
Ca2+ Microdomains, Calcineurin and the Regulation of Gene Transcription


Abstract
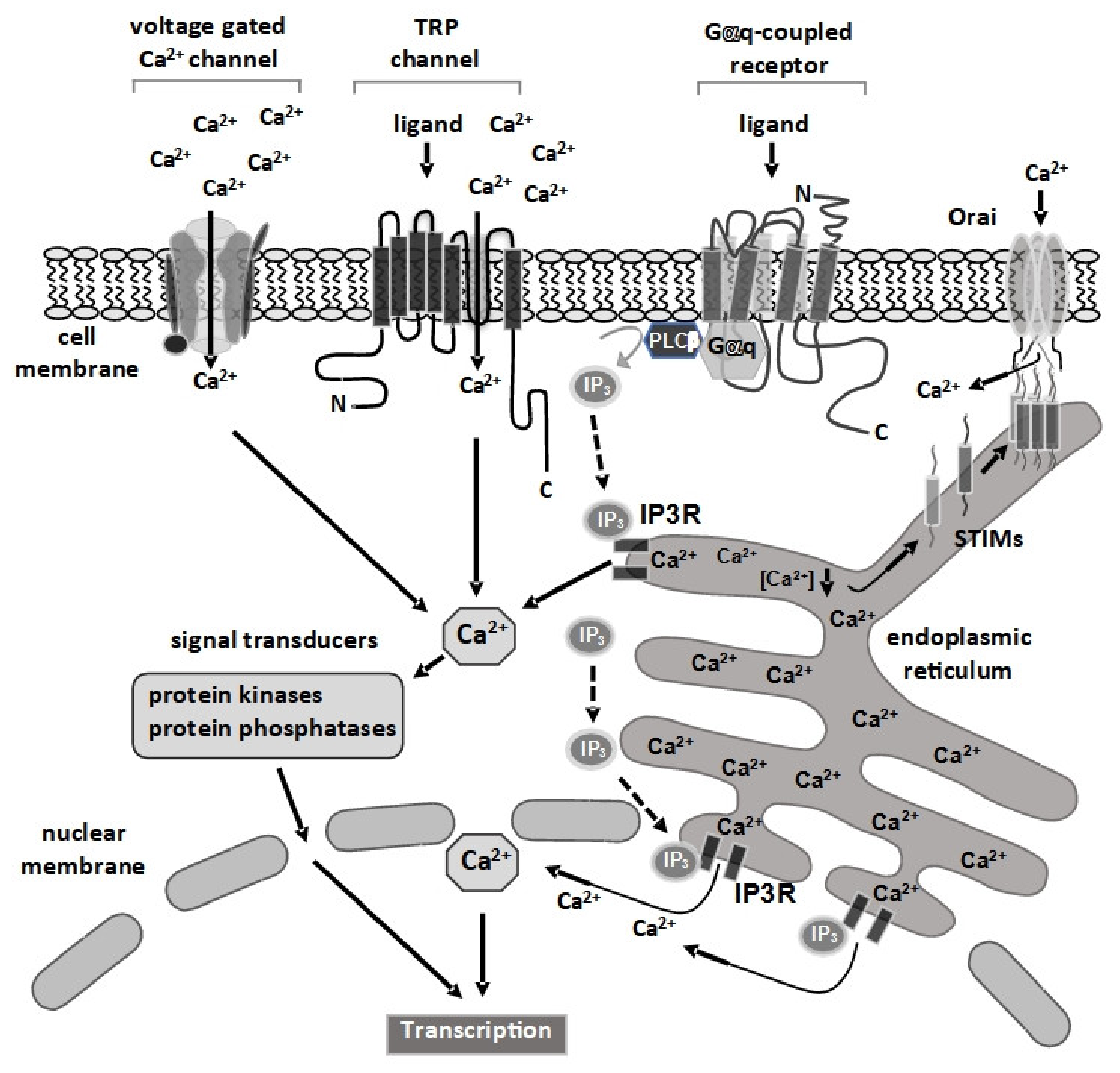
:1. Introduction
2. Signal Transducers Required for Ca2+-Induced Signaling from the Plasma Membrane to the Nucleus
3. Ca2+-Responsive Transcription Factors
4. Ca2+-Induced Gene Transcription: Role of Nuclear Ca2+
5. Calcineurin
6. Calcineurin Regulates Gene Transcription
6.1. Calcineurin-Catalyzed Dephosphorylation Activates NFAT
6.2. Calcineurin Modulates CREB Activity
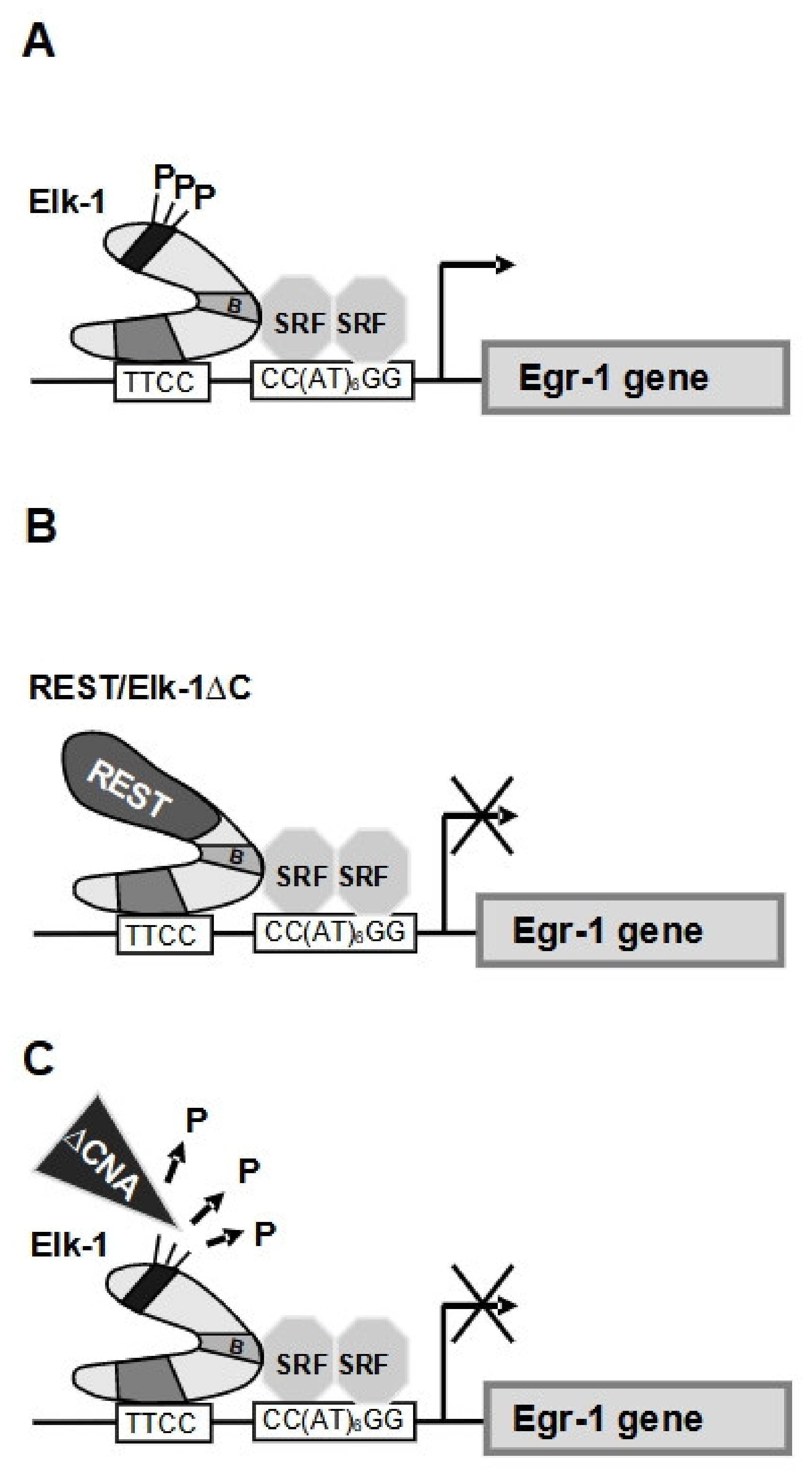
6.3. Calcineurin Dephosphorylates Elk-1 and Inhibits SRE-Mediated Transcription
7. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Morgan, A.J.; Platt, F.M.; Lloyd-Evans, E.; Galione, A. Molecular mechanisms of endolysosomal Ca2+ signaling in health and disease. Biochem. J. 2011, 439, 349–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavoie, H.; Gagnon, J.; Therrien, M. ERK signalling: A master regulator of cell behaviour, life and fate. Nat. Rev. Mol. Biol. 2020, 21, 607–632. [Google Scholar] [CrossRef]
- Rubil, S.; Rössler, O.G.; Thiel, G. CREB, AP-1, ternary complex factors and MAP kinases connect transient receptor potential melastatin-3 (TRPM3) channel stimulation with increased c-Fos expression. Brit. J. Pharmacol. 2016, 173, 305–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolmetsch, R.E.; Pajvani, U.; Fife, K.; Spotts, J.M.; Greenberg, M.E. Signaling to the nucleus by an L-type calcium channel-calmodulin complex through the MAP kinase pathway. Science 2001, 294, 333–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnette, D.; Gibson, T.B.; Lawrence, M.C.; January, B.; Khoo, S.; McGlynn, K.; Vanderbilt, C.A.; Cobb, M.H. Regulation of ERK1 and ERK2 by glucose and peptide hormones in pancreatic β cells. J. Biol. Chem. 2003, 278, 32517–32525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayer, S.I.; Thiel, G. Calcium influx into MIN6 insulinoma cells induces expression of Egr-1 involving extracellular signal-regulated protein kinase and the transcription factors Elk-1 and CREB. Eur. J. Cell Biol. 2009, 88, 19–33. [Google Scholar] [CrossRef]
- Stefano, L.; Rössler, O.G.; Griesemer, D.; Hoth, M.; Thiel, G. P2X7 receptor stimulation upregulates Egr-1 biosynthesis involving a cytosolic Ca2+ rise, transactivation of the EGF receptor and phosphorylation of ERK and Elk-1. J. Cell Physiol. 2007, 213, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Mayer, S.I.; Müller, I.; Mannebach, S.; Endo, T.; Thiel, G. 2011. Signal transduction of pregnenolone sulfate in insulinoma cells. Activation of Egr-1 expression involving TRPM3, voltage-gated calcium channels, ERK, and ternary complex factors. J. Biol. Chem. 2011, 286, 10084–10096. [Google Scholar] [CrossRef] [Green Version]
