
Post-Translational Modification Networks of Contractile and Cellular Stress Response Proteins in Bladder Ischemia

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bladder Ischemia Model
2.2. Sample Preparation for Proteomic Analysis
2.3. Sodium Dodecyl Sulfate Polyacrylamide Gel Electrophoresis (SDS-PAGE) Fractionation and Trypsin Digestion
2.4. Liquid Chromatography–Tandem Mass Spectrometry (LC-MS/MS) Analysis
2.5. The Clustered Delta Masses
2.6. Gene Ontology, Protein–Protein Interaction Network and Pathway Analysis
2.7. Statistical Analysis
3. Results
3.1. Validation of Bladder Ischemia in the Rat Model
3.2. Delta Mass Clusters in the Bladder Proteome
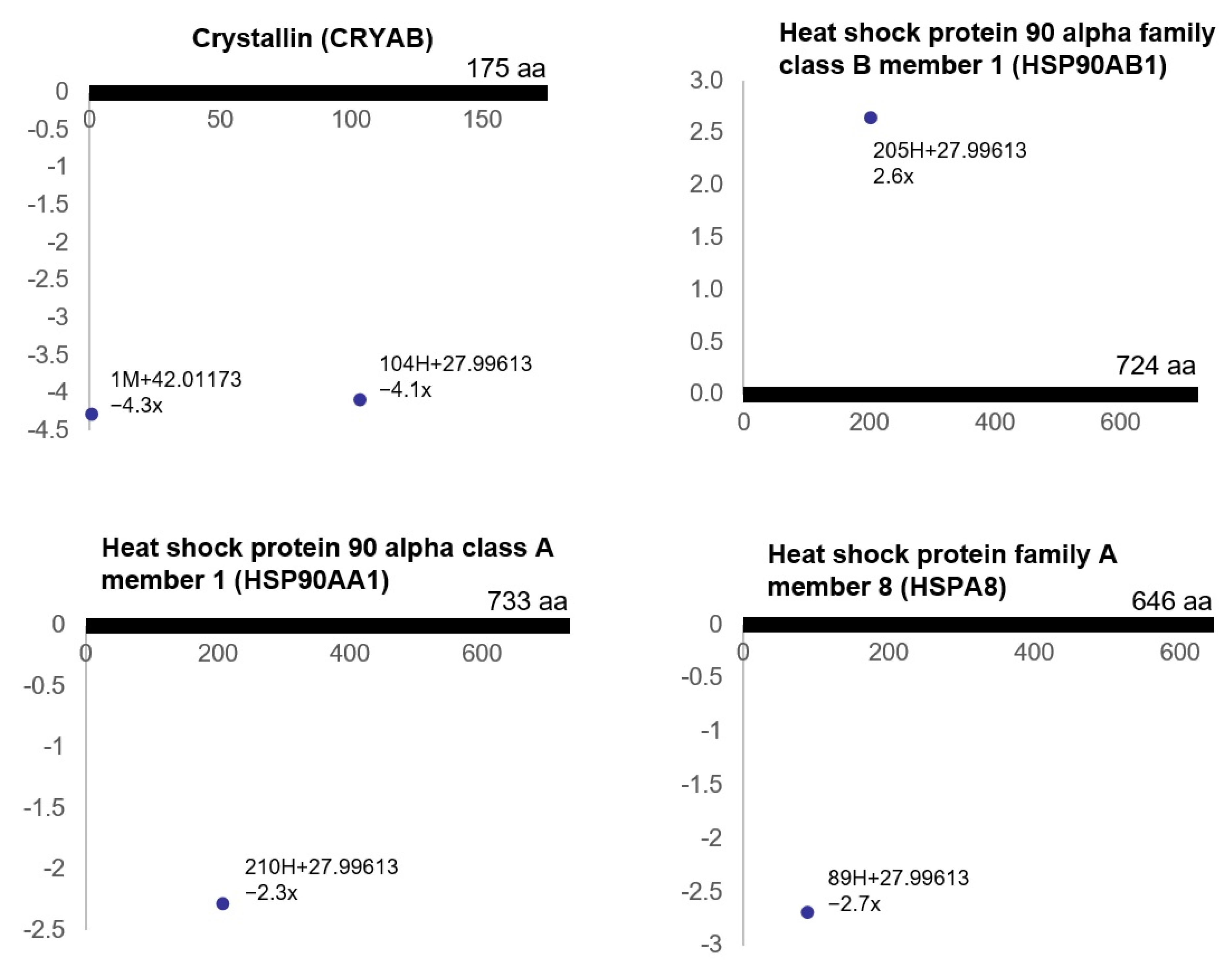
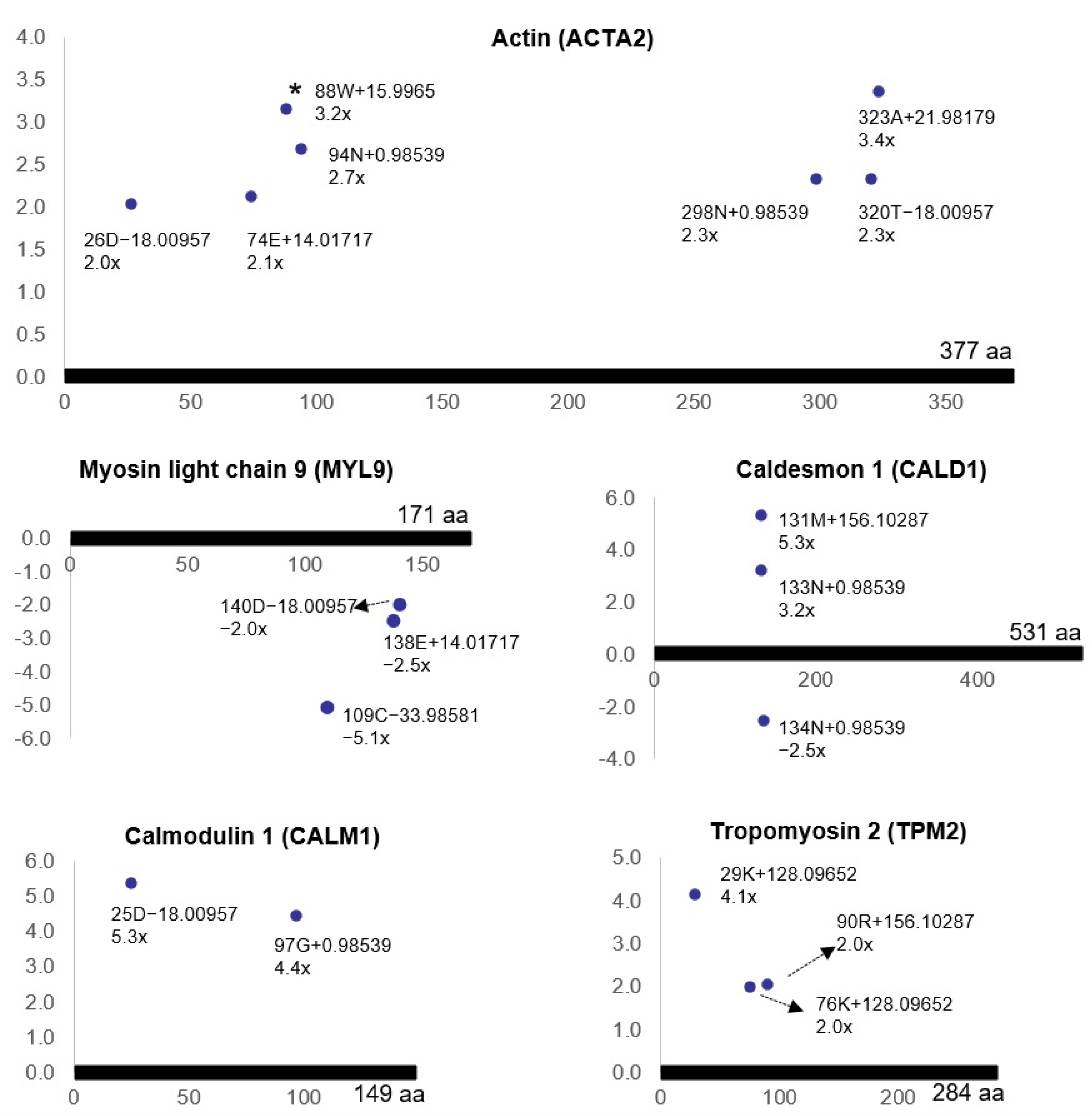
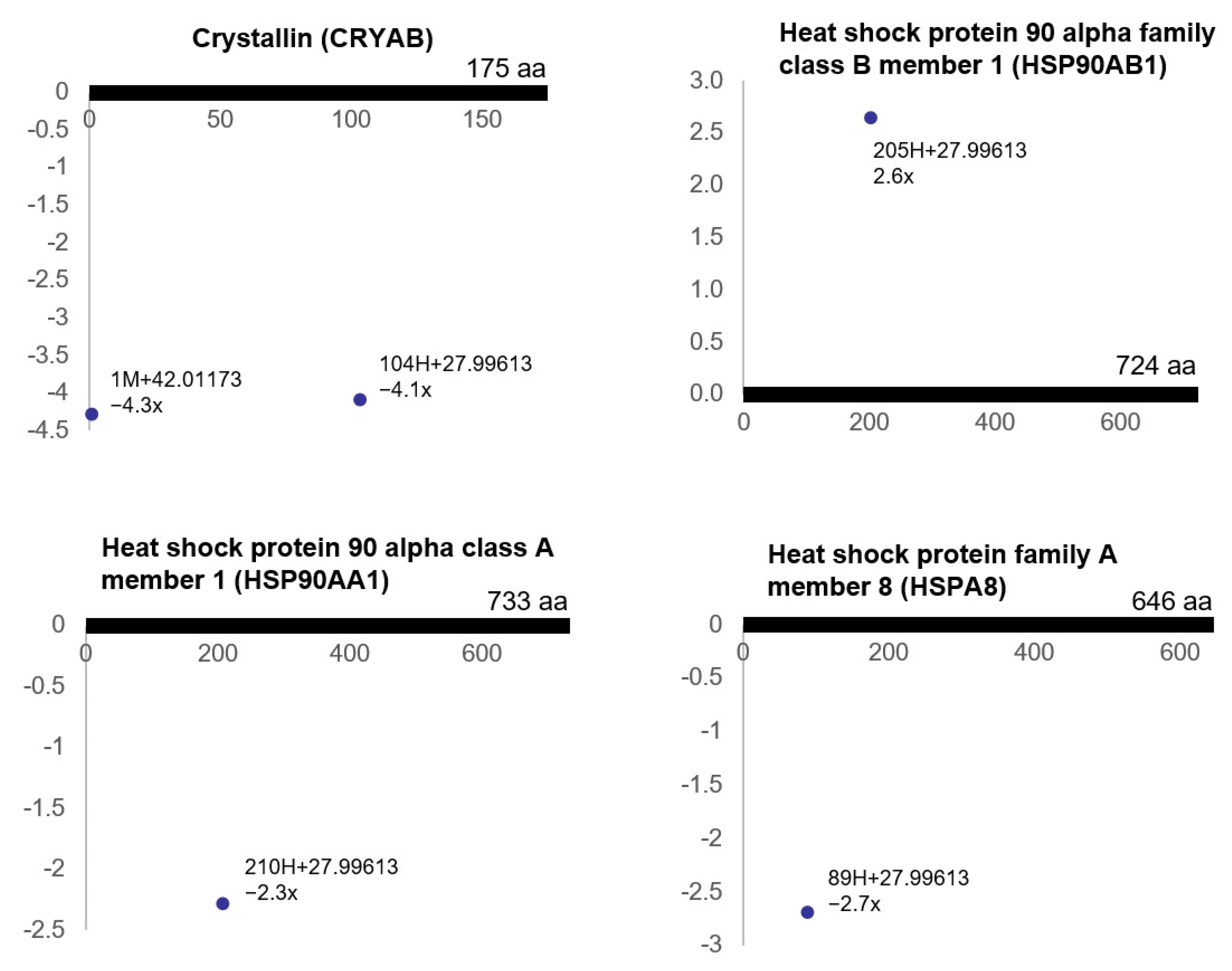
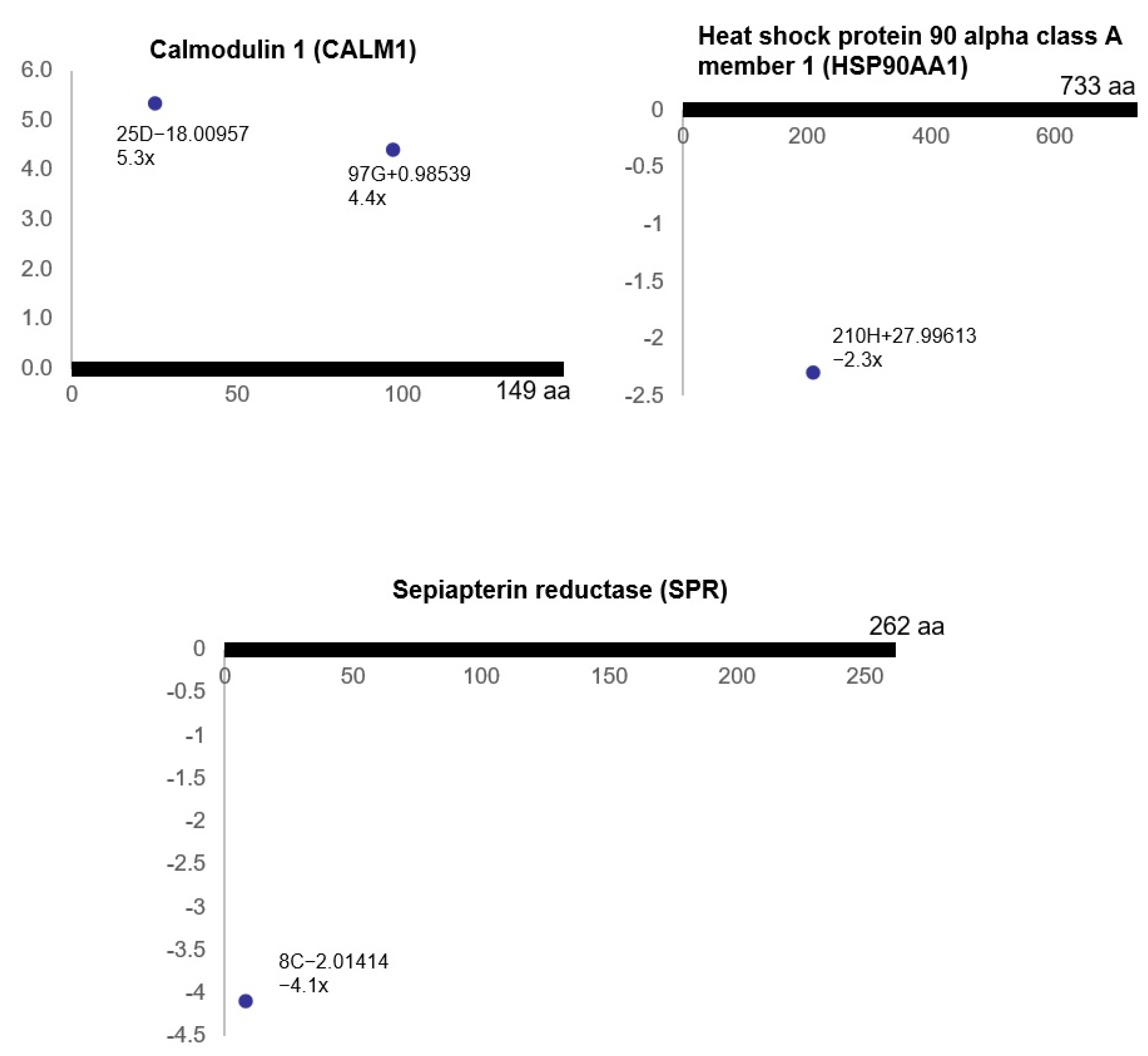
3.3. Ischemia-Regulated Amino Acid Substitutions in the Bladder Proteome
3.4. Pathway Analysis of the Ischemia-Associated ncAA-Containing Proteins
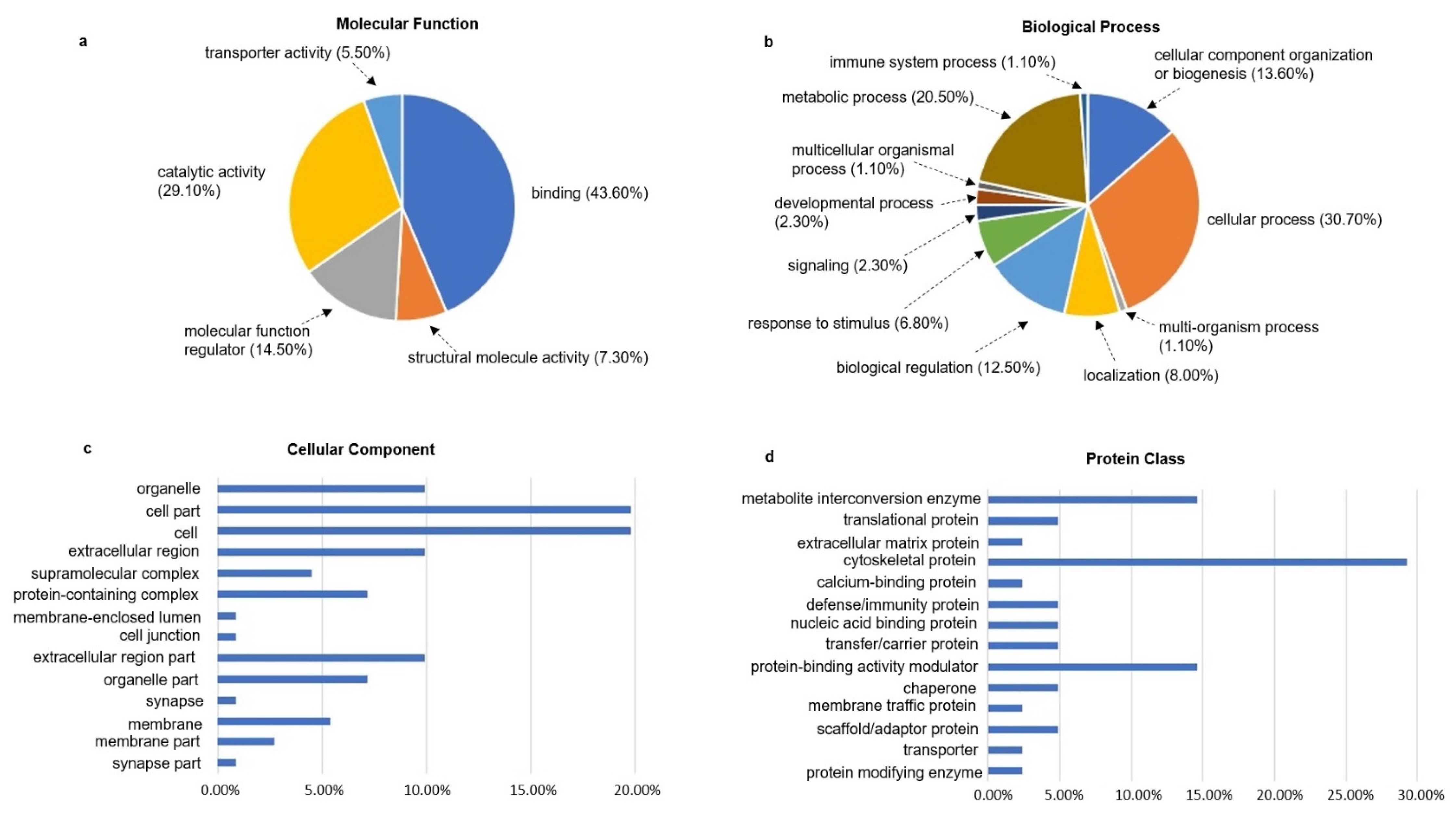
3.5. Gene Ontology Analysis of the Ischemia-Associated ncAA-Containing Proteins
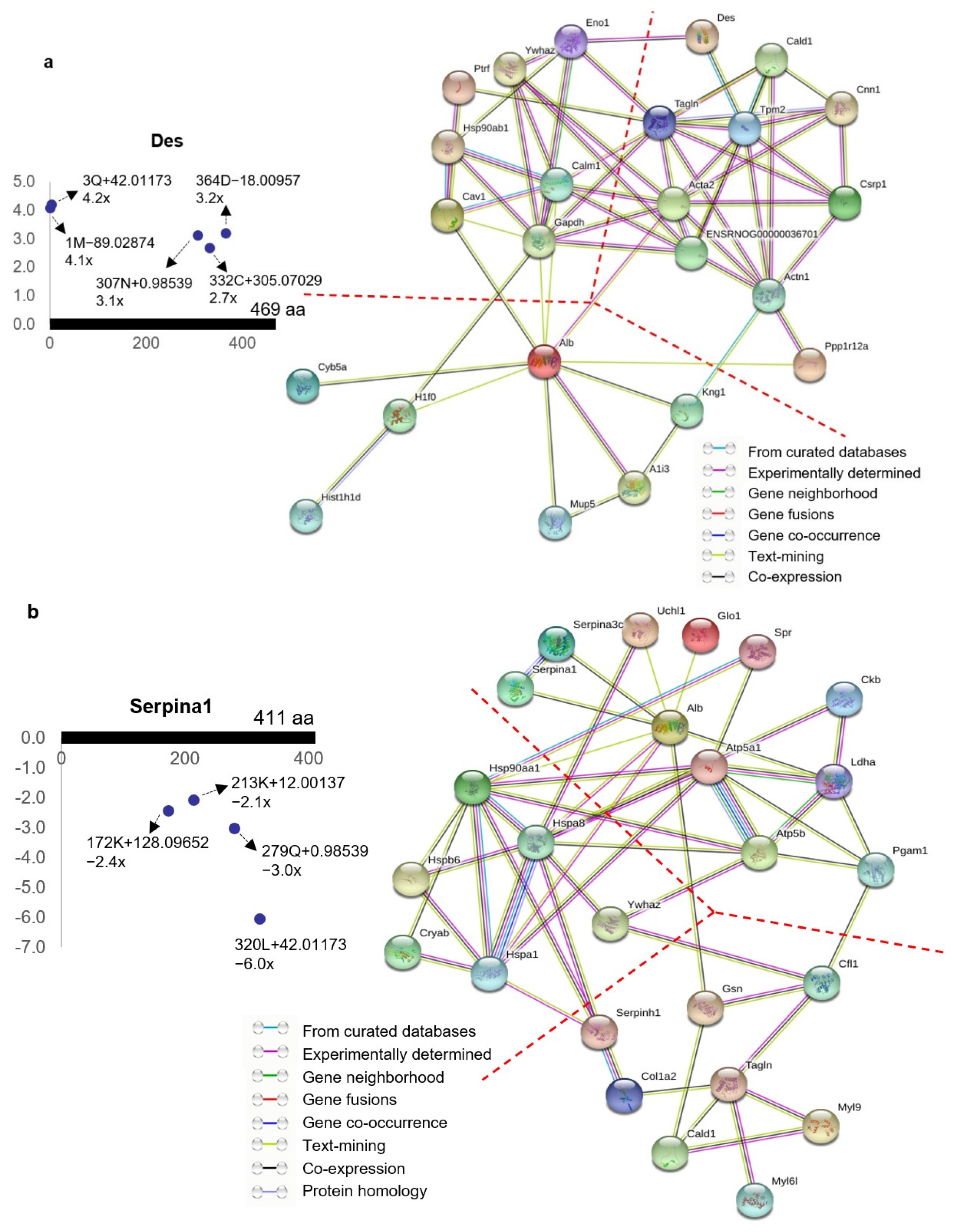
3.6. Protein–Protein Interaction Network Analysis of the Ischemia-Associated ncAA-Containing Proteins
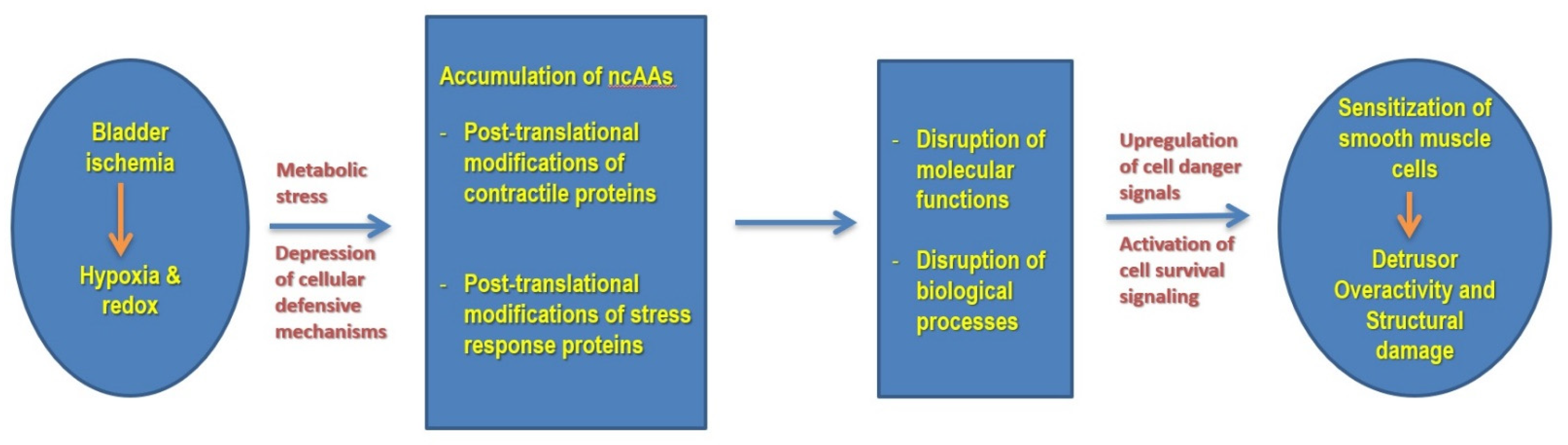
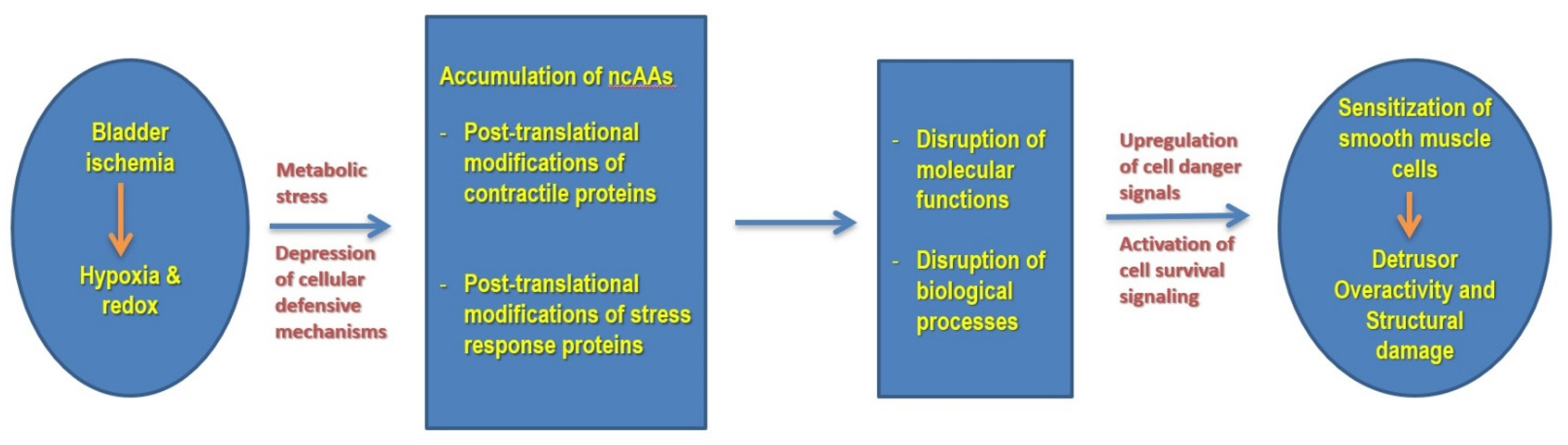
