
Impaired Mitochondrial Function in iPSC-Retinal Pigment Epithelium with the Complement Factor H Polymorphism for Age-Related Macular Degeneration

 , , and
, , and
Abstract
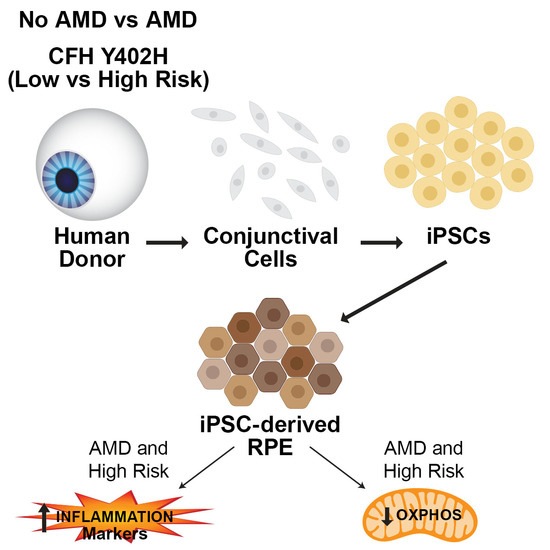
1. Introduction
2. Materials and Methods
2.1. Human Tissue Procurement and Grading
2.2. Genotyping
2.3. Culturing iPSC-RPE Cells
2.4. Immunofluorescence
2.5. Enzyme-Linked Immunosorbent Assay (ELISA)
2.6. Western Blotting
2.7. RNA Extraction, cDNA Synthesis and Quantitative RT-PCR
2.8. Measurement of Metabolic Function Using XFe96 Extracellular Flux Analyzer
2.9. Statistical Analysis
3. Results
3.1. Donor Demographics for iPSC-RPE
3.2. Characterization of iPSC-RPE
3.3. Metabolic Dysfunction in AMD and High-Risk iPSC-RPE
3.4. No Significant Difference in Mitochondrial Proteins
3.5. Altered Expression of Complement Pathway and Markers of Inflammation in AMD and High-Risk iPSC-RPE
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Friedman, D.S.; O’Colman, B.J.; Munoz, B. Prevalence of age-related macular degeneration in the United States. Arch. Ophthalomol. 2004, 122, 564–572. [Google Scholar]
- Buschini, E.; Fea, A.M.; Lavia, C.A.; Nassisi, M.; Pignata, G.; Zola, M.; Grignolo, F.M. Recent developments in the management of dry age-related macular degeneration. Clin. Ophthalmol. 2015, 9, 563–574. [Google Scholar] [CrossRef] [PubMed]
- Fritsche, L.G.; Igl, W.; Bailey, J.N.; Grassman, F.; Sengupta, S.; Bragg-Gresham, J.L.; Burdon, K.P.; Hebbring, S.J.; Wen, C.; Gorski, M.; et al. A large genome-wide association study of age-related macular degeneration highlights contributions of rare and common variants. Nat. Genet. 2016, 48, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Shaumberg, D.A.; Hankinson, S.E.; Guo, Q.; Rimm, E.; Hunter, D.J. A prospective study of 2 major age-related macular degeneration susceptibility alleles and interactions with modifiable risk factors. Arch. Ophthalmol. 2007, 125, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, V.P.; Pangburn, M.K.; Cortes, C. Complement control protein factor H: The good, the bad, and the inadequate. Mol. Immunol. 2010, 47, 2187–2197. [Google Scholar] [CrossRef] [PubMed]
- Anderson, D.H.; Radeke, M.J.; Gallo, N.B.; Chapin, E.A.; Johnson, P.T.; Curletti, C.R.; Hancox, L.S.; Hu, J.; Ebright, J.N.; Malek, G.; et al. The pivotal role of the complement system in aging and age-related macular degeneration: Hypothesis revisited. Prog. Retin. Eye Res. 2010, 29, 95–112. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.J.; Perveen, R.; Hakobyan, S.; Morgan, B.P.; Sim, R.B.; Bishop, P.N.; Day, A.J. Impaired binding of age-related macular degeneration-associated complement factor H 402H allotype to Brich’s membrane in human retina. J. Biol. Chem. 2010, 285, 30192–30202. [Google Scholar] [CrossRef]
- Sofat, R.; Casas, J.P.; Webster, A.R.; Bird, A.C.; Mann, S.S.; Yates, J.R.; Moore, A.T.; Sepp, T.; Cipriani, V.; Bunce, C.; et al. Complement Factor H genetic variant and age-related macular degeneration: Effect size, modifiers and relationship to disease subtype. Int. J. Epidemiol. 2012, 41, 250–262. [Google Scholar] [CrossRef]