- Kaufmann, A.; Keim, A.; Thiel, G. Regulation of immediate-early gene transcription following activation of Gαq-coupled designer receptors. J. Cell Biochem. 2013, 114, 681–696. [Google Scholar] [CrossRef]
- Lesch, A.; Rössler, O.G.; Thiel, G. Extracellular signal-regulated protein kinase, c-Jun N-terminal protein kinase, and calcineurin regulate transient receptor potential M3 (TRPM3) induced activation of AP-1. J. Cell Biochem. 2017, 118, 2409–2419. [Google Scholar] [CrossRef]
- Duan, L.; Cobb, M.H. Calcineurin increases glucose activation of ERK1/2 by reversing negative feedback. Proc. Nat. Acad. Sci. USA 2010, 107, 22314–22319. [Google Scholar] [CrossRef] [Green Version]
- Backes, T.M.; Rössler, O.G.; Hui, X.; Grötzinger, C.; Lipp, P.; Thiel, G. Stimulation of TRPV1 channels activates the AP-1 transcription factor. Biochem. Pharmacol. 2018, 150, 160–169. [Google Scholar] [CrossRef]
- Rubil, S.; Lesch, A.; Mukaida, N.; Thiel, G. Stimulation of transient receptor potential M3 (TRPM3) channels increases interleukin-8 gene promoter activity involving AP-1 and extracellular signal-regulated protein kinase. Cytokine 2018, 103, 133–141. [Google Scholar] [CrossRef]
- Rubil, S.; Thiel, G. Stimulation of TRPM3 channels increases the transcriptional activation potential of Elk-1 involving cytosolic Ca2+, extracellular signal-regulated protein kinase, and calcineurin. Eur. J. Pharmacol. 2019, 844, 225–230. [Google Scholar] [CrossRef]
- Thiel, G.; Backes, T.M.; Welck, J.; Steinhausen, S.; Fischer, A.-L.; Langfermann, D.S.; Ulrich, M.; Wissenbach, U.; Rössler, O.G. Pharmacological inhibition of TRPM8-induced gene transcription. Biochem. Pharmacol. 2019, 170, 113678. [Google Scholar] [CrossRef] [PubMed]
- Müller, I.; Endo, T.; Thiel, G. 2010 Regulation of AP-1 activity in glucose-stimulated insulinoma cells. J. Cell Biochem. 2010, 110, 1481–1494. [Google Scholar] [CrossRef]
- Thiel, G.; Lesch, A.; Keim, A. Transcriptional response to calcium-sensing receptor stimulation. Endocrinology 2012, 153, 4716–4728. [Google Scholar] [CrossRef]
- Samak, G.; Narayanan, D.; Jaggar, J.H.; Rao, R. CaV1.3 channels and intracellular calcium mediate osmotic stress-induced N-terminal c-Jun kinase activation and disruption of tight junctions in Caco-2 cell monolayers. J. Biol. Chem. 2011, 286, 30232–30243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.-Y.; Hour, M.-J.; Lin, W.-C.; Wong, K.-L.; Shiao, L.-R.; Cheng, K.-S.; Chan, P.; Leung, Y.