
Perspective: Treatment for Disease Modification in Chronic Neurodegeneration


Abstract
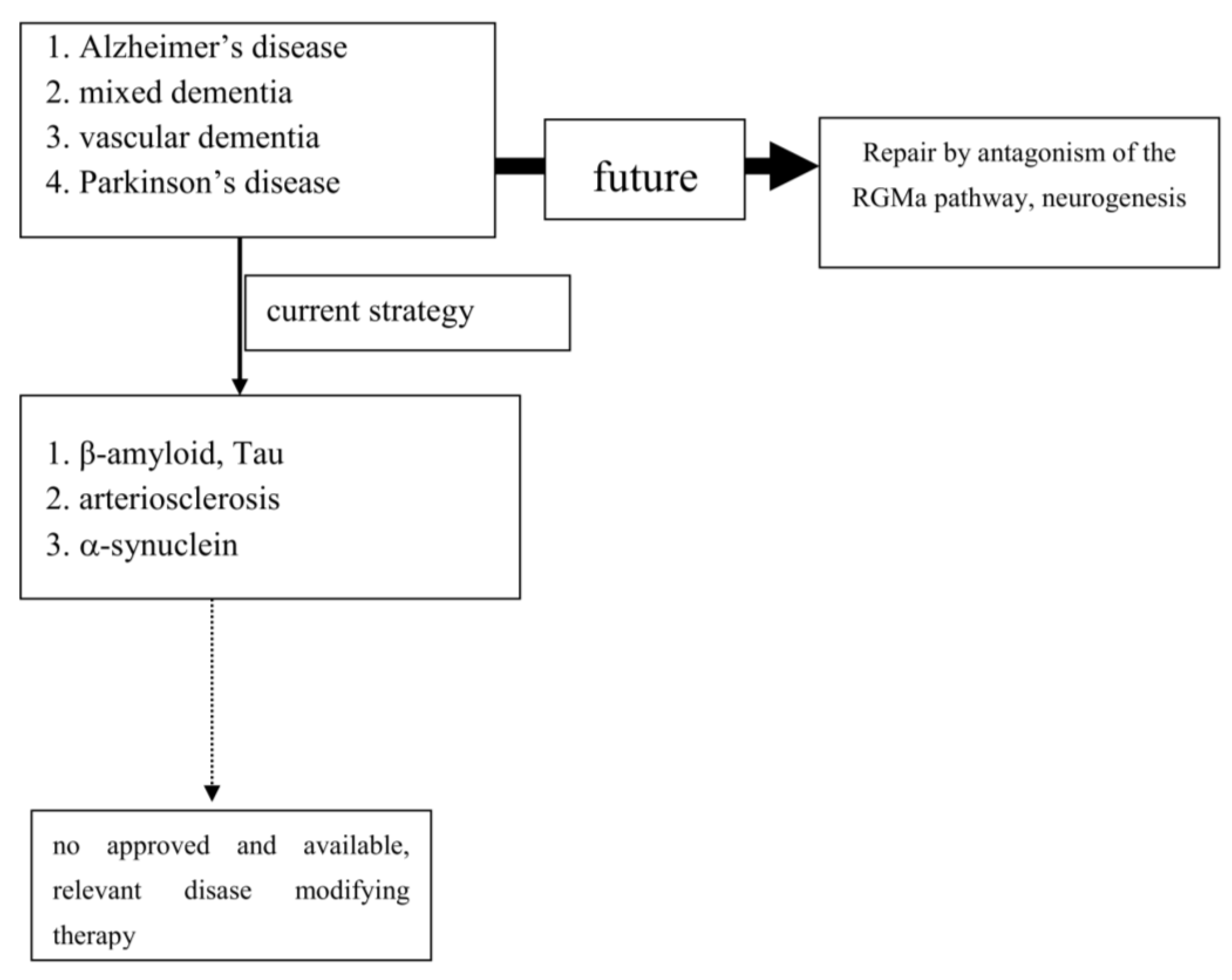
:1. Introduction
The Current Situation and Unmet Needs
2. Pitfalls of Translational Concepts in Clinical Research
3. Dementia Syndromes
Pragmatism of Clincians
4. Parkinson’s Disease
4.1. Excurs: Clinical Research on Disease Modification in PD with MAO-B Inhibition
4.2. Current Ongoing Clinical Research Strategies on Disease Modification in PD
5. Conclusions
6. Outlook
Funding
Conflicts of Interest
References
- Dorsey, E.R.; Elbaz, A.; Nichols, E.; Abd-Allah, F.; Abdelalim, A.; Adsuar, J.C.; Ansha, M.G.; Brayne, C.; Choi, J.-Y.J.; Collado-Mateo, D.; et al. Global, regional, and national burden of Parkinson’s disease, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2018, 17, 939–953. [Google Scholar] [CrossRef] [Green Version]
- Nichols, E.; Szoeke, C.E.; Vollset, S.E.; Abbasi, N.; Abd-Allah, F.; Abdela, J.; Aichour, M.T.E.; Akinyemi, R.O.; Alahdab, F.; Asgedom, S.W.; et al. Global, regional, and national burden of Alzheimer’s disease and other dementias, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 88–106. [Google Scholar] [CrossRef] [Green Version]
- Deuschl, G.; Beghi, E.; Fazekas, F.; Varga, T.; Christoforidi, K.A.; Sipido, E.; Bassetti, C.L.; Vos, T.; Feigin, V.L. The burden of neurological diseases in Europe: An analysis for the Global Burden of Disease Study 2017. Lancet Public Health 2020, 5, e551–e567. [Google Scholar] [CrossRef]
- Chaplot, K.; Jarvela, T.S.; Lindberg, I. Secreted Chaperones in Neurodegeneration. Front. Aging Neurosci. 2020, 12, 268. [Google Scholar] [CrossRef]
- Gracia, P.; Camino, J.D.; Volpicelli-Daley, L.; Cremades, N. Multiplicity of α-Synuclein Aggregated Species and Their Possible Roles in Disease. Int. J. Mol. Sci. 2020, 21, 8043. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, M.J.; Okun, M.S. Diagnosis and Treatment of Parkinson Disease: A Review. JAMA 2020, 323, 548–560. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, H.; Abushouk, A.I.; Gabr, M.; Negida, A.; Abdel-Daim, M.M. Parkinson’s disease and pesticides: A meta-analysis of disease connection and genetic alterations. Biomed. Pharmacother. 2017, 90, 638–649. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Ma, T.; Qu, B.; Ji, Y.; Liu, Z. Pesticide-Induced Gene Mutations and Parkinson Disease Risk: A Meta-Analysis. Genet. Test. Mol. Biomark. 2013, 17, 826–832. [Google Scholar] [CrossRef] [PubMed]
- Riederer, P.; Ter Meulen, V. Coronaviruses: A challenge of today and a call for extended human postmortem brain analyses. J. Neural Transm. 2020, 127, 1217–1228. [Google Scholar] [CrossRef]
- Birkmayer, W.; Hornykiewicz, O. The L-3,4-dioxyphenylalanine (DOPA)-effect in Parkinson-akinesia. Wien. Klin. Wochenschr. 1961, 73, 787–788. [Google Scholar] [PubMed]
- Cotzias, G.C.; Papavasiliou, P.S.; Gellene, R. Modification of Parkinsonism—Chronic treatment with L-dopa. N. Engl. J. Med. 1969, 280, 337–345. [Google Scholar] [CrossRef]
- Carlsson, A. Biochemical and pharmacological aspects of Parkinsonism. Acta Neurol. Scand. Suppl. 1972, 51, 11–42. [Google Scholar] [PubMed]
- Tolosa, E.; Marti, M.J.; Valldeoriola, F.; Molinuevo, J.L. History of levodopa and dopamine agonists in Parkinson’s disease treatment. Neurology 1998, 50 (Suppl. 6), S2–S10. [Google Scholar] [CrossRef]
- De Bie, R.M.A.; Clarke, C.E.; Espay, A.J.; Fox, S.H.; Lang, A.E. Initiation of pharmacological therapy in Parkinson’s disease: When, why, and how. Lancet Neurol. 2020, 19, 452–461. [Google Scholar] [CrossRef]
- Leal, R.M.; Rascol, O.; Ferreira, J.J. The “long and winding road” of the disease-modifying effects of levodopa has not ended yet. Mov. Disord. 2020, 35, 397–399. [Google Scholar] [CrossRef] [PubMed]
- Müller, T. Detoxification and antioxidative therapy for levodopa-induced neurodegeneration in Parkinson’s disease. Expert Rev. Neurother. 2013, 13, 707–718. [Google Scholar] [CrossRef]
- Verschuur, C.V.; Suwijn, S.R.; Boel, J.A.; Post, B.; Bloem, B.R.; van Hilten, J.J.; van Laar, T.; Tissingh, G.; Munts, A.G.; Deuschl, G.; et al. Randomized Delayed-Start Trial of Levodopa in Parkinson’s Disease. N. Engl. J. Med. 2019, 380, 315–324. [Google Scholar] [CrossRef]
- Müller, T. Pharmacokinetics and pharmacodynamics of levodopa/carbidopa cotherapies for Parkinson’s disease. Expert Opin. Drug Metab. Toxicol. 2020, 16, 403–414. [Google Scholar] [CrossRef] [PubMed]
- Ramot, Y.; Nyska, A.; Maronpot, R.R.; Shaltiel-Karyo, R.; Tsarfati, Y.; Manno, R.A.; Sacco, G.; Yacoby-Zeevi, O. Ninety-day Local Tolerability and Toxicity Study of ND0612, a Novel Formulation of Levodopa/Carbidopa, Administered by Subcutaneous Continuous Infusion in Minipigs. Toxicol. Pathol. 2017, 45, 764–773. [Google Scholar] [CrossRef]
