
Cockayne Syndrome Group B (CSB): The Regulatory Framework Governing the Multifunctional Protein and Its Plausible Role in Cancer

 ,
,  , ,
, , 
{kind=link}
{kind=link}
Abstract
:1. Introduction
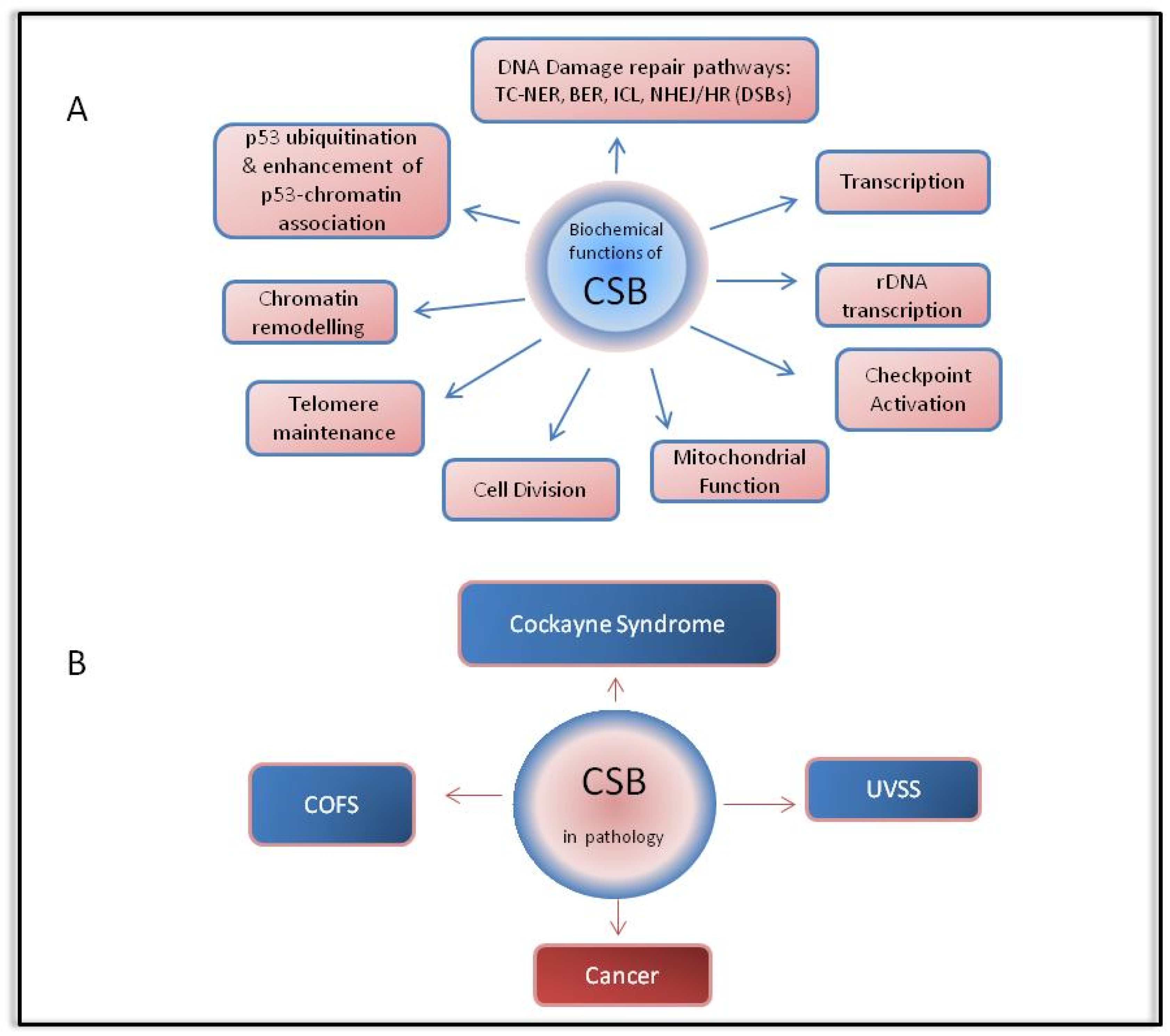
2. Cockayne Syndrome Protein B (CSB)
3. Regulatory Framework of CSB
3.1. Structural Regulatory Elements
3.2. Post-Translational Modifications
3.2.1. CSB Phosphorylation
3.2.2. CSB Ubiquitination
3.2.3. CSB Poly-ADP-Ribosylation
3.2.4. CSB SUMOylation
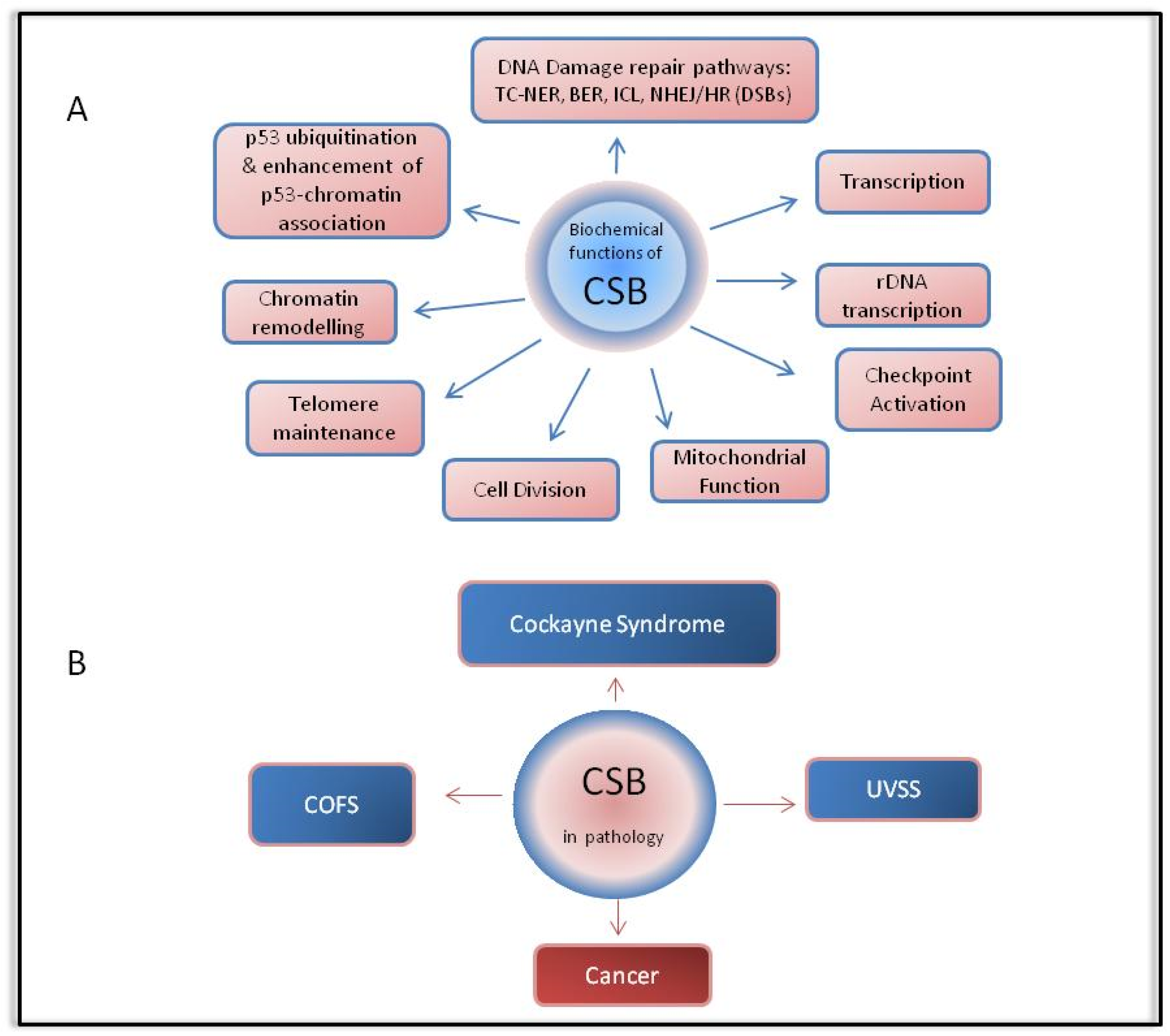
4. CSB in Pathology
4.1. Cockayne Syndrome
4.2. Models of Cockayne Syndrome
4.3. Other CSB-Related Pathologies
4.4. The Role of CSB in Cancer
5. Future Prospects—Potential Therapeutic Targeting of CSB
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kraemer, K.H.; Patronas, N.J.; Schiffmann, R.; Brooks, B.P.; Tamura, D.; DiGiovanna, J.J. Xeroderma pigmentosum, trichothiodystrophy and Cockayne syndrome: A complex genotype-phenotype relationship. Neuroscience 2007, 145, 1388–1396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henning, K.A.; Li, L.; Iyer, N.; McDaniel, L.D.; Reagan, M.S.; Legerski, R.; Schultz, R.A.; Stefanini, M.; Lehmann, A.R.; Mayne, L.V.; et al. The Cockayne syndrome group A gene encodes a WD repeat protein that interacts with CSB protein and a subunit of RNA polymerase II TFIIH. Cell 1995, 82, 555–564. [Google Scholar] [CrossRef] [Green Version]
- Troelstra, C.; van Gool, A.; de Wit, J.; Vermeulen, W.; Bootsma, D.; Hoeijmakers, J.H. ERCC6, a member of a subfamily of putative helicases, is involved in Cockayne’s syndrome and preferential repair of active genes. Cell 1992, 71, 939–953. [Google Scholar] [CrossRef] [Green Version]
- Tiwari, V.; Baptiste, B.A.; Okur, M.N.; Bohr, V.A. Current and emerging roles of Cockayne syndrome. Nucleic Acids Res. 2021, 49, 2418–2434. [Google Scholar] [CrossRef]
- Eisen, J.A.; Sweder, K.S.; Hanawalt, P.C. Evolution of the SNF2 family of proteins: Subfamilies with distinct sequences and functions. Nucleic Acids Res. 1995, 23, 2715–2723. [Google Scholar] [CrossRef] [Green Version]
- Selby, C.P.; Sancar, A. Human transcription-repair coupling factor CSB/ERCC6 is a DNA-stimulated ATPase but is not a helicase and does not disrupt the ternary transcription complex of stalled RNA polymerase II. J. Biol. Chem. 1997, 272, 1885–1890. [Google Scholar] [CrossRef] [Green Version]
- Citterio, E.; Rademakers, S.; van der Horst, G.T.; van Gool, A.J.; Hoeijmakers, J.H.; Vermeulen, W. Biochemical and biological characterization of wild-type and ATPase-deficient Cockayne syndrome B repair protein. J. Biol. Chem. 1998, 273, 11844–11851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muftuoglu, M.; Sharma, S.; Thorslund, T.; Stevnsner, T.; Soerensen, M.M.; Brosh, R.M., Jr.; Bohr, V.A. Cockayne syndrome group B protein has novel strand annealing and exchange activities. Nucleic Acids Res. 2006, 34, 295–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beerens, N.; Hoeijmakers, J.H.; Kanaar, R.; Vermeulen, W.; Wyman, C. The CSB protein actively wraps DNA. J. Biol. Chem. 2005, 280, 4722–4729. [Google Scholar] [CrossRef] [Green Version]
- Kumar, N.; Moreno, N.C.; Feltes, B.C.; Menck, C.F.; Van Houten, B.V. Cooperation and interplay between base and nucleotide excision repair pathways: From DNA lesions to proteins. Genet. Mol. Biol. 2020, 43, e20190104. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.H.; Kang, T.H. DNA Oxidation and Excision Repair Pathways. Int. J. Mol. Sci. 2019, 20, 6092. [Google Scholar] [CrossRef] [Green Version]