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Azadzoi, K.M.; Tarcan, T.; Kozlowski, R.; Krane, R.J.; Siroky, M.B. Overactivity and structural changes in the chronically ischemic bladder. J. Urol. 1999, 162, 1768–1778. [Google Scholar] [CrossRef]
- Azadzoi, K.M.; Chen, B.G.; Radisavljevic, Z.M.; Siroky, M.B. Molecular reactions and ultrastructural damage in the chronically ischemic bladder. J. Urol. 2011, 186, 2115–2122. [Google Scholar] [CrossRef]
- Zhao, Z.; Azad, R.; Yang, J.H.; Siroky, M.B.; Azadzoi, K.M. Progressive changes in detrusor function and micturition patterns with chronic bladder ischemia. Investig. Clin. Urol. 2016, 57, 249–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersson, K.E.; Boedtkjer, D.B.; Forman, A. The link between vascular dysfunction, bladder ischemia, and aging bladder dysfunction. Ther. Adv. Urol. 2017, 9, 11–27. [Google Scholar] [CrossRef] [PubMed]
- Pinggera, G.M.; Mitterberger, M.; Steiner, E.; Pallwein, L.; Frauscher, F.; Aigner, F.; Bartsch, G.; Strasser, H. Association of lower urinary tract symptoms and chronic ischaemia of the lower urinary tract in elderly women and men: Assessment using colour Doppler ultrasonography. BJU Int. 2008, 102, 470–474. [Google Scholar] [CrossRef]