- Gannon, M.; Che, P.; Chen, Y.; Jiao, K.; Roberson, E.D.; Wang, Q. Noradrenergic dysfunction in Alzheimer’s disease. Front. Neurosci. 2015, 9, 220. [Google Scholar] [CrossRef] [Green Version]
- Moll, G.; Gsell, W.; Wichart, I.; Jellinger, K.; Riederer, P. Cholinergic and monoaminergic neuromediator systems in DAT. Neuropathological and neurochemical findings. In Alzheimer’s Disease. Epidemiology, Neuropathology, Neurochemistry, and Clinics; Maurer, K., Riederer, P., Beckmann, H., Eds.; Springer: Vienna, Austria, 1990; pp. 235–243. [Google Scholar]
- Gilhus, N.E.; Deuschl, G. Neuroinflammation—A common thread in neurological disorders. Nat. Rev. Neurol. 2019, 15, 429–430. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, E.C.; Standaert, D.G. Ten Unsolved Questions about Neuroinflammation in Parkinson’s Disease. Mov. Disord. 2021, 36, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Lane, C.A.; Hardy, J.; Schott, J.M. Alzheimer’s disease. Eur. J. Neurol. 2018, 25, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Marucci, G.; Buccioni, M.; Ben, D.D.; Lambertucci, C.; Volpini, R.; Amenta, F. Efficacy of acetylcholinesterase inhibitors in Alzheimer’s disease. Neuropharmacology 2020, 108352. [Google Scholar] [CrossRef]
- Koola, M.M. Galantamine-Memantine combination in the treatment of Alzheimer’s disease and beyond. Psychiatry Res. 2020, 293, 113409. [Google Scholar] [CrossRef] [PubMed]
- Petrazzuoli, F.; Vinker, S.; Palmqvist, S.; Midlöv, P.; De Lepeleire, J.; Pirani, A.; Frese, T.; Buono, N.; Ahrensberg, J.; Asenova, R.; et al. Unburdening dementia—A basic social process grounded theory based on a primary care physician survey from 25 countries. Scand. J. Prim. Health Care 2020, 38, 253–264. [Google Scholar] [CrossRef]
- Viel, T.A.; Toricelli, M.; Pereira, A.A.R.; Abrao, G.S.; Malerba, H.N.; Maia, J.; Buck, H.S. Mechanisms of neuroplasticity and brain degeneration: Strategies for protection during the aging process. Neural Regen. Res. 2021, 16, 58–67. [Google Scholar] [CrossRef]
- Boonman, Z.; Isacson, O. Apoptosis in Neuronal Development and Transplantation: Role of Caspases and Trophic Factors. Exp. Neurol. 1999, 156, 1–15. [Google Scholar] [CrossRef]
- Demicheva, E.; Cui, Y.-F.; Bardwell, P.; Barghorn, S.; Kron, M.; Meyer, A.H.; Schmidt, M.; Gerlach, B.; Leddy, M.; Barlow, E.; et al. Targeting Repulsive Guidance Molecule A to Promote Regeneration and Neuroprotection in Multiple Sclerosis. Cell Rep. 2015, 10, 1887–1898. [Google Scholar] [CrossRef] [Green Version]
- Saitoh, Y.; Takahashi, Y. Riluzole for the treatment of amyotrophic lateral sclerosis. Neurodegener. Dis. Manag. 2020, 10, 343–355. [Google Scholar] [CrossRef]
- Gross, R.E.; Watts, R.L.; Hauser, R.A.; Bakay, R.A.; Reichmann, H.; von Kummer, R.; Ondo, W.G.; Reissig, E.; Eisner, W.; Steiner-Schulze, H.; et al. Intrastriatal transplantation of microcarrier-bound human retinal pigment epithelial cells versus sham surgery in patients with advanced Parkinson’s disease: A double-blind, randomised, controlled trial. Lancet Neurol. 2011, 10, 509–519. [Google Scholar] [CrossRef]
- Lang, A.E.; Gill, S.; Patel, N.K.; Lozano, A.; Nutt, J.G.; Penn, R.; Brooks, D.J.; Hotton, G.; Moro, E.; Heywood, P.; et al. Randomized controlled trial of intraputamenal glial cell line-derived neurotrophic factor infusion in Parkinson disease. Ann. Neurol. 2006, 59, 459–466. [Google Scholar] [CrossRef]
- Guarnieri, G.; Sarchielli, E.; Vannelli, G.B.; Morelli, A. Cell-based therapy in Alzheimer’s disease: Can human fetal cholinergic neurons “untangle the skein”? Neural Regen. Res. 2018, 13, 2105–2107. [Google Scholar]
- Liu, Z.; Cheung, H.-H. Stem Cell-Based Therapies for Parkinson Disease. Int. J. Mol. Sci. 2020, 21, 8060. [Google Scholar] [CrossRef]
- Allen, S.J.; Watson, J.J.; Shoemark, D.K.; Barua, N.U.; Patel, N.K. GDNF, NGF and BDNF as therapeutic options for neurodegeneration. Pharmacol. Ther. 2013, 138, 155–175. [Google Scholar] [CrossRef]
- Sampaio, T.B.; Savall, A.S.; Gutierrez, M.E.Z.; Pinton, S. Neurotrophic factors in Alzheimer’s and Parkinson’s diseases: Implications for pathogenesis and therapy. Neural Regen. Res. 2017, 12, 549–557. [Google Scholar] [PubMed]
- Barker, R.A.; Mason, S.L.; Harrower, T.P.; Swain, R.A.; Ho, A.K.; Sahakian, B.J.; Mathur, R.; Elneil, S.; Thornton, S.; Hurrelbrink, C.; et al. The long-term safety and efficacy of bilateral transplantation of human fetal striatal tissue in patients with mild to moderate Huntington’s disease. J. Neurol. Neurosurg. Psychiatry 2013, 84, 657–665. [Google Scholar] [CrossRef]
