Abstract
Synaptic plasticity events, including long-term potentiation (LTP), are often regarded as correlates of brain functions of memory and cognition. One of the central players in these plasticity-related phenomena is the α-amino-3-hydroxy-5-methylisoxazole-4-propionate receptor (AMPAR). Increased levels of AMPARs on postsynaptic membranes thus constitute a biochemical measure of LTP. Isolated synaptic terminals (synaptosomes) are an excellent ex vivo tool to monitor synaptic physiology in healthy and diseased brains, particularly in human research. We herein describe three protocols for chemically-induced LTP (cLTP) in synaptosomes from both rodent and human brain tissues. Two of these chemical stimulation protocols are described for the first time in synaptosomes. A pharmacological block of synaptosomal actin dynamics confirmed the efficiency of the cLTP protocols. Furthermore, the study prototypically evaluated the deficiency of cLTP in cortical synaptosomes obtained from human cases of early-onset Alzheimer’s disease (EOAD) and frontotemporal lobar degeneration (FLTD), as well as an animal model that mimics FLTD.
Keywords:
glycine; KCl; NMDAR; depolarization; rolipram; forskolin; tetraethyl ammonium; PS19 mice; latrunculin A; radio-labelled 1. Introduction
Synapses, the communication points between neurons, elicit tremendous plasticity and undergo dynamic short- and long-term modifications. Activity-dependent alterations in synaptic plasticity, such as long-term potentiation (LTP) and long-term depression (LTD), are thought to represent the cellular basis of learning and memory [1,2,3]. Among the most well-characterized molecular targets at the center of synaptic plasticity-related phenomena are the α-amino-3-hydroxy-5-methylisoxazole-4-propionate (AMPA) types of glutamatergic receptors. AMPA receptors (AMPARs) are rapidly cycled between intracellular pools and synaptic surfaces during LTP and LTD [4,5,6,7]. In fact, both electrical and chemical (chemical LTP or cLTP) means of LTP induction have been shown to result in rapid trafficking of AMPARs at the plasmalemmal surfaces of the synaptic terminals [8,9,10].
Synaptosomes are biochemically isolated subcellular preparations of presynaptic terminals in close apposition to postsynaptic densities [11,12,13]. These preparations can be conveniently obtained from post-mortem brain tissues, and are excellent ex vivo models to recapitulate multiple aspects of basal and activity-induced synaptic phenomena. In fact, because of the preservation of most enzymatic and metabolic activities, synaptosomes serve as efficient “halfway house” tools between neurochemistry and electrophysiology [11,12,13,14,15]. It is worth noting that synaptosomes are being established as a prominent research tool to understand the mechanisms of synaptic dysfunction in diseased human brains, wherein the acquisition of acute slices and cultures are technically challenging [11,13,15]. Importantly, viable synaptosomes can be obtained from post-mortem human brain tissues with significant post-mortem delays and storage times [16,17,18,19]. In recent years, synaptosomes from AD brains have been utilized to study different aspects of synaptic physiology, such as neurotransmitter release and uptake [20,21], disease pathogenesis [22,23,24,25], protein translation [26,27], actin dynamics [28,29], protein and lipid signaling cascades [26,30,31,32,33] and bioenergetic functions [34,35,36].
The present study aimed to further establish the utility of synaptosomal preparations from human and rodent brain tissues in studying cLTP events using three different chemical stimulation protocols with AMPAR insertion at postsynaptic sites as a common readout. The first chemical stimulation protocol involved depolarization mediated by high extracellular K+ following the priming of N-methyl-D-aspartate receptors (NMDARs) with glycine. This cLTP protocol has been employed in ex vivo synaptosomes coupled with various methods for the assessment of surface-expressed AMPARs, including liquid scintillation [37], surface biotinylation followed by immunoblotting [38] and immunolabelling followed by fluorescence-based sorting [39,40,41]. The second stimulation protocol is based upon the coincubation of rolipram and forskolin, a phosphodiesterase 4 (PDE4) inhibitor and an adenylate cyclase activator respectively, to assess NMDAR-dependent LTP [42,43,44,45,46,47]. The third protocol utilizes a potassium channel blocker tetraethyl ammonium (TEA), which induces membrane depolarization and a consequent burst of calcium spikes resulting in an NMDAR-independent form of LTP [48,49,50,51,52]. Although the latter two protocols have been employed in cell cultures and acute slices, for the first time, the present study applied both methods to synaptosomes obtained from either rodent and human brain tissues for the induction of cLTP. It should be noted, however, that there are reports of employing rolipram and/or forskolin for the stimulation of presynaptic neurotransmitter release in isolated synaptosomes [53,54,55,56].
The second aim of this study was to obtain a proof-of-principle of the cLTP protocols in ex vivo synaptosomes, by evaluating the efficacy of the three protocols via a pharmacological blockade of synaptosomal actin dynamics. Actin is a cytoskeletal protein forming the integral framework maintaining both structural and functional aspects of synaptic physiology [57,58,59]. Actin dynamics, the interconversion between its monomeric globular form (G-actin) and its polymerized filamentous form (F-actin), plays a fundamental role in controlling the size, number and morphology of neuronal dendritic spines. It has been well documented that both pre- and post-synaptic functions are critically dependent on actin dynamics [60,61,62,63,64]. We therefore determined the effects of blocking actin dynamics on cLTP-induced changes in surface-exposed AMPARs in synaptosomes using latrunculin A [65,66,67,68].
Synapse loss and synaptic dysfunction closely correlate with cognitive deficits in pathophysiological conditions across the whole developmental spectrum of the central nervous system (CNS), ranging from early life insults [69,70,71] to neurodegenerative disorders such as Alzheimer’s disease (AD) and frontotemporal lobar degeneration (FTLD) [72,73,74,75]. Moreover, there is recent evidence indicating the dysfunction of synaptic cytoskeletal actin dynamics in neurodegeneration [28,29,76,77,78,79,80]. The third aim of the study was therefore to obtain the proof-of-concept of the utility of synaptosomes in the determination of cLTP changes in neurodegenerative disorders using post-mortem brain tissues from human subjects of early-onset AD (EOAD) and frontotemporal lobar degeneration (FTLD), and PS19 mice bearing human microtubule-associated protein tau P301S mutation, which mimics human FTLD [81,82].
2. Materials and Methods
2.1. Human Tissues
Human brain tissues were obtained from the Neurological Tissue Bank of Hospital Clínic-IDIBAPS BioBank in Barcelona, Spain. All tissue collection protocols were approved by the Ethics Committee of Hospital Clínic, Barcelona (October 2018) and informed consent was obtained from all families. The unfixed snap-frozen superior frontal gyrus was obtained from EOAD, FTLD and neurologically normal (NOR) cases (n = 5/group). The neurologically normal cases were defined by the absence of any history of neurological and psychiatric diseases and the absence of any neuropathological findings from a detailed analysis of the brain by an independent neuropathologist. All diseased cases fulfilled the neuropathological consensus criteria for AD or FTLD. EOAD was defined as AD with age at onset before 65 years. There were no significant differences between groups in terms of the age and post-mortem delay (Table 1). The experimenters were blind to the grouping information.

Table 1.
Details of the human cases used for the study.
2.2. Animals and Tissue Collection
Male P301S tau transgenic (PS19) mice (B6;C3-Tg(Prnp-MAPT*P301S)PS19Vle/J; stock number: 008169) and C57BL/6J female mice were crossed to produce the offspring of PS19 mice and wildtype (WT) littermates that were confirmed by tail tip genotyping. The present study used 8 months old male PS19 and WT mice, as well as 5 months old male Sprague-Dawley rats. All animals were maintained on a 12-h light/dark cycle with ad libitum access to food chow and water. All animals were anesthetized with sodium pentobarbitone and transcardially perfused with ice-cold saline. The brain from each animal was rapidly removed and transferred to saline on ice. Brain tissues (cortices) were then freshly dissected on ice from each hemisphere and immediately snap-frozen and stored at −80 °C [83,84]. All experimental procedures were carried out in accordance with the regulations of the University of Otago Committee on Ethics in the Care and Use of Laboratory Animals and New Zealand legislature (Ethics Protocol No. AUP-95-18 and AUP-80-17).