-M. Antagonism of Ca2+-sensing receptor by NPS 2143 is transiently masked by p38 activation in mouse brain bEND.3 endothelial cells. Naunyn Schmiedebergs Arch. Pharmacol. 2019, 392, 823–832. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, M.; Hata, K.; Nagayama, T.; Sakurai, T.; Nishisho, T.; Wakabayashi, H.; Hiraga, T.; Ebisu, S.; Yoneda, T. Acid activation of Trpv1 leads to an up-regulation of calcitonin gene-related peptide expression in dorsal root ganglion neurons via the CamK-CREB cascade: A potential mechanism of inflammatory pain. Mol. Biol. Cell 2010, 21, 2568–2577. [Google Scholar] [CrossRef] [Green Version]
- Sun, P.; Enslen, H.; Myung, P.S.; Maurer, R.A. Differential activation of CREB by Ca2+/calmodulin-dependent protein kinases type II and type IV involves phosphorylation of a site that negatively regulates activity. Genes Dev. 1994, 8, 2527–2539. [Google Scholar] [CrossRef] [Green Version]
- Gallin, W.J.; Greenberg, M.E. Calcium regulation of gene expression in neurons: The mode of entry matters. Curr. Opin. Neurobiol. 1995, 5, 367–374. [Google Scholar] [CrossRef]
- Wagner, T.F.J.; Loch, S.; Lambert, S.; Straub, I.; Mannebach, S.; Mathar, I.; Düfer, M.; Lis, A.; Flockerzi, V.; Philipp, S.E.; et al. Transient receptor potential M3 channels are ionotropic steroid receptors in pancreatic beta cells. Nat. Cell Biol. 2008, 10, 1421–1430. [Google Scholar] [CrossRef] [PubMed]
- Lesch, A.; Rubil, S.; Thiel, G. Activation and inhibition of transient receptor potential TRPM3-induced gene transcription. Br. J. Pharmacol. 2014, 171, 2645–2658. [Google Scholar] [CrossRef] [Green Version]
- Gallo, E.M.; Cante-Barett, K.; Crabtree, G.R. Lymphocyte calcium signaling from membrane to nucleus. Nat. Immunol. 2006, 7, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.-P.; Bakowski, D.; Mirams, G.R.; ParekH, A.B. Selective recruitment of different Ca2+-dependent transcription factors by STIM-Orai channel cluster. Nat. Commun. 2019, 10, 2516. [Google Scholar] [CrossRef]
- Dolmetsch, R.E.; Lewis, R.S.; Goodnow, C.C.; Healy, J.I. Differential activation of transcription factors induced by Ca2+ response amplitude and duration. Nature 1997, 386, 855–858. [Google Scholar] [CrossRef]
- Kar, P.; Parekh, A.B. Distinct spatial Ca2+ signatures selectively activate different NFAT transcription factor isoforms. Mol. Cell 2015, 58, 232–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parekh, A.B.; Muallem, S. Ca2+ signalling and gene regulation. Cell Calcium 2011, 49, 279. [Google Scholar] [CrossRef]
- Sheng, M.; McFadden, G.; Greenberg, M.E. Membrane depolarization and calcium induce c-fos transcription via phosphorylation of transcription factor CREB. Neuron 1990, 4, 571–582. [Google Scholar] [CrossRef]