- Brooks, P.J. The 8,5′-Cyclopurine-2′-Deoxynucleosides: Candidate Neurodegenerative DNA Lesions in Xeroderma Pigmentosum, and Unique Probes of Transcription and Nucleotide Excision Repair. DNA Repair 2008, 7, 1168–1179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van den Boom, V.; Citterio, E.; Hoogstraten, D.; Zotter, A.; Egly, J.M.; van Cappellen, W.A.; Hoeijmakers, J.H.; Houtsmuller, A.B.; Vermeulen, W. DNA damage stabilizes interaction of CSB with the transcription elongation machinery. J. Cell Biol. 2004, 166, 27–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vermeulen, W.; Fousteri, M. Mammalian transcription-coupled excision repair. Cold Spring Harb. Perspect. Biol. 2013, 5, a012625. [Google Scholar] [CrossRef]
- van den Heuvel, D.; Spruijt, C.G.; González-Prieto, R.; Kragten, A.; Paulsen, M.T.; Zhou, D.; Wu, H.; Apelt, K.; van der Weegen, Y.; Yang, K.; et al. A CSB-PAF1C axis restores processive transcription elongation after DNA damage repair . Nat. Commun. 2021, 12, 1342. [Google Scholar]
- Licht, C.L.; Stevnsner, T.; Bohr, V.A. Cockayne Syndrome Group B Cellular and Biochemical Functions. Am. J. Hum. Genet. 2003, 73, 1217–1239. [Google Scholar] [CrossRef] [Green Version]
- Stevnsner, T.; Muftuoglu, M.; Aamann, M.D.; Bohr, V.A. The role of Cockayne Syndrome group B (CSB) protein in base excision repair and aging. Mech. Ageing Dev. 2008, 129, 441–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iyama, T.; Lee, S.Y.; Berquist, B.R.; Gileadi, O.; Bohr, V.A.; Seidman, M.M.; McHugh, P.J.; Wilson, D.M., 3rd. CSB interacts with SNM1A and promotes DNA interstrand crosslink processing. Nucleic Acids Res. 2015, 43, 247–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batenburg, N.L.; Walker, J.R.; Noordermeer, S.M.; Moatti, N.; Durocher, D.; Zhu, X.D. ATM and CDK2 control chromatin remodeler CSB to inhibit RIF1 in DSB repair pathway choice. Nat. Commun. 2017, 8, 1021. [Google Scholar] [CrossRef] [PubMed]
- Batenburg, N.L.; Thompson, E.L.; Hendrickson, E.A.; Zhu, X.D. Cockayne syndrome group B protein regulates DNA double-strand break repair and checkpoint activation. EMBO J. 2015, 34, 1399–1416. [Google Scholar] [CrossRef] [Green Version]
- Galanos, P.; Pappas, G.; Polyzos, A.; Kotsinas, A.; Svolaki, I.; Giakoumakis, N.N.; Glytsou, C.; Pateras, I.S.; Swain, U.; Souliotis, V.L.; et al. Mutational signatures reveal the role of RAD52 in p53-independent p21-driven genomic instability. Genome Biol. 2018, 19, 37. [Google Scholar] [CrossRef] [Green Version]
- Proietti-De-Santis, L.; Drané, P.; Egly, J.M. Cockayne syndrome B protein regulates the transcriptional program after UV irradiation. EMBO J. 2006, 25, 1915–1923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vélez-Cruz, R.; Egly, J.M. Cockayne syndrome group B (CSB) protein: At the crossroads of transcriptional networks. Mech. Ageing Dev. 2013, 134, 234–242. [Google Scholar] [CrossRef] [PubMed]
- Citterio, E.; Van Den Boom, V.; Schnitzler, G.; Kanaar, R.; Bonte, E.; Kingston, R.E.; Hoeijmakers, J.H.; Vermeulen, W. ATP-dependent chromatin remodeling by the Cockayne syndrome B DNA repair-transcription-coupling factor. Mol. Cell. Biol. 2000, 20, 7643–7653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradsher, J.; Auriol, J.; Proietti de Santis, L.; Iben, S.; Vonesch, J.L.; Grummt, I.; Egly, J.M. CSB is a component of RNA pol I transcription. Mol. Cell 2002, 10, 819–829. [Google Scholar] [CrossRef]
- Okur, M.N.; Lee, J.H.; Osmani, W.; Kimura, R.; Demarest, T.G.; Croteau, D.L.; Bohr, V.A. Cockayne syndrome group A and B proteins function in rRNA transcription through nucleolin regulation. Nucleic Acids Res. 2020, 48, 2473–2485. [Google Scholar] [CrossRef]
- Aamann, M.D.; Sorensen, M.M.; Hvitby, C.; Berquist, B.R.; Muftuoglu, M.; Tian, J.; de Souza-Pinto, N.C.; Scheibye-Knudsen, M.; Wilson, D.M., 3rd; Stevnsner, T.; et al. Cockayne syndrome group B protein promotes mitochondrial DNA stability by supporting the DNA repair association with the mitochondrial membrane. FASEB J. 2010, 24, 2334–2346. [Google Scholar] [CrossRef] [Green Version]