2.3. Drugs, Reagents and Antibodies
Phosphatase inhibitor cocktail IV (ab201115) and EDTA-free protease inhibitor cocktail (4693159001) were from Abcam (Cambridge, MA, USA) and Roche Diagnostics (Mannheim, Germany), respectively. [3H]-AMPA (DL-α-[5-methy-3H)) was obtained from Perkin Elmer (Boston, MA, USA; NET833250UC; 58.1 Ci/mmol). Potassium thiocyanate (KSCN; 207799), tetraethyl ammonium (TEA; T-2265) and latrunculin A (L5163) were procured from Sigma-Aldrich (St. Louis, MO, USA). Forskolin (HB1348) was from HelloBio. Rolipram (ab120029) and primary antibodies against PSD-95 (rabbit; ab18258) and GAPDH (mouse; ab8245) were from Abcam (Cambridge, MA, USA). Fluorescent secondary antibodies (IRDye® 800 CW donkey anti-rabbit IgG and IRDye® 680 RD goat anti-mouse IgG) were from LI-COR Biosciences (Lincoln, NE, USA). Other regents used in the study were of analytical grade and purchased either from Thermo Fisher Scientific or Sigma-Aldrich.
2.4. Preparation of Synaptosomes
Synaptosomes from either human or rodent brain tissues were prepared as described in our previous studies [26,30,85]. Briefly, tissues were homogenized using a Potter-Elvehjem tissue grinder (Duran Wheaton Kimble, Mexico; 358034) in 10 volumes of ice-cold homogenization buffer containing 5 mM HEPES (pH 7.4) and 0.32 M sucrose supplemented with protease and phosphatase inhibitor cocktails. The homogenate was centrifuged at 1500× g for 10 min at 4 °C to obtain a crude nuclear pellet (containing nuclei and cellular debris), which was discarded. The supernatant obtained was further centrifuged at 12,000× g for 15 min at 4 °C to obtain a crude synaptosomal pellet containing mitochondria and myelin. This crude synaptosomal pellet was resuspended in 5 mM Tris buffer at pH 7.4 containing 0.32 M sucrose supplemented with protease and phosphatase inhibitor cocktails. The resuspended synaptosomal pellet was then subjected to fractionation on a discontinuous sucrose gradient (equal volumes of 0.85 M, 1.0 M and 1.2 M sucrose) at 85,000× g for 1.5 hr at 4 °C to separate out the contaminating mitochondria and myelin fractions from the synaptosomes. The purified synaptosomes were collected from the interface between the 1.0 and 1.2 M sucrose layers, and then subjected to two washes, first with 5 mM Tris buffer (pH 8.1) and second with homogenization buffer, at 12,000× g for 15 min at 4 °C. The washed synaptosomes were resuspended in homogenization buffer for further analyses. An aliquot of synaptosomes from each sample was used for the quantification of the protein content using Bradford assay. Of note, discontinuous gradient-based subcellular fractionation has been shown to generate intact and enriched synaptic terminals from both human and rodent brain tissues by us and others [19,26,30,85,86,87].
2.5. Immunoblotting
Immunoblotting was performed to assess the purity of the synaptosomes by evaluating the enrichment of PSD-95, a synaptic protein marker. For this, homogenates and synaptosomal samples were mixed with gel loading buffer (containing 50 mM Tris-HCl at pH 6.8, 10% SDS, 10% glycerol, 10% 2-mercaptoethanol and 2 mg/mL bromophenol blue) and were heated at 95 °C for 5 min. Ten μg protein per sample was loaded on a CriterionTM XT 4–12% gradient SDS-PAGE gel (BioRad; 3450124), and electroblotted onto a nitrocellulose membrane as detailed in our publication [88]. Following blocking with 5% BSA in Tris-buffered saline at pH 7.4 containing 1% Tween 20 (TBST), the membrane was immunostained with primary antibodies against PSD-95 and housekeeping protein GAPDH. Fluorescently labelled secondary antibodies were used to generate immunoreactive signals. The signals were detected using the Odyssey Infrared System (LI-COR Biosciences; Lincoln, NE, USA) at 700 and 800 nm, and the analysis of immunoreactive signals was performed using ImageStudioLite (LI-COR Biosciences; Lincoln, NE, USA).
2.6. cLTP Protocol 1: Glycine Priming—KCl Depolarization (gK)
Synaptosomal NMDAR priming and stimulation were carried out based upon the methodologies from previous publications with modifications [37,38,39,40,41,89]. Briefly, synaptosomes (200 μg protein) were resuspended in duplicates with one set in buffer A1 (containing in mM; 120 NaCl, 3 KCl, 2 CaCl2, 2 MgCl2, 15 glucose, 15 HEPES; pH 7.4) and the other set in buffer B1 (containing in mM; 125 NaCl, 2 CaCl2, 5 KCl, 30 glucose, 10 HEPES at pH 7.4, 0.02 bicuculline and 0.001 strychnine), and pre-incubated at 37 °C for 10 min. A1 synaptosomes constituted the synaptosomes with basal activity, while B1 synaptosomes underwent cLTP. Glycine (500 μM) was added to B1 synaptosomes along with fresh bicuculline (0.02 mM) and strychnine (0.001 mM) to prime NMDARs for 15 min at 37 °C. An equal volume of buffer A1 was added to A1 synaptosomes for 15 min at 37 °C. Depolarization-induced stimulation of NMDARs was performed by a steep increase in KCl to a final [K+]extracellular of 50 mM, via the addition of buffer C1 (containing in mM; 50 NaCl, 2 CaCl2, 100 KCl, 30 glucose, 0.5 glycine and 10 HEPES at pH 7.4, 0.02 bicuculine and 0.001 strychnine). Depolarization was proceeded for a duration of 60 min at 37 °C. A1 Synaptosomes (mock-stimulated) were supplemented with an equal volume of buffer A1 and incubated for the same duration of 60 min at 37 °C. cLTP in synaptosomes was measured as changes in the surface expression of AMPARs, evaluated by [3H]-AMPA binding (Section 2.10).
2.7. cLTP Protocol 2: Rolipram—Forskolin (RF)
This protocol takes advantage of the fact that a sustained increase of cyclic adenosine monophosphate (cAMP) level in the presence of forskolin (an adenylate cyclase activator) and rolipram (PDE4 inhibitor) results in the induction of cLTP, as shown earlier in acute slices [42,43,44,90], primary neurons [45,46,47,91,92] and organotypic slice cultures [93,94]. Here, we extended this cLTP protocol to ex vivo stimulation of synaptosomes with minor modifications. Briefly, synaptosomes (200 μg protein) were resuspended in duplicates, one set in buffer A2 (containing in mM; 120 NaCl, 3 KCl, 2 CaCl2, 2 MgCl2, 15 glucose and 15 HEPES at pH 7.4) and the other in buffer B2 (containing in mM; 125 NaCl, 5 CaCl2, 5 KCl, 30 glucose and 15 HEPES at pH 7.4, 0.02 bicuculline and 0.001 strychnine) and pre-incubated at 37 °C for 10 min. B2 synaptosomes (that underwent cLTP) were treated with 50 μM forskolin and 0.1 μM rolipram for 30 min at 37 °C. An equal volume of buffer A2 was added to control mock-stimulated A2 synaptosomes for the same duration of 30 min at 37 °C. cLTP was calculated as increases in the surface expression of AMPARs, measured by [3H]-AMPA binding on synaptosomal membranes (Section 2.10).
2.8. cLTP Protocol 3: Tetraethyl Ammonium (TEA)
The third chemical stimulation of synaptosomes was carried out in the presence of TEA, a non-specific voltage-gated potassium channel blocker that has been used to induce NMDAR-independent cLTP in acute slices [44,48,49,50,51,52], primary neurons [66,95,96] and organotypic slice cultures [97,98]. Briefly, synaptosomes (200 μg protein) was resuspended in duplicates with one set in buffer A3 and the other in buffer B3, and then pre-incubated at 37 °C for 10 min. cLTP was induced in B3 synaptosomes by addition of 50 mM TEA at 37 °C for 15 min, while control mock-stimulated A3 synaptosomes were supplemented with an equal volume of buffer A3 for the same duration of incubation. cLTP was quantitated as changes in the levels of surface-exposed AMPARs, which was evaluated by [3H]-AMPA binding (Section 2.10).