- Camoes, J.; Coelho, A.; Castro-Diaz, D.; Cruz, F. Lower Urinary Tract Symptoms and Aging: The Impact of Chronic Bladder Ischemia on Overactive Bladder Syndrome. Urol. Int. 2015, 95, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Azadzoi, K.M.; Yalla, S.V.; Siroky, M.B. Oxidative stress and neurodegeneration in the ischemic overactive bladder. J. Urol. 2007, 178, 710–715. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Siroky, M.; Yang, J.H.; Zhao, Z.; Azadzoi, K. Effects of ischemia and oxidative stress on bladder purinoceptors expression. Urology 2014, 84, 1249.e1–1249.e7. [Google Scholar] [CrossRef] [PubMed]
- Minhas, G.; Sharma, J.; Khan, N. Cellular Stress Response and Immune Signaling in Retinal Ischemia-Reperfusion Injury. Front. Immunol. 2016, 7, 444. [Google Scholar] [CrossRef] [Green Version]
- Martindale, J.J.; Metzger, J.M. Uncoupling of increased cellular oxidative stress and myocardial ischemia reperfusion injury by directed sarcolemma stabilization. J. Mol. Cell. Cardiol. 2014, 67, 26–37. [Google Scholar] [CrossRef] [Green Version]
- Swan, C.L.; Sistonen, L. Cellular stress response cross talk maintains protein and energy homeostasis. EMBO J. 2015, 34, 267–269. [Google Scholar] [CrossRef] [Green Version]
- Mo, J.S.; Meng, Z.; Kim, Y.C.; Park, H.W.; Hansen, C.G.; Kim, S.; Lim, D.S.; Guan, K.L. Cellular energy stress induces AMPK-mediated regulation of YAP and the Hippo pathway. Nat. Cell Biol. 2015, 17, 500–510. [Google Scholar] [CrossRef]
- Leavy, O. Signalling: New roles for cell stress sensor. Nat. Rev. Immunol. 2014, 14, 135. [Google Scholar] [CrossRef] [PubMed]