- Müller, I.; Rössler, O.G.; Thiel, G. Pregnenolone sulfate activates basic region leucine zipper transcription factors in insulinoma cells: Role of voltage-gated Ca2+ channels and transient receptor potential melastatin 3 channels. Mol. Pharmacol. 2011, 80, 1179–1189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayr, B.; Montminy, M. Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat. Rev. Mol. Cell Biol. 2001, 2, 599–609. [Google Scholar] [CrossRef]
- Johannessen, M.; Delghandi, M.P.; Moens, U. What turns CREB on? Cell. Signal. 2004, 16, 1211–1227. [Google Scholar] [CrossRef]
- Barco, A.; Kandel, E.R. The Role of CREB and CBP in Brain Function. In Transcription Factors in the Nervous System—Development, Brain Function, and Diseases; Thiel, G., Ed.; Wiley-VCH Verlag: Weinheim, Germany, 2006; pp. 207–241. [Google Scholar]
- Steven, A.; Friedrich, M.; Jank, P.; Heimer, N.; Budczies, J.; Denkert, C.; Selinger, B. What turns CREB on? And off? And why does it matter? Cell. Mol. Life Sci. 2020, 77, 4049–4067. [Google Scholar] [CrossRef] [PubMed]
- Sharrocks, A.D. The ETS-domain transcription factor family. Nat. Mol. Cell Biol. 2001, 2, 827–837. [Google Scholar] [CrossRef] [PubMed]
- Shaw, P.E.; Saxton, J. Ternary complex factors: Prime targets for mitogen-activated protein kinases. Int. J. Biochem. Cell. Biol. 2003, 35, 1210–1226. [Google Scholar] [CrossRef]
- Cavigelli, M.; Dolfi, F.; Claret, F.X.; Karin, M. Induction of c-fos expression through JNK-mediated TCF/Elk-1 phosphorylation. EMBO J. 1995, 14, 5957–5964. [Google Scholar] [CrossRef]
- Whitmarsh, A.J.; Yang, S.H.; Su, M.S.S.; Sharrocks, A.D.; Davis, R.J. Role of p38 and JNK mitogen-activated protein kinases in the activation of ternary complex factors. Mol. Cell. Biol. 1997, 17, 2360–2371. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.H.; Whitmarsh, A.J.; Davis, R.J.; Sharrocks, A.D. Differential targeting of MAP kinases to the ETS-domain transcription factor Elk-1. EMBO J. 1998, 17, 1740–1749. [Google Scholar] [CrossRef] [Green Version]
- Cruzalegui, F.H.; Cano, E.; Treisman, R. ERK activation induces phosphorylation of Elk-1 at multiple S/T-P motifs to high stoichiometry. Oncogene 1999, 18, 7948–7957. [Google Scholar] [CrossRef] [Green Version]
- Müller, M.R.; Rao, A. NFAT, immunity and cancer: A transcription factor comes of age. Nat. Rev. Immunol. 2010, 10, 645–656. [Google Scholar] [CrossRef]