2.9. Pharmacological Block of Actin Polymerization
Pharmacological blockade of actin polymerization has often been employed to evaluate its role in mediating plastic changes at the synapses induced by neuronal activity. Latrunculin A, a pharmacological agent that depolymerizes actin, has been commonly used to abolish LTP [66,68,99,100,101]. Of note, low concentrations of latrunculin A prevent actin polymerization without leading to substantial depolymerization of existing actin filaments, hence preserving the basal synaptic organization [66,101,102]. Latrunculin A at a concentration of 200 nM was used in the present study to depolymerize actin filaments without affecting the basal surface-exposed AMPAR levels. Pre-incubation with latrunculin A was carried out both for synaptosomes undergoing cLTP (B1, B2 or B3) and control unstimulated synaptosomes (A1, A2 or A3) at 37 °C for 30 min. Induction of cLTP (in B1, B2 or B3 synaptosomes) or mock-cLTP stimulation (for A1, A2 or A3 synaptosomes) was also performed in the presence of latrunculin A. Effects of latrunculin A on cLTP was again assessed by evaluating the changes in synaptosomal [3H]-AMPA binding (Section 2.10).
2.10. [3H]-AMPA Labelling
Exogenous radiolabeled [3H]-AMPA has been conveniently employed to evaluate changes in the surface-expressed AMPARs using both autoradiography and liquid scintillation spectroscopy [37,103,104,105]. [3H]-AMPA labelling of synaptosomes in the presence of KSCN was carried out as described previously [37,106,107,108]. KSCN was used because this thiocyanate chaotropic anion increases the efficiency of AMPA binding [106]. First, ex vivo cLTP (or mock-cLTP) in synaptosomes was culminated by addition of 1 mL of ice-cold binding buffer (containing in mM; 100 Tris-acetate at pH 7.4, 0.1 EGTA and 100 KSCN). Following centrifugation at 12,000× g for 15 min at 4 °C, the synaptosomal pellets were resuspended and incubated in binding buffer on ice for 30 min. Synaptosomal AMPA-labeling was carried in binding buffer containing 25 nM [3H]-AMPA on ice for 60 min. Binding of radiolabeled AMPA was terminated by centrifugation at 18,000× g at 4 °C for 10 min, followed by a superficial wash of the pellet with ice-cold binding buffer. Synaptosomal pellets were then solubilized in 0.2 N NaOH and the bound radioactivity was counted by liquid scintillation spectrometry on a Perkin Elmer TriCarb 2910 TR liquid scintillator (Downers Grove, IL, USA). Non-specific binding of AMPA was determined in the presence of 100 mM glutamate [107,108]. To account for any variable retention of stimulated and unstimulated paired sets of synaptosomes during the washing steps, protein content of the AMPA-labelled synaptosomes were evaluated using Bradford assay.
2.11. Statistical Analyses
[3H]-AMPA binding is represented as femtomoles per mg protein. Data were presented as mean ± standard error of mean (SEM). Statistically significant differences in [3H]-AMPA binding between cLTP-stimulated synaptosomes and their respective mock-stimulated control samples were evaluated using two-tailed paired Student’s t-test with a p < 0.05 considered to be significant. All analyses and representation of the data were performed using GraphPad Prism 9 software (San Diego, CA, USA).
3. Results
3.1. Biochemical Preparations of Synaptosomes Are Enriched in PSD-95
We determined the protein levels of PSD-95 (a synaptic marker protein) in the rat cortical homogenates and synaptosomes isolated using a discontinuous sucrose gradient fractionation. In consistence with our previous studies [30,85], a robust 3-fold increase of PSD-95 was observed in synaptosomal samples when compared to the starting homogenates (p = 0.025; Figure 1), confirming appreciable enrichment of vesicular synaptic terminals in the our synaptosomal preparations.
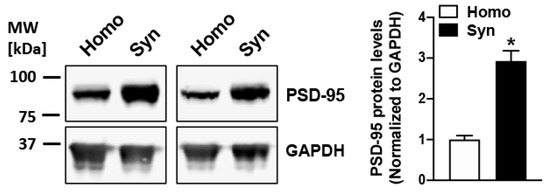
Figure 1.
PSD-95 is enriched in rat cortical synaptosomes obtained by sucrose density gradient-based subcellular fractionation. Robust enrichment of synaptic marker protein PSD-95 is observed in synaptosomes (Syn) as compared to the respective starting homogenate (Homo) samples (n = 3), indicating a sturdy enhancement of isolated synaptic terminals in the synaptosomal fraction. * represents a statistical significance at p < 0.05.
3.2. Stimulation of Synaptosomes Results in AMPAR Mobilization at the Surface
Changes in plasmalemmal AMPARs induced by the three protocols of cLTP in ex vivo synaptosomes were evaluated using exogenous [3H]-AMPA binding. Synaptosomes from rat brain cortices primed with glycine and depolarized with high extracellular K+ elicited a significant increase (23%; p = 0.004) in surface-exposed AMPARs compared to the respective mock-stimulated (with no glycine and KCl treatments) controls (Figure 2A). Appreciable increases in surface-expressed AMPARs were also observed upon stimulations of synaptosomes with rolipram-forskolin (21%; p = 0.001 Figure 2B) when compared to the respective unstimulated synaptosomes (without treatment with rolipram-forskolin). Lastly, cLTP induction with TEA also resulted in increments in synaptosomal binding of [3H]-AMPA (15%; p = 0.005; Figure 2C) when compared to the respective mock-stimulated control synaptosomes (without treatment with TEA).
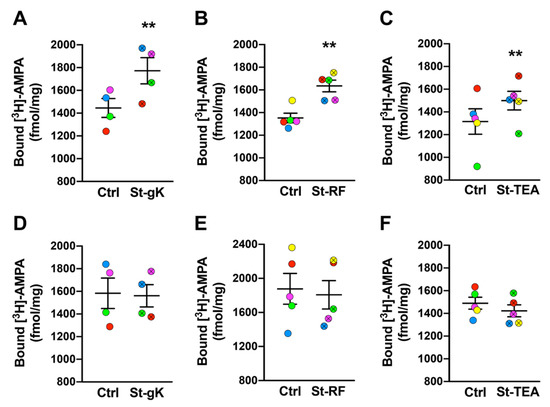
Figure 2.
Chemically induced long-term potentiation (cLTP) in rat cortical synaptosomes (A–C) is abolished by latrunculin A (D–F). All three chemical stimulation protocols, glycine-KCl (St-gK; A), rolipram-forskolin (St-RF; B) and TEA (St-TEA; C), resulted in elevated surface AMPAR expression in rat cortical synaptosomes relative to their corresponding non-stimulation matched controls (Ctrl). However, latrunculin A (200 nM; a blocker of actin polymerization) completely abolished cLTP-induced insertion of AMPARs on rat synaptosomal membranes following the stimulations by glycine-KCl (D), rolipram-forskolin (E) and TEA (F). Results are expressed as mean (±SEM) bound [3H]-AMPA (n = 4–6 for each experiment). ** represents a statistical significance at p < 0.01. The respective pairs of stimulated and unstimulated synaptosomes are color coded in each panel.
3.3. Latrunculin A Abolishes cLTP-Induced Stimulation in AMPAR Surface Expression
Actin dynamics is a primary event in mediating plasticity-induced responses at the synapses. Hence, we proceeded to determine the effects of blocking actin polymerization on the changes in surface-exposed AMPARs induced by cLTP via all three chemical protocols. Synaptosomes were pre-treated with latrunculin A (200 nM) and their stimulation (or mock-stimulation) was also performed in the presence of latrunculin A. Importantly, the sustained presence of latrunculin A (and consequent failure to stimulate new actin depolarization) prevented the increases in surface-exposed AMPARs following the cLTP induction by all the three protocols; glycine-KCl (Figure 2D), rolipram-forskolin (Figure 2E) and TEA (Figure 2F).