- Lige, L.; Zengmin, T. Transplantation of neural precursor cells in the treatment for parkinson disease: An efficacy and safety analysis. Turk. Neurosurg. 2015, 26, 378–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olanow, C.W.; Goetz, C.G.; Kordower, J.H.; Stoessl, A.J.; Sossi, V.; Brin, M.F.; Shannon, K.M.; Nauert, G.M.; Perl, D.P.; Godbold, J.; et al. A double-blind controlled trial of bilateral fetal nigral transplantation in Parkinson’s disease. Ann. Neurol. 2003, 54, 403–414. [Google Scholar] [CrossRef] [PubMed]
- Reddy, A.P.; Ravichandran, J.; Carkaci-Salli, N. Neural regeneration therapies for Alzheimer’s and Parkinson’s disease-related disorders. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165506. [Google Scholar] [CrossRef] [PubMed]
- Russ, K.; Flores, J.; Brudek, T.; Doudet, D. Neonatal human retinal pigment epithelial cells secrete limited trophic factors in vitro and in vivo following striatal implantation in parkinsonian rats. J. Neural Transm. 2015, 123, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Jha, N.K.; Jha, S.K.; Ramani, K.; Ambasta, R.K. Tau Phosphorylation, Molecular Chaperones, and Ubiquitin E3 Ligase: Clinical Relevance in Alzheimer’s Disease. J. Alzheimers Dis. 2015, 43, 341–361. [Google Scholar] [CrossRef] [PubMed]
- Liedhegner, E.A.; Steller, K.M.; Mieyal, J.J. Levodopa activates apoptosis signaling kinase 1 (ASK1) and promotes apoptosis in a neuronal model: Implications for the treatment of Parkinson’s disease. Chem. Res. Toxicol. 2011, 24, 1644–1652. [Google Scholar] [CrossRef] [Green Version]
- Naoi, M.; Maruyama, W.; Yi, H.; Inaba, K.; Akao, Y.; Shamoto-Nagai, M. Mitochondria in neurodegenerative disorders: Regulation of the redox state and death signaling leading to neuronal death and survival. J. Neural Transm. 2009, 116, 1371–1381. [Google Scholar] [CrossRef] [PubMed]
- Mothe, A.J.; Coelho, M.; Huang, L.; Monnier, P.P.; Cui, Y.-F.; Mueller, B.K.; Jacobson, P.B.; Tator, C.H. Delayed administration of the human anti-RGMa monoclonal antibody elezanumab promotes functional recovery including spontaneous voiding after spinal cord injury in rats. Neurobiol. Dis. 2020, 143, 104995. [Google Scholar] [CrossRef]
- Beal, M.F.; Oakes, D.; Shoulson, I.; Henchcliffe, C.; Galpern, W.R.; Haas, R.; Juncos, J.L.; Nutt, J.G.; Voss, T.S.; Ravina, B.; et al. A randomized clinical trial of high-dosage coenzyme Q10 in early Parkinson disease: No evidence of benefit. JAMA Neurol. 2014, 71, 543–552. [Google Scholar]
- Schapira, A.H.; McDermott, M.P.; Barone, P.; Comella, C.L.; Albrecht, S.; Hsu, H.H.; Massey, D.H.; Mizuno, Y.; Poewe, W.; Rascol, O.; et al. Pramipexole in patients with early Parkinson’s disease (PROUD): A randomised delayed-start trial. Lancet Neurol. 2013, 12, 747–755. [Google Scholar] [CrossRef] [Green Version]
- Whone, A.L.; Watts, R.L.; Stoessl, A.J.; Davis, M.; Reske, S.; Nahmias, C.; Lang, A.E.; Rascol, O.; Ribeiro, M.J.; Remy, P.; et al. Slower progression of Parkinson’s disease with ropinirole versus levodopa: The REAL-PET study. Ann Neurol. 2003, 54, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Oertel, W.H.; Wolters, E.; Sampaio, C.; Gimenez-Roldan, S.; Bergamasco, B.; Dujardin, M.; Grosset, D.G.; Arnold, G.; Leenders, K.L.; Hundemer, H.P.; et al. Pergolide versus levodopa monotherapy in early Parkinson’s disease patients: The PELMOPET study. Mov. Disord. 2006, 21, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Parkinson Study Group. A Controlled, Randomized, Delayed-Start Study of Rasagiline in Early Parkinson Disease. Arch. Neurol. 2004, 61, 561–566. [Google Scholar] [CrossRef]
- Rascol, O.; Fitzer-Attas, C.J.; Hauser, R.; Jankovic, J.; Lang, A.; Langston, J.W.; Melamed, E.; Poewe, W.; Stocchi, F.; Tolosa, E.; et al. A double-blind, delayed-start trial of rasagiline in Parkinson’s disease (the ADAGIO study): Prespecified and post-hoc analyses of the need for additional therapies, changes in UPDRS scores, and non-motor outcomes. Lancet Neurol. 2011, 10, 415–423. [Google Scholar] [CrossRef]
- Parkinson Study Group. A controlled trial of rasagiline in early Parkinson disease: The TEMPO Study. Arch. Neurol. 2002, 59, 1937–1943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parkinson Study Group. Pramipexole vs levodopa as initial treatment for Parkinson disease: A randomized controlled trial. JAMA 2000, 284, 1931–1938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pålhagen, S.; Heinonen, E.; Hägglund, J.; Kaugesaar, T.; Kontants, H.; Mäki-Ikola, O.; Palm, R.; Turunen, J.; Swedish Parkinson Study Group. Selegiline delays the onset of disability in de novo parkinsonian patients. Neurology 1998, 51, 520–525. [Google Scholar] [CrossRef] [PubMed]