- Soboloff, J.; Madesh, M.; Gill, D.L. Sensing cellular stress through STIM proteins. Nat. Chem. Biol. 2011, 7, 488–492. [Google Scholar] [CrossRef]
- Galluzzi, L.; Yamazaki, T.; Kroemer, G. Linking cellular stress responses to systemic homeostasis. Nat. Rev. Mol. Cell. Biol. 2018, 19, 731–745. [Google Scholar] [CrossRef] [PubMed]
- Kultz, D. Molecular and evolutionary basis of the cellular stress response. Annu. Rev. Physiol. 2005, 67, 225–257. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.H.; Siroky, M.B.; Yalla, S.V.; Azadzoi, K.M. Mitochondrial stress and activation of PI3K and Akt survival pathway in bladder ischemia. Res. Rep. Urol. 2017, 9, 93–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lothrop, A.P.; Torres, M.P.; Fuchs, S.M. Deciphering post-translational modification codes. FEBS Lett. 2013, 587, 1247–1257. [Google Scholar] [CrossRef] [Green Version]
- Holmgren, M.; Wagg, J.; Bezanilla, F.; Rakowski, R.F.; De Weer, P.; Gadsby, D.C. Three distinct and sequential steps in the release of sodium ions by the Na+/K+-ATPase. Nature 2000, 403, 898–901. [Google Scholar] [CrossRef]
- Zentner, G.E.; Henikoff, S. Regulation of nucleosome dynamics by histone modifications. Nat. Struct. Mol. Biol. 2013, 20, 259–266. [Google Scholar] [CrossRef]