- Lake, R.J.; Basheer, A.; Fan, H.Y. Reciprocally regulated chromatin association of Cockayne syndrome protein B and p53 protein. J. Biol. Chem. 2011, 286, 34951–34958. [Google Scholar] [CrossRef] [Green Version]
- Paccosi, E.; Costanzo, F.; Costantino, M.; Balzerano, A.; Monteonofrio, L.; Soddu, S.; Prantera, G.; Brancorsini, S.; Egly, J.; Proietti-De-Santis, L. The Cockayne syndrome group A and B proteins are part of a ubiquitin–proteasome degradation complex regulating cell division. Proc. Natl. Acad. Sci. USA 2020, 117, 30498–30508. [Google Scholar] [CrossRef]
- Batenburg, N.L.; Mitchell, T.R.; Leach, D.M.; Rainbow, A.J.; Zhu, X.D. Cockayne Syndrome group B protein interacts with TRF2 and regulates telomere length and stability. Nucleic Acids Res. 2012, 40, 9661–9674. [Google Scholar] [CrossRef]
- Brosh, R.M., Jr.; Balajee, A.S.; Selzer, R.R.; Sunesen, M.; Proietti De Santis, L.; Bohr, V.A. The ATPase domain but not the acidic region of Cockayne syndrome group B gene product is essential for DNA repair. Mol. Biol. Cell 1999, 10, 3583–3594. [Google Scholar] [CrossRef] [Green Version]
- Anindya, R.; Mari, P.O.; Kristensen, U.; Kool, H.; Giglia-Mari, G.; Mullenders, L.H.; Fousteri, M.; Vermeulen, W.; Egly, J.M.; Svejstrup, J.Q. A ubiquitin-binding domain in Cockayne syndrome B required for transcription-coupled nucleotide excision repair. Mol. Cell 2010, 38, 637–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Weegen, Y.; Golan-Berman, H.; Mevissen, T.E.T.; Apelt, K.; González-Prieto, R.; Goedhart, J.; Heilbrun, E.E.; Vertegaal, A.C.O.; van den Heuvel, D.; Walter, J.C.; et al. The cooperative action of CSB, CSA, and UVSSA target TFIIH to DNA damage-stalled RNA polymerase II. Nat. Commun. 2020, 11, 2104. [Google Scholar] [CrossRef] [PubMed]
- Iyama, T.; Okur, M.N.; Golato, T.; Mc Neill, D.R.; Lu, H.; Hamilton, R.; Raja, A.; Bohr, V.A.; Wilson, D.M., 3rd. Regulation of the Intranuclear Distribution of the Cockayne Syndrome Proteins. Sci. Rep. 2018, 8, 17490. [Google Scholar] [CrossRef] [PubMed]
- Cho, I.; Tsai, P.F.; Lake, R.J.; Basheer, A.; Fan, H.Y. ATP-dependent chromatin remodeling by Cockayne syndrome protein B and NAP1-like histone chaperones is required for efficient transcription-coupled DNA repair. PLoS Genet. 2013, 9, e1003407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sin, Y.; Tanaka, K.; Saijo, M. The C-terminal Region and SUMOylation of Cockayne Syndrome Group B Protein Play Critical Roles in Transcription-coupled Nucleotide Excision Repair. J. Biol. Chem. 2016, 291, 1387–1397. [Google Scholar] [CrossRef] [Green Version]
- Batenburg, N.L.; Cui, S.; Walker, J.R.; Schellhorn, H.E.; Zhu, X.-D. The Winged Helix Domain of CSB Regulates RNAPII Occupancy at Promoter Proximal Pause Sites. Int. J. Mol. Sci. 2021, 22, 3379. [Google Scholar] [CrossRef]
- Batenburg, N.L.; Qin, J.; Walker, J.R.; Zhu, X.D. Efficient UV repair requires disengagement of the CSB winged helix domain from the CSB ATPase domain. DNA Repair (Amst) 2018, 68, 58–67. [Google Scholar] [CrossRef] [PubMed]
- van den Heuvel, D.; van der Weegen, Y.; Boer, D.E.C.; Ogi, T.; Luijsterburg, M.S. Transcription-Coupled DNA Repair: From Mechanism to Human Disorder. Trends Cell Biol. 2021. [Google Scholar] [CrossRef]
- Lake, R.J.; Geyko, A.; Hemashettar, G.; Zhao, Y.; Fan, H.Y. UV-induced association of the CSB remodeling protein with chromatin requires ATP-dependent relief of N-terminal autorepression. Mol. Cell 2010, 37, 235–246. [Google Scholar] [CrossRef] [Green Version]
- Iyama, T.; Wilson, D.M., 3rd. Elements That Regulate the DNA Damage Response of Proteins Defective in Cockayne Syndrome. J. Mol. Biol. 2016, 428, 62–78. [Google Scholar] [CrossRef] [Green Version]