- Parkinson Study Group. Impact of deprenyl and tocopherol treatment on Parkinson’s disease in DATATOP patients requiring levodopa. Ann. Neurol. 1996, 39, 37–45. [Google Scholar] [CrossRef]
- Olanow, C.; Hauser, R.; Gauguster, L.; Malapira, T.; Koller, W.; Hubble, J.; Bushenbark, K.; Lilienfeld, D.; Esterlitz, J. The effect of deprenyl and levodopa on the progression of Parkinson’s disease. Ann. Neurol. 1995, 38, 771–777. [Google Scholar] [CrossRef]
- Emamzadeh, F.N.; Surguchov, A. Parkinson’s Disease: Biomarkers, Treatment, and Risk Factors. Front. Neurosci. 2018, 12, 612. [Google Scholar] [CrossRef]
- Sian-Hulsmann, J.; Monoranu, C.; Strobel, S.; Riederer, P. Lewy Bodies: A Spectator or Salient Killer? CNS Neurol. Disord. Drug Targets 2015, 14, 947–955. [Google Scholar] [CrossRef]
- Müller, T. Investigational agents for the management of Huntington’s disease. Expert Opin. Investig. Drugs 2016, 26, 175–185. [Google Scholar] [CrossRef]
- Guarnieri, G.; Sarchielli, E.; Comeglio, P.; Herrera-Puerta, E.; Piaceri, I.; Nacmias, B.; Benelli, M.; Kelsey, G.; Maggi, M.; Gallina, P.; et al. Tumor Necrosis Factor α Influences Phenotypic Plasticity and Promotes Epigenetic Changes in Human Basal Forebrain Cholinergic Neuroblasts. Int. J. Mol. Sci. 2020, 21, 6128. [Google Scholar] [CrossRef]
- Kreisl, W.C.; Kim, M.-J.; Coughlin, J.M.; Henter, I.D.; Owen, D.R.; Innis, R.B. PET imaging of neuroinflammation in neurological disorders. Lancet Neurol. 2020, 19, 940–950. [Google Scholar] [CrossRef]
- Avila, J.; Pallas, N.; Bolos, M.; Sayas, C.L.; Hernandez, F. Intracellular and extracelleular microtubule associated protein tau as a therapeutic target in Alzheimer disease and other tauopathies. Expert Opin. Ther. Targets 2015, 20, 653–661. [Google Scholar] [CrossRef]
- Awasthi, M.; Singh, S.; Pandey, V.P.; Dwivedi, U.N. Alzheimer’s disease: An overview of amyloid beta dependent pathogenesis and its therapeutic implications along with in silico approaches emphasizing the role of natural products. J. Neurol. Sci. 2016, 361, 256–271. [Google Scholar] [CrossRef]
- Qiu, Y.; Li, L.; Zhou, T.Y.; Lu, W. Alzheimer’s disease progression model based on integrated biomarkers and clinical measures. Acta Pharmacol. Sin. 2014, 35, 1111–1120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez-Vieitez, E.; Nielsen, H.M. Associations between APOE Variants, Tau and α-Synuclein. Adv. Exp. Med. Biol. 2019, 1184, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Pozo, A.; Das, S.; Hyman, B.T. APOE and Alzheimer’s disease: Advances in genetics, pathophysiology, and therapeutic approaches. Lancet Neurol. 2021, 20, 68–80. [Google Scholar] [CrossRef]
- Ballard, C.; Aarsland, D.; Cummings, J.; O’Brien, J.; Mills, R.; Molinuevo, J.L.; Fladby, T.; Williams, G.; Doherty, P.; Corbett, A.; et al. Drug repositioning and repurposing for Alzheimer disease. Nat. Rev. Neurol. 2020, 16, 661–673. [Google Scholar] [CrossRef]
- Livingston, G.; Sommerlad, A.; Orgeta, V.; Costafreda, S.G.; Huntley, J.; Ames, D.; Ballard, C.; Banerjee, S.; Burns, A.; Cohen-Mansfield, J.; et al. Dementia prevention, intervention, and care. Lancet 2017, 390, 2673–2734. [Google Scholar] [CrossRef] [Green Version]
- Giordani, B.; Boivin, M.; Hall, A.; Foster, N.; Lehtinen, S.; Bluemlein, L.; Berent, S. The utility and generality of Mini-Mental State Examination scores in Alzheimer’s disease. Neurology 1990, 40, 1894–1896. [Google Scholar] [CrossRef] [PubMed]
- Hoops, S.; Nazem, S.; Siderowf, A.D.; Duda, J.E.; Xie, S.X.; Stern, M.B.; Weintraub, D. Validity of the MoCA and MMSE in the detection of MCI and dementia in Parkinson disease. Neurology 2009, 73, 1738–1745. [Google Scholar] [CrossRef]
- Walter, S.; Dufouil, C.; Gross, A.L.; Jones, R.N.; Mungas, D.; Filshtein, T.J.; Manly, J.J.; Arpawong, T.E.; Glymour, M.M. Neuropsychological Test Performance and MRI Markers of Dementia Risk: Reducing Education Bias. Alzheimer Dis. Assoc. Disord. 2019, 33, 179–185. [Google Scholar] [CrossRef]
- Zhou, A.; Jia, J. The value of the clock drawing test and the mini-mental state examination for identifying vascular cognitive impairment no dementia. Int. J. Geriatr. Psychiatry 2008, 23, 422–426. [Google Scholar] [CrossRef]
- Scarmeas, N.; Albert, M.; Brandt, J.; Blacker, D.; Hadjigeorgiou, G.; Papadimitriou, A.; Dubois, B.; Sarazin, M.; Wegesin, D.; Marder, K.; et al. Motor signs predict poor outcomes in Alzheimer disease. Neurology 2005, 64, 1696–1703. [Google Scholar] [CrossRef] [Green Version]
- Pohanka, M. Vaccination to Alzheimer Disease. Is it a Promising Tool or a Blind Way? Curr. Med. Chem. 2016, 23, 1432–1441. [Google Scholar] [CrossRef]