- Eder, A.; Bading, H. Calcium signals can freely cross the nuclear envelope in hippocampal neurons: Somatic calcium increases generate nuclear calcium transient. BMC Neurosci. 2007, 8, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Echevarría, W.; Leite, M.F.; Guerra, M.T.; Zipfel, W.R.; Nathanson, M. Regulation of calcium signals in the nucleus by a nucleoplasmic reticulum. Nat. Cell Biol. 2003, 5, 440–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kar, P.; Mirams, G.R.; Christian, H.C.; Parekh, A.B. Control of NFAT isoform activation and NFAT-dependent gene expression through two coincident and spatially segregated intracellular Ca2+ signals. Mol. Cell 2016, 64, 746–759. [Google Scholar] [CrossRef] [Green Version]
- Bading, H. Nuclear calcium signalling in the regulation of brain function. Nat. Rev. Neurosci. 2013, 14, 593–608. [Google Scholar] [CrossRef] [PubMed]
- Hardingham, G.E.; Chawla, S.; Johnson, C.M.; Bading, H. Distinct functions of nuclear and cytoplasmic calcium in the control of gene expression. Nature 1997, 385, 260–265. [Google Scholar] [CrossRef]
- Müller, I.; Lipp, P.; Thiel, G. Ca2+ signaling and gene transcription in glucose-stimulated insulinoma cells. Cell Calcium 2012, 52, 137–151. [Google Scholar] [CrossRef] [PubMed]
- Pusl, T.; Wu, J.J.; Zimmerman, T.L.; Zhang, L.; Ehrlich, B.E.; Berchtold, M.W.; Hoek, J.B.; Karpen, S.J.; Nathanson, M.H.; Bennett, A.M. Epidermal growth factor-mediated activation of the ETS domain transcription factor Elk-1 requires nuclear calcium. J. Biol. Chem. 2002, 277, 27517–27527. [Google Scholar] [CrossRef] [Green Version]
- Arif, S.H. A Ca2+-binding protein with numerous roles and uses: Parvalbumin in molecular biology and physiology. BioEssays 2009, 31, 410–421. [Google Scholar] [CrossRef]
- Mayer, S.I.; Rössler, O.G.; Endo, T.; Charnay, P.; Thiel, G. Epidermal growth factor-induced proliferation of astrocytes requires Egr transcription factors. J. Cell Sci. 2009, 122, 3340–3350. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Trebak, M.; Gill, D.L. Calcium signals tune the fidelity of transcriptional responses. Mol. Cell 2015, 58, 197–199. [Google Scholar] [CrossRef] [Green Version]
- Klee, C.B.; Ren, H.; Wang, X. Regulation of the calmodulin-stimulated protein phosphatase, calcineurin. J. Biol. Chem. 1998, 273, 13367–13370. [Google Scholar] [CrossRef] [Green Version]
- Aramburo, J.; Rao, A.; Klee, C.B. Calcineurin: From structure to function. Curr. Top. Cell. Reg. 2000, 36, 237–295. [Google Scholar]