- Fousteri, M.; Vermeulen, W.; van Zeeland, A.A.; Mullenders, L.H. Cockayne syndrome A and B proteins differentially regulate recruitment of chromatin remodeling and repair factors to stalled RNA polymerase II in vivo. Mol. Cell 2006, 23, 471–482. [Google Scholar] [CrossRef]
- Wei, L.; Nakajima, S.; Böhm, S.; Bernstein, K.A.; Shen, Z.; Tsang, M.; Levine, A.S.; Lan, L. DNA damage during the G0/G1 phase triggers RNA-templated, Cockayne syndrome B-dependent homologous recombination. Proc. Natl. Acad. Sci. USA 2015, 112, E3495–E3504. [Google Scholar]
- Selzer, R.R.; Nyaga, S.; Tuo, J.; May, A.; Muftuoglu, M.; Christiansen, M.; Citterio, E.; Brosh, R.M., Jr.; Bohr, V.A. Differential requirement for the ATPase domain of the Cockayne syndrome group B gene in the processing of UV-induced DNA damage and 8-oxoguanine lesions in human cells. Nucleic Acids Res. 2002, 30, 782–793. [Google Scholar]
- Maddukuri, L.; Speina, E.; Christiansen, M.; Dudzińska, D.; Zaim, J.; Obtułowicz, T.; Kabaczyk, S.; Komisarski, M.; Bukowy, Z.; Szczegielniak, J.; et al. Cockayne syndrome group B protein is engaged in processing of DNA adducts of lipid peroxidation product trans-4-hydroxy-2-nonenal. Mutat. Res. 2009, 666, 23–31. [Google Scholar]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 2021, 30, 70–82. [Google Scholar] [CrossRef] [PubMed]
- Goddard, T.D.; Huang, C.C.; Meng, E.C.; Pettersen, E.F.; Couch, G.S.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci. 2018, 27, 14–25. [Google Scholar] [CrossRef]
- Karve, T.M.; Cheema, A.K. Small changes huge impact: The role of protein posttranslational modifications in cellular homeostasis and disease. J. Amino Acids 2011, 2011, 207691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imam, S.Z.; Indig, F.E.; Cheng, W.H.; Saxena, S.P.; Stevnsner, T.; Kufe, D.; Bohr, V.A. Cockayne syndrome protein B interacts with and is phosphorylated by c-Abl tyrosine kinase. Nucleic Acids Res. 2007, 35, 4941–4951. [Google Scholar] [CrossRef] [PubMed]
- Christiansen, M.; Stevnsner, T.; Modin, C.; Martensen, P.M.; Brosh, R.M., Jr.; Bohr, V.A. Functional consequences of mutations in the conserved SF2 motifs and post-translational phosphorylation of the CSB protein. Nucleic Acids Res. 2003, 31, 963–973. [Google Scholar] [CrossRef] [Green Version]
- Hornbeck, P.V.; Zhang, B.; Murray, B.; Kornhauser, J.M.; Latham, V.; Skrzypek, E. PhosphoSitePlus, 2014: Mutations, PTMs and recalibrations. Nucleic Acids Res. 2015, 43, D512–D520. [Google Scholar]
- Takahashi, T.S.; Sato, Y.; Yamagata, A.; Goto-Ito, S.; Saijo, M.; Fukai, S. Structural basis of ubiquitin recognition by the winged-helix domain of Cockayne syndrome group B protein. Nucleic Acids Res. 2019, 47, 3784–3794. [Google Scholar] [CrossRef] [Green Version]
- Groisman, R.; Kuraoka, I.; Chevallier, O.; Gaye, N.; Magnaldo, T.; Tanaka, K.; Kisselev, A.F.; Harel-Bellan, A.; Nakatani, Y. CSA-dependent degradation of CSB by the ubiquitin-proteasome pathway establishes a link between complementation factors of the Cockayne syndrome. Genes Dev. 2006, 20, 1429–1434. [Google Scholar]
- Wei, L.; Lan, L.; Yasui, A.; Tanaka, K.; Saijo, M.; Matsuzawa, A.; Kashiwagi, R.; Maseki, E.; Hu, Y.; Parvin, J.D.; et al. BRCA1 contributes to transcription-coupled repair of DNA damage through polyubiquitination and degradation of Cockayne syndrome B protein. Cancer Sci. 2011, 102, 1840–1847. [Google Scholar]
- Ranes, M.; Boeing, S.; Wang, Y.; Wienholz, F.; Menoni, H.; Walker, J.; Encheva, V.; Chakravarty, P.; Mari, P.O.; Stewart, A.; et al. A ubiquitylation site in Cockayne syndrome B required for repair of oxidative DNA damage, but not for transcription-coupled nucleotide excision repair. Nucleic Acids Res. 2016, 44, 5246–5255. [Google Scholar]