3.4. cLTP Can Be Induced in Human Cortical Synaptosomes
We next sought to extend the chemical stimulation protocols to synaptosomes from superior frontal gyri of cognitively normal human cases. Again, significant increases in surface AMPAR expression levels were induced by glycine-KCl (14%; Figure 3A), rolipram-forskolin (13%; Figure 3B) and TEA (15%; Figure 3C). Among the three cLTP protocols, the most consistent stimulation was achieved by glycine priming and KCl depolarization.
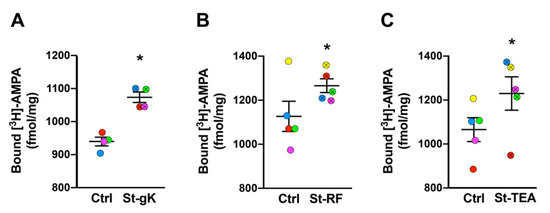
Figure 3.
Long-term potentiation (LTP) is chemically induced in synaptosomes from superior frontal gyri of cognitively normal cases. All three chemical stimulation protocols, glycine-KCl (St-gK; A), rolipram-forskolin (St-RF; B) and TEA (St-TEA; C), resulted in increased levels of surface AMPARs relative to their corresponding non-stimulation matched controls (Ctrl). Results are expressed as mean (±SEM) bound [3H]-AMPA (n = 4–5 for each experiment). * represents a statistical significance at p < 0.05. The respective stimulated and unstimulated synaptosome pairs are color coded in each panel.
3.5. cLTP Is Impaired in Frontocortical Synaptosomes of EOAD and FTD Cases
Using the glycine priming and KCl depolarization protocol, the present study determined the cLTP changes in synaptosomes from tissue samples of superior frontal gyri obtained from EOAD and FTLD cases relative to cognitively normal (NOR) subjects. Upon the evaluation of [3H]-AMPA labelling following priming with glycine and subsequent stimulation with KCl, a significant increase of surface AMPAR expression was observed in the synaptosomes from the normal cases (17%, p = 0.009; Figure 4A). However, the same protocol elicited a small but insignificant increase in surface AMPAR levels of the synaptosomes obtained from the EOAD (6%, p = 0.41; Figure 4B) or FTLD (7%, p = 0.11; Figure 4C) cases, indicating synaptic cLTP deficits in patients with neurodegenerative disorders. Of note, the basal (unstimulated) levels of surface-exposed AMPARs were not found to be significantly altered between the three groups.
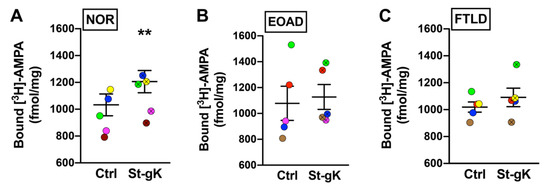
Figure 4.
Chemically induced long-term potentiation (cLTP) is deficient in human cortical synaptosomes from patients with neurodegeneration. Surface AMPAR levels in cortical synaptosomes obtained from the normal (NOR), early-onset Alzheimer’s disease (EOAD) and frontotemporal lobar degeneration (FTLD) cases were assessed following glycine priming and KCl stimulation (St-gK). A robust cLTP was induced by St-gK relative to non-stimulation control (Ctrl) in synaptosomes from the normal cases (A), but not the EOAD (B) or FTLD (C) cases. Results are expressed as mean (±SEM) bound [3H]-AMPA (n = 5 for each experiment). ** represents a statistical significance at p < 0.01. The respective stimulated and unstimulated synaptosome pairs are color coded in each panel.
3.6. cLTP Is Impaired in Cortical Synaptosomes of PS19 Mice
Next, we proceeded to evaluate cLTP-induced changes in surface-exposed AMPAR levels in cortical synaptosomes obtained from 8 months old male PS19 mice (expressing the human tau P301S mutation) relative to the age- and sex-matched WT control mice using the protocol of glycine priming and subsequent KCl depolarization. As expected, the basal levels of surface-exposed AMPARs in unstimulated synaptosomes were not found to be significantly altered in PS19 mice when compared to WT mice. However, while glycine priming-KCl stimulation resulted in a 19% increase in [3H]-AMPA binding in WT synaptosomes (p = 0.03; Figure 5A), cortical synaptosomes from PS19 mice failed to elicit a similar increase in surface expression of AMPARs (p = 0.14; Figure 5B).
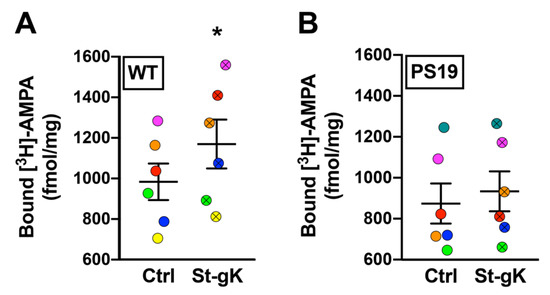
Figure 5.
Chemically induced long-term potentiation (cLTP) is abolished in cortical synaptosomes of 8 months old PS19 mice. Surface AMPAR levels in cortical synaptosomes obtained from wildtype (WT) and PS19 mice were assessed following glycine priming and KCl stimulation (St-gK). A robust cLTP was induced in synaptosomes from WT mice (A), but not in PS19 mice (B). Results are expressed as mean (±SEM) bound [3H]-AMPA (n = 6 for each experiment). * represents a statistical significance at p < 0.05. The respective stimulated and unstimulated synaptosome pairs are color coded in each panel.
4. Discussion
Synaptosomes are fast being recognized as efficient tools to characterize multiple aspects of mammalian synaptic physiology. These ex vivo biochemical preparations of pinched-off and resealed synaptic terminals are disconnected from the cell bodies. However, they retain much of the metabolic and enzymatic activities [11,14]. Synaptosomes are particularly of relevance in the perspective of human pathological cases, as they can be efficiently obtained from long-term stored post-mortem brain tissues with significant post-mortem delays. Not surprisingly, human synaptosomes have been employed for biochemical profiling of a wide range of synapse-specific phenomena [11,15].
The present study utilized synaptosomes obtained from both rodent and human cryopreserved brains to evaluate the postsynaptic responses upon the induction of cLTP by three different ways of chemical stimulation. Increases or decreases in surface expression of AMPARs at synaptic sites relate to the enhancements or repressions in synaptic strength in LTP and LTD, respectively. In fact, the increment of surface-expressed AMPAR levels is often regarded as a biochemical measure of LTP [5,6,7,9]. Taking advantage of this, we chose the quantification of surface AMPARs as a common readout for all the three different cLTP protocols (Figure 2). Noteworthy, surface expression of AMPARs in acute slices, primary neurons and synaptosomes have relied on membrane-impermeant radiolabeled AMPA with subsequent quantitation of surface expressed AMPARs by liquid scintillation spectroscopy [37] or autoradiography [103]. There are other methods such as surface biotinylation [45], fluorescent in situ hybridization [92] and immunolabelling followed by fluorescence- and size- sorting [39] to assess plasmalemmal AMPARs. In cell cultures, exogenous expression of fluorescently tagged AMPAR subunit GluR1 has also been employed to directly evaluate changes in its surface expression [96]. However, time and equipment constraints involved with these methods means that radiolabeling with [3H]-AMPA followed by liquid scintillation is still widely used for the assessment of surface AMPARs [105,109,110,111,112]. With this method, importantly, we observed similar levels of increases in AMPAR surface expression as reported in previous studies using different protocols of electrically or chemically induced LTP that employed liquid scintillation spectroscopy [37], autoradiography [103], surface biotinylation [45], fluorescent in situ hybridization using anti-AMPAR subunit antibodies [113] and live cell image of cells expressing fluorescently tagged AMPAR subunits [66].
There are many novel aspects of our study. While cLTP induction by rolipram-forskolin and TEA have been routinely performed in acute and organotypic slices and primary neurons, for the first time, this study extends the two chemical stimulations to ex vivo synaptosomes obtained from both rodent (Figure 2 A-C) and human (Figure 3) brain tissues. It should be pointed out that while we did observe statistically significant increases of synaptosomal surface expression of AMPARs using all three chemical stimulation protocols, the effects of TEA stimulation were slightly varied relative to the other two methods, particularly for human brain tissues. Nevertheless, as an NMDAR-independent LTP induction protocol (relative to the NMDAR-dependent rolipram/forskolin and glycine-KCl protocols), the TEA protocol might serve as a useful addition to the tools employed to characterize postsynaptic responses in ex vivo synaptosomes.