- Geerlings, M.J.; de Jong, E.K.; den Hollander, A.I. The complement system in age-related macular degeneration: A review of rare genetic variants and implications for personalized treatment. Mol. Immunol. 2017, 84, 65–76. [Google Scholar] [CrossRef]
- Strauss, O. The retinal pigment epithelium in visual function. Physiol. Rev. 2005, 85, 845–881. [Google Scholar] [CrossRef]
- Feher, J.; Kovacs, I.; Artico, M.; Cavallotti, C.; Papale, A.; Balacco Gabrieli, C. Mitochondrial alterations of retinal pigment epithelium in age-related macular degeneration. Neurobiol. Aging 2006, 27, 983–993. [Google Scholar] [CrossRef]
- Nordgaard, C.L.; Karunadharma, P.P.; Feng, X.; Olsen, T.W.; Ferrington, D.A. Mitochondrial proteomics of the retinal pigment epithelium at progressive stages of AMD. Investig. Ophthalmol. Vis. Sci. 2008, 49, 2848–2855. [Google Scholar] [CrossRef]
- Karunadhrama, P.P.; Nordgaard, C.L.; Olsen, T.W.; Ferrington, D.A. Mitochondrial DNA damage as a potential mechanism for age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2010, 51, 5470–5479. [Google Scholar] [CrossRef] [PubMed]
- Terluk, M.R.; Kapphahn, R.J.; Soukup, L.M.; Gong, H.; Gallardo, C.; Montezuma, S.R.; Ferrington, D.A. Investigating mitochondria as a target for treating age-related macular degeneration. J. Neurosci. 2015, 35, 7304–7311. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Xu, H.; Liang, F.Q.; Liang, H.; Gupta, P.; Havey, A.N.; Boulton, M.E.; Godley, B.F. Mitochondrial DNA damage and repair in RPE associated with aging and age-related macular degeneration. Investig. Opthalmol. Vis. Sci. 2011, 52, 3251–3259. [Google Scholar] [CrossRef] [PubMed]
- Ferrington, D.A.; Ebeling, M.C.; Kapphahn, R.J.; Terluk, M.R.; Fisher, C.R.; Polanco, J.R.; Roehrich, H.; Leary, M.M.; Geng, Z.; Dutton, J.R.; et al. Altered bioenergetics and enhanced resistance to oxidative stress in human retinal pigment epithelial cells from donors with age-related macular degeneration. Redox Biol. 2017, 13, 255–265. [Google Scholar] [CrossRef]
- Ferrington, D.A.; Kapphahn, R.J.; Leary, M.M.; Atilano, S.R.; Terluk, M.R.; Karunadharma, P.; Chen, G.K.; Ratnapriya, R.; Swaroop, A.; Montezuma, S.R.; et al. Increased retinal mtDNA damage in the CFH variant associated with age-related macular degeneration. Exp. Eye Res. 2016, 145, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Kanow, M.A.; Giarmarco, M.M.; Jankowski, C.S. Biochemical adaptations of the retina and retinal pigment epithelium support a metabolic ecosystem in the vertebrate eye. eLife 2017, 6, e28899. [Google Scholar] [CrossRef] [PubMed]
- Fisher, C.R.; Ferrington, D.A. Perspective on AMD pathobiology: A bioenergetic crisis in the RPE. Investig. Ophthalmol. Vis. Sci. 2018, 59, AMD41–AMD47. [Google Scholar] [CrossRef]
