1. Introduction
Cardiovascular diseases (CVD) resulting in heart failure are the leading cause of mortality worldwide. Currently, CVD poses an economic burden of more than
$215 billion annually in the United States alone which is projected to rise to
$1208 billion each year by 2030 [
1]. Dilated cardiomyopathy (DCM) is one of the most prevalent non-ischemic cardiomyopathies, originating due to multiple causes including mutations in cardiac sarcomeric protein-encoding genes [
2]. Moreover, DCM is the most common lethal form of pediatric cardiomyopathy; often without any known cause [
3]. The hallmark of DCM comprises enlargement of ventricular chambers with successive wall thinning due to cardiomyocyte death followed by fibrosis leading to myocardial dysfunction. This results in systolic pump impairment and arrhythmia leading to congestive heart failure. Global research trials are in progress to reverse such abnormal cardiac remodeling and restore normal heart function post-DCM, although none of these have transitioned into routine clinical use. Expensive and aggressive surgeries like cardiac transplants remain the only viable option to date. Thus, considering the high cost, a limited number of donors, and an ever-increasing demand for organ transplants, there is an immediate and essential need for the development of the least invasive and cost-effective ideal therapy for DCM.
Angiogenesis plays key roles during tissue development and homeostasis and impaired angiogenesis is closely related to several diseases, including CVD [
4]. Preliminary studies in animal models and human patients suggest the key roles played by tissue angiogenesis in pathologic cardiac remodeling during ischemic injury [
5] and DCM [
6,
7,
8,
9]. Hence, angiogenesis intervention strategies have been explored clinically for DCM therapeutics [
10]. Studies with transplant patients with DCM have reported a significant decrease in cardiac angiogenic factors [such as vascular endothelial growth factor A (VEGF-A) and vascular endothelial growth factor receptor 1 (VEGFR1)] expression and lower capillary density [
11], suggesting downregulation of cardiac angiogenesis during DCM phenotype. Promoting tissue angiogenesis during DCM has been shown to have an anti-fibrotic and cardioprotective effect [
12,
13,
14]. Overall, depending on this information, we hypothesized that augmenting angiogenesis in the DCM heart could be a possible novel strategy that could be translated into promising therapy. Earlier reports have shown that microRNAs, a group of small non-coding RNAs, could play key regulatory roles during various physiological and pathological processes, including angiogenesis [
15,
16]. Likewise, our previous study has revealed the beneficial effects of microRNA-210 (miR-210) mediated upregulated angiogenesis during cardiac injury [
16]. Thus, based on reported beneficial effects of miR-210 via promoting cardiac tissue angiogenesis, we raised the question, could miR-210 mediated increase in cardiac angiogenesis modulate the DCM phenotype? To test this notion, we overexpressed miR-210 in DCM mice to improve cardiac angiogenesis to reverse pathologic cardiac remodeling. However, miR-210 mediated upregulated angiogenesis was found to be insufficient to rescue DCM phenotype and heart function in miR-210 overexpressing DCM mice.
2. Materials and Methods
Chemicals and reagents were purchased from Sigma-Aldrich, Saint Louis, MO, USA unless otherwise mentioned. Antibodies used in the current study were obtained from Sigma-Aldrich, MO, USA (Rabbit anti-GAPDH, Cat. G9545; mouse anti-α-smooth muscle actin, Cat. A5228); Santa Cruz Biotechnology, TX, USA (Rabbit anti-cardiac troponin-I, Cat. sc-15368; rabbit anti-VEGF-A, Cat. sc-152); GE Healthcare, IL, USA (ECL anti-rabbit-HRP, Cat. NA9340V); Life Technologies, Carlsbad, CA, USA (Goat anti-mouse Alexa Fluor 488, Cat. A11029; goat anti-mouse Alexa Fluor 594, Cat. A11005; goat anti-rabbit Alexa Fluor 488, Cat. A11008; goat anti-rabbit Alexa Fluor 594, Cat. A11037); VWF (von Willebrand factor) (Dako, Carpinteria, CA, USA, Cat. GA527).
Animals were maintained at Laboratory Animal Medical Services (LAMS), University of Cincinnati, USA. The animals were fed ad libitum standard feed and were utilized per the protocol approved by the Institutional Animal Care and Use Committee (IACUC).
