
S6K1 Is Indispensible for Stress-Induced Microtubule Acetylation and Autophagic Flux



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cell Lines and Cell Culture
2.3. Transient Transfection with Plasmid DNA or siRNA
2.4. Stress Conditions for the Induction of Autophagy
2.5. Immunoblotting
2.6. Immunofluorescence
2.7. Cathepsin L Activity Assay
2.8. Flow Cytometry
2.9. pH Measurements Using LysoSensor Yellow/Blue
2.10. Statistical Analysis
3. Results
3.1. Impaired Autophagic Flux under Stress Conditions Is a General Phenotype of S6K DKO MEF
3.2. Absence of S6K Kinases Does Not Perturb Lysosomal Function under SFN-Induced Stress
3.3. Autophagosome-Lysosome Fusion Is Impaired in S6K DKO MEF under Stress Conditions
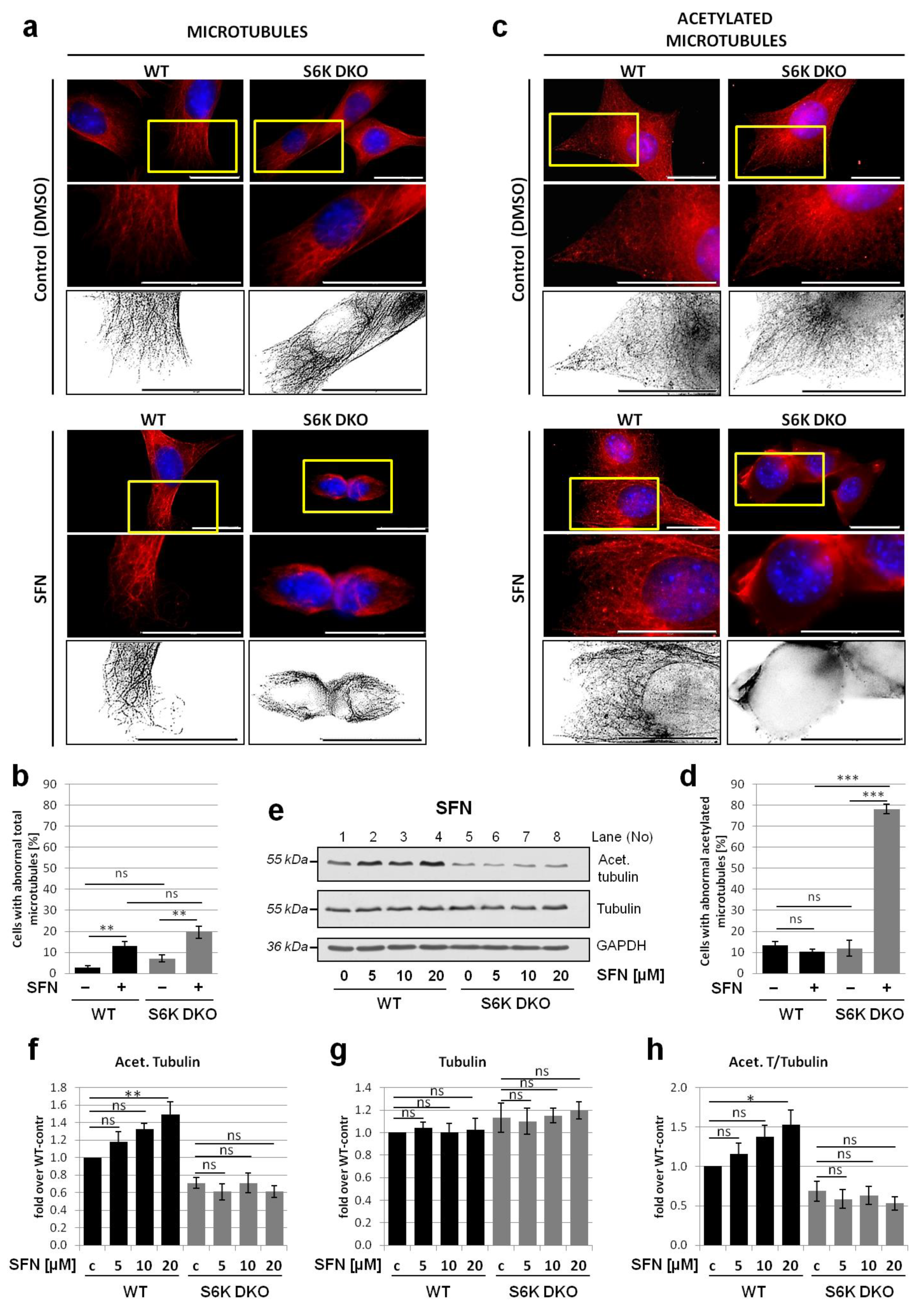
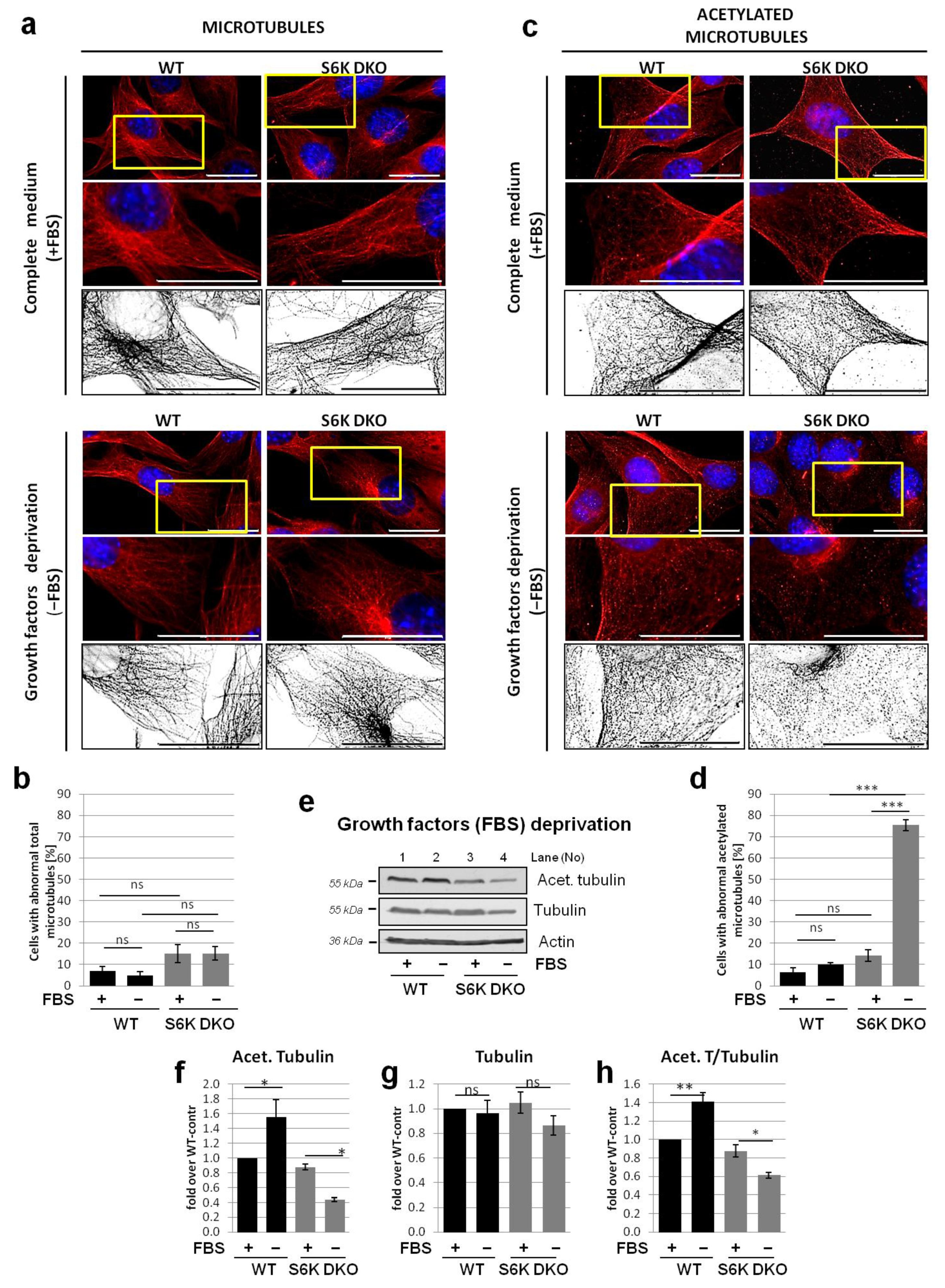
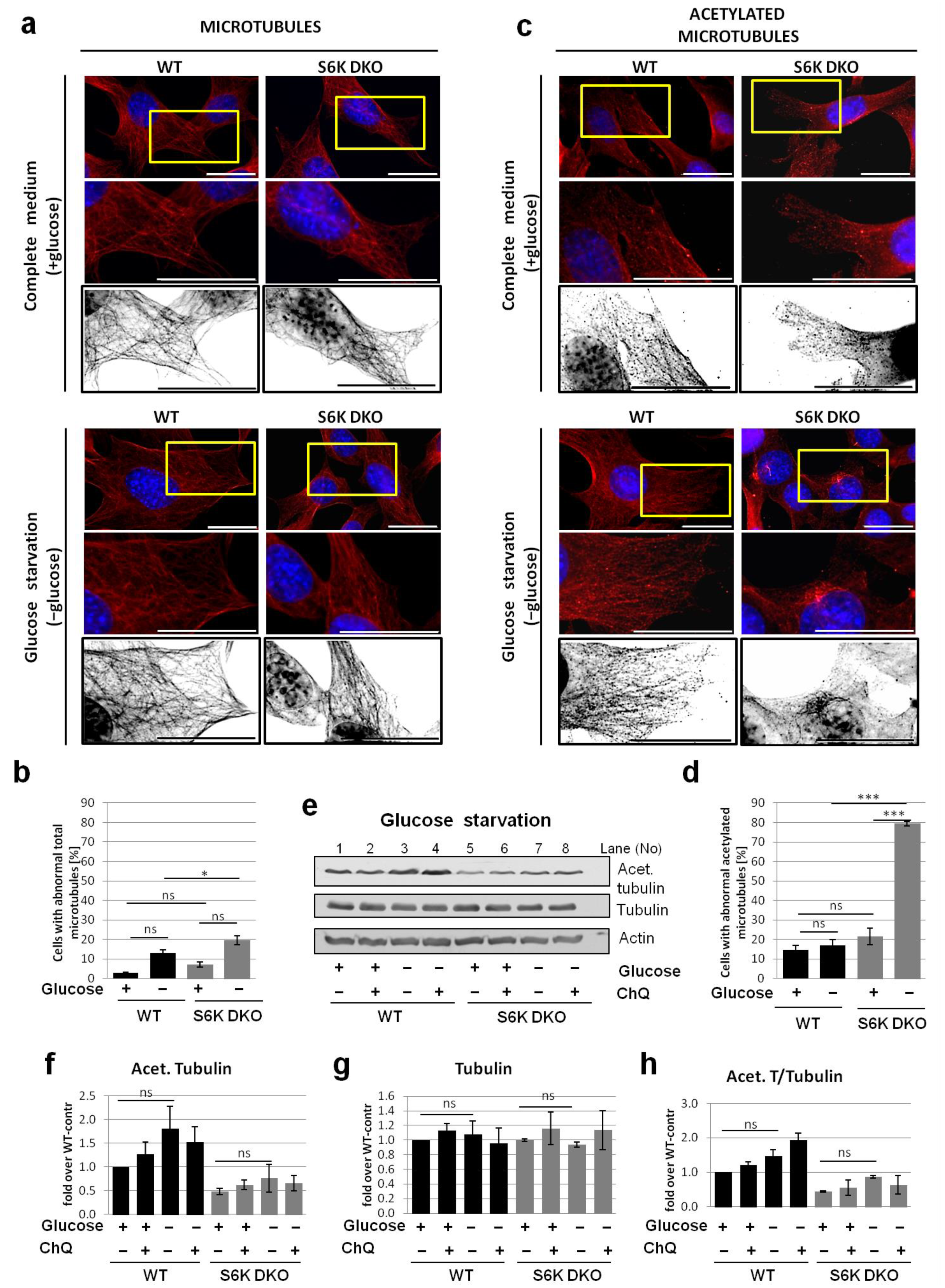
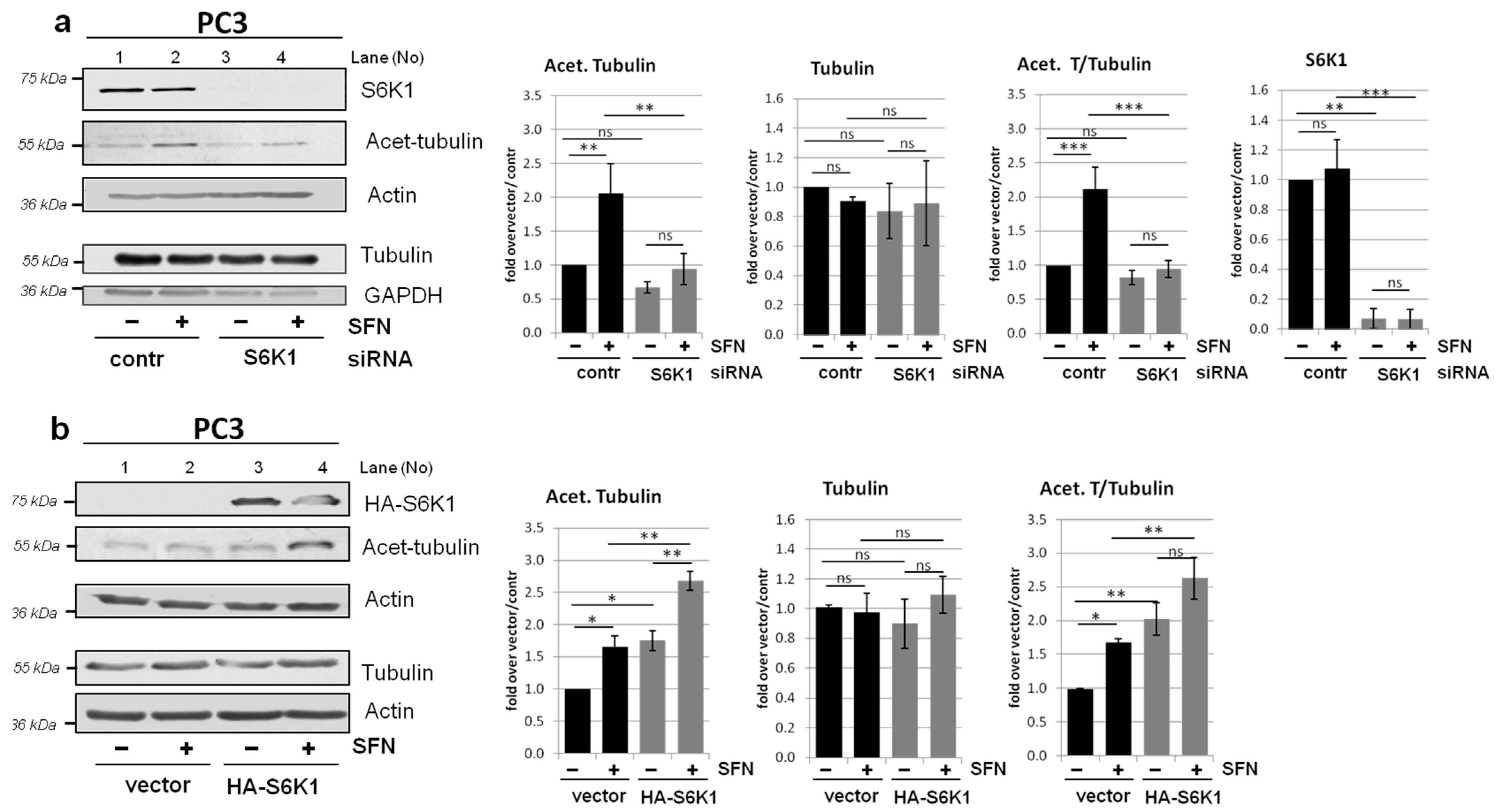
3.4. Lack of S6K1 Kinase Affects Acetylated Microtubules but Not Total Microtubule Network in Cells Exposed to Stress
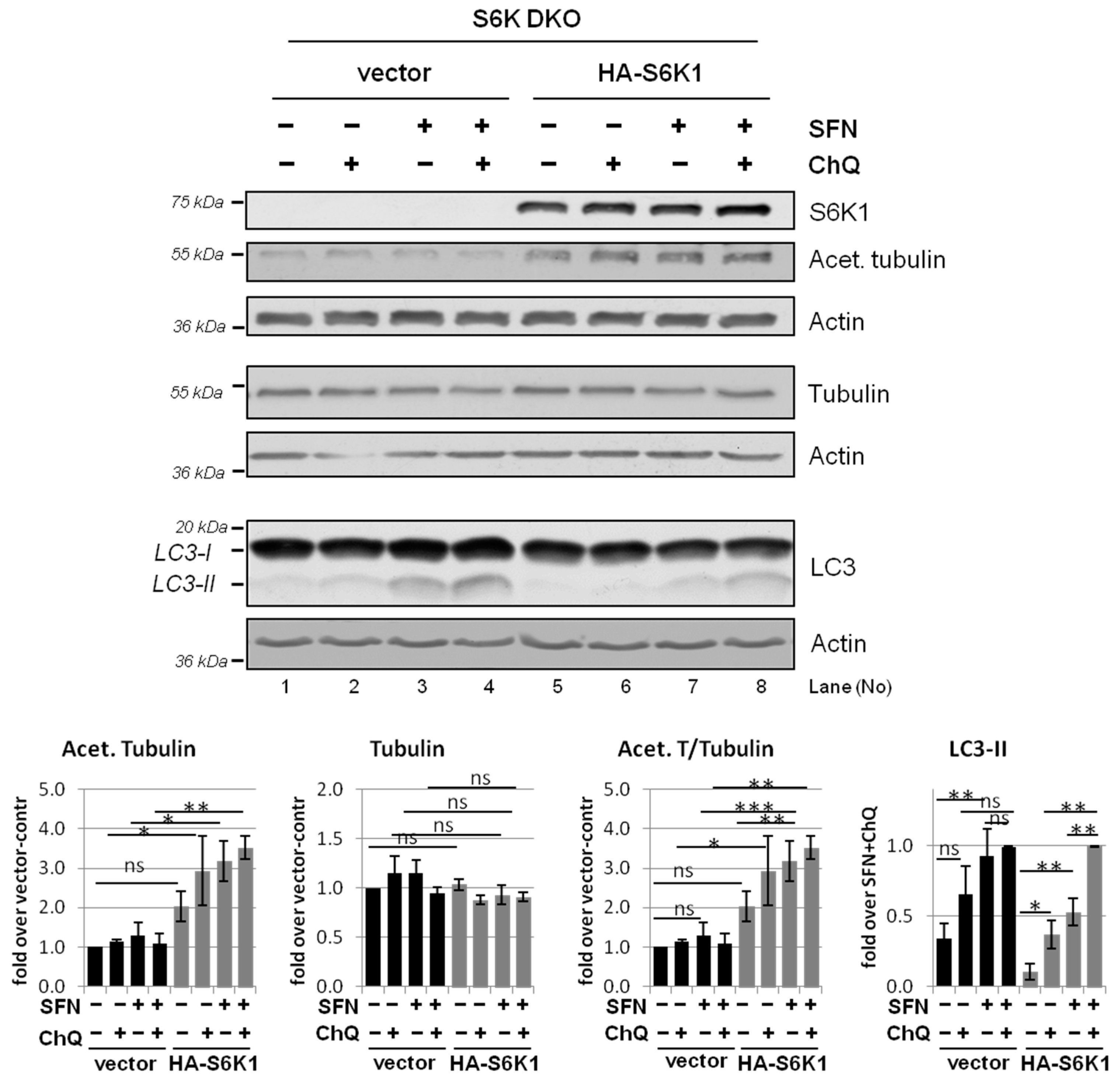
3.5. Overexpression of Exogenous S6K1 Restores Tubulin Acetylation and Lysosomal LC3-II Degradation in S6K DKO MEF Exposed to SFN
3.6. Modulation of Tubulin Acetylation by S6K1 Is Conserved in Different Cells
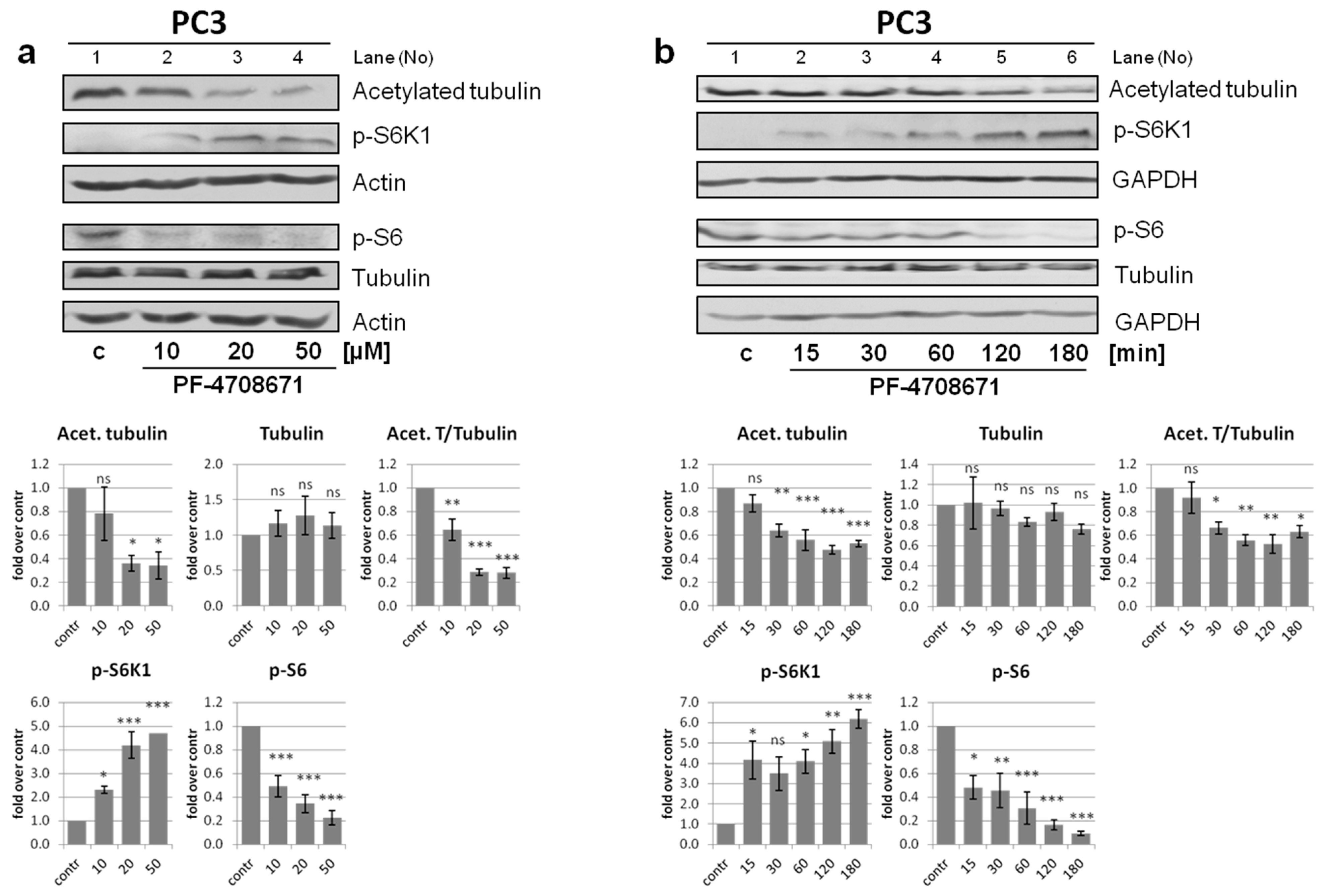
3.7. Role of S6K1 in Tubulin Acetylation Is Regulated by Its Phosphorylation Status
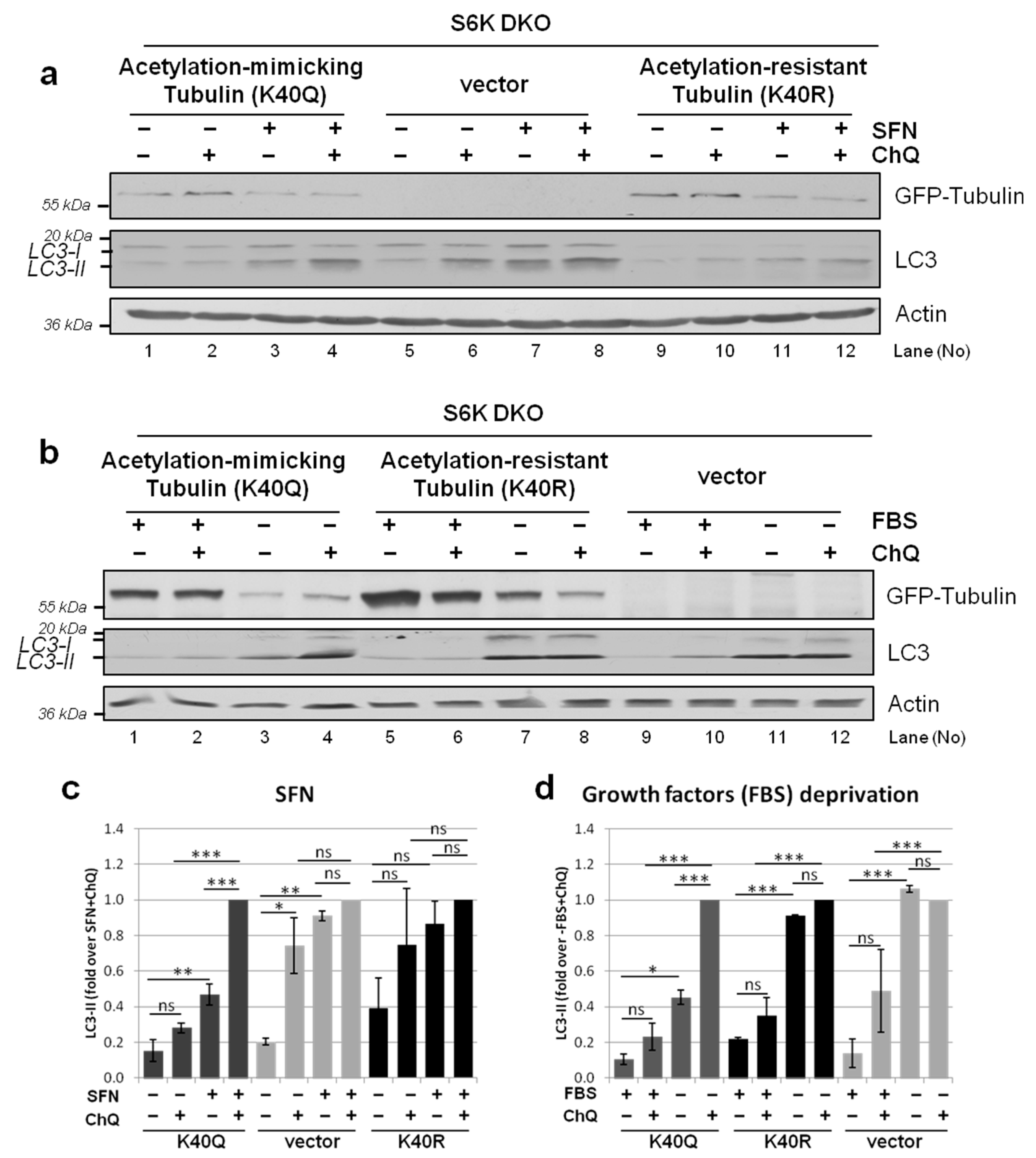
3.8. Overexpression of Acetylation-Mimicking but Not Acetylation-Resistant Tubulin Variant Restores Autophagic Flux in S6K DKO MEF
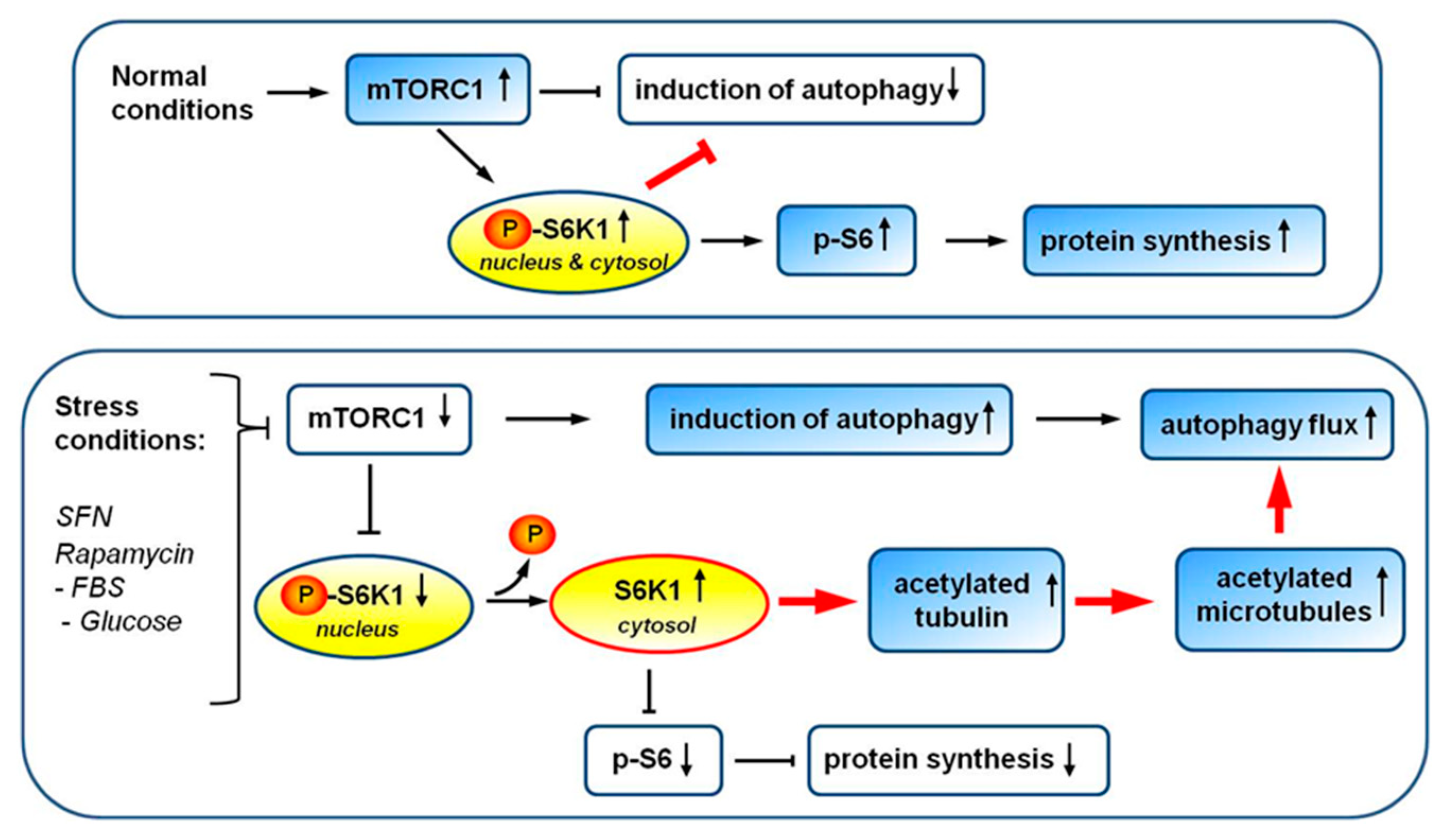
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Mizushima, N. The pleiotropic role of autophagy: From protein metabolism to bactericide. Cell Death Differ. 2005, 12 (Suppl. 2), 1535–1541. [Google Scholar] [CrossRef] [PubMed]
- Meijer, A.J.; Codogno, P. Regulation and role of autophagy in mammalian cells. Int. J. Biochem. Cell Biol. 2004, 36, 2445–2462. [Google Scholar] [CrossRef]
- Shintani, T.; Klionsky, D.J. Autophagy in health and disease: A double-edged sword. Science 2004, 306, 990–995. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N.; Noda, T.; Yoshimori, T.; Tanaka, Y.; Ishii, T.; George, M.D.; Klionsky, D.J.; Ohsumi, M.; Ohsumi, Y. A protein conjugation system essential for autophagy. Nature 1998, 395, 395–398. [Google Scholar] [CrossRef] [PubMed]
- Tanida, I.; Minematsu-Ikeguchi, N.; Ueno, T.; Kominami, E. Lysosomal turnover, but not a cellular level, of endogenous LC3 is a marker for autophagy. Autophagy 2005, 1, 84–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.J.; Chen, S.; Huang, K.X.; Le, W.D. Why should autophagic flux be assessed? Acta Pharmacol. Sin. 2013, 34, 595–599. [Google Scholar] [CrossRef] [Green Version]