Another major conclusion from our study is the further evidence for the critical role of dynamic actin polymerization in mediating postsynaptic changes upon the induction of cLTP (Figure 2D-F). As the major component of synaptic cytoskeleton, actin is a key player in regulating the size and structure of the synapses. Moreover, dynamic alterations in actin polymerization status are critical for plasticity-induced remodeling of morpho-functional attributes of the synapses [58,59,63,64]. For example, actin polymerization and depolymerization are associated with spine enlargement and shrinkage during LTP and LTD, respectively [60,99,114,115]. During LTP, calcium influx through NMDARs induces a chain of molecular events resulting in fast actin polymerization, that is, the conversion of G-actin to F-actin. Activity-dependent plastic modulation of AMPAR expression on synaptic surfaces relies on such fast changes in cytoskeletal actin polymerization status [66,68,99,114,116]. Of note, ex vivo depolarization of synaptosomes has been shown to results in rapid actin polymerization [117,118]. Not surprisingly, pharmacological block of actin polymerization abolishes LTP-induced synaptic changes and AMPAR dynamics [66,68,99,101,102]. Consistent with these data, our results showing the abolishment of cLTP-induced surface expression of AMPARs by latrunculin A (Figure 2D-F) demonstrates the robustness of the synaptosomal cLTP methodologies.
Finally, we obtained the proof-of-concept of the utility of synaptosomes in the determination of cLTP changes in neurodegenerative disorders using post-mortem brain tissues from both the EOAD and FTLD cases and the PS19 mice bearing human tau P301S mutation. It should be noted here that cytoskeletal dysfunction of actin has long been associated with neurodegenerative disorders such as AD [76,77,119] and FLTD models [78,79,80]. Interestingly, widespread alterations in actin polymerization status have also been observed recently in synaptosomes obtained from AD brains [28,29]. In addition, LTP deficits are a common feature of FLTD as observed by deficiency of electrically-induced LTP in ex vivo brain slices from PS19 mice [81,120,121]. It is worth noting that only a few studies have evaluated cLTP responses in ex vivo synaptosomes obtained from animal models of AD [41,122]. Interestingly, Prieto et al. (2017) reported abnormal cLTP (using the glycine priming and KCl depolarization protocol) in synaptosomes from AD brains [41]. The present study, for the first time, demonstrated that ex vivo synaptosomes from the EOAD and FTLD cases and PS19 mice elicit deficits in cLTP responses even when the sample size was very small.
5. Conclusions
In conclusion, by validating two novel chemical stimulation protocols (rolipram-forskolin and TEA) in synaptosomes from rodent and human synaptosomes, we build upon the evidence for the utility of synaptosomes as an efficient tool in the apprehension of synaptic physiology and the associated molecular and cellular mechanisms associated with them, particularly in the study of clinical cases. Moreover, the elucidation of cLTP defects in both human subjects with neurodegenerative conditions of EOAD and FLTD and an animal model of FLTD suggests that this approach can be applied efficiently to study synaptic plasticity in neuropathologies. It should be noted that the clinically diagnosed FTLD cases were a mixture of FTLD-CBD (with tau inclusions) and FTLD-TDP43 (with TDP-43 inclusions). The present study is a preliminary work as part of the proof-of-concept for evaluating cLTP in synaptosomes from human and rodent brains. Additional studies with a larger sample size and multiple brain regions (for human tissue work) and age points (for animal work) are warranted to further evaluate the details of synaptic dysfunction in neurodegenerative diseases and are currently ongoing in our laboratory.
Author Contributions
Conceptualization, F.A. and P.L.; methodology, F.A.; validation, F.A., formal analysis, F.A. and P.L.; investigation, F.A. and Y.J.; resources, P.L. and A.L.; data curation, F.A.; writing—original draft preparation, F.A.; writing—review and editing, F.A. and P.L.; visualization, F.A. and P.L.; supervision, P.L.; project administration, P.L.; funding acquisition, P.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Neurological Foundation of New Zealand, grant number 1835-PG, the New Zealand Health Research Council, grant number 16-597 and the Department of Anatomy, University of Otago, New Zealand.
Institutional Review Board Statement
The study was conducted according to the guidelines of the Declaration of Helsinki and approved by the University of Otago Committee on Ethics in the Care and Use of Laboratory Animals, University of Otago (protocol code AUP-95-18, approved on 14.09.2018; AUP-80-17, approved on 14.11.2017) and the Ethics Committee of Hospital Clinic, Barcelona, Spain (approved on 30 October 2018).
Informed Consent Statement
Informed consent was obtained from families of all subjects involved in the post-mortem study.
Data Availability Statement
The data presented in this study are available in the article.
Acknowledgments
We are indebted to the Neurological Tissue Bank of HCB-IDIBAPS BioBank (Spain) for human sample and data procurement. We thank Jiaxian Zhang, Cliff Abraham, Owen Jones, Stephanie Hughes for their assistance.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
References
- Bannerman, D.M.; Sprengel, R.; Sanderson, D.J.; Mchugh, S.B.; Rawlins, J.N.P.; Monyer, H.; Seeburg, P.H. Hippocampal synaptic plasticity, spatial memory and anxiety. Nat. Rev. Neurosci. 2014, 15, 181–192. [Google Scholar] [CrossRef] [PubMed]
- Mayford, M.; Siegelbaum, S.A.; Kandel, E.R. Synapses and memory storage. Cold Spring Harb. Perspect. Biol. 2012, 4, a005751. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, T.; Duszkiewicz, A.J.; Morris, R.G.M. The synaptic plasticity and memory hypothesis: Encoding, storage and persistence. Philos. Trans. R. Soc. B Biol. Sci. 2014, 369, 20130288. [Google Scholar] [CrossRef]
- Diering, G.H.; Huganir, R.L. The AMPA receptor code of synaptic plasticity. Neuron 2018, 100, 314–329. [Google Scholar] [CrossRef]
- Henley, J.M.; Wilkinson, K.A. Synaptic AMPA receptor composition in development, plasticity and disease. Nat. Rev. Neurosci. 2016, 17, 337–350. [Google Scholar] [CrossRef] [PubMed]
- Huganir, R.L.; Nicoll, R.A. AMPARs and synaptic plasticity: The last 25 years. Neuron 2013, 80, 704–717. [Google Scholar] [CrossRef] [PubMed]
- Kessels, H.W.; Malinow, R. Synaptic AMPA Receptor Plasticity and Behavior. Neuron 2009, 61, 340–350. [Google Scholar] [CrossRef]
- Moretto, E.; Passafaro, M. Recent findings on AMPA receptor recycling. Front. Cell. Neurosci. 2018, 12, 286. [Google Scholar] [CrossRef]