- Schwertman, P.; Lagarou, A.; Dekkers, D.H.; Raams, A.; van der Hoek, A.C.; Laffeber, C.; Hoeijmakers, J.H.; Demmers, J.A.; Fousteri, M.; Vermeulen, W.; et al. UV-sensitive syndrome protein UVSSA recruits USP7 to regulate transcription-coupled repair. Nat. Genet. 2012, 44, 598–602. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Q.; Ding, N.; Wei, S.; Li, P.l; Wani, G.; He, J.; Wani, A.A. USP7-mediated deubiquitination differentially regulates CSB but not UVSSA upon UV radiation-induced DNA damage. Cell Cycle 2020, 19, 124–141. [Google Scholar]
- Thorslund, T.; von Kobbe, C.; Harrigan, J.A.; Indig, F.E.; Christiansen, M.; Stevnsner, T.; Bohr, V.A. Cooperation of the Cockayne syndrome group B protein and poly (ADP-ribose) polymerase 1 in the response to oxidative stress. Mol. Cell Biol. 2005, 25, 7625–7636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayne, L.V.; Lehmann, A.R. Failure of RNA synthesis to recover after UV irradiation: An early defect in cells from individuals with Cockayne’s syndrome and xeroderma pigmentosum. Cancer Res. 1982, 42, 1473–1478. [Google Scholar]
- Liebelt, F.; Schimmel, J.; Verlaan-de Vries, M.; Klemann, E.; van Royen, M.E.; van der Weegen, Y.; Luijsterburg, M.S.; Mullenders, L.H.; Pines, A.; Vermeulen, W.; et al. Transcription-coupled nucleotide excision repair is coordinated by ubiquitin and SUMO in response to ultraviolet irradiation. Nucleic Acids Res. 2020, 48, 231–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karikkineth, A.C.; Scheibye-Knudsen, M.; Fivenson, E.; Croteau, D.L.; Bohr, V.A. Cockayne syndrome: Clinical features, model systems and pathways. Ageing Res. Rev. 2017, 33, 3–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calmels, N.; Botta, E.; Jia, N.; Fawcett, H.; Nardo, T.; Nakazawa, Y.; Lanzafame, M.; Moriwaki, S.; Sugita, K.; Kubota, M.; et al. Functional and clinical relevance of novel mutations in a large cohort of patients with Cockayne syndrome. J. Med. Genet. 2018, 55, 329–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vessoni, A.T.; Guerra, C.C.C.; Kajitani, G.S.; Nascimento, L.L.S.; Garcia, C.C.M. Cockayne Syndrome: The many challenges and approaches to understand a multifaceted disease. Genet. Mol. Biol. 2020, 43, e20190085. [Google Scholar] [CrossRef]
- Kashiyama, K.; Nakazawa, Y.; Pilz, D.T.; Guo, C.; Shimada, M.; Sasaki, K.; Fawcett, H.; Wing, J.F.; Lewin, S.O.; Carr, L.; et al. Malfunction of nuclease ERCC1-XPF results in diverse clinical manifestations and causes Cockayne syndrome, xeroderma pigmentosum, and Fanconi anemia. Am. J. Hum. Genet. 2013, 92, 807–819. [Google Scholar]
- van der Horst, G.T.; van Steeg, H.; Berg, R.J.; van Gool, A.J.; de Wit, J.; Weeda, G.; Morreau, H.; Beems, R.B.; van Kreijl, C.F.; de Gruijl, F.R.; et al. Defective transcription-coupled repair in Cockayne syndrome B mice is associated with skin cancer predisposition. Cell 1997, 89, 425–435. [Google Scholar] [CrossRef] [Green Version]
- Itoh, M.; Hayashi, M.; Shioda, K.; Minagawa, M.; Isa, F.; Tamagawa, K.; Morimatsu, Y.; Oda, M. Neurodegeneration in hereditary nucleotide repair disorders. Brain Dev. 1999, 21, 326–333. [Google Scholar] [CrossRef]
- Hayashi, M.; Miwa-Saito, N.; Tanuma, N.; Kubota, M. Brain vascular changes in Cockayne syndrome. Neuropathology 2012, 32, 113–117. [Google Scholar]
- Murai, M.; Enokido, Y.; Inamura, N.; Yoshino, M.; Nakatsu, Y.; van der Horst, G.T.; Hoeijmakers, J.H.; Tanaka, K.; Hatanaka, H. Early postnatal ataxia and abnormal cerebellar development in mice lacking Xeroderma pigmentosum Group A and Cockayne syndrome Group B DNA repair genes. Proc. Natl. Acad. Sci. USA 2001, 98, 13379–13384. [Google Scholar]
- Laposa, R.R.; Huang, E.J.; Cleaver, J.E. Increased apoptosis, p53 up-regulation, and cerebellar neuronal degeneration in repair-deficient Cockayne syndrome mice. Proc. Natl. Acad. Sci. USA 2007, 104, 1389–1394. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.H.; Ahn, B.; Choi, I.S.; Koo, H.S. The gene expression and deficiency phenotypes of Cockayne syndrome B protein in Caenorhabditis elegans. FEBS Lett. 2002, 522, 47–51. [Google Scholar]
