT5G52530 | dentin sialophosphoprotein-related |
| 18 | 2848835 | 146 | 86 | AT5G08740 | NAD(P)H dehydrogenase C1 |
| 19 | 15546833 | 13 | 0 | AT4G32190 | Myosin heavy chain-related protein |
| 20 | 3392918 | 144 | 14 | AT2G07709 | - |
| 21 | 21319578 | 0 | 5 | AT5G52530 | dentin sialophosphoprotein-related |
| 22 | 21077241 | 0 | 2 | AT1G56290 | CwfJ-like family protein |
| 23 | 7622202 | 0 | 2 | AT4G13070 | RNA-binding CRS1/YhbY (CRM) domain protein |
| 24 | 17692876 | 29 | 0 | AT5G43970 | translocase of outer membrane 22-V |
| 25 | 10266697 | 46 | 6 | AT1G29340 | plant U-box 17 |
| 26 | 7869982 | 17 | 0 | AT5G23380 | Protein of unknown function (DUF789) |
| 27 | 7836325 | 19 | 0 | AT1G22190 | Integrase-type DNA-binding superfamily protein |
| 28 | 19998466 | 36 | 0 | AT3G54000 | Unknown |
| 29 | 603074 | 13 | 5 | AT5G02670 | Unknown |
| 30 | 22561577 | 0 | 2 | AT3G60970 | multidrug resistance-associated protein 15 |
| 31 | 6025041 | 0 | 27 | AT4G09520 | Cofactor-independent phosphoglycerate mutase |
| 32 | 909133 | 0 | 2 | AT4G02070 | MUTS homolog 6 |
| 33 | 7797368 | 0 | 4 | AT4G13420 | high affinity K+ transporter 5 |
| 34 | 8662474 | 0 | 3 | AT4G15180 | SET domain protein 2 |
| 35 | 12669828 | 0 | 2 | AT4G24530 | O-fucosyltransferase family protein |
| 36 | 15653919 | 0 | 2 | AT4G32430 | Pentatricopeptide repeat (PPR) superfamily protein |
| 37 | 5075516 | 0 | 2 | AT2G12490 | - |
| 38 | 17587422 | 0 | 2 | AT2G42200 | squamosa promoter binding protein-like 9 |
| 39 | 17958701 | 0 | 2 | AT2G43200 | S-adenosyl-L-methionine-dependent methyltransferases superfamily protein |
| 40 | 526197 | 0 | 5 | AT3G02515 | - |
| 41 | 20795012 | 69 | 64 | AT3G56040 | UDP-glucose pyrophosphorylase 3 |
| 42 | 3264804 | 0 | 2 | AT5G10370 | helicase domain-containing protein/IBR domain-containing protein/zinc finger protein-related |
| 43 | 9633752 | 0 | 2 | AT5G27330 | Prefoldin chaperone subunit family protein |
| 44 | 12108844 | 0 | 9 | AT5G32481 | - |
| 45 | 15644809 | 0 | 4 | AT5G39090 | HXXXD-type acyl-transferase family protein |
| 46 | 3332097 | 0 | 2 | AT1G10160 | - |
| 47 | 3564739 | 0 | 2 | AT1G10720 | BSD domain-containing protein |
| 48 | 9825469 | 0 | 6 | AT1G28130 | Auxin-responsive GH3 family protein |
| 49 | 9997031 | 0 | 2 | AT1G28440 | HAESA-like 1 |
| 50 | 4006628 | 0 | 13 | AT5G12370 | exocyst complex component sec10 |
| 51 | 5097198 | 0 | 5 | AT2G12505 | - |
| 52 | 11465954 | 0 | 11 | AT1G31930 | extra-large GTP-binding protein 3 |
| 53 | 7014676 | 0 | 2 | AT3G20087 | N/A |
| 54 | 15766171 | 0 | 2 | AT2G37585 | Core-2/I-branching beta-1,6-N-acetylglucosaminyltransferase family protein |
| 55 | 7191297 | 249 | 171 | AT2G16586 | Unknown |
| 56 | 17908527 | 0 | 2 | AT1G48450 | Protein of unknown function (DUF760) |
| S.No. | Position | Edited Site | Gene ID | RNA Editing Efficiency (in %) | Encoded Protein | |
|---|---|---|---|---|---|---|
| 12 Days | 20 Days | |||||
| 1. | 14198871 | 5′ UTR | AT2G16586 | 77.30 | 65.74 | Transmembrane protein |
| 2. | 16918673 | CDS | AT5G42320 | 24.20 | 0 | Zn-dependent exopeptidase superfamily protein |
| 3. | 603074 | 5′ UTR | AT5G02670 | 0 | 22.80 | Hypothetical protein |
| 4. | 7191297 | 3′ UTR | AT3G41768 | 45.54 | 49.65 | Ribosomal RNA |
| 5. | 15653919 | 3′ UTR | AT4G32430 | 0 | 20.43 | PPR-like superfamily protein |
| 6. | 17708862 | 3′ UTR | AT3G47965 | 24.54 | 22.48 | Hypothetical protein |
| 7. | 21320395 | CDS | AT5G52530 | 20.65 | 0 | Dentin sialophosphoprotein-like protein |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruchika; Okudaira, C.; Sakari, M.; Tsukahara, T. Genome-Wide Identification of U-To-C RNA Editing Events for Nuclear Genes in Arabidopsis thaliana. Cells 2021, 10, 635. https://doi.org/10.3390/cells10030635
Ruchika, Okudaira C, Sakari M, Tsukahara T. Genome-Wide Identification of U-To-C RNA Editing Events for Nuclear Genes in Arabidopsis thaliana. Cells. 2021; 10(3):635. https://doi.org/10.3390/cells10030635
Chicago/Turabian StyleRuchika, Chisato Okudaira, Matomo Sakari, and Toshifumi Tsukahara. 2021. "Genome-Wide Identification of U-To-C RNA Editing Events for Nuclear Genes in Arabidopsis thaliana" Cells 10, no. 3: 635. https://doi.org/10.3390/cells10030635
APA StyleRuchika, Okudaira, C., Sakari, M., & Tsukahara, T. (2021). Genome-Wide Identification of U-To-C RNA Editing Events for Nuclear Genes in Arabidopsis thaliana. Cells, 10(3), 635. https://doi.org/10.3390/cells10030635