- Song, G.; Yang, H.; Shen, N.; Pham, P.; Brown, B.; Lin, X.; Hong, Y.; Sinu, P.; Cai, J.; Li, X.; et al. An Immunomodulatory Therapeutic Vaccine Targeting Oligomeric Amyloid-beta. J. Alzheimers Dis. 2020, 77, 1639–1653. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.J.; Zhou, H.D.; Zhou, X.F. Modified immunotherapies against Alzheimer’s disease: Toward safer and effective amyloid clearance. J. Alzheimers Dis. 2010, 21, 1065–1075. [Google Scholar] [CrossRef]
- Yang, C.; Xiao, S. New developments of clinical trial in immunotherapy for Alzheimer’s disease. Curr. Pharm. Biotechnol. 2015, 16, 484–491. [Google Scholar] [CrossRef] [PubMed]
- Joly-Amado, A.; Davtyan, H.; Serraneau, K.; Jules, P.; Zitnyar, A.; Pressman, E.; Zagorski, K.; Antonyan, T.; Hovakimyan, A.; Paek, H.; et al. Active immunization with tau epitope in a mouse model of tauopathy induced strong antibody response together with improvement in short memory and pSer396-tau pathology. Neurobiol. Dis. 2020, 134, 104636. [Google Scholar] [CrossRef] [PubMed]
- Schneider, F.; Horowitz, A.; Lesch, K.-P.; Dandekar, T. Delaying memory decline: Different options and emerging solutions. Transl. Psychiatry 2020, 10, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; He, Z.; Xing, Z.; Zuo, Z.; Yuan, L.; Wu, Y.; Jiang, M.; Qi, F.; Yao, Z. Influenza vaccination in early Alzheimer’s disease rescues amyloidosis and ameliorates cognitive deficits in APP/PS1 mice by inhibiting regulatory T cells. J. Neuroinflamm. 2020, 17, 65. [Google Scholar] [CrossRef]
- Liang, Z.; Zhao, Y.; Ruan, L.; Zhu, L.; Jin, K.; Zhuge, Q.; Su, D.-M.; Zhao, Y. Impact of aging immune system on neurodegeneration and potential immunotherapies. Prog. Neurobiol. 2017, 157, 2–28. [Google Scholar] [CrossRef]
- Lisko, I.; Kulmala, J.; Annetorp, M.; Ngandu, T.; Mangialasche, F.; Kivipelto, M. How can dementia and disability be prevented in older adults: Where are we today and where are we going? J. Intern. Med. 2020. [Google Scholar] [CrossRef]
- Nugent, S.; Potvin, O.; Cunnane, S.C.; Chen, T.-H.; Duchesne, S. Associating Type 2 Diabetes Risk Factor Genes and FDG-PET Brain Metabolism in Normal Aging and Alzheimer’s Disease. Front. Aging Neurosci. 2020, 12, 580633. [Google Scholar] [CrossRef]
- Cheng, H.-C.; Ulane, C.M.; Burke, R.E. Clinical progression in Parkinson disease and the neurobiology of axons. Ann. Neurol. 2010, 67, 715–725. [Google Scholar] [CrossRef]
- Berg, D.; Godau, J.; Seppi, K.; Behnke, S.; Liepelt-Scarfone, I.; Lerche, S.; Stockner, H.; Gaenslen, A.; Mahlknecht, P.; Huber, H.; et al. The PRIPS study: Screening battery for subjects at risk for Parkinson’s disease. Eur. J. Neurol. 2013, 20, 102–108. [Google Scholar] [CrossRef]
- Mahlknecht, P.; Seppi, K.; Poewe, W. The Concept of Prodromal Parkinson’s Disease. J. Parkinson’s Dis. 2015, 5, 681–697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braak, H.; Rub, U.; Gai, W.P.; Del, T.K. Idiopathic Parkinson’s disease: Possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J. Neural Transm. 2003, 110, 517–536. [Google Scholar] [CrossRef] [PubMed]
- Halliday, G.; McCann, H.; Shepherd, C. Evaluation of the Braak hypothesis: How far can it explain the pathogenesis of Parkinson’s disease? Expert Rev. Neurother. 2012, 12, 673–686. [Google Scholar] [CrossRef]
- Kingsbury, A.E.; Bandopadhyay, R.; Silveira-Moriyama, L.; Ayling, H.; Kallis, C.; Sterlacci, W.; Maeir, H.; Poewe, W.; Lees, A.J. Brain stem pathology in Parkinson’s disease: An evaluation of the Braak staging model. Mov. Disord. 2010, 25, 2508–2515. [Google Scholar] [CrossRef] [PubMed]
- Jellinger, K.A. Is Braak staging valid for all types of Parkinson’s disease? J. Neural Transm. 2019, 126, 423–431. [Google Scholar] [CrossRef] [PubMed]
- Brooks, D.J. Examining Braak’s hypothesis by imaging Parkinson’s disease. Mov. Disord. 2010, 25 (Suppl. 1), S83–S88. [Google Scholar] [CrossRef]
- Patterson, L.; Rushton, S.P.; Attems, J.; Thomas, A.J.; Morris, C.M. Degeneration of dopaminergic circuitry influences depressive symptoms in Lewy body disorders. Brain Pathol. 2018, 29, 544–557. [Google Scholar] [CrossRef]
- Kalia, L.V.; Lang, A.E.; Hazrati, L.-N.; Fujioka, S.; Wszolek, Z.K.; Dickson, D.W.; Ross, O.A.; Van Deerlin, V.M.; Trojanowski, J.Q.; Hurtig, H.I.; et al. Clinical Correlations With Lewy Body Pathology inLRRK2-Related Parkinson Disease. JAMA Neurol. 2015, 72, 100–105. [Google Scholar] [CrossRef] [Green Version]
- Weiner, W.J. There Is No Parkinson Disease. Arch. Neurol. 2008, 65, 705–708. [Google Scholar] [CrossRef] [Green Version]