- Rusnak, F.; Mertz, P. Calcineurin: Form and function. Physiol. Rev. 2000, 80, 1483–1521. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Rao, A.; Hogan, P.G. Interaction of calcineurin with substrates and targeting proteins. Trends. Cell Biol. 2011, 21, 91–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hallhuber, M.; Burkhard, N.; Wu, R.; Buch, M.H.; Engelhardt, S.; Hein, L.; Neyes, L.; Schuh, K.; Ritter, O. Inhibition of nuclear import of calcineurin prevents myocardial hypertrophy. Circ. Res. 2006, 99, 626–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Farmer, J.D., Jr.; Lane, W.S.; Friedman, J.; Weissman, I.; Schreiber, S.L. Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell 1991, 66, 807–815. [Google Scholar] [CrossRef]
- Helekar, S.A.; Char, D.; Neff, S.; Patrick, J. Prolyl isomerase requirement for the expression of functional homo-oligomeric ligand-gated ion channels. Neuron 1994, 12, 179–189. [Google Scholar] [CrossRef]
- Marks, A.R. Cellular functions of immunophilins. Physiol. Rev. 1996, 76, 631–649. [Google Scholar] [CrossRef] [PubMed]
- Montero, M.; Lobatón, C.D.; Gutierrez-Fernández, S.; Moreno, A.; Alvarez, J. Calcineurin-independent inhibition of mitochondrial Ca2+ uptake by cyclosporin A. Br. J. Pharmacol. 2004, 141, 263–268. [Google Scholar] [CrossRef] [Green Version]
- Bernardi, P.; Di Lisa, F. The mitochondrial permeability transition pore: Molecular nature and role as a target in cardioprotection. J. Mol. Cell Cardiol. 2015, 78, 100–106. [Google Scholar] [CrossRef]
- Liddicoat, A.M.; Lavelle, E.C. Modulation of innate immunity by cyclosporine A. Biochem. Pharmacol. 2019, 163, 472–480. [Google Scholar] [CrossRef]
- Arcas, J.M.; González, A.; Gers-Barlag, K.; González-González, O.; Bech, F.; Demirkhanyan, L.; Zakharian, E.; Belmonte, C.; Gomis, A.; Viana, F. The immunosuppressant macrolide tacrolimus activates cold-sensing TRPM8 channels. J. Neurosci. 2019, 39, 949–969. [Google Scholar] [CrossRef] [PubMed]
- Fuentes, J.J.; Genescà, L.; Kingbury, T.J.; Cunningham, K.W.; Pérez-Riba, M.; Estivill, X.; de la Luna, S. DSCR1, overexpressed in Down syndrome, is an inhibitor of calcineurin-mediated signaling pathways. Hum. Mol. Genet. 2000, 9, 1681–1690. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.-K.; Ahnn, J. Regulator of calcineurin (RCAN): Beyond down syndrome critical region. Mol. Cells 2020, 43, 671–685. [Google Scholar] [PubMed]
- Boss, V.; Talpade, D.J.; Murthy, T.J. Induction of NFAT-mediated transcription by Gq-coupled receptors in lymphoid and non-lymphoid cells. J. Biol. Chem. 1996, 271, 10429–10432. [Google Scholar] [CrossRef] [Green Version]
- Langfermann, D.S.; Schmidt, T.; Rössler, O.G.; Thiel, G. Calcineurin controls gene transcription following stimulation of a Gaq-coupled designer receptor. Exp. Cell Res. 2019, 383, 111553. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.-S.; Usachev, Y.M. Mitochondrial Ca2+ cycling facilitates activation of the transcription factor NFAT in sensory neurons. J. Neurosci. 2009, 29, 12101–12114. [Google Scholar] [CrossRef]