- Kaarniranta, K.; Uusitalo, H.; Blasiak, J.; Felszeghy, S.; Kannan, R.; Kauppinen, A.; Salminen, A.; Sinha, D.; Ferrington, D.A. Mechanisms of mitochondrial dysfunction and their impact on age-related macular degeneration. Prog. Retin. Eye Res. 2020. [Google Scholar] [CrossRef]
- Golestaneh, N.; Chu, Y.; Cheng, S.K.; Cao, H.; Poliakov, E.; Berinstein, D.M. Repressed SIRT1/PGC-1α pathway and mitochondrial disintegration in iPSC-derived RPE disease model of age-related macular degeneration. J. Trans. Med. 2016, 14, 344. [Google Scholar] [CrossRef] [PubMed]
- Fields, M.A.; Bowrey, H.E.; Gong, J.; Moreira, E.F.; Cai, H.; Del Priore, L.V. Extracellular matrix nitration alters growth factor release and activates bioactive complement in human retinal pigment epithelial cells. PLoS ONE 2017, 15, e0177763. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Cai, H.; NYSCF Global Stem Cell Array Team; Noggle, S.; Paull, D.; Rizzolo, L.J.; Del Priore, L.V.; Fields, M.A. Stem cell-derived retinal pigment epithelium from patients with age-related macular degeneration exhibit reduced metabolism and matrix interactions. Stem Cells Transl. Med. 2020, 9, 364–376. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Li, Y.; Chan, L.; Tsai, Y.T.; Wu, W.H.; Nguyen, H.V.; Hsu, C.W.; Li, X.; Brown, L.M.; Egli, D.; et al. Validation of genome-wide association study (GWAS)-identified disease risk alleles with patient-specific stem cell lines. Hum. Mol. Genet. 2014, 23, 3445–3455. [Google Scholar] [CrossRef] [PubMed]
- Saini, J.S.; Corneo, B.; Miller, J.D.; Kiehl, T.R.; Wang, Q.; Boles, N.C.; Blenkinsop, T.A.; Stern, J.H.; Temple, S. Nicotinamide ameliorates disease phenotypes in a human iPSC model of age-related macular degeneration. Cell Stem Cell 2017, 20, 635–647. [Google Scholar] [CrossRef] [PubMed]
- Hallam, D.; Collin, J.; Bojic, S.; Chichagova, V.; Buskin, A.; Xu, Y.; Lafage, L.; Otten, E.G.; Anyfantis, G.; Mellough, C.; et al. An induced pluripotent stem cell patient specific model of complement factor H (Y402H) polymorphism displays characteristic features of age-related macular degeneration and indicates a beneficial role for UV light exposure. Stem Cells 2017, 35, 2305–2320. [Google Scholar] [CrossRef] [PubMed]
- Cerniauskas, E.; Kurwana-Akanbi, M.; Xie, L.; Hallam, D.; Moya-Molina, M.; White, K.; Steel, D.; Doherty, M.; Whitfield, P.; Al-Aama, J.; et al. Complement modulation reverses pathology in Y402H-retinal pigment epithelium cell model of age-related macular degeneration by restoring lysosomal function. Stem Cells Transl. Med. 2020, 9, 1585–1603. [Google Scholar] [CrossRef]
- Olson, T.W.; Feng, X. The Minnesota Grading System of eye bank eyes for age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2004, 45, 4484–4490. [Google Scholar] [CrossRef]
- Geng, Z.; Walsh, P.J.; Troung, V.; Hill, C.; Ebeling, M.; Kapphahn, R.J.; Montezuma, S.R.; Yuan, C.; Roehrich, H.; Ferrington, D.A.; et al. Generation of retinal pigmented epithelium from iPSCs derived from conjunctiva of donors with and without age related macular degeneration. PLoS ONE 2017, 12, e0173575. [Google Scholar] [CrossRef]