- Poewe, W. Non-motor symptoms in Parkinson’s disease. Eur. J. Neurol. 2008, 15 (Suppl. 1), 14–20. [Google Scholar] [CrossRef]
- Przuntek, H.; Riederer, P. Diagnostic staging of Parkinson?s disease: Conceptual aspects. J. Neural Transm. 2004, 111, 201–216. [Google Scholar] [CrossRef]
- Shabir, O.; Moll, T.A.; Matuszyk, M.M.; Eyre, B.; Dake, M.D.; Berwick, J.; Francis, S.E. Preclinical models of disease and multimorbidity with focus upon cardiovascular disease and dementia. Mech. Ageing Dev. 2020, 192, 111361. [Google Scholar] [CrossRef]
- Parkinson Study Group. Dopamine Transporter Brain Imaging to Assess the Effects of Pramipexole vs Levodopa on Parkinson Disease Progression. JAMA 2002, 287, 1653–1661. [Google Scholar] [CrossRef] [PubMed]
- Fahn, S.; Oakes, D.; Shoulson, I.; Kieburtz, K.; Rudolph, A.; Lang, A.; Olanow, C.W.; Tanner, C.; Schifitto, G.; Zhao, H.; et al. Levodopa and the progression of Parkinson’s disease. N. Engl. J. Med. 2004, 351, 2498–2508. [Google Scholar]
- Przuntek, H.; Conrad, B.; Dichgans, J.; Kraus, P.; Krauseneck, P.; Pergande, G.; Rinne, U.; Schimrigk, K.; Schnitker, J.; Vogel, H. SELEDO: A 5-year long-term trial on the effect of selegiline in early parkinsonian patients treated with levodopa. Eur. J. Neurol. 1999, 6, 141–150. [Google Scholar] [CrossRef]
- Olanow, C.W.; Hauser, R.A.; Jankovic, J.; Langston, W.; Lang, A.; Poewe, W.; Tolosa, E.; Stocchi, F.; Melamed, E.; Eyal, E.; et al. A randomized, double-blind, placebo-controlled, delayed start study to assess rasagiline as a disease modifying therapy in Parkinson’s disease (the ADAGIO study): Rationale, design, and baseline characteristics. Mov. Disord. 2008, 23, 2194–2201. [Google Scholar] [CrossRef] [PubMed]
- Shoulson, I.; Oakes, D.; Fahn, S.; Lang, A.; Langston, J.W.; LeWitt, P.; Olanow, C.W.; Penney, J.B.; Tanner, C.; Kieburtz, K.; et al. Impact of sustained deprenyl (selegiline) in levodopa-treated Parkinson’s disease: A randomized placebo-controlled extension of the deprenyl and tocopherol antioxidative therapy of parkinsonism trial. Ann. Neurol. 2002, 51, 604–612. [Google Scholar] [CrossRef]
- Balestrino, R.; Tunesi, S.; Tesei, S.; Lopiano, L.; Zecchinelli, A.L.; Goldwurm, S. Penetrance of Glucocerebrosidase (GBA) Mutations in Parkinson’s Disease: A Kin Cohort Study. Mov. Disord. 2020, 35, 2111–2114. [Google Scholar] [CrossRef]
- Greuel, A.; Trezzi, J.P.; Glaab, E.; Ruppert, M.C.; Maier, F.; Jäger, C.; Hodak, Z.; Lohmann, K.; Ma, Y.; Eidelberg, D.; et al. GBA Variants in Parkinson’s Disease: Clinical, Metabolomic, and Multimodal Neuroimaging Phenotypes. Mov. Disord. 2020, 35, 2201–2210. [Google Scholar] [CrossRef]
- Mullin, S.; Stokholm, M.G.; Hughes, D.; Mehta, A.; Parbo, P.; Hinz, R.; Pavese, N.; Brooks, D.J.; Schapira, A.H. Brain Microglial Activation Increased in Glucocerebrosidase (GBA) Mutation Carriers without Parkinson’s disease. Mov. Disord. 2021, 36, 774–779. [Google Scholar] [CrossRef]
- Straniero, L.; Asselta, R.; Bonvegna, S.; Rimoldi, V.; Melistaccio, G.; Soldà, G.; Aureli, M.; Della Porta, M.; Lucca, U.; Di Fonzo, A.; et al. The SPID-GBA study: Sex distribution, Penetrance, Incidence, and Dementia in GBA-PD. Neurol. Genet. 2020, 6, e523. [Google Scholar] [CrossRef]
- Thaler, A.; Shenhar-Tsarfaty, S.; Shaked, Y.; Gurevich, T.; Omer, N.; Bar-Shira, A.; Gana-Weisz, M.; Goldstein, O.; Kestenbaum, M.; Cedarbaum, J.M.; et al. Metabolic syndrome does not influence the phenotype of LRRK2 and GBA related Parkinson’s disease. Sci. Rep. 2020, 10, 9329. [Google Scholar] [CrossRef] [PubMed]
- Castonguay, A.-M.; Gravel, C.; Lévesque, M. Treating Parkinson’s Disease with Antibodies: Previous Studies and Future Directions. J. Parkinson’s Dis. 2021, 11, 71–92. [Google Scholar] [CrossRef] [PubMed]
- Jamal, F. Immunotherapies Targeting α-Synuclein in Parkinson Disease. Fed. Pract. 2020, 37, 375–379. [Google Scholar] [CrossRef] [PubMed]
- Fields, C.R.; Bengoa-Vergniory, N.; Wade-Martins, R. Targeting Alpha-Synuclein as a Therapy for Parkinson’s Disease. Front. Mol. Neurosci. 2019, 12, 299. [Google Scholar] [CrossRef] [Green Version]
- Kuhn, W.; Muller, T.; Nastos, I.; Poehlau, D. The neuroimmune hypothesis in Parkinson’s disease. Rev. Neurosci. 1997, 8, 29–34. [Google Scholar] [CrossRef]
- Sian-Hulsmann, J.; Riederer, P. The role of alpha-synuclein as ferrireductase in neurodegeneration associated with Parkinson’s disease. J. Neural Transm. 2020, 127, 749–754. [Google Scholar] [CrossRef]
- Jellinger, K.A. Interaction between α-Synuclein and Other Proteins in Neurodegenerative Disorders. Sci. World J. 2011, 11, 1893–1907. [Google Scholar] [CrossRef] [Green Version]