- Crabtree, G.R. Calcium, calcineurin, and the control of transcription. J. Biol. Chem. 2001, 276, 2313–2316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hogan, P.G.; Chen, L.; Nardone, J.; Rao, A. Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev. 2003, 17, 2205–2232. [Google Scholar] [CrossRef] [Green Version]
- Aramburo, J.; García-Cózar, F.; Raghavan, A.; Okamura, H.; Rao, A.; Hogan, P. Selective inhibition of NFAT activation by a peptide spanning the calcineurin targeting site of NFAT. Mol. Cell 1998, 1, 627–637. [Google Scholar] [CrossRef]
- Zhu, J.; Shibasaki, F.; Price, R.; Guillemot, J.C.; Yano, T.; Dotsch, V.; Wagner, G.; Ferrara, P.; McKeon, F. Intramolecular masking of nuclear import signal on NF-AT4 by casein kinase I and MEKK1. Cell 1998, 93, 851–861. [Google Scholar] [CrossRef] [Green Version]
- Aramburo, J.; Yaffe, M.B.; López-Rodríguez, C.; Cantley, L.C.; Hogan, P.; Rao, A. Affinity-driven peptide selection of an NFAT inhibitor more selective than cyclosporin A. Science 1999, 285, 2129–2133. [Google Scholar] [CrossRef]
- Hudry, E.; Wu, H.-Y.; Arbel-Ornath, M.; Hashimoto, T.; Matsouaka, R.; Fan, Z.; Spires-Jones, T.L.; Betensky, R.A.; Bacskai, B.J.; Hyman, B.T. Inhibition of the NFAT pathway alleviates amyloid beta neurotoxicity in a mouse model of Alzheimer’s disease. J. Neurosci. 2012, 32, 3176–3192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibasaki, F.; Price, E.R.; Milan, D.; McKeon, F. Role of kinases and the phosphatase calcineurin in the nuclear shuttling of transcription factor NF-AT4. Nature 1996, 382, 370–373. [Google Scholar] [CrossRef]
- Kim, S.S.; Seo, S.R. The regulator of calcineurin 1 (RCAN1/DSCR1) activates the cAMP response element-binding protein (CREB) pathway. J. Biol. Chem. 2011, 286, 37841–37848. [Google Scholar] [CrossRef] [Green Version]
- Bito, H.; Deisseroth, K.; Tsien, R.W. CREB phosphorylation and dephosphorylation: A Ca2+- and stimulus duration-dependent switch for hippocampal gene expression. Cell 1998, 87, 1203–1214. [Google Scholar] [CrossRef] [Green Version]
- Screaton, R.A.; Conkright, M.D.; Katoh, Y.; Best, J.L.; Canettieri, G.; Jeffries, S.; Guzman, E.; Niessen, S.; Yates, J.R., III; Takemori, H.; et al. The CREB coactivator TORC2 functions as a calcium- and cAMP-sensitive coincidence detector. Cell 2004, 119, 61–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugimoto, T.; Stewart, S.; Guan, K.-L. The calcium/calmodulin-dependent protein phosphatase calcineurin is the major Elk-1 phosphatase. J. Biol. Chem. 1997, 272, 29415–29418. [Google Scholar] [CrossRef] [Green Version]