- Terluk, M.R.; Ebeling, M.C.; Fisher, C.R.; Kapphahn, R.J.; Yuan, C.; Kartha, R.V.; Montezuma, S.R.; Ferrington, D.A. N-Acetyl-L-cysteine protects human retinal pigment epithelial cells from oxidative damage: Implications for age-related macular degeneration. Oxid. Med. Cell Longev. 2019, 2019, 5174957. [Google Scholar] [CrossRef]
- Chacko, B.K.; Kramer, P.A.; Ravi, S.; Benavides, G.A.; Mitchell, T.; Dranka, B.P.; Ferrick, D.; Singal, A.K.; Ballinger, S.W.; Bailey, S.M.; et al. The Bioenergetic Health Index: A new concept in mitochondrial translational research. Clin. Sci. (Lond) 2014, 127, 367–373. [Google Scholar] [CrossRef]
- Marmorstein, A.D.; Marmorstein, L.Y.; Rayborn, M.; Wang, X.; Hollyfield, J.G.; Petrukhin, K. Bestrophin, the product of the Best vitelliform macular dystrophy gene (VMD2), localizes to the basolateral plasma membrane of the retinal pigment epithelium. Proc. Natl. Acad. Sci. USA 2000, 97, 12758–12763. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Zheng, S.G. Hall of Fame among Pro-inflammatory cytokines: Interleukin-6 gene and its transcriptional regulation mechanisms. Front. Immunol. 2016, 7, 604. [Google Scholar] [CrossRef] [PubMed]
- Bent, R.; Moll, L.; Grabbe, S.; Bros, M. Interleukin-1 Beta—A friend or foe in malignancies? Int. J. Mol. Sci. 2018, 19, 2155. [Google Scholar] [CrossRef] [PubMed]
- Revelo, N.H.; ter Beest, M.; van den Bogaart, G. Membrane trafficking as an active regulator of constitutively secreted cytokines. J. Cell Sci. 2020, 133, jcs234781. [Google Scholar] [CrossRef]
- Ebeling, M.C.; Polanco, J.R.; Qu, J.; Tu, C.; Montezuma, S.R.; Ferrington, D.A. Improving retinal mitochondrial function as treatment for age-related macular degeneration. Redox Biol. 2020, 34, 101552. [Google Scholar] [CrossRef] [PubMed]
- Golestaneh, N.; Chu, Y.; Xiao, Y.Y.; Stoleru, G.L.; Theos, A.C. Dysfunctional autophagy in RPE, a contributing factor in age-related macular degeneration. Cell Death Dis. 2017, 8, e2537. [Google Scholar] [CrossRef] [PubMed]
- Miyagishima, K.J.; Wan, Q.; Corneo, B.; Sharma, R.; Lotfi, M.R.; Boles, N.C.; Hua, F.; Maminishkis, A.; Zhang, C.; Blenkinsop, T.A.; et al. In pursuit of authenticity: Induced pluripotent stem cell-derived retinal pigment epithelium for clinical applications. Stem Cells Transl. Med. 2016, 11, 1562–1574. [Google Scholar] [CrossRef]
- Chang, Y.-C.; Chang, W.-C.; Hung, K.-H.; Yang, D.-M.; Cheng, Y.-H.; Liao, Y.-W.; Woung, L.-C.; Tsai, C.-Y.; Hsu, C.-C.; Lin, T.-C.; et al. The generation of induced pluripotent stem cells for macular degeneration as a drug screening platform: Identification of curcumin as a protective agent for retinal pigment epithelial cells against oxidative stress. Front. Aging Neurosci. 2014, 6, 191. [Google Scholar] [CrossRef]
- Calaza, K.C.; Hoh Kam, J.; Hogg, C.; Jeffery, G. Mitochondrial decline precedes phenotype development in the complement factor H mouse model of retinal degeneration but can be corrected by near infrared light. Neurobiol. Aging 2015, 36, 2869–2876. [Google Scholar] [CrossRef]