- He, Y.; Korboukh, I.; Jin, J.; Huang, J. Targeting protein lysine methylation and demethylation in cancers. Acta Biochim. Biophys. Sin. (Shanghai) 2012, 44, 70–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popovic, D.; Vucic, D.; Dikic, I. Ubiquitination in disease pathogenesis and treatment. Nat. Med. 2014, 20, 1242–1253. [Google Scholar] [CrossRef]
- Smith, L.E.; White, M.Y. The role of post-translational modifications in acute and chronic cardiovascular disease. Proteom. Clin. Appl. 2014, 8, 506–521. [Google Scholar] [CrossRef]
- Kristian, T.; Hu, B. The Protein Modification and Degradation Pathways after Brain Ischemia. Transl. Stroke Res. 2018, 9, 199–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, L.J. Protein redox modification as a cellular defense mechanism against tissue ischemic injury. Oxid. Med. Cell. Longev. 2014, 2014, 343154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tadeusiewicz, J.; Nowak, P. The role of post-translational modification of fibrinogen in the pathogenesis of thrombosis. Pol. Merkur. Lekarski 2015, 38, 107–112. [Google Scholar]
- Hwang, N.R.; Yim, S.H.; Kim, Y.M.; Jeong, J.; Song, E.J.; Lee, Y.; Lee, J.H.; Choi, S.; Lee, K.J. Oxidative modifications of glyceraldehyde-3-phosphate dehydrogenase play a key role in its multiple cellular functions. Biochem. J. 2009, 423, 253–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, X.; Sidoli, S.; Wang, L.; Wang, W.; Guo, L.; Jensen, O.N.; Zheng, L. Comparative