- Isaksen, T.J.; Yamashita, T. Repulsive Guidance Molecule A Regulates Adult Neurogenesis via the Neogenin Receptor. Neurosci. Insights 2020, 15. [Google Scholar] [CrossRef]
- Korecka, J.A.; Moloney, E.B.; Eggers, R.; Hobo, B.; Scheffer, S.; Ras-Verloop, N.; Pasterkamp, R.J.; Swaab, D.F.; Smit, A.B.; van Kesteren, R.E.; et al. Repulsive Guidance Molecule a (RGMa) Induces Neuropathological and Behavioral Changes That Closely Resemble Parkinson’s Disease. J. Neurosci. 2017, 37, 9361–9379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, T.; Barghorn, S.; Lütge, S.; Haas, T.; Mueller, R.; Gerlach, B.; Öhm, G.; Eilert, K.; Trommer, I.; Mueller, B.K. Decreased levels of repulsive guidance molecule A in association with beneficial effects of repeated intrathecal triamcinolone acetonide application in progressive multiple sclerosis patients. J. Neural Transm. 2014, 122, 841–848. [Google Scholar] [CrossRef]
- Oda, W.; Fujita, Y.; Baba, K.; Mochizuki, H.; Niwa, H.; Yamashita, T. Inhibition of repulsive guidance molecule-a protects dopaminergic neurons in a mouse model of Parkinson’s disease. Cell Death Dis. 2021, 12, 1–15. [Google Scholar] [CrossRef]
- Robinson, R.A.; Griffiths, S.C.; van de Haar, L.L.; Malinauskas, T.; van Battum, E.Y.; Zelina, P.; Schwab, R.A.; Karia, D.; Malinauskaite, L.; Brignani, S.; et al. Simultaneous binding of Guidance Cues NET1 and RGM blocks extracellular NEO1 signaling. Cell 2021. [Google Scholar] [CrossRef]
- Satoh, J.; Tabunoki, H.; Ishida, T.; Saito, Y.; Arima, K. Accumulation of a repulsive axonal guidance molecule RGMa in amyloid plaques: A possible hallmark of regenerative failure in Alzheimer’s disease brains. Neuropathol. Appl. Neurobiol. 2013, 39, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Müller, T.; Trommer, I.; Muhlack, S.; Mueller, B.K. Levodopa increases oxidative stress and repulsive guidance molecule A levels: A pilot study in patients with Parkinson’s disease. J. Neural Transm. 2016, 123, 401–406. [Google Scholar] [CrossRef]
- Babitt, J.L.; Zhang, Y.; Samad, T.A.; Xia, Y.; Tang, J.; Campagna, J.A.; Schneyer, A.L.; Woolf, C.J.; Lin, H.Y. Repulsive Guidance Molecule (RGMa), a DRAGON Homologue, Is a Bone Morphogenetic Protein Co-receptor. J. Biol. Chem. 2005, 280, 29820–29827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Key, B.; Lah, G.J. Repulsive guidance molecule A (RGMa): A molecule for all seasons. Cell Adh. Migr. 2012, 6, 85–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malinauskas, T.; Peer, T.V.; Bishop, B.; Mueller, T.D.; Siebold, C. Repulsive guidance molecules lock growth differentiation factor 5 in an inhibitory complex. Proc. Natl. Acad. Sci. USA 2020, 117, 15620–15631. [Google Scholar] [CrossRef]
- Kubo, T.; Tokita, S.; Yamashita, T. Repulsive Guidance Molecule-a and Demyelination: Implications for Multiple Sclerosis. J. Neuroimmune Pharmacol. 2011, 7, 524–528. [Google Scholar] [CrossRef] [PubMed]
- Mothe, A.J.; Tassew, N.G.; Shabanzadeh, A.P.; Penheiro, R.; Vigouroux, R.J.; Huang, L.; Grinnell, C.; Cui, Y.-F.; Fung, E.; Monnier, P.P.; et al. RGMa inhibition with human monoclonal antibodies promotes regeneration, plasticity and repair, and attenuates neuropathic pain after spinal cord injury. Sci. Rep. 2017, 7, 10529. [Google Scholar] [CrossRef]
- Charish, J.; Shabanzadeh, A.P.; Chen, D.; Mehlen, P.; Sethuramanujam, S.; Harada, H.; Bonilha, V.L.; Awatramani, G.; Bremner, R.; Monnier, P.P. Neogenin neutralization prevents photoreceptor loss in inherited retinal degeneration. J. Clin. Investig. 2020, 130, 2054–2068. [Google Scholar] [CrossRef]
- Shabanzadeh, A.P.; Tassew, N.G.; Szydlowska, K.; Tymianski, M.; Banerjee, P.; Vigouroux, R.J.; Eubanks, J.H.; Huang, L.; Geraerts, M.; Koeberle, P.D.; et al. Uncoupling Neogenin association with lipid rafts promotes neuronal survival and functional recovery after stroke. Cell Death Dis. 2015, 6, e1744. [Google Scholar] [CrossRef] [Green Version]
- Song, M.; Tian, F.; Xia, H.; Xie, Y. Repulsive guidance molecule a suppresses seizures and mossy fiber sprouting via the FAK‑p120RasGAP‑Ras signaling pathway. Mol. Med. Rep. 2019, 19, 3255–3262. [Google Scholar] [CrossRef]
- Tanabe, S.; Yamashita, T. Repulsive Guidance Molecule-a Is Involved in Th17-Cell-Induced Neurodegeneration in Autoimmune Encephalomyelitis. Cell Rep. 2014, 9, 1459–1470. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Shifman, M.I. Inhibition of neogenin promotes neuronal survival and improved behavior recovery after spinal cord injury. Neuroscience 2019, 408, 430–447. [Google Scholar] [CrossRef]