- Mackeh, R.; Lorin, S.; Ratier, A.; Mejdoubi-Charef, N.; Baillet, A.; Bruneel, A.; Hamai, A.; Codogno, P.; Pous, C.; Perdiz, D. Reactive oxygen species, AMP-activated protein kinase, and the transcription cofactor p300 regulate alpha-tubulin acetyltransferase-1 (alphaTAT-1/MEC-17)-dependent microtubule hyperacetylation during cell stress. J. Biol. Chem. 2014, 289, 11816–11828. [Google Scholar] [CrossRef] [Green Version]
- Hubbert, C.; Guardiola, A.; Shao, R.; Kawaguchi, Y.; Ito, A.; Nixon, A.; Yoshida, M.; Wang, X.F.; Yao, T.P. HDAC6 is a microtubule-associated deacetylase. Nature 2002, 417, 455–458. [Google Scholar] [CrossRef] [PubMed]
- North, B.J.; Marshall, B.L.; Borra, M.T.; Denu, J.M.; Verdin, E. The human Sir2 ortholog, SIRT2, is an NAD+-dependent tubulin deacetylase. Mol. Cell 2003, 11, 437–444. [Google Scholar] [CrossRef]
- Piperno, G.; LeDizet, M.; Chang, X.J. Microtubules containing acetylated alpha-tubulin in mammalian cells in culture. J. Cell Biol. 1987, 104, 289–302. [Google Scholar] [CrossRef]
- Cambray-Deakin, M.A.; Burgoyne, R.D. Posttranslational modifications of alpha-tubulin: Acetylated and detyrosinated forms in axons of rat cerebellum. J. Cell Biol. 1987, 104, 1569–1574. [Google Scholar] [CrossRef]
- Matsuyama, A.; Shimazu, T.; Sumida, Y.; Saito, A.; Yoshimatsu, Y.; Seigneurin-Berny, D.; Osada, H.; Komatsu, Y.; Nishino, N.; Khochbin, S.; et al. In Vivo destabilization of dynamic microtubules by HDAC6-mediated deacetylation. EMBO J. 2002, 21, 6820–6831. [Google Scholar] [CrossRef] [Green Version]
- Perdiz, D.; Mackeh, R.; Pous, C.; Baillet, A. The ins and outs of tubulin acetylation: More than just a post-translational modification? Cell Signal. 2011, 23, 763–771. [Google Scholar] [CrossRef] [PubMed]
- Geeraert, C.; Ratier, A.; Pfisterer, S.G.; Perdiz, D.; Cantaloube, I.; Rouault, A.; Pattingre, S.; Proikas-Cezanne, T.; Codogno, P.; Pous, C. Starvation-induced hyperacetylation of tubulin is required for the stimulation of autophagy by nutrient deprivation. J. Biol. Chem. 2010, 285, 24184–24194. [Google Scholar] [CrossRef] [Green Version]
- Kimura, S.; Noda, T.; Yoshimori, T. Dynein-dependent movement of autophagosomes mediates efficient encounters with lysosomes. Cell Struct. Funct. 2008, 33, 109–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boyault, C.; Sadoul, K.; Pabion, M.; Khochbin, S. HDAC6, at the crossroads between cytoskeleton and cell signaling by acetylation and ubiquitination. Oncogene 2007, 26, 5468–5476. [Google Scholar] [CrossRef] [Green Version]
- Clarke, J.D.; Hsu, A.; Yu, Z.; Dashwood, R.H.; Ho, E. Differential effects of sulforaphane on histone deacetylases, cell cycle arrest and apoptosis in normal prostate cells versus hyperplastic and cancerous prostate cells. Mol. Nutr. Food Res. 2011, 55, 999–1009. [Google Scholar] [CrossRef] [Green Version]