- Armento, A.; Honisch, S.; Panagiotakopoulou, V.; Sonntag, I.; Jacob, A.; Bolz, S.; Kilger, E.; Deleidi, M.; Clark, S.; Ueffing, M. Loss of complement factor H impairs antioxidant capacity and energy metabolism of human RPE cells. Sci. Rep. 2020, 10, 10320. [Google Scholar] [CrossRef]
- Sivapathasuntharam, C.; Hayes, M.J.; Shinhmar, H.; Kam, J.H.; Sivaprasad, S.; Jeffery, G. Complement factor H regulates retinal development and its absence may establish a footprint for age related macular degeneration. Sci. Rep. 2019, 9, 1082. [Google Scholar] [CrossRef] [PubMed]
- Coffey, P.J.; Gias, C.; McDermott, C.J.; Lundh, P.; Pickering, M.C.; Sethi, C.; Bird, A.; Fitzke, F.W.; Maass, A.; Chen, L.L.; et al. Complement factor H deficiency in aged mice causes retinal abnormalities and visual dysfunction. Proc. Natl. Acad. Sci. USA 2007, 104, 16651–16656. [Google Scholar] [CrossRef] [PubMed]
- Landowski, M.; Kelly, U.; Klingeborn, M.; Groelle, M.; Ding, J.D.; Grigsby, D.; Bowes Rickman, C. Human complement factor H Y402H polymorphism causes an age-related macular degeneration phenotype and lipoprotein dysregulation in mice. Proc. Natl. Acad. Sci. USA 2019, 116, 3703–3711. [Google Scholar] [CrossRef] [PubMed]
- Reichhardt, M.P.; Meri, S. Intracellular complement activation—An alarm raising mechanism? Semin. Immunol. 2018, 38, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.J.; Bishop, P.N. The eye as a complement dysregulation hotspot. Semin. Immunopathol. 2018, 40, 65–74. [Google Scholar] [CrossRef]
- Luo, C.; Chen, M.; Xu, H. Complement gene expression and regulation in mouse retina and retinal pigment epithelium/choroid. Mol. Vis. 2011, 17, 1588–1597. [Google Scholar]
- Kaur, G.; Tan, L.X.; Rathnasamy, G.; La Cunza, N.; Germer, C.J.; Toops, K.A.; Fernandes, M.; Blenkinsop, T.A.; Lakkaraju, A. Aberrant early endosome biogenesis mediates complement activation in the retinal pigment epithelium in models of macular degeneration. Proc. Natl. Acad. Sci. USA 2018, 115, 9014–9019. [Google Scholar] [CrossRef]
- Martin, M.; Leffler, J.; Smolag, K.I.; Mytych, J.; Bjork, A.; Chaves, L.D.; Alexander, J.J.; Quigg, R.J.; Blom, A.M. Factor H uptake regulates intracellular C3 activation during apoptosis and decreases the inflammatory potential of nucleosomes. Cell Death Diff. 2016, 23, 903–911. [Google Scholar] [CrossRef]
- Ramos de Carvalho, J.E.; Klassen, I.; Vogels, I.M.; Schipper-Krom, S.; van Noorden, C.J.; Reits, E.; Gorgels, T.G.; Bergen, A.A.; Schlingemann, R.O. Complement Factor C3a alters proteasome function in human RPE cells and in an animal model of age-related RPE degeneration. Investig. Ophthalmol. Vis. Sci. 2013, 54, 6489–6501. [Google Scholar] [CrossRef]
- Borras, C.; Canonica, J.; Jorieux, S.; Abache, T.; El Sanharawi, M.; Klein, C.; Delaunay, K.; Jonet, L.; Salovodelli, M.; Naud, M.C.; et al. CFH exerts anti-oxidant effects on retinal pigment epithelial cells independently from protecting against membrane attack complex. Sci. Rep. 2019, 9, 13873. [Google Scholar] [CrossRef]