proteomic analysis of histone post-translational modifications upon ischemia/reperfusion-induced retinal injury. J. Proteome Res. 2014, 13, 2175–2186. [Google Scholar] [CrossRef]
- Iwabuchi, M.; Sheng, H.; Thompson, J.W.; Wang, L.; Dubois, L.G.; Gooden, D.; Moseley, M.; Paschen, W.; Yang, W. Characterization of the ubiquitin-modified proteome regulated by transient forebrain ischemia. J. Cereb. Blood Flow Metab. 2014, 34, 425–432. [Google Scholar] [CrossRef]
- Shinmura, K. Post-translational modification of mitochondrial proteins by caloric restriction: Possible involvement in caloric restriction-induced cardioprotection. Trends Cardiovasc. Med. 2013, 23, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Walsh, C.T.; Garneau-Tsodikova, S.; Gatto, G.J., Jr. Protein posttranslational modifications: The chemistry of proteome diversifications. Angew. Chem. Int. Ed. Engl. 2005, 44, 7342–7372. [Google Scholar] [CrossRef]
- Bern, M.; Kil, Y.J.; Becker, C. Byonic: Advanced peptide and protein identification software. Curr. Protoc. Bioinform. 2012, 40, 13–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraley, C.; Raftery, A. mclust Version 4 for R: Normal Mixture Modeling for Model-Based Clustering, Classification, and Density Estimation. Available online: https://mclust-org.github.io/mclust (accessed on 23 February 2021).