- Laplante, M.; Sabatini, D.M. mTOR signaling at a glance. J. Cell Sci. 2009, 122, 3589–3594. [Google Scholar] [CrossRef] [Green Version]
- Dunlop, E.A.; Tee, A.R. mTOR and autophagy: A dynamic relationship governed by nutrients and energy. Semin Cell Dev. Biol. 2014, 36, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Shima, H.; Pende, M.; Chen, Y.; Fumagalli, S.; Thomas, G.; Kozma, S.C. Disruption of the p70(s6k)/p85(s6k) gene reveals a small mouse phenotype and a new functional S6 kinase. EMBO J. 1998, 17, 6649–6659. [Google Scholar] [CrossRef] [Green Version]
- Pende, M.; Um, S.H.; Mieulet, V.; Sticker, M.; Goss, V.L.; Mestan, J.; Mueller, M.; Fumagalli, S.; Kozma, S.C.; Thomas, G. S6K1(−/−)/S6K2(−/−) mice exhibit perinatal lethality and rapamycin-sensitive 5′-terminal oligopyrimidine mRNA translation and reveal a mitogen-activated protein kinase-dependent S6 kinase pathway. Mol. Cell Biol. 2004, 24, 3112–3124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Hage, J.A.; van den Broek, L.J.; Legrand, C.; Clahsen, P.C.; Bosch, C.J.; Robanus-Maandag, E.C.; van de Velde, C.J.; van de Vijver, M.J. Overexpression of P70 S6 kinase protein is associated with increased risk of locoregional recurrence in node-negative premenopausal early breast cancer patients. Br. J. Cancer 2004, 90, 1543–1550. [Google Scholar] [CrossRef]
- Amaral, C.L.; Freitas, L.B.; Tamura, R.E.; Tavares, M.R.; Pavan, I.C.; Bajgelman, M.C.; Simabuco, F.M. S6Ks isoforms contribute to viability, migration, docetaxel resistance and tumor formation of prostate cancer cells. BMC Cancer 2016, 16, 602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filonenko, V.V.; Tytarenko, R.; Azatjan, S.K.; Savinska, L.O.; Gaydar, Y.A.; Gout, I.T.; Usenko, V.S.; Lyzogubov, V.V. Immunohistochemical analysis of S6K1 and S6K2 localization in human breast tumors. Exp. Oncol. 2004, 26, 294–299. [Google Scholar]
- Blommaart, E.F.; Luiken, J.J.; Blommaart, P.J.; van Woerkom, G.M.; Meijer, A.J. Phosphorylation of ribosomal protein S6 is inhibitory for autophagy in isolated rat hepatocytes. J. Biol. Chem. 1995, 270, 2320–2326. [Google Scholar] [CrossRef] [Green Version]
- Klionsky, D.J.; Meijer, A.J.; Codogno, P. Autophagy and p70S6 kinase. Autophagy 2005, 1, 59–60, discussion 51–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.B.; Kim, S.; Lee, J.; Park, J.; Lee, G.; Kim, Y.; Kim, J.M.; Chung, J. ATG1, an autophagy regulator, inhibits cell growth by negatively regulating S6 kinase. EMBO Rep. 2007, 8, 360–365. [Google Scholar] [CrossRef]