- Malinow, R.; Malenka, R.C. AMPA receptor trafficking and synaptic plasticity. Annu. Rev. Neurosci. 2002, 25, 103–126. [Google Scholar] [CrossRef] [PubMed]
- Hanley, J.G. Actin-dependent mechanisms in AMPA receptor trafficking. Front. Cell. Neurosci. 2014, 8, 381. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, F.; Liu, P. Synaptosome as a tool in Alzheimer’s disease research. Brain Res. 2020, 1746, 147009. [Google Scholar] [CrossRef] [PubMed]
- Evans, G.J.O. The synaptosome as a model system for studying synaptic physiology. Cold Spring Harb. Protoc. 2015, 2015, 421–424. [Google Scholar] [CrossRef]
- Murphy, K.M. Synaptosomes (Neuromethods 141); Humana Press: Totowa, NJ, USA, 2018; ISBN 9781493987382. [Google Scholar]
- Erecinska, M.; Nelson, D.; Silver, I.A. Metabolic and energetic properties of isolated nerve ending particles (synaptosomes). Biochim. Biophys. Acta Bioenergy 1996, 1277, 13–34. [Google Scholar] [CrossRef]
- Jhou, J.F.; Tai, H.C. The Study of Postmortem Human Synaptosomes for Understanding Alzheimer’s Disease and Other Neurological Disorders: A Review. Neurol. Ther. 2017, 6, 57–68. [Google Scholar] [CrossRef] [PubMed]
- Dodd, P.R.; Hardy, J.A.; Baig, E.B.; Kidd, A.M.; Bird, E.D.; Watson, W.E.J.; Johnston, G.A.R. Optimization of freezing, storage, and thawing conditions for the preparation of metabolically active synaptosomes from frozen rat and human brain. Neurochem. Pathol. 1986, 4, 177–198. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.A.; Dodd, P.R. CHAPTER 2—Metabolic and functional studies on post-mortem human brain. In Selected Topics from Neurochemistry; Elsevier: Amsterdam, The Netherlands, 1985; pp. 25–41. [Google Scholar]
- Hardy, J.A.; Dodd, P.R.; Oakley, A.E.; Kidd, A.M.; Perry, R.; Edwardson, J.A. Use of post-mortem human synaptosomes for studies of metabolism and transmitter amino acid release. Neurosci. Lett. 1982, 33, 317–322. [Google Scholar] [CrossRef]
- Hardy, J.A.; Dodd, P.R.; Oakley, A.E.; Perry, R.H.; Edwardson, J.A.; Kidd, A.M. Metabolically active synaptosomes can be prepared from frozen rat and human brain. J. Neurochem. 1983, 40, 608–614. [Google Scholar] [CrossRef]
- Slotkin, T.A.; Seidler, F.J.; Crain, B.J.; Bell, J.M.; Bissette, G.; Nemeroff, C.B. Regulatory changes in presynaptic cholinergic function assessed in rapid autopsy material from patients with Alzheimer disease: Implications for etiology and therapy. Proc. Natl. Acad. Sci. USA 1990, 87, 2452–2455. [Google Scholar] [CrossRef]
- Westphalen, R.I.; Scott, H.L.; Dodd, P.R. Synaptic vesicle transport and synaptic membrane transporter sites in excitatory amino acid nerve terminals in Alzheimer disease. J. Neural Transm. 2003, 110, 1013–1027. [Google Scholar] [CrossRef] [PubMed]
- Berrocal, M.; Corbacho, I.; Gutierrez-Merino, C.; Mata, A.M. Methylene blue activates the PMCA activity and cross-interacts with amyloid β-peptide, blocking Aβ-mediated PMCA inhibition. Neuropharmacology 2018, 139, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Bilousova, T.; Miller, C.A.; Poon, W.W.; Vinters, H.V.; Corrada, M.; Kawas, C.; Hayden, E.Y.; Teplow, D.B.; Glabe, C.; Albay, R.; et al. Synaptic amyloid-β oligomers precede p-Tau and differentiate high pathology control cases. Am. J. Pathol. 2016, 186, 185–198. [Google Scholar] [CrossRef] [PubMed]
- Tai, H.C.; Serrano-Pozo, A.; Hashimoto, T.; Frosch, M.P.; Spires-Jones, T.L.; Hyman, B.T. The synaptic accumulation of hyperphosphorylated tau oligomers in alzheimer disease is associated with dysfunction of the ubiquitin-proteasome system. Am. J. Pathol. 2012, 181, 1426–1435. [Google Scholar] [CrossRef] [PubMed]
- Sokolow, S.; Henkins, K.M.; Bilousova, T.; Gonzalez, B.; Vinters, H.V.; Miller, C.A.; Cornwell, L.; Poon, W.W.; Gylys, K.H. Pre-synaptic C-terminal truncated tau is released from cortical synapses in Alzheimer’s disease. J. Neurochem. 2015, 133, 368–379. [Google Scholar] [CrossRef]
- Ahmad, F.; Singh, K.; Das, D.; Gowaikar, R.; Shaw, E.; Ramachandran, A.; Rupanagudi, K.V.; Kommaddi, R.P.; Bennett, D.A.; Ravindranath, V. Reactive Oxygen Species-Mediated Loss of Synaptic Akt1 Signaling Leads to Deficient Activity-Dependent Protein Translation Early in Alzheimer’s Disease. Antioxid. Redox Signal. 2017, 27, 1269–1280. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.; Shai, R.M.; Wu, Y.; Hsu, Y.H.; Sitzer, T.; Spann, B.; McCleary, C.; Mo, Y.; Miller, C.A. Transcriptome analysis of synaptoneurosomes identifies neuroplasticity genes overexpressed in incipient Alzheimer’s disease. PLoS ONE 2009, 4, e4936. [Google Scholar] [CrossRef]
- Kommaddi, R.P.; Das, D.; Karunakaran, S.; Nanguneri, S.; Bapat, D.; Ray, A.; Shaw, E.; Bennett, D.A.; Nair, D.; Ravindranath, V. Aβ mediates F-actin disassembly in dendritic spines leading to cognitive deficits in alzheimer’s disease. J. Neurosci. 2018, 38, 1085–1099. [Google Scholar] [CrossRef] [PubMed]
- Rush, T.; Martinez-Hernandez, J.; Dollmeyer, M.; Frandemiche, M.L.; Borel, E.; Boisseau, S.; Jacquier-Sarlin, M.; Buisson, A. Synaptotoxicity in Alzheimer’s disease involved a dysregulation of actin cytoskeleton dynamics through cofilin 1 phosphorylation. J. Neurosci. 2018, 38, 10349–10361. [Google Scholar] [CrossRef]
- Ahmad, F.; Das, D.; Kommaddi, R.P.; Diwakar, L.; Gowaikar, R.; Rupanagudi, K.V.; Bennett, D.A.; Ravindranath, V. Isoform-specific hyperactivation of calpain-2 occurs presymptomatically at the synapse in Alzheimer’s disease mice and correlates with memory deficits in human subjects. Sci. Rep. 2018, 8, 13119. [Google Scholar] [CrossRef] [PubMed]
- Bader Lange, M.L.; Cenini, G.; Piroddi, M.; Mohmmad Abdul, H.; Sultana, R.; Galli, F.; Memo, M.; Butterfield, D.A. Loss of phospholipid asymmetry and elevated brain apoptotic protein levels in subjects with amnestic mild cognitive impairment and Alzheimer disease. Neurobiol. Dis. 2008, 29, 456–464. [Google Scholar] [CrossRef] [PubMed]
- Bothmer, J.; Markerink, M.; Jolles, J. Phosphoinositide kinase activities in synaptosomes prepared from brains of patients with Alzheimer’s disease and controls. Neurosci. Lett. 1994, 176, 169–172. [Google Scholar] [CrossRef]
- Krishnan, B.; Kayed, R.; Taglialatela, G. Elevated phospholipase D isoform 1 in Alzheimer’s disease patients’ hippocampus: Relevance to synaptic dysfunction and memory deficits. Alzheimers Dement. Transl. Res. Clin. Interv. 2018, 4, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Amadoro, G.; Corsetti, V.; Atlante, A.; Florenzano, F.; Capsoni, S.; Bussani, R.; Mercanti, D.; Calissano, P. Interaction between NH2-tau fragment and Aβ in Alzheimer’s disease mitochondria contributes to the synaptic deterioration. Neurobiol. Aging 2012, 33, 833.e1–833.e25. [Google Scholar] [CrossRef]