- Fraley, C.; Raftery, A.E. How Many Clusters? Which Clustering Method? Answers Via Model-Based Cluster Analysis. Comput. J. 1998, 41, 578–588. [Google Scholar] [CrossRef]
- Carter, R.N.; Morton, N.M. Cysteine and hydrogen sulphide in the regulation of metabolism: Insights from genetics and pharmacology. J. Pathol. 2016, 238, 321–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gooley, A.A.; Packer, N.H. The Importance of Protein Co- and Post-Translational Modifications in Proteome Projects. In Proteome Research: New Frontiers in Functional Genomics; Wilkins, M.R., Williams, K.L., Appel, R.D., Hochstrasser, D.F., Eds.; Springer: Berlin/Heidelberg, Germany, 1997; pp. 65–91. [Google Scholar] [CrossRef]
- Kim, Y.E.; Hipp, M.S.; Bracher, A.; Hayer-Hartl, M.; Hartl, F.U. Molecular chaperone functions in protein folding and proteostasis. Annu. Rev. Biochem. 2013, 82, 323–355. [Google Scholar] [CrossRef] [PubMed]
- Azadzoi, K.M.; Yalla, S.V.; Siroky, M.B. Human bladder smooth muscle cell damage in disturbed oxygen tension. Urology 2011, 78, 967.e9–967.e15. [Google Scholar] [CrossRef]
- Powell, S.R.; Gurzenda, E.M.; Wahezi, S.E. Actin is oxidized during myocardial ischemia. Free Radic. Biol. Med. 2001, 30, 1171–1176. [Google Scholar] [CrossRef]
- Eaton, P.; Byers, H.L.; Leeds, N.; Ward, M.A.; Shattock, M.J. Detection, quantitation, purification, and identification of cardiac proteins S-thiolated during ischemia and reperfusion. J. Biol. Chem. 2002, 277, 9806–9811. [Google Scholar] [CrossRef] [Green Version]
- DalleDonne, I.M.; Milzani, A.; Colombo, R. The tert-butyl hydroperoxide-induced oxidation of actin Cys-374 is coupled with structural changes in distant regions of the protein. Biochemistry 1999, 38, 12471–12480. [Google Scholar] [CrossRef]
- Wang, J.; Boja, E.S.; Tan, W.; Tekle, E.; Fales, H.M.; English, S.; Mieyal, J.J.; Chock, P.B. Reversible glutathionylation regulates actin polymerization in A431 cells. J. Biol. Chem. 2001, 276, 47763–47766. [Google Scholar] [CrossRef] [Green Version]
- Stournaras, C.; Drewes, G.; Blackholm, H.; Merkler, I.; Faulstich, H. Glutathionyl (cysteine-374) actin forms filaments of low mechanical stability. Biochim. Biophys. Acta 1990, 1037, 86–91. [Google Scholar] [CrossRef]
- Varland, S.; Vandekerckhove, J.; Drazic, A. Actin Post-translational Modifications: The Cinderella of Cytoskeletal Control. Trends Biochem. Sci. 2019, 44, 502–516. [Google Scholar] [CrossRef] [Green Version]
- Xu, Q.; Huff, L.P.; Fujii, M.; Griendling, K.K. Redox regulation of the actin cytoskeleton and its role in the vascular system. Free Radic. Biol. Med. 2017, 109, 84–107. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo-Freitas, C.; Dulce, R.A.; Foster, M.W.; Liang, J.; Yamashita, A.M.S.; Lima-Rosa, F.L.; Thompson, J.W.; Moseley, M.A.; Hare, J.M.; Nogueira, L.; et al. S-nitrosylation of sarcomeric proteins depresses myofilament Ca2+ sensitivity in intact cardiomyocytes. Antioxid. Redox Signal 2015, 23, 1017–1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moldovan, L.; Moldovan, N.I.; Sohn, R.H.; Parikh, S.A.; Goldschmidt-Clermont, P.J. Redox changes of cultured endothelial cells and actin dynamics. Circ. Res. 2000, 86, 549–557. [Google Scholar] [CrossRef] [Green Version]
- Zou, Y.; Zhu, W.; Sakamoto, M.; Qin, Y.; Akazawa, H.; Toko, H.; Mizukami, M.; Takeda, N.; Minamino, T.; Takano, H.; et al. Heat shock transcription factor 1 protects cardiomyocytes from ischemia/reperfusion injury. Circulation 2003, 108, 3024–3030. [Google Scholar] [CrossRef]
- Westfall, M.V.; Solaro, R.J. Alterations in myofibrillar function and protein profiles after complete global ischemia in rat hearts. Circ. Res. 1992, 70, 302–313. [Google Scholar] [CrossRef] [Green Version]
- Hartl, F.U. Molecular chaperones in cellular protein folding. Nature 1996, 381, 571–579. [Google Scholar] [CrossRef] [PubMed]
- Hightower, L.E. Heat shock, stress proteins, chaperones, and proteotoxicity. Cell 1991, 66, 191–197. [Google Scholar] [CrossRef]
- Cloutier, P.; Coulombe, B. Regulation of molecular chaperones through post-translational modifications: Decrypting the chaperone code. Biochim. Biophys. Acta 2013, 1829, 443–454. [Google Scholar] [CrossRef] [Green Version]
- Cloutier, P.; Lavallée-Adam, M.; Faubert, D.; Blanchette, M.; Coulombe, B. A newly uncovered group of distantly related lysine methyltransferases preferentially interact with molecular chaperones to regulate their activity. PLoS Genet. 2013, 9, e1003210. [Google Scholar] [CrossRef] [Green Version]
- Nitika; Truman, A.W. Cracking the chaperone code: Cellular roles for Hsp70 phosphorylation. Trends Biochem. Sci. 2017, 42, 932–935. [Google Scholar] [CrossRef]
- Backe, S.J.; Sager, R.A.; Woodford, M.R.; Makedon, A.M.; Mollapour, M. Post-translational modifications of Hsp90 and translating the chaperone code. J. Biol. Chem. 2020, 295, 11099–11117. [Google Scholar] [CrossRef]
- Gourdin, M.; Dubois, P. Impact of ischemia on cellular metabolism. In Artery Bypass; Aronow, W.S., Ed.; IntechOpen: London, UK, 2013. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| a Delta Mass Clusters (Da) | Calculated Mass (Da) | b Error | c Possible Chemistry | d Freq | Counts |