- Tian, J.; Karin, M. Stimulation of Elk1 transcriptional activity by mitogen-activated protein kinases is negatively regulated by protein phosphatase 2B (calcineurin). J. Biol. Chem. 1999, 274, 15173–15180. [Google Scholar] [CrossRef] [Green Version]
- Lam, B.Y.H.; Zhang, W.; Enticknap, N.; Haggis, E.; Cader, M.Z.; Chawla, S. Inverse regulation of plasticity-related immediate early genes by calcineurin in hippocampal neurons. J. Biol. Chem. 2009, 284, 12562–12571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernal-Mizrachi, E.; Cras-Méneur, C.; Ye, B.R.; Johnson, J.D.; Permutt, M.A. Transgenic overexpression of active calcineurin in β-cells results in decreased β-cell mass and hyperglycemia. PLoS ONE 2010, 5, e11969. [Google Scholar] [CrossRef] [Green Version]
- Lesch, A.; Backes, T.M.; Langfermann, D.S.; Rössler, O.G.; Laschke, M.W.; Thiel, G. Ternary complex factor regulates pancreatic islet size and blood glucose homeostasis in transgenic mice. Pharmacol. Res. 2020, 159, 104983. [Google Scholar] [CrossRef] [PubMed]
- Müller, I.; Rössler, O.G.; Wittig, C.; Menger, M.D.; Thiel, G. Critical role of Egr transcription factors in regulating insulin biosynthesis, blood glucose homeostasis and islet size. Endocrinology 2012, 153, 3040–3053. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mouse Model | β-cell ↓ Mass | Hyperglycemia | ↑ Apoptosis | Reference |
|---|---|---|---|---|
| Expression of a constitutively active mutant of calcineurin in β-cells | √ | √ | √ | [83] |
| Expression of a dominant negative mutant of Elk-1 in β-cells | √ | √ | √ | [84] |
| Expression of a dominant negative mutant of Egr-1 in β-cells | √ | √ | √ | [85] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thiel, G.; Schmidt, T.; Rössler, O.G. Ca2+ Microdomains, Calcineurin and the Regulation of Gene Transcription. Cells 2021, 10, 875. https://doi.org/10.3390/cells10040875
Thiel G, Schmidt T, Rössler OG. Ca2+ Microdomains, Calcineurin and the Regulation of Gene Transcription. Cells. 2021; 10(4):875. https://doi.org/10.3390/cells10040875
Chicago/Turabian StyleThiel, Gerald, Tobias Schmidt, and Oliver G. Rössler. 2021. "Ca2+ Microdomains, Calcineurin and the Regulation of Gene Transcription" Cells 10, no. 4: 875. https://doi.org/10.3390/cells10040875
APA StyleThiel, G., Schmidt, T., & Rössler, O. G. (2021). Ca2+ Microdomains, Calcineurin and the Regulation of Gene Transcription. Cells, 10(4), 875. https://doi.org/10.3390/cells10040875