- Scott, R.C.; Schuldiner, O.; Neufeld, T.P. Role and regulation of starvation-induced autophagy in the Drosophila fat body. Dev. Cell 2004, 7, 167–178. [Google Scholar] [CrossRef] [Green Version]
- Armour, S.M.; Baur, J.A.; Hsieh, S.N.; Land-Bracha, A.; Thomas, S.M.; Sinclair, D.A. Inhibition of mammalian S6 kinase by resveratrol suppresses autophagy. Aging 2009, 1, 515–528. [Google Scholar] [CrossRef] [Green Version]
- Hac, A.; Domachowska, A.; Narajczyk, M.; Cyske, K.; Pawlik, A.; Herman-Antosiewicz, A. S6K1 controls autophagosome maturation in autophagy induced by sulforaphane or serum deprivation. Eur. J. Cell Biol. 2015, 94, 470–481. [Google Scholar] [CrossRef]
- Herman-Antosiewicz, A.; Johnson, D.E.; Singh, S.V. Sulforaphane causes autophagy to inhibit release of cytochrome C and apoptosis in human prostate cancer cells. Cancer Res. 2006, 66, 5828–5835. [Google Scholar] [CrossRef] [Green Version]
- Schalm, S.S.; Blenis, J. Identification of a conserved motif required for mTOR signaling. Curr. Biol. 2002, 12, 632–639. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.S.; Hubbert, C.C.; Yao, T.P. The microtubule-associated histone deacetylase 6 (HDAC6) regulates epidermal growth factor receptor (EGFR) endocytic trafficking and degradation. J. Biol. Chem. 2010, 285, 11219–11226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- N’Diaye, E.N.; Kajihara, K.K.; Hsieh, I.; Morisaki, H.; Debnath, J.; Brown, E.J. PLIC proteins or ubiquilins regulate autophagy-dependent cell survival during nutrient starvation. EMBO Rep. 2009, 10, 173–179. [Google Scholar] [CrossRef] [Green Version]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [Green Version]
- Ge, J.N.; Huang, D.; Xiao, T.; Wang, Z.; Li, X.L.; Xiao, H.; Tao, D.D.; Gong, J.P. Effect of starvation-induced autophagy on cell cycle of tumor cells. Ai Zheng 2008, 27, 788–794. [Google Scholar]
- Steiger-Barraissoul, S.; Rami, A. Serum deprivation induced autophagy and predominantly an AIF-dependent apoptosis in hippocampal HT22 neurons. Apoptosis 2009, 14, 1274–1288. [Google Scholar] [CrossRef]
- Zhao, S.; Li, L.; Wang, S.; Yu, C.; Xiao, B.; Lin, L.; Cong, W.; Cheng, J.; Yang, W.; Sun, W.; et al. H2O2 treatment or serum deprivation induces autophagy and apoptosis in naked mole-rat skin fibroblasts by inhibiting the PI3K/Akt signaling pathway. Oncotarget 2016, 7, 84839–84850. [Google Scholar] [CrossRef]
- Wilcox, A.; Murphy, M.; Tucker, D.; Laprade, D.; Roussel, B.; Chin, C.; Hallisey, V.; Kozub, N.; Brass, A.; Austriaco, N. Sulforaphane alters the acidification of the yeast vacuole. Microb. Cell 2020, 7, 129–138. [Google Scholar] [CrossRef]
- Mundy, D.I.; Li, W.P.; Luby-Phelps, K.; Anderson, R.G. Caveolin targeting to late endosome/lysosomal membranes is induced by perturbations of lysosomal pH and cholesterol content. Mol. Biol. Cell 2012, 23, 864–880. [Google Scholar] [CrossRef]