- Nakagawa, H.; Ninomiya, T.; Yamashita, T.; Takada, M. Treatment with the Neutralizing Antibody against Repulsive Guidance Molecule-a Promotes Recovery from Impaired Manual Dexterity in a Primate Model of Spinal Cord Injury. Cereb. Cortex 2018, 29, 561–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.; Sun, P. Promoting functions of microRNA-29a/199B in neurological recovery in rats with spinal cord injury through inhibition of the RGMA/STAT3 axis. J. Orthop. Surg. Res. 2020, 15, 427. [Google Scholar] [CrossRef]
- Isaksen, T.J.; Fujita, Y.; Yamashita, T. Repulsive Guidance Molecule A Suppresses Adult Neurogenesis. Stem Cell Rep. 2020, 14, 677–691. [Google Scholar] [CrossRef] [PubMed]
- Tian, C.; Shi, H.; Xiong, S.; Hu, F.; Xiong, W.-C.; Liu, J. The neogenin/DCC homolog UNC-40 promotes BMP signaling via the RGM protein DRAG-1 in C. elegans. Development 2013, 140, 4070–4080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schweyer, K.; Rüschoff-Steiner, C.; Arias-Carrión, O.; Oertel, W.H.; Rösler, T.W.; Höglinger, G.U. Neuronal precursor cells with dopaminergic commitment in the rostral migratory stream of the mouse. Sci. Rep. 2019, 9, 13359. [Google Scholar] [CrossRef] [Green Version]
- Desplats, P.; Spencer, B.; Crews, L.; Pathel, P.; Morvinski-Friedmann, D.; Kosberg, K.; Roberts, S.; Patrick, C.; Winner, B.; Winkler, J.; et al. α-Synuclein Induces Alterations in Adult Neurogenesis in Parkinson Disease Models via p53-mediated Repression of Notch1*. J. Biol. Chem. 2012, 287, 31691–31702. [Google Scholar] [CrossRef] [Green Version]
- Winner, B.; Regensburger, M.; Schreglmann, S.; Boyer, L.; Prots, I.; Rockenstein, E.; Mante, M.; Zhao, C.; Winkler, J.; Masliah, E.; et al. Role of α-Synuclein in Adult Neurogenesis and Neuronal Maturation in the Dentate Gyrus. J. Neurosci. 2012, 32, 16906–16916. [Google Scholar] [CrossRef]
- Winner, B.; Marchetto, M.C.; Winkler, J.; Gage, F.H. Human-induced pluripotent stem cells pave the road for a better understanding of motor neuron disease. Hum. Mol. Genet. 2014, 23, R27–R34. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| PD | Lewy Bodies (α-synuclein) |
| DLB | Lewy bodies plus β-amyloid |
| DLB + AD | Lewy bodies plus β-amyloid plus neurofibrillary tangles |
| PDD | Lewy bodies plus β-amyloid plus neurofibrillary tangles (tau-protein) |
| mixed variants (PSP + LB) | Lewy bodies plus neurofibrillary tangles |
| neurofibrillary tangles dementia (PSP, CBD) | neurofibrillary tangles |
| AD | β-amyloid plus neurofibrillary tangles |
| pathological ageing | β-amyloid |
| FIT | Scale | Comparison | Result | ||
|---|---|---|---|---|---|
| LEAP-Study | No | MDS-UPDRS | Early vs. later application of L-dopa | faster PD progression with longer L-dopa exposure | [17] |
| Coenzyme Q 10 | No | UPDRS | Coenzyme Q 10 vs. placebo | Negative | [47] |
| PROUD-Study | Yes | UPDRS | delayed start design; Pramipexole vs. Placebo | no difference | [48] |
| REAL-PET | Yes | UPDRS | Ropinirole vs. levodopa | positive in favor of ropinirole in terms of PET outcomes | [49] |
| PELMOPET | Yes | UPDRS | Pergolide vs. Levodopa | no difference due to use of different PET machines for intraindividual comparisons | [50] |
| ADAGIO | No | UPDRS | Delayed start design Rasagiline 1 mg or 2 mg vs. placebo | positive for 1 mg, but not 2 mg | [51,52] |
| TEMPO | No | UPDRS | Rasagiline | Positive effect of rasagiline | [51,53] |
| Pramipexole vs. levodopa as initial treatment for Parkinson disease: double blind trial. | Yes | UPDRS | Pramipexole vs. Levodopa | Tendency in favor of pramipexole (Pramipexole: 20.0% (14.2%) vs. LD: 24.8% (14.4%) mean (SD) p = 0.1) | [54] |
| Swedish selegiline study | No | UPDRS | Selegiline vs. placebo | Positive, but after 8 weeks of washout no difference | [55] |
| DATATOP | No | UPDRS | Selegiline vs. tocopherol | positive after 9 months. Endpoint was need for L-dopa therapy | [56] |
| SINDEPAR | No | UPDRS | Selegiline plus bromocriptine plus L-dopa | Positive effect of selegiline | [57] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Müller, T.; Mueller, B.K.; Riederer, P. Perspective: Treatment for Disease Modification in Chronic Neurodegeneration. Cells 2021, 10, 873. https://doi.org/10.3390/cells10040873
Müller T, Mueller BK, Riederer P. Perspective: Treatment for Disease Modification in Chronic Neurodegeneration. Cells. 2021; 10(4):873. https://doi.org/10.3390/cells10040873
Chicago/Turabian StyleMüller, Thomas, Bernhard Klaus Mueller, and Peter Riederer. 2021. "Perspective: Treatment for Disease Modification in Chronic Neurodegeneration" Cells 10, no. 4: 873. https://doi.org/10.3390/cells10040873
APA StyleMüller, T., Mueller, B. K., & Riederer, P. (2021). Perspective: Treatment for Disease Modification in Chronic Neurodegeneration. Cells, 10(4), 873. https://doi.org/10.3390/cells10040873