- Leffler, J.; Herbert, A.P.; Norstrom, E.; Schmidt, C.Q.; Barlow, P.N.; Blom, A.M.; Martin, M. Annexin-II, DNA, and histones serve as factor H ligands on the surface of apoptotic cells. J. Biol. Chem. 2010, 285, 3766–3776. [Google Scholar] [CrossRef] [PubMed]
- Liszewski, M.K.; Kolev, M.; Le Friec, G.; Leung, M.; Bertram, P.G.; Fara, A.F.; Subias, M.; Pickering, M.C.; Drouet, C.; Meri, S.; et al. Intracellular complement activation sustains T cell homeostasis and mediates effector differentiation. Immunity 2013, 39, 1143–1157. [Google Scholar] [CrossRef] [PubMed]
- Kolev, M.; Kemper, C. Keeping it all going—Complement meets metabolism. Front. Immunol. 2017, 8, 1. [Google Scholar] [CrossRef] [PubMed]
- Arbore, G.; Kemper, C. A novel “complement-metabolism-inflammasome axis” as a key regulator of immune cell effector function. Eur. J. Immunol. 2016, 46, 1563–1573. [Google Scholar] [CrossRef]
- Cao, S.; Wang, J.C.C.; Gao, J.; Wong, M.; To, E.; White, V.A.; Cui, J.Z.; Matsubara, J.A. CFH Y402H polymorphism and the complement activation product C5a: Effects on NF-κB activation and inflammasome gene regulation. Br. J. Ophthamol. 2016, 100, 713–718. [Google Scholar] [CrossRef] [PubMed]
- Hess, C.; Kemper, C. Complement-mediated regulation of metabolism and basic cellular processes. Immunity 2016, 45, 240–254. [Google Scholar] [CrossRef] [PubMed]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.; Lee, S.J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 2012, 36, 401–414. [Google Scholar] [CrossRef]
- Zhang, Q.; Raoof, M.; Chen, Y.; Sumi, Y.; Sursal, T.; Junger, W.; Brohi, K.; Itagaki, K.; Hauser, C.J. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 2010, 464, 104–109. [Google Scholar] [CrossRef]
- Samanta, A.; Aziz, A.A.; Jhingan, M.; Singh, S.R.; Khanani, A.M.; Chhablani, J. Emerging Therapies in Nonexudative Age-Related Macular Degeneration in 2020. Asia Pac. J. Ophthalmol. (Phila) 2021. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| iPSC-RPE Line ID | Line # | Age a/Gender b | MGS Stage c | CFH Genotype d | Figures Using Data from Specific Lines |
|---|---|---|---|---|---|
| MGS1-1473-1D3 | 1 | 71/M | MGS1 | TT | 1B–D, 2B–F, 4C–E, 5C–E |
| MGS1-1473-2B6 | 2 | 71/M | MGS1 | TT | 1A–D, 2A–F, 3A–D, 4A–E, 5A–E |
| MGS1-0237-2C3 | 3 | 77/F | MGS1 | TT | 1B–D, 2B–F, 4A–E, 5A–D |
| MGS1-0237-3A3 | 4 | 77/F | MGS1 | TT | 1B–D, 2B–F, 4A–B, 5A–D |
| MGS1-0698-1A2 | 5 | 73/F | MGS1 | TT | 2A, 3A–D, 4A–B, 5A–B |
| MGS1-0698-1A3 | 6 | 73/F | MGS1 | TT | 4A–B, 5A–B |
| MGS1-1418-1A2 | 7 | 54/M | MGS1 | TT | 1B–D, 2B–F, 4C–E, 5C–E |
| MGS1-1580-1A4 | 8 | 57/M | MGS1 | TT | 1B–D, 2B–F, 3A–D, 4C–E, 5C–E |
| MGS1-1686-C | 9 | 73/M | MGS1 | TT | 5A–B,E |
| MGS1-1345-1C4 | 10 | 66/M | MGS1 | CT | 1B–D, 2B–F, 3A–D, 4C–E, 5C–D |
| MGS1-0027-1A3 | 11 | 80/M | MGS1 | CT | 1B–D, 2A–F, 3A–D, 4A–E, 5A–E |
| MGS1-0027-1B3 | 12 | 80/M | MGS1 | CT | 1B–D, 2B–F, 3A–D, 4A–E, 5A–E |
| MGS1-0553-2 | 13 | 68/M | MGS1 | CT | 1A–D, 2A–F, 3A–D, 4A–E, 5A–E |