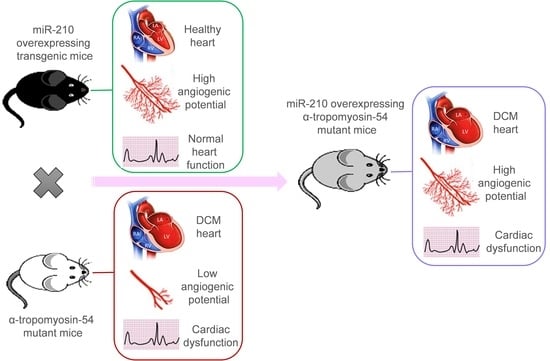
2.1. Dilated Cardiomyopathy (DCM), MicroRNA-210 (MiR-210) Transgenic, and α-TM54 Mutant (TMx210) Mice Models
As reported earlier [
17], here we used a DCM mice model having a point mutation within α-tropomyosin at Glu54Lys (
Figure S1), which is similar to the human counterpart. These DCM mice were produced in the laboratory of Dr. David F. Wieczorek who also donated these mice to us; they were further maintained at the University of Cincinnati animal facility. Also, miR-210 overexpressing (global) transgenic mice (210-TG) were generated at the University of Cincinnati Transgenic core using the C57BL/6 mice strain as mentioned elsewhere [
16] (
Figure S2). Similarly, DCM (white coat) and 210xTG (black coat) mice were crossed to get TMx210 (grey coat) mice.
2.2. Mice Genotyping
Tail tissues from all weaned mice were digested in 100 µL digestion buffer (10 µL 10× polymerase chain reaction (PCR) buffer, 25 mM MgCl
2, 10 µL 10mg/mL proteinase K, 61 µL nuclease-free water, 4.5 µL 10% Tween 20, 4.5 µL 10% NP-40) for 5 h at 60 °C. Samples were boiled for 10 min followed by a 5-s vortex and centrifuged at 10,000 RPM for 5 min. Supernatant was used to set up the Multiplex PCR using the PCR reaction mix and custom-designed primers (5 µL genomic DNA sample, 5 µL 10× PCR buffer, 10 µL Q-solution, 1 µL test gene and Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) forward and reverse primers, 1 µL 10 mM dNTP, 22.5 µL water, 0.5 µL Taq Polymerase) (
Table S2). PCR conditions were set as 4 min 94 °C, 34 cycles of 30 s 94 °C, 30 s 55 °C and 30 s 70 °C, and 10 min 72 °C followed by 25 min 4 °C. PCR product was resolved via 1.5% agarose gel electrophoresis and DNA bands were analyzed under the ultraviolet (UV) gel doc system (BioRad).
2.3. Histological Examination and Immunohistochemistry
Before isolation, hearts were arrested in the diastolic phase with 1 M KCl and perfused and fixed in 10% buffered formalin solution for 48 h at room temperature (RT). Tissue samples were fixed in paraffin blocks and 5 micron thick sections were cut using a microtome. Formalin-fixed tissue slides were de-paraffinized via incubation at 70 °C for 30 min followed by immediately dissolving paraffin in Xylene, and re-hydrating through sequential 5 min incubations in graded ethanol (100%, 90%, and 80%) and water. Antigen retrieval was performed via heating the slides in a pressure cooker in 0.1 M citrate buffer (pH-6.0) for 3 min followed by cooling in the same buffer for 45 min. Tissue sections were 3× washed with PBS (phosphate buffered saline, pH-7.3), blocked with 3% BSA (bovine serum albumin) for 1 hr. Then they were washed with PBS and slides were incubated with the primary antibody in 3% BSA against analyzing antigen for 1–3 h at 37 °C followed by incubation with appropriate secondary antibody linked with Alexa fluor for 1 h at 37 °C. Slides were incubated with DAPI for 10 min, washed with PBS twice, and mounted with Fluoromount-G (Southern Biotech, AL, USA). Slides were examined using a confocal microscope (Olympus). Cell area/size was defined by the staining of cardiac tissue with wheat germ agglutinin (WGA) (Invitrogen, CA, USA). To analyze angiogenesis, blood vessels were stained using anti-smooth muscle actin and anti-troponin I antibodies.