- Devi, L.; Prabhu, B.M.; Galati, D.F.; Avadhani, N.G.; Anandatheerthavarada, H.K. Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer’s disease brain is associated with mitochondrial dysfunction. J. Neurosci. 2006, 26, 9057–9068. [Google Scholar] [CrossRef]
- Reddy, P.H.; Oliver, D.M. Amyloid beta and phosphorylated tau-induced defective autophagy and mitophagy in Alzheimer’s disease. Cells 2019, 8, 488. [Google Scholar] [CrossRef] [PubMed]
- Corera, A.T.; Doucet, G.; Fon, E.A. Long-term potentiation in isolated dendritic spines. PLoS ONE 2009, 4, e6021. [Google Scholar] [CrossRef] [PubMed]
- Mitroi, D.N.; Pereyra-Gómez, G.; Soto-Huelin, B.; Senovilla, F.; Kobayashi, T.; Esteban, J.A.; Ledesma, M.D. NPC 1 enables cholesterol mobilization during long-term potentiation that can be restored in Niemann–Pick disease type C by CYP 46A1 activation. EMBO Rep. 2019, 20, e48143. [Google Scholar] [CrossRef]
- Prieto, G.A.; Snigdha, S.; Baglietto-Vargas, D.; Smith, E.D.; Berchtold, N.C.; Tong, L.; Ajami, D.; Laferla, F.M.; Rebek, J.; Cotman, C.W. Synapse-specific IL-1 receptor subunit reconfiguration augments vulnerability to IL-1β in the aged hippocampus. Proc. Natl. Acad. Sci. USA 2015, 112, E5078–E5087. [Google Scholar] [CrossRef] [PubMed]
- Prieto, G.A.; Tong, L.; Smith, E.D.; Cotman, C.W. TNFα and IL-1β but not IL-18 suppresses hippocampal long-term potentiation directly at the synapse. Neurochem. Res. 2019, 44, 49–60. [Google Scholar] [CrossRef]
- Prieto, G.A.; Trieu, B.H.; Dang, C.T.; Bilousova, T.; Gylys, K.H.; Berchtold, N.C.; Lynch, G.; Cotman, C.W. Pharmacological rescue of long-term potentiation in Alzheimer diseased synapses. J. Neurosci. 2017, 37, 1197–1212. [Google Scholar] [CrossRef] [PubMed]
- Vitolo, O.V.; Sant’Angelo, A.; Costanzo, V.; Battaglia, F.; Arancio, O.; Shelanski, M. Amyloid β-peptide inhibition of the PKA/CREB pathway and long-term potentiation: Reversibility by drugs that enhance cAMP signaling. Proc. Natl. Acad. Sci. USA 2002, 99, 13217–13221. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, T.; Kunii, M.; Jitsuki, S.; Sano, A.; Kuroiwa, Y.; Takahashi, T. Social isolation perturbs experience-driven synaptic glutamate receptor subunit 4 delivery in the developing rat barrel cortex. Eur. J. Neurosci. 2013, 37, 1602–1609. [Google Scholar] [CrossRef]
- Matsumoto-Miyai, K.; Sokolowska, E.; Zurlinden, A.; Gee, C.E.; Lüscher, D.; Hettwer, S.; Wölfel, J.; Ladner, A.P.; Ster, J.; Gerber, U.; et al. Coincident pre- and postsynaptic activation induces dendritic filopodia via neurotrypsin-dependent agrin cleavage. Cell 2009, 136, 1161–1171. [Google Scholar] [CrossRef]
- Oh, M.C.; Derkach, V.A.; Guire, E.S.; Soderling, T.R. Extrasynaptic membrane trafficking regulated by GluR1 serine 845 phosphorylation primes AMPA receptors for long-term potentiation. J. Biol. Chem. 2006, 281, 752–758. [Google Scholar] [CrossRef]
- Yasuda, S.; Tanaka, H.; Sugiura, H.; Okamura, K.; Sakaguchi, T.; Tran, U.; Takemiya, T.; Mizoguchi, A.; Yagita, Y.; Sakurai, T.; et al. Activity-induced protocadherin arcadlin regulates dendritic spine number by triggering N-cadherin endocytosis via TAO2β and p38 MAP kinases. Neuron 2007, 56, 456–471. [Google Scholar] [CrossRef]
- Kim, Y.; Jang, Y.N.; Kim, J.Y.; Kim, N.; Noh, S.; Kim, H.; Queenan, B.N.; Bellmore, R.; Mun, J.Y.; Park, H.; et al. Microtubule-associated protein 2 mediates induction of long-term potentiation in hippocampal neurons. FASEB J. 2020, 34, 6965–6983. [Google Scholar] [CrossRef] [PubMed]
- Aniksztejn, L.; Ben-Ari, Y. Novel form of long-term potentiation produced by a K+ channel blocker in the hippocampus. Nature 1991, 349, 67–69. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, E.; Okada, T. TEA-induced long-term potentiation at hippocampal mossy fiber-CA3 synapses: Characteristics of its induction and expression. Brain Res. 2009, 1247, 21–27. [Google Scholar] [CrossRef]
- Ramakers, G.M.J.; Pasinelli, P.; Van Beest, M.; Van Der Slot, A.; Gispen, W.H.; De Graan, P.N.E. Activation of pre- and postsynaptic protein kinase C during tetraethylammonium-induced long-term potentiation in the CA1 field of the hippocampus. Neurosci. Lett. 2000, 286, 53–56. [Google Scholar] [CrossRef]
- Jagodzinski, P.; Hess, G. Tetraethylammonium-induced long-term potentiation in layer V horizontal connections of rat motor cortex. Neurosci. Lett. 2001, 298, 37–40. [Google Scholar] [CrossRef]
- Pelletier, M.R.; Hablitz, J.J. Tetraethylammonium-induced synaptic plasticity in rat neocortex. Cereb. Cortex 1996, 6, 771–780. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, D.; White, T.D.; Sawynok, J. Forskolin and phosphodiesterase inhibitors release adenosine but inhibit morphine-evoked release of adenosine from spinal cord synaptosomes. Can. J. Physiol. Pharmacol. 1991, 69, 877–885. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Villafuertes, R.; Pintor, J.; Gualix, J.; Miras-Portugal, M.T. GABAB receptor-mediated presynaptic potentiation of ATP ionotropic receptors in rat midbrain synaptosomes. Neuropharmacology 2003, 44, 311–323. [Google Scholar] [CrossRef]
- Schoffelmeer, A.N.M.; Hogenboom, F.; Mulder, A.H. Evidence for a presynaptic adenylate cyclase system facilitating [3H]norepinephrine release from rat brain neocortex slices and synaptosomes. J. Neurosci. 1985, 5, 2685–2689. [Google Scholar] [CrossRef]
- García-Font, N.; Martín, R.; Torres, M.; Oset-Gasque, M.J.; Sánchez-Prieto, J. The loss of β adrenergic receptor mediated release potentiation in a mouse model of fragile X syndrome. Neurobiol. Dis. 2019, 130, 104482. [Google Scholar] [CrossRef] [PubMed]
- Dillon, C.; Goda, Y. The actin cytoskeleton: Integrating form and function at the synapse. Annu. Rev. Neurosci. 2005, 28, 25–55. [Google Scholar] [CrossRef] [PubMed]
- Penzes, P.; Rafalovich, I. Regulation of the actin cytoskeleton in dendritic spines. Adv. Exp. Med. Biol. 2012, 970, 81–95. [Google Scholar] [CrossRef] [PubMed]
- Bellot, A.; Guivernau, B.; Tajes, M.; Bosch-Morató, M.; Valls-Comamala, V.; Muñoz, F.J. The structure and function of actin cytoskeleton in mature glutamatergic dendritic spines. Brain Res. 2014, 1573, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, K.; Bosch, M.; Hayashi, Y. The roles of CaMKII and F-Actin in the structural plasticity of dendritic Spines: A potential molecular identity of a synaptic tag? Physiology 2009, 24, 357–366. [Google Scholar] [CrossRef]
- Stefen, H.; Chaichim, C.; Power, J.; Fath, T. Regulation of the postsynaptic compartment of excitatory synapses by the actin cytoskeleton in health and its disruption in disease. Neural Plast. 2016, 2016, 2371970. [Google Scholar] [CrossRef]
- Hotulainen, P.; Hoogenraad, C.C. Actin in dendritic spines: Connecting dynamics to function. J. Cell Biol. 2010, 189, 619–629. [Google Scholar] [CrossRef] [PubMed]