|---|---|---|---|---|---|
| 14.0172 | 14.0156 | 0.0016 | Methylation, D > E, N > Q, G > A, S > T, V > I/L | 2 | 418 |
| 12.0014 | 12.0000 | 0.0014 | +Carbon | 2 | 47 |
| 183.0368 | 183.0354 | 0.0014 | Aminoethylbenzene-sulfonylation | 5 | 45 |
| −33.9858 | −33.9844/−33.9877 | 0.0014/0.0019 | F > L/I, C > deHA, M > P | 5 | 33 |
| 27.9961 | 27.9949 | 0.0012 | Formylation, S > D, T > E | 6 | 31 |
| 156.1029 | 156.1011 | 0.0018 | Arg addition | 4 | 20 |
| −17.0252 | −17.0265 | 0.0014 | Deamination, pyroglutamate | 2 | 15 |
| 305.0703 | 305.0682 | 0.0021 | glutathione disulfide | 1 | 15 |
| 42.0117 | 42.0106 | 0.0012 | Acetylation, S > E | 1 | 6 |
| 31.9913 | 31.9898 | 0.0015 | Dioxidation, P > E | 1 | 5 |
| −2.0141 | −2.0157 | 0.0015 | Disulfidelation, V > P, −2H(didehydro) | 1 | 4 |
| 43.9912 | 43.9898 | 0.0013 | Carboxylation, A > D | 1 | 3 |
| Upregulated ncAA-Containing Proteins | Downregulated ncAA-Containing Proteins | ||
|---|---|---|---|
| Protein Name | Protein ID | Protein Name | Protein ID |
| Hemoglobin subunit alpha-1/2 | P01946 | Heat shock 70 kDa protein 1A | P0DMW0 |
| Actin, cytoplasmic 2 | P63259 | Oligoribonuclease, mitochondrial | Q5U1X1 |
| Protein phosphatase 1 regulatory subunit 12A | Q10728 | Phosphoglycerate mutase 1 | P25113 |
| Serum albumin | P02770 | Serum albumin | P02770 |
| Tropomyosin beta chain | P58775 | Serine protease inhibitor A3K | P05545 |
| Actin, aortic smooth muscle | P62738 | Desmin | P48675 |
| Desmin | P48675 | Collagen alpha-2(I) chain | P02466 |
| Alpha-actinin-1 | Q9Z1P2 | Transgelin | P31232 |
| Cysteine and glycine-rich protein 1 | P47875 | L-lactate dehydrogenase A chain | P04642 |
| Prothymosin alpha | P06302 | Serpin H1 | P29457 |
| Complement component C9 | Q62930 | Alpha-1-antiproteinase | P17475 |
| Glutathione S-transferase P | P04906 | Creatine kinase B-type | P07335 |
| Hemoglobin subunit beta-1 | P02091 | Ubiquitin carboxyl-terminal hydrolase isozyme L1 | Q00981 |
| Histone H1.0 | P43278 | Alpha-crystallin B chain | P23928 |
| Calmodulin-1 | P0DP29 | ATP synthase subunit beta, mitochondrial | P10719 |
| 14-3-3 protein zeta/delta | P63102 | Elongation factor 1-delta | Q68FR9 |
| Glyceraldehyde-3-phosphate dehydrogenase | P04797 | 40S ribosomal protein S4, X isoform | P62703 |
| Alpha-1-inhibitor 3 | P14046 | Lactoylglutathione lyase | Q6P7Q4 |
| Non-muscle caldesmon | Q62736 | ATP synthase subunit alpha, mitochondrial | P15999 |
| Calponin-1 | Q08290 | Sepiapterin reductase | P18297 |
| Polymerase I and transcript release factor | P85125 | Ig gamma-2B chain C region | P20761 |
| Transgelin | P31232 | Myosin regulatory light polypeptide 9 | Q64122 |
| Alpha-enolase | P04764 | Four and a half LIM domains protein 1 | Q9WUH4 |
| Histone H1.4 | P15865 | Myosin light polypeptide 6 | Q64119 |
| T-kininogen 2 | P08932 | Gelsolin | Q68FP1 |
| Cytochrome b5 | P00173 | 14-3-3 protein zeta/delta | P63102 |
| Tubulin alpha-1B chain | Q6P9V9 | Non-muscle caldesmon | Q62736 |
| Caveolin-1 | P41350 | Selenium-binding protein 1 | Q8VIF7 |
| Major urinary protein | P02761 | Heat shock protein HSP 90-alpha | P82995 |
| Heat shock protein HSP 90-beta | P34058 | Hemoglobin subunit beta-2 | P11517 |
| Heat shock protein beta-6 | P97541 | ||
| Cofilin-1 | P45592 | ||
| Heat shock cognate 71 kDa protein | P63018 | ||
| Function | Count | p-Value | Protein Name |
|---|---|---|---|
| Smooth Muscle Contraction | 5 | 1.4 × 10−5 | ACTA2, MYL9, CALD1, CALM1, TPM2 |
| Scavenging of heme from plasma | 4 | 9.4 × 10−5 | ALB, HBB2, HBB1, HBA1 |
| HSF1-dependent transactivation | 4 | 1.1 × 10−4 | CRYAB, HSP90AB1, HSP90AA1, HSPA8 |
| Tetrahydrobiopterin (BH4) synthesis, recycling, salvage and regulation | 3 | 1.9 × 10−3 | CALM1, HSP90AA1, SPR |
| Attenuation phase | 3 | 1.9 × 10−3 | HSP90AB1, HSP90AA1, HSPA8 |
| Erythrocytes take up oxygen and release carbon dioxide | 3 | 2.2 × 10−3 | HBB2, HBB1, HBA1 |
| eNOS activation | 3 | 2.2 × 10−3 | CALM1, CAV1, HSP90AA1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, J.-H.; Choi, H.-P.; Yang, A.; Azad, R.; Chen, F.; Liu, Z.; Azadzoi, K.M. Post-Translational Modification Networks of Contractile and Cellular Stress Response Proteins in Bladder Ischemia. Cells 2021, 10, 1031. https://doi.org/10.3390/cells10051031
Yang J-H, Choi H-P, Yang A, Azad R, Chen F, Liu Z, Azadzoi KM. Post-Translational Modification Networks of Contractile and Cellular Stress Response Proteins in Bladder Ischemia. Cells. 2021; 10(5):1031. https://doi.org/10.3390/cells10051031
Chicago/Turabian StyleYang, Jing-Hua, Han-Pil Choi, Annie Yang, Roya Azad, Fengmei Chen, Zhangsuo Liu, and Kazem M. Azadzoi. 2021. "Post-Translational Modification Networks of Contractile and Cellular Stress Response Proteins in Bladder Ischemia" Cells 10, no. 5: 1031. https://doi.org/10.3390/cells10051031
APA StyleYang, J.-H., Choi, H.-P., Yang, A., Azad, R., Chen, F., Liu, Z., & Azadzoi, K. M. (2021). Post-Translational Modification Networks of Contractile and Cellular Stress Response Proteins in Bladder Ischemia. Cells, 10(5), 1031. https://doi.org/10.3390/cells10051031