- McGuire, C.M.; Forgac, M. Glucose starvation increases V-ATPase assembly and activity in mammalian cells through AMP kinase and phosphatidylinositide 3-kinase/Akt signaling. J. Biol. Chem. 2018, 293, 9113–9123. [Google Scholar] [CrossRef] [Green Version]
- Ueno, T.; Takahashi, K. A cathepsin L-specific inhibitor preferentially inhibits degradation of autophagosomal LC3 and GABARAP in HeLa and Huh-7 cells. Autophagy 2009, 5, 878–879. [Google Scholar] [CrossRef] [Green Version]
- Yadati, T.; Houben, T.; Bitorina, A.; Shiri-Sverdlov, R. The Ins and outs of cathepsins: Physiological function and role in disease management. Cells 2020, 9, 1679. [Google Scholar] [CrossRef]
- Hansen, T.E.; Johansen, T. Following autophagy step by step. BMC Biol. 2011, 9, 39. [Google Scholar] [CrossRef] [Green Version]
- Cardoso, C.M.; Groth-Pedersen, L.; Hoyer-Hansen, M.; Kirkegaard, T.; Corcelle, E.; Andersen, J.S.; Jaattela, M.; Nylandsted, J. Depletion of kinesin 5B affects lysosomal distribution and stability and induces peri-nuclear accumulation of autophagosomes in cancer cells. PLoS ONE 2009, 4, e4424. [Google Scholar] [CrossRef] [Green Version]
- Olenieva, V.; Lytvyn, D.; Yemets, A.; Bergounioux, C.; Blume, Y. Tubulin acetylation accompanies autophagy development induced by different abiotic stimuli in Arabidopsis thaliana. Cell Biol. Int. 2019, 43, 1056–1064. [Google Scholar] [CrossRef]
- Pihan, G.A.; Purohit, A.; Wallace, J.; Knecht, H.; Woda, B.; Quesenberry, P.; Doxsey, S.J. Centrosome defects and genetic instability in malignant tumors. Cancer Res. 1998, 58, 3974–3985. [Google Scholar] [PubMed]
- Rosner, M.; Schipany, K.; Hengstschlager, M. p70 S6K1 nuclear localization depends on its mTOR-mediated phosphorylation at T389, but not on its kinase activity towards S6. Amino Acids 2012, 42, 2251–2256. [Google Scholar] [CrossRef] [PubMed]
- Pearce, L.R.; Alton, G.R.; Richter, D.T.; Kath, J.C.; Lingardo, L.; Chapman, J.; Hwang, C.; Alessi, D.R. Characterization of PF-4708671, a novel and highly specific inhibitor of p70 ribosomal S6 kinase (S6K1). Biochem. J. 2010, 431, 245–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akella, J.S.; Wloga, D.; Kim, J.; Starostina, N.G.; Lyons-Abbott, S.; Morrissette, N.S.; Dougan, S.T.; Kipreos, E.T.; Gaertig, J. MEC-17 is an alpha-tubulin acetyltransferase. Nature 2010, 467, 218–222. [Google Scholar] [CrossRef] [Green Version]