- Wu, Z.; Zhu, X.; Yu, Q.; Xu, Y.; Wang, Y. Multisystem analyses of two Cockayne syndrome associated proteins CSA and CSB reveal shared and unique functions. DNA Repair (Amst) 2019, 83, 102696. [Google Scholar]
- Xu, Y.; Wu, Z.; Liu, L.; Liu, J.; Wang, Y. Rat Model of Cockayne Syndrome Neurological Disease. Cell Rep. 2019, 29, 800–809. [Google Scholar]
- Pacak, C.A.; Brooks, P.J. The past, present, and future of modeling Cockayne Syndrome—A commentary on “Rat Model of Cockayne Syndrome Neurological Disease”. DNA Repair (Amst) 2020, 88, 102788. [Google Scholar] [CrossRef] [PubMed]
- Spivak, G.; Itoh, T.; Matsunaga, T.; Nikaido, O.; Hanawalt, P.; Yamaizumi, M. Ultraviolet-sensitive syndrome cells are defective in transcription-coupled repair of cyclobutane pyrimidine dimers. DNA Repair (Amst) 2002, 1, 629–643. [Google Scholar] [CrossRef]
- Spivak, G. UV-sensitive syndrome. Mutat. Res. 2005, 577, 162–169. [Google Scholar] [CrossRef]
- Spivak, G.; Hanawalt, P.C. Host cell reactivation of plasmids containing oxidative DNA lesions is defective in Cockayne syndrome but normal in UV-sensitive syndrome fibroblasts. DNA Repair (Amst) 2006, 5, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Horibata, K.; Iwamoto, Y.; Kuraoka, I.; Jaspers, N.G.; Kurimasa, A.; Oshimura, M.; Ichihashi, M.; Tanaka, K. Complete absence of Cockayne syndrome group B gene product gives rise to UV-sensitive syndrome but not Cockayne syndrome. Proc. Natl. Acad. Sci. USA 2004, 101, 15410–15415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laugel, V.; Dalloz, C.; Tobias, E.S.; Tolmie, J.L.; Martin-Coignard, D.; Drouin-Garraud, V.; Valayannopoulos, V.; Sarasin, A.; Dollfus, H. Cerebro-oculo-facio-skeletal syndrome: Three additional cases with CSB mutations, new diagnostic criteria and an approach to investigation. J. Med. Genet. 2008, 45, 564–571. [Google Scholar]
- Suzumura, H.; Arisaka, O. Cerebro-oculo-facio-skeletal syndrome. Adv. Exp. Med. Biol. 2010, 685, 210–214. [Google Scholar]
- Meira, L.B.; Graham, J.M., Jr.; Greenberg, C.R.; Busch, D.B.; Doughty, A.T.; Ziffer, D.W.; Coleman, D.M.; Savre-Train, I.; Friedberg, E.C. Manitoba aboriginal kindred with original cerebro-oculo- facio-skeletal syndrome has a mutation in the Cockayne syndrome group B (CSB) gene. Am. J. Hum. Genet. 2000, 66, 1221–1228. [Google Scholar]
- Graham, J.M., Jr.; Anyane-Yeboa, K.; Raams, A.; Appeldoorn, E.; Kleijer, W.J.; Garritsen, V.H.; Busch, D.; Edersheim, T.G.; Jaspers, N.G. Cerebro-oculo-facio-skeletal syndrome with a nucleotide excision-repair defect and a mutated XPD gene, with prenatal diagnosis in a triplet pregnancy. Am. J. Hum. Genet. 2001, 69, 291–300. [Google Scholar]
- Hamel, B.C.; Raams, A.; Schuitema-Dijkstra, A.R.; Simons, P.; van der Burgt, I.; Jaspers, N.G.; Kleijer, W. Xeroderma pigmentosum–Cockayne syndrome complex: A further case. J. Med. Genet. 1996, 33, 607–610. [Google Scholar] [CrossRef] [PubMed]
- Jaspers, N.G.; Raams, A.; Silengo, M.C.; Wijgers, N.; Niedernhofer, L.J.; Robinson, A.R.; Giglia-Mari, G.; Hoogstraten, D.; Kleijer, W.J.; Hoeijmakers, J.H.; et al. First reported patient with human ERCC1 deficiency has cerebro-oculo-facio-skeletal syndrome with a mild defect in nucleotide excision repair and severe developmental failure. Am. J. Hum. Genet. 2007, 80, 457–466. [Google Scholar] [CrossRef] [Green Version]
- Colella, S.; Nardo, T.; Botta, E.; Lehmann, A.R.; Stefanini, M. Identical mutations in the CSB gene associated with either Cockayne syndrome or the DeSanctis-cacchione variant of xeroderma pigmentosum. Hum. Mol. Genet. 2000, 9, 1171–1175. [Google Scholar] [CrossRef] [Green Version]