2.4. Echocardiography
Indices of left ventricular function and chamber dimensions during systole and diastole were examined via transthoracic echocardiography (Echo-), performed on one-, three-, and six-month-old mice using a Vevo 2100 Imaging digital ultrasound system (VisualSonics) with a 22–55 MHz (MS550D) transducer probe. Before echocardiography, mice were anesthetized with oxygen mixed with isoflurane (2–2.5%). Mice chest hairs were shaved and then it was placed on a warm platform in the supine position. Data acquisition was initiated with the parasternal cardiac long axis view followed by a transition to the short axis view, at the level of mid-papillary muscles. Measurements were obtained from long-axis M-mode images, and graphically represented as % LVEF (left ventricular ejection fraction), % LVFS (left ventricular fraction shortening), LVID;d (left ventricular internal diameter end diastole) (mm), LVID;s (left ventricular internal diameter end-systole) (mm), LVPW;d (left ventricular posterior wall diastole) (mm), and LVPW;s (left ventricular posterior wall systole) (mm).
2.5. In Silico Analysis
DCM-associated genes were pulled out using the DisGeNET database (
http://www.disgenet.org/search, accessed on 20 August 2020). These genes were further evaluated using Cytoscape 3.6.1 (ClueGo plugin) for signaling pathway analysis. Common genes related to DCM and angiogenesis were filtered and illustrated in the form of gene networks for Gene Ontology (GO): biological process, Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways, and Reactome pathways.
2.6. Tube Formation Assay: Adult Rat Cardiomyocyte (ARCM) Isolation, and Cell Culture
Primary cultures of ventricular cardiomyocytes were obtained from 12 weeks old Fisher rats. Briefly, rats were euthanized, hearts were dissected and perfused using solution A (118 mM NaCl, 4.8 mM KCl, 25 mM HEPES, 1.25 mM MgSO4, 1.25 mM K2HPO4, 10 mM glucose, 4.95 mM taurine, 9.89 mM 2,3-butanedione monoxime pH-7.35). Hearts were then adjusted on the Langendorff system and digested using digestion buffer (solution A with 0.1% BSA, 0.05 mM CaCl2, 0.07% collagenase type II, 0.02% hyaluronidase type I). Then ventricular tissue was taken and minced in digestion buffer. The cell suspension was filtered using a 100-micron cell strainer (BD Biosciences, CA, USA) and the filtrate was centrifuged at 300 rpm for 3 min at RT. The cell pellet was suspended in solution B (solution A with 1% BSA, 0.1 mM CaCl2) and cells were allowed to settle under gravity. The cell pellet was resuspended in Dulbecco’s modified Eagle medium (DMEM)/high glucose media (GE Healthcare, UK) containing 10% fetal bovine serum (FBS) (Fisher Scientific, MA) and 1% Penicillin/Streptomycin (Fisher Scientific, MA, USA) and seeded on culture plates. One day post-seeding, cells were transfected with Caenorhabditis elegans specific miR, cel-miR-67 (50 nM) (Dharmacon, CO, USA) (negative control), and rno-miR-210 mimic (50 nM) (Dharmacon, CO, USA), using Lipofectamine RNAiMAX reagent (Life Technologies, CA, USA). Post 24 h following transfection, media was removed and fresh culture media was added. Cells were cultured for one-week post-transfection and conditioned media was collected. In another parallel setup, seeded HUVEC (human umbilical vein endothelial cells) were cultured for 6–8 h using 50% conditioned media +50% endothelial growth media (Corning, NY, USA) containing 2.5% fetal bovine serum. In a positive control setup, VEGF (Vascular endothelial growth factor; 750 ng/mL medium) and bFGF (basic fibroblast growth factor; 500 ng/mL medium) were added in the HUVEC culture medium. Then, tube formation analysis was performed by evaluating the total tube length, and total branching points.
2.7. RNA Extraction and Quantitative Polymerase Chain Reaction (qPCR)
Total RNA (including miRNAs) was extracted from mice hearts using the PAXgene kit (Qiagen, Cat. 763134) following the manufacturer protocol. The RNA concentration was estimated using nanodrop and purity was checked by A260/280 ratio >1.8. Purified RNA was further used for cDNA synthesis using miScript II RT kit (Qiagen, Cat. 218161) followed by qPCR for miR-210 using custom-designed primers (
Table S3), miScript Primer assay kit (Qiagen, Cat. 218300), and miScript SYBER Green PCR kit (Qiagen, Cat. 218075) on a Bio-Rad Real-Time PCR Detection System. Results were normalized with an internal control (such as miR-U6) and analyzed via the comparative C
t method.