- Shida, T.; Cueva, J.G.; Xu, Z.; Goodman, M.B.; Nachury, M.V. The major alpha-tubulin K40 acetyltransferase alphaTAT1 promotes rapid ciliogenesis and efficient mechanosensation. Proc. Natl. Acad. Sci. USA 2010, 107, 21517–21522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morley, S.J.; Qi, Y.; Iovino, L.; Andolfi, L.; Guo, D.; Kalebic, N.; Castaldi, L.; Tischer, C.; Portulano, C.; Bolasco, G.; et al. Acetylated tubulin is essential for touch sensation in mice. Elife 2016, 5. [Google Scholar] [CrossRef]
- Li, L.; Wei, D.; Wang, Q.; Pan, J.; Liu, R.; Zhang, X.; Bao, L. MEC-17 deficiency leads to reduced alpha-tubulin acetylation and impaired migration of cortical neurons. J. Neurosci. 2012, 32, 12673–12683. [Google Scholar] [CrossRef]
- Deakin, N.O.; Turner, C.E. Paxillin inhibits HDAC6 to regulate microtubule acetylation, Golgi structure, and polarized migration. J. Cell Biol. 2014, 206, 395–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawai, A.; Uchiyama, H.; Takano, S.; Nakamura, N.; Ohkuma, S. Autophagosome-lysosome fusion depends on the pH in acidic compartments in CHO cells. Autophagy 2007, 3, 154–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Ueno, T.; Tanida, I.; Minematsu-Ikeguchi, N.; Murata, M.; Kominami, E. Characterization of CAA0225, a novel inhibitor specific for cathepsin L, as a probe for autophagic proteolysis. Biol. Pharm. Bull. 2009, 32, 475–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haupts, U.; Maiti, S.; Schwille, P.; Webb, W.W. Dynamics of fluorescence fluctuations in green fluorescent protein observed by fluorescence correlation spectroscopy. Proc. Natl. Acad. Sci. USA 1998, 95, 13573–13578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kneen, M.; Farinas, J.; Li, Y.; Verkman, A.S. Green fluorescent protein as a noninvasive intracellular pH indicator. Biophys. J. 1998, 74, 1591–1599. [Google Scholar] [CrossRef] [Green Version]
- Llopis, J.; McCaffery, J.M.; Miyawaki, A.; Farquhar, M.G.; Tsien, R.Y. Measurement of cytosolic, mitochondrial, and Golgi pH in single living cells with green fluorescent proteins. Proc. Natl. Acad. Sci. USA 1998, 95, 6803–6808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Groth-Pedersen, L.; Ostenfeld, M.S.; Hoyer-Hansen, M.; Nylandsted, J.; Jaattela, M. Vincristine induces dramatic lysosomal changes and sensitizes cancer cells to lysosome-destabilizing siramesine. Cancer Res. 2007, 67, 2217–2225. [Google Scholar] [CrossRef] [Green Version]
- Broker, L.E.; Huisman, C.; Span, S.W.; Rodriguez, J.A.; Kruyt, F.A.; Giaccone, G. Cathepsin B mediates caspase-independent cell death induced by microtubule stabilizing agents in non-small cell lung cancer cells. Cancer Res. 2004, 64, 27–30. [Google Scholar] [CrossRef] [Green Version]
- Scheel, J.; Matteoni, R.; Ludwig, T.; Hoflack, B.; Kreis, T.E. Microtubule depolymerization inhibits transport of cathepsin D from the Golgi apparatus to lysosomes. J. Cell Sci. 1990, 96 (Pt 4), 711–720. [Google Scholar]