| MGS1-1230-C | 14 | 84/F | MGS1 | CT | 5A–B,E |
| MGS2-1747-1A1 | 15 | 76/M | MGS2 | CT | 1B–D, 4A–E, 5A–E |
| MGS2-1747-2A4 | 16 | 76/M | MGS2 | CT | 1B–D, 2B–F, 4A–E, 5A–E |
| MGS2-1759-1E3 | 17 | 63/F | MGS2 | TT | 2B–F, 4A–E, 5A–B |
| MGS2-1759-1E6 | 18 | 63/F | MGS2 | TT | 2B–F, 4A–E, 5A–B |
| MGS2-1826-1C2 | 19 | 80/M | MGS2 | CT | 1B–D, 2B–F, 3A–D, 4A–B, 5A–E |
| MGS2-2360-3 | 20 | 58/M | MGS2 | CT | 1B–D, 2A–F, 3A–D, 4A–E, 5A–E |
| MGS2-0024-2 | 21 | 75/F | MGS2 | CC | 1B–D, 2B–F, 3A–D, 4A–E, 5A–E |
| MGS2-0024-3 | 22 | 75/F | MGS2 | CC | 1B–D, 2B–F, 3A–D, 4A–E, 5A–E |
| MGS2-0590-1 | 23 | 66/M | MGS2 | CT | 1A–D, 2B–F, 3A–D, 4A–E, 5A–E |
| MGS2-0590-2 | 24 | 66/M | MGS2 | CT | 2A–F, 3A–D, 4A–E, 5A–B |
| MGS2-1935-C | 25 | 80/F | MGS2 | TT | 5A–B,E |
| MGS2-1825-3 | 26 | 70/F | MGS2 | CC | 5A–B,E |
| MGS3-1775-6B4 | 27 | 85/F | MGS3 | CC | 1B–D, 2B–F, 4C–E, 5C–D |
| MGS3-0878-1B1 | 28 | 79/M | MGS3 | CT | 1B–D, 2B–F, 4A–E, 5A–D |
| MGS3-0878-1C4 | 29 | 79/M | MGS3 | CT | 1B–D, 2B–F, 3A–D, 4A–E, 5A–D |
| MGS3-1424-1A4 | 30 | 72/F | MGS3 | CT | 1B–D, 2A–F, 3A–D, 4A–E, 5A–D |
| MGS3-1424-1A5 | 31 | 72/F | MGS3 | CT | 1B–D, 2B–F, 4C–E, 5C–D |
| MGS3-2020-1A6 | 32 | 84/F | MGS3 | TT | 1B–D, 2A–F, 3A–D, 4A–E, 5A–E |
| MGS3-0277-1C1 | 33 | 80/F | MGS3 | TT | 1B–D, 2B–F, 3A–D, 4A–E, 5A–E |
| MGS3-0277-1C5 | 34 | 80/F | MGS3 | TT | 1B–D, 2B–F, 4A–E, 5A–E |
| MGS3-0824-2 | 35 | 85/M | MGS3 | TT | 1A, 2A–F, 3A–D, 4A–E, 5A–E |
| MGS3-0011-1 | 36 | 83/F | MGS3 | TT | 2B–F, 3A–D, 4A–E, 5A–B |
| MGS3-0011-3 | 37 | 83/F | MGS3 | TT | 1B–D, 2A–F, 3A–D, 4A–E, 5A–E |
| MGS3-0276-1 | 38 | 75/F | MGS3 | CC | 1B–D, 2B–F, 3A–D, 4A–E, 5A–E |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ebeling, M.C.; Geng, Z.; Kapphahn, R.J.; Roehrich, H.; Montezuma, S.R.; Dutton, J.R.; Ferrington, D.A. Impaired Mitochondrial Function in iPSC-Retinal Pigment Epithelium with the Complement Factor H Polymorphism for Age-Related Macular Degeneration. Cells 2021, 10, 789. https://doi.org/10.3390/cells10040789
Ebeling MC, Geng Z, Kapphahn RJ, Roehrich H, Montezuma SR, Dutton JR, Ferrington DA. Impaired Mitochondrial Function in iPSC-Retinal Pigment Epithelium with the Complement Factor H Polymorphism for Age-Related Macular Degeneration. Cells. 2021; 10(4):789. https://doi.org/10.3390/cells10040789
Chicago/Turabian StyleEbeling, Mara C., Zhaohui Geng, Rebecca J. Kapphahn, Heidi Roehrich, Sandra R. Montezuma, James R. Dutton, and Deborah A. Ferrington. 2021. "Impaired Mitochondrial Function in iPSC-Retinal Pigment Epithelium with the Complement Factor H Polymorphism for Age-Related Macular Degeneration" Cells 10, no. 4: 789. https://doi.org/10.3390/cells10040789
APA StyleEbeling, M. C., Geng, Z., Kapphahn, R. J., Roehrich, H., Montezuma, S. R., Dutton, J. R., & Ferrington, D. A. (2021). Impaired Mitochondrial Function in iPSC-Retinal Pigment Epithelium with the Complement Factor H Polymorphism for Age-Related Macular Degeneration. Cells, 10(4), 789. https://doi.org/10.3390/cells10040789