2.8. Aortic Ring Assay
To study angiogenic potential in mice groups, an aortic ring assay was performed according to a published protocol [
18] with minor changes. In short, six-month-old mice were euthanized, and aorta, located along with the spine, were dissected, cleaned in Opti-MEM media, and cut into rings of 1 mm width. Rings were serum-starved overnight in Opti-MEM at 37 °C and 5% CO
2 followed by embedding in Matrigel matrix (Sigma, MO, USA). Rings were fed with 199 media (Sigma-Aldrich, MO, USA) containing 2.5% FBS, 1% Penicillin/Streptomycin. Post one week, rings were fixed with 4% paraformaldehyde in PBS for 20 min followed by 3× washes with PBS. Under bright field imaging, neovascularization area and number of microvessel sprouts were counted in each aortic ring.
2.9. Tissue Lysate Preparation, Sodium Dodecyl Sulfate (SDS) Gel Electrophoresis, and Western Blotting
Mice hearts were collected, perfused with PBS, and homogenized in Laemmli buffer with β-mercaptoethanol followed by immediate boiling in a hot water bath for 20 min. Subsequently, the lysate was centrifuged at 12,000× g for 30 min at 4 °C and the supernatant was stored at −80 °C until use. Protein content was estimated using the Pierce 660 nm reagent kit (Thermo Scientific, Waltham, MA, USA). Linear slab gel electrophoresis under the denaturing condition (in the presence of 0.1% sodium dodecyl sulfate (SDS)) was performed. Protein samples were electrophoresed using 10–12% polyacrylamide gel (Invitrogen, CA, USA) and running buffer (containing 0.025 M Tris-base, 0.192 M glycine pH 8.3, and 0.1% SDS) in Novex apparatus (Life Technologies, CA, USA). The standard molecular weight marker was electrophoresed alongside to observe the subunit size of the analyzed protein. Electrophoresed protein samples were electroblotted onto PVDF membrane by applying 0.8 mA/h current in a wet transfer unit (Bio-Rad, Hercules, CA, USA), and were detected using rabbit/mouse primary antibody and anti-rabbit/mouse conjugated horseradish peroxidase (HRP) second antibody. Blots were incubated for 2–3 min with SuperSignal W. Femto max sensitivity (Thermo Fisher, MA, USA) enhanced chemiluminescence (ECL) substrate. Blots were kept in a cassette; a photographic film was exposed to it and developed to visualize the bands for analyte proteins.
2.10. Statistical Analysis
Student’s t-test was used to identify significant differences in quantitative values between the two test groups. For more than two groups one way analysis of variance (ANOVA) was performed. All studies or experiments were performed independently at least three times (n = 3), to achieve statistical power and t-test significance (where * p < 0.05; ** p < 0.01; *** p < 0.001, two-tailed distribution, paired). Wherever needed, the graphical approach was done for further analysis and presentation of data by using the GraphPad Prism 7.04 program.
4. Discussion
DCM is one of the most common causes of end-stage heart failure with limited treatment options.
Pathologic cardiac remodeling, consisting of abnormal ventricular geometry, abnormal cardiomyocyte morphology, and function, altered extracellular matrix composition, are the hallmarks of DCM hearts [
19,
20,
21,
22,
23,
24]. Such cardiac changes might result in cardiac cell damage, tissue fibrosis, ventricular dilatation, and systolic dysfunction, leading to heart failure. In line with other reports [
6,
9], our in silico analysis advised that the DCM involves dysregulation in angiogenesis and associated signaling pathways. Interestingly, earlier reports reveal the remedial effects of angiogenesis intervention during the DCM phenotype [
12,
13,
14]. Therefore, more studies are needed to explore such a beneficial effect that can be utilized to develop therapeutic strategies for DCM.