- Hamai, A.; Codogno, P. New targets for acetylation in autophagy. Sci. Signal. 2012, 5, pe29. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, B.; Acevedo-Arozena, A.; Imarisio, S.; Berger, Z.; Vacher, C.; O’Kane, C.J.; Brown, S.D.; Rubinsztein, D.C. Dynein mutations impair autophagic clearance of aggregate-prone proteins. Nat. Genet. 2005, 37, 771–776. [Google Scholar] [CrossRef] [PubMed]
- Korolchuk, V.I.; Rubinsztein, D.C. Regulation of autophagy by lysosomal positioning. Autophagy 2011, 7, 927–928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, M.; Li, X.X.; Chen, Y.; Pitzer, A.L.; Zhang, Y.; Li, P.L. Enhancement of dynein-mediated autophagosome trafficking and autophagy maturation by ROS in mouse coronary arterial myocytes. J. Cell. Mol. Med. 2014, 18, 2165–2175. [Google Scholar] [CrossRef] [PubMed]
- Du, W.; Su, Q.P.; Chen, Y.; Zhu, Y.; Jiang, D.; Rong, Y.; Zhang, S.; Zhang, Y.; Ren, H.; Zhang, C.; et al. Kinesin 1 Drives Autolysosome Tubulation. Dev. Cell 2016, 37, 326–336. [Google Scholar] [CrossRef] [PubMed]
- Dennis, P.B.; Pullen, N.; Kozma, S.C.; Thomas, G. The principal rapamycin-sensitive p70(s6k) phosphorylation sites, T-229 and T-389, are differentially regulated by rapamycin-insensitive kinase kinases. Mol. Cell. Biol. 1996, 16, 6242–6251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burnett, P.E.; Barrow, R.K.; Cohen, N.A.; Snyder, S.H.; Sabatini, D.M. RAFT1 phosphorylation of the translational regulators p70 S6 kinase and 4E-BP1. Proc. Natl. Acad. Sci. USA 1998, 95, 1432–1437. [Google Scholar] [CrossRef] [Green Version]
- Scott, R.C.; Juhasz, G.; Neufeld, T.P. Direct induction of autophagy by Atg1 inhibits cell growth and induces apoptotic cell death. Curr. Biol. 2007, 17, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Kamada, Y.; Funakoshi, T.; Shintani, T.; Nagano, K.; Ohsumi, M.; Ohsumi, Y. Tor-mediated induction of autophagy via an Apg1 protein kinase complex. J. Cell Biol. 2000, 150, 1507–1513. [Google Scholar] [CrossRef] [Green Version]
- Noda, T.; Ohsumi, Y. Tor, a phosphatidylinositol kinase homologue, controls autophagy in yeast. J. Biol. Chem. 1998, 273, 3963–3966. [Google Scholar] [CrossRef] [Green Version]
- Hong, S.; Zhao, B.; Lombard, D.B.; Fingar, D.C.; Inoki, K. Cross-talk between sirtuin and mammalian target of rapamycin complex 1 (mTORC1) signaling in the regulation of S6 kinase 1 (S6K1) phosphorylation. J. Biol. Chem. 2014, 289, 13132–13141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben-Hur, V.; Denichenko, P.; Siegfried, Z.; Maimon, A.; Krainer, A.; Davidson, B.; Karni, R. S6K1 alternative splicing modulates its oncogenic activity and regulates mTORC1. Cell Rep. 2013, 3, 