- Natale, V.; Raquer, H. Xeroderma pigmentosum-Cockayne syndrome complex. Orphanet J. Rare Dis. 2017, 12, 65. [Google Scholar] [CrossRef]
- Zhang, W.R.; Garrett, G.L.; Cleaver, J.E.; Arron, S.T. Absence of skin cancer in the DNA repair-deficient disease Cockayne Syndrome (CS): A survey study. J. Am. Acad. Dermatol. 2016, 74, 1270–1272. [Google Scholar] [CrossRef] [Green Version]
- Cleaver, J.E.; Lam, E.T.; Revet, I. Disorders of nucleotide excision repair: The genetic and molecular basis of heterogeneity. Nat. Rev. Genet. 2009, 10, 756–768. [Google Scholar] [CrossRef] [PubMed]
- Nance, M.A.; Berry, S.A. Cockayne syndrome: Review of 140 cases. Am. J. Med. Genet. 1992, 42, 68–84. [Google Scholar] [CrossRef]
- Reid-Bayliss, K.S.; Arron, S.T.; Loeb., L.A.; Bezrookove, V.; Cleaver, J.E. Why Cockayne syndrome patients do not get cancer despite their DNA repair deficiency. Proc. Natl. Acad. Sci. USA 2016, 113, 10151–10156. [Google Scholar] [CrossRef] [Green Version]
- Caputo, M.; Frontini, M.; Velez-Cruz, R.; Nicolai, S.; Prantera, G.; Proietti-De-Santis, L. The CSB repair factor is overexpressed in cancer cells, increases apoptotic resistance, and promotes tumor growth. DNA Repair (Amst) 2013, 12, 293–299. [Google Scholar] [CrossRef] [Green Version]
- Proietti-De-Santis, L.; Balzerano, A.; Prantera, G. CSB: An Emerging Actionable Target for Cancer Therapy. Trends Cancer 2018, 4, 172–175. [Google Scholar] [CrossRef]
- Meek, D.W. The p53 response to DNA damage. DNA Repair (Amst) 2004, 3, 1049–1056. [Google Scholar] [CrossRef] [PubMed]
- Gorgoulis, V.G.; Pefani, D.E.; Pateras, I.S.; Trougakos, I.P. Integrating the DNA damage and protein stress responses during cancer development and treatment. J. Pathol. 2018, 246, 12–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halazonetis, T.D.; Gorgoulis, V.G.; Bartek, J. An oncogene-induced DNA damage model for cancer development. Science 2008, 319, 1352–1355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amelio, I.; Melino, G. The p53 family and the hypoxia-inducible factors (HIFs): Determinants of cancer progression. Trends Biochem. Sci. 2015, 40, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Lagopati, N.; Belogiannis, K.; Angelopoulou, A.; Papaspyropoulos, A.; Gorgoulis, V. Non-Canonical Functions of the ARF Tumor Suppressor in Development and Tumorigenesis. Biomolecules 2021, 11, 86. [Google Scholar] [CrossRef]
- Latini, P.; Frontini, M.; Caputo, M.; Gregan, J.; Cipak, L.; Filippi, S.; Kumar, V.; Vélez-Cruz, R.; Stefanini, M.; Proietti-De-Santis, L. CSA and CSB proteins interact with p53 and regulate its Mdm2-dependent ubiquitination. Cell Cycle 2011, 10, 3719–3730. [Google Scholar] [CrossRef] [Green Version]
- Paccosi, E.; Proietti-De-Santis, L. The emerging role of Cockayne group A and B proteins in ubiquitin/proteasome-directed protein degradation. Mech. Ageing Dev. 2021, 195, 111466. [Google Scholar] [CrossRef] [PubMed]
- Filippi, S.; Latini, P.; Frontini, M.; Palitti, F.; Egly, J.M.; Proietti-De-Santis, L. CSB protein is (a direct target of HIF-1 and) a critical mediator of the hypoxic response. EMBO J. 2008, 27, 2545–2556. [Google Scholar] [CrossRef]
- Harris, A.L. Hypoxia—A key regulatory factor in tumour growth. Nat. Rev. Cancer 2002, 2, 38–47. [Google Scholar] [CrossRef]
- Storz, P. Reactive oxygen species in tumor progression. Front. Biosci. 2005, 10, 1881–1896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yadav, R.K.; Chae, S.W.; Kim, H.R.; Chae, H.J. Endoplasmic reticulum stress and cancer. J. Cancer Prev. 2014, 19, 75–88. [Google Scholar] [CrossRef]
- Pascucci, B.; Lemma, T.; Iorio, E.; Giovannini, S.; Vaz, B.; Iavarone, I.; Calcagnile, A.; Narciso, L.; Degan, P.; Podo, F.; et al. An altered redox balance mediates the hypersensitivity of Cockayne syndrome primary fibroblasts to oxidative stress. Aging Cell 2012, 11, 520–529. [Google Scholar] [CrossRef]