Clinical studies in heart transplant patients with DCM have reported a significant decrease in cardiac proangiogenic factors (such as VEGF-A and VEGFR1) and lower blood capillary density [
11], suggesting downregulation of angiogenesis during DCM phenotype. Likewise, we observed decreased angiogenic potential in DCM mice, as also supported by earlier reports in rodents [
6]. Thus, these reports suggest the alternative mechanism of angiogenesis signaling in DCM pathogenesis in humans and rodents. Similarly, promoting angiogenesis during DCM has been shown to have an anti-fibrotic and cardioprotective effect [
12,
13,
14]. Overall, this information suggests that angiogenesis could be a crucial target for DCM therapeutics. Therefore, we hypothesized that augmenting angiogenesis in the DCM heart might act as a trigger to reverse pathologic cardiac remodeling, to restore cardiac function.
Evidence from previous studies suggests that miRNAs play significant roles in cardiac remodeling [
25]. Also, we have previously reported that the constitutive overexpression of miR-210 can induce cardiac angiogenesis following ischemic injury [
16]. Similarly, our in vitro studies using miR-210 transfected rat cardiomyocytes culture conditioned media treated HUVEC showed a significant increase in tube formation, proposing the angiogenesis inducer properties of miR-210.
Taken together, we postulated that miR-210-mediated upregulated angiogenesis might rescue the DCM phenotype, as also advocated by earlier reports [
12,
13,
14]. Therefore, in the present study, we used α-TM 54 mutant mice as a DCM model, which illustrates molecular, structural, and functional abnormalities similar to DCM phenotype such as myofibrillar disarray, interstitial collagen deposition indicating fibrosis, bigger hearts with dilated ventricles, and systolic and diastolic dysfunction. To evaluate the effect of angiogenesis intervention during the DCM phenotype, previously reported cardiac angiogenesis inducer miR-210 [
16] was overexpressed in the DCM mice model via crossbreeding miR-210 transgenic mice (210-TG) and mutated α-TM 54 expressing DCM mice to achieve miR-210 overexpressing DCM phenotype mice (TMx210). Our results showed a significant increase in heart weight to body weight ratio in six-month-old mixed-gender TMx210 and DCM mice when compared to WT and 210-TG groups individually. Histopathological analysis, using heart tissue sections, demonstrated signs of myofibrillar loss (as indicated by myocardial disarray, and loss of cardiac tissue integrity), and interstitial fibrosis in DCM and TMx210 mice. However, by contrast with DCM, comparatively lesser interstitial fibrosis in TMx210 hearts was observed which may be due to the protective effect of miR-210 overexpression as reported earlier [
16]. Also, cardiomyocyte size remained comparable in all groups, suggesting the absence of cardiac cell hypertrophy in DCM and TMx210 mice, although there were trends for bigger cell size in both of these groups. These observations suggest that a bigger cardiac size in DCM and TMx210 mice might be due to the additive effect of interstitial collagen deposition (tissue fibrosis) and uptrends in cardiomyocyte size which may also one of the reasons for cardiac dysfunction in these groups. Also, an elevated angiogenic potential was observed by ex-situ aortic culture neovascularization in TMx210 mice when compared to DCM and WT. Interestingly, there is a significant downregulation in the angiogenic potential of DCM mice aorta which is in line with previous studies in DCM patients [
11]. Our in vivo study indicated an elevated cardiac blood vessel density and upregulated proangiogenic factor VEGF-A expression in TMx210 hearts as compared to DCM, suggesting improved angiogenesis in the former. Also, upregulation of VEGF, a potent angiogenic factor, in 210-TG mice heart is suggestive of miR-210 mediated VEGF pathway activation which is one of the major pathways in tissue angiogenesis.
The echocardiographic assessment showed no significant restoration of cardiac chamber dilation and heart function in TMx210 as compared to DCM. However, in contrast to DCM, there is an inhibition of LV posterior wall thinning in TMx210, indicating the partial beneficial effect of miR-210 mediated angiogenesis in TMx210 hearts.
One possible explanation for these observations may be the diverse effect of miR-210 on overall gene expression profile and associated pathways including angiogenesis signaling. In other words, miR-210-mediated upregulated angiogenesis might helpful in restoring the cardiac phenotype and physiological function in DCM mice, however, the miR-210 mediated activation of several other related genes and signaling pathways may nullify such favorable effects. Thus, more studies are needed to selectively intervene in the angiogenic signaling to examine the eventual effects during DCM development and pathogenesis.
Overall, based on these results, we conclude that miR-210-mediated upregulated angiogenesis is insufficient to rescue the DCM phenotype and cardiac function in mice.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