103–115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, E.Y.; Longatti, A.; McKnight, N.C.; Tooze, S.A. Kinase-inactivated ULK proteins inhibit autophagy via their conserved C-terminal domains using an Atg13-independent mechanism. Mol. Cell. Biol. 2009, 29, 157–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, D.J.; Tan-Sah, V.P.; Ding, E.Y.; Smith, J.M.; Miyamoto, S. Hexokinase-II positively regulates glucose starvation-induced autophagy through TORC1 inhibition. Mol. Cell 2014, 53, 521–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ip, C.K.; Cheung, A.N.; Ngan, H.Y.; Wong, A.S. p70 S6 kinase in the control of actin cytoskeleton dynamics and directed migration of ovarian cancer cells. Oncogene 2011, 30, 2420–2432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pavan, I.C.; Yokoo, S.; Granato, D.C.; Meneguello, L.; Carnielli, C.M.; Tavares, M.R.; do Amaral, C.L.; de Freitas, L.B.; Paes Leme, A.F.; Luchessi, A.D.; et al. Different interactomes for p70-S6K1 and p54-S6K2 revealed by proteomic analysis. Proteomics 2016, 16, 2650–2666. [Google Scholar] [CrossRef]
- Castro-Castro, A.; Janke, C.; Montagnac, G.; Paul-Gilloteaux, P.; Chavrier, P. ATAT1/MEC-17 acetyltransferase and HDAC6 deacetylase control a balance of acetylation of alpha-tubulin and cortactin and regulate MT1-MMP trafficking and breast tumor cell invasion. Eur. J. Cell Biol. 2012, 91, 950–960. [Google Scholar] [CrossRef]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, E.; DiPaola, R.S. The double-edged sword of autophagy modulation in cancer. Clin Cancer Res. 2009, 15, 5308–5316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saera-Vila, A.; Kish, P.E.; Louie, K.W.; Grzegorski, S.J.; Klionsky, D.J.; Kahana, A. Autophagy regulates cytoplasmic remodeling during cell reprogramming in a zebrafish model of muscle regeneration. Autophagy 2016, 12, 1864–1875. [Google Scholar] [CrossRef] [PubMed]
- Pennock, R.; Bray, E.; Pryor, P.; James, S.; McKeegan, P.; Sturmey, R.; Genever, P. Human cell dedifferentiation in mesenchymal condensates through controlled autophagy. Sci. Rep. 2015, 5, 13113. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hać, A.; Pierzynowska, K.; Herman-Antosiewicz, A. S6K1 Is Indispensible for Stress-Induced Microtubule Acetylation and Autophagic Flux. Cells 2021, 10, 929. https://doi.org/10.3390/cells10040929
Hać A, Pierzynowska K, Herman-Antosiewicz A. S6K1 Is Indispensible for Stress-Induced Microtubule Acetylation and Autophagic Flux. Cells. 2021; 10(4):929. https://doi.org/10.3390/cells10040929
Chicago/Turabian StyleHać, Aleksandra, Karolina Pierzynowska, and Anna Herman-Antosiewicz. 2021. "S6K1 Is Indispensible for Stress-Induced Microtubule Acetylation and Autophagic Flux" Cells 10, no. 4: 929. https://doi.org/10.3390/cells10040929