- D’Errico, M.; Pascucci, B.; Iorio, E.; Van Houten, B.; Dogliotti, E. The role of CSA and CSB protein in the oxidative stress response. Mech. Ageing Dev. 2013, 134, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Cleaver, J.E.; Brennan-Minnella, A.M.; Swanson, R.A.; Fong, K.W.; Chen, J.; Chou, K.M.; Chen, Y.W.; Revet, I.; Bezrookove, V. Mitochondrial reactive oxygen species are scavenged by Cockayne syndrome B protein in human fibroblasts without nuclear DNA damage. Proc. Natl. Acad. Sci. USA 2014, 111, 13487–13492. [Google Scholar] [CrossRef] [Green Version]
- Caputo, M.; Balzerano, A.; Arisi, I.; D’Onofrio, M.; Brandi, R.; Bongiorni, S.; Brancorsini, S.; Frontini, M.; Proietti-De-Santis, L. CSB ablation induced apoptosis is mediated by increased endoplasmic reticulum stress response. PLoS ONE 2017, 12, e0172399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stubbert, L.J.; Smith, J.M.; McKay, B.C. Decreased transcription-coupled nucleotide excision repair capacity is associated with increased p53- and MLH1-independent apoptosis in response to cisplatin. BMC Cancer 2010, 10, 207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Z.; Zhang, X.; Tuo, J.; Guo, Y.; Green, B.; Chan, C.C.; Tan, W.; Huang, Y.; Ling, W.; Kadlubar, F.F.; et al. A variant of the Cockayne syndrome B gene ERCC6 confers risk of lung cancer. Hum. Mutat. 2008, 29, 113–122. [Google Scholar] [CrossRef] [Green Version]
- Song, X.; Wang, S.; Hong, X.; Li, X.; Zhao, X.; Huai, C.; Chen, H.; Gao, Z.; Qian, J.; Wang, J.; et al. Single nucleotide polymorphisms of nucleotide excision repair pathway are significantly associated with outcomes of platinum-based chemotherapy in lung cancer. Sci. Rep. 2017, 7, 11785. [Google Scholar] [CrossRef] [PubMed]
- Ceccaldi, R.; O’Connor, K.W.; Mouw, K.W.; Li, A.Y.; Matulonis, U.A.; D’Andrea, A.D.; Konstantinopoulos, P.A. A unique subset of epithelial ovarian cancers with platinum sensitivity and PARP inhibitor resistance. Cancer Res. 2015, 75, 628–634. [Google Scholar] [CrossRef] [Green Version]
- Chae, Y.K.; Anker, J.F.; Carneiro, B.A.; Chandra, S.; Kaplan, J.; Kalyan, A.; Santa-Maria, C.A.; Platanias, L.C.; Giles, F.J. Genomic landscape of DNA repair genes in cancer. Oncotarget 2016, 7, 23312–23321. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spyropoulou, Z.; Papaspyropoulos, A.; Lagopati, N.; Myrianthopoulos, V.; Georgakilas, A.G.; Fousteri, M.; Kotsinas, A.; Gorgoulis, V.G. Cockayne Syndrome Group B (CSB): The Regulatory Framework Governing the Multifunctional Protein and Its Plausible Role in Cancer. Cells 2021, 10, 866. https://doi.org/10.3390/cells10040866
Spyropoulou Z, Papaspyropoulos A, Lagopati N, Myrianthopoulos V, Georgakilas AG, Fousteri M, Kotsinas A, Gorgoulis VG. Cockayne Syndrome Group B (CSB): The Regulatory Framework Governing the Multifunctional Protein and Its Plausible Role in Cancer. Cells. 2021; 10(4):866. https://doi.org/10.3390/cells10040866
Chicago/Turabian StyleSpyropoulou, Zoi, Angelos Papaspyropoulos, Nefeli Lagopati, Vassilios Myrianthopoulos, Alexandros G. Georgakilas, Maria Fousteri, Athanassios Kotsinas, and Vassilis G. Gorgoulis. 2021. "Cockayne Syndrome Group B (CSB): The Regulatory Framework Governing the Multifunctional Protein and Its Plausible Role in Cancer" Cells 10, no. 4: 866. https://doi.org/10.3390/cells10040866
APA StyleSpyropoulou, Z., Papaspyropoulos, A., Lagopati, N., Myrianthopoulos, V., Georgakilas, A. G., Fousteri, M., Kotsinas, A., & Gorgoulis, V. G. (2021). Cockayne Syndrome Group B (CSB): The Regulatory Framework Governing the Multifunctional Protein and Its Plausible Role in Cancer. Cells, 10(4), 866. https://doi.org/10.3390/cells10040866