- Hlushchenko, I.; Koskinen, M.; Hotulainen, P. Dendritic spine actin dynamics in neuronal maturation and synaptic plasticity. Cytoskeleton 2016, 73, 435–441. [Google Scholar] [CrossRef]
- Dent, E.W.; Merriam, E.B.; Hu, X. The dynamic cytoskeleton: Backbone of dendritic spine plasticity. Curr. Opin. Neurobiol. 2011, 21, 175–181. [Google Scholar] [CrossRef]
- Fischer, M.; Kaech, S.; Knutti, D.; Matus, A. Rapid actin-based plasticity in dendritic spines. Neuron 1998, 20, 847–854. [Google Scholar] [CrossRef]
- Gu, J.; Lee, C.W.; Fan, Y.; Komlos, D.; Tang, X.; Sun, C.; Yu, K.; Hartzell, H.C.; Chen, G.; Bamburg, J.R.; et al. ADF/cofilin-mediated actin dynamics regulate AMPA receptor trafficking during synaptic plasticity. Nat. Neurosci. 2010, 13, 1208–1215. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, K.I.; Nagai, T.; Miyawaki, A.; Hayashi, Y. Rapid and persistent modulation of actin dynamics regulates postsynaptic reorganization underlying bidirectional plasticity. Nat. Neurosci. 2004, 7, 1104–1112. [Google Scholar] [CrossRef]
- Star, E.N.; Kwiatkowski, D.J.; Murthy, V.N. Rapid turnover of actin in dendritic spines and its regulation by activity. Nat. Neurosci. 2002, 5, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Schaefers, A.T.U.; Teuchert-Noodt, G. Developmental neuroplasticity and the origin of neurodegenerative diseases. World J. Biol. Psychiatry 2016, 17, 587–599. [Google Scholar] [CrossRef]
- Vester, A.; Caudle, W.M. The synapse as a central target for neurodevelopmental susceptibility to pesticides. Toxics 2016, 4, 18. [Google Scholar] [CrossRef] [PubMed]
- Fenoglio, K.A.; Brunson, K.L.; Baram, T.Z. Hippocampal neuroplasticity induced by early-life stress: Functional and molecular aspects. Front. Neuroendocrinol. 2006, 27, 180–192. [Google Scholar] [CrossRef]
- Spires-Jones, T.L.; Stoothoff, W.H.; de Calignon, A.; Jones, P.B.; Hyman, B.T. Tau pathophysiology in neurodegeneration: A tangled issue. Trends Neurosci. 2009, 32, 150–159. [Google Scholar] [CrossRef]
- Spires-Jones, T.; Knafo, S. Spines, plasticity, and cognition in Alzheimer’s model mice. Neural Plast. 2012, 2012, 319836. [Google Scholar] [CrossRef] [PubMed]
- Tönnies, E.; Trushina, E. Oxidative stress, synaptic dysfunction, and Alzheimer’s disease. J. Alzheimers Dis. 2017, 57, 1105–1121. [Google Scholar] [CrossRef]
- Chen, Y.; Fu, A.K.Y.; Ip, N.Y. Synaptic dysfunction in Alzheimer’s disease: Mechanisms and therapeutic strategies. Pharmacol. Ther. 2019, 195, 186–198. [Google Scholar] [CrossRef] [PubMed]
- Bamburg, J.R.; Bernstein, B.W. Actin dynamics and cofilin-actin rods in Alzheimer disease. Cytoskeleton 2016, 73, 477–497. [Google Scholar] [CrossRef] [PubMed]
- Penzes, P.; VanLeeuwen, J.E. Impaired regulation of synaptic actin cytoskeleton in Alzheimer’s disease. Brain Res. Rev. 2011, 67, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Bardai, F.H.; Ordonez, D.G.; Bailey, R.M.; Hamm, M.; Lewis, J.; Feany, M.B. Lrrk promotes tau neurotoxicity through dysregulation of actin and mitochondrial dynamics. PLoS Biol. 2018, 16, e2006265. [Google Scholar] [CrossRef] [PubMed]
- Fulga, T.A.; Elson-Schwab, I.; Khurana, V.; Steinhilb, M.L.; Spires, T.L.; Hyman, B.T.; Feany, M.B. Abnormal bundling and accumulation of F-actin mediates tau-induced neuronal degeneration in vivo. Nat. Cell Biol. 2007, 9, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Tracy, T.E.; Sohn, P.D.; Minami, S.S.; Wang, C.; Min, S.W.; Li, Y.; Zhou, Y.; Le, D.; Lo, I.; Ponnusamy, R.; et al. Acetylated tau obstructs KIBRA-mediated signaling in synaptic plasticity and promotes tauopathy-related memory loss. Neuron 2016, 90, 245–260. [Google Scholar] [CrossRef]
- Yoshiyama, Y.; Higuchi, M.; Zhang, B.; Shu-Ming, H.; Nobuhisa, I.; Saido, T.C.; Maeda, J.; Suhara, T.; Trojanowski, J.Q.; Lee, V.M.-Y. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 2008, 53, 337–351. [Google Scholar] [CrossRef]
- López-González, I.; Aso, E.; Carmona, M.; Armand-Ugon, M.; Blanco, R.; Naudí, A.; Cabré, R.; Portero-Otin, M.; Pamplona, R.; Ferrer, I. Neuroinflammatory gene regulation, mitochondrial function, oxidative stress, and brain lipid modifications with disease progression in tau P301S transgenic mice as a model of frontotemporal lobar degeneration-tau. J. Neuropathol. Exp. Neurol. 2015, 74, 975–999. [Google Scholar] [CrossRef]
- Vemula, P.; Jing, Y.; Zhang, H.; Hunt, J.B.; Sandusky-Beltran, L.A.; Lee, D.C.; Liu, P. Altered brain arginine metabolism in a mouse model of tauopathy. Amino Acids 2019, 51, 513–528. [Google Scholar] [CrossRef]
- Bergin, D.; Jing, Y.; Mockett, B.; Zhang, H.; Abraham, W.; Liu, P. Altered plasma arginine metabolome precedes behavioural and brain arginine metabolomic profile changes in the APPswe/PS1ΔE9 mouse model of Alzheimer’s disease. Transl. Psychiatry 2018, 8, 108. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, F.; Haque, S.; Ravinayagam, V.; Ahmad, A.; Kamli, M.R.; Barreto, G.E.; Ashraf, G.M. Developmental lead (Pb)-induced deficits in redox and bioenergetic status of cerebellar synapses are ameliorated by ascorbate supplementation. Toxicology 2020, 440, 152492. [Google Scholar] [CrossRef]
- Wijasa, T.S.; Sylvester, M.; Brocke-Ahmadinejad, N.; Kummer, M.P.; Brosseron, F.; Gieselmann, V.; Heneka, M.T. Proteome profiling of S-nitrosylated synaptosomal proteins by isobaric mass tags. J. Neurosci. Methods 2017, 291, 95–100. [Google Scholar] [CrossRef]
- Plum, S.; Eggers, B.; Helling, S.; Stepath, M.; Theiss, C.; Leite, R.E.P.; Molina, M.; Grinberg, L.T.; Riederer, P.; Gerlach, M.; et al. Proteomic characterization of synaptosomes from human substantia nigra indicates altered mitochondrial translation in Parkinson’s disease. Cells 2020, 9, 2580. [Google Scholar] [CrossRef] [PubMed]
- Hariharan, A.; Jing, Y.; Collie, N.D.; Zhang, H.; Liu, P. Altered neurovascular coupling and brain arginine metabolism in endothelial nitric oxide synthase deficient mice. Nitric Oxide Biol. Chem. 2019, 87, 60–72. [Google Scholar] [CrossRef]
- Forner, S.; Martini, A.C.; Prieto, G.A.; Dang, C.T.; Rodriguez-Ortiz, C.J.; Reyes-Ruiz, J.M.; Trujillo-Estrada, L.; da Cunha, C.; Andrews, E.J.; Phan, J.; et al. Intra- and extracellular β-amyloid overexpression via adeno-associated virus-mediated gene transfer impairs memory and synaptic plasticity in the hippocampus. Sci. Rep. 2019. [Google Scholar] [CrossRef]
- Otmakhov, N.; Khibnik, L.; Otmakhova, N.; Carpenter, S.; Riahi, S.; Asrican, B.; Lisman, J. Forskolin-induced LTP in the CA1 hippocampal region is NMDA receptor dependent. J. Neurophysiol. 2004, 91, 1955–1962. [Google Scholar] [CrossRef]
- Lu, W.; Fang, W.; Li, J.; Zhang, B.; Yang, Q.; Yan, X.; Peng, L.; Ai, H.; Wang, J.J.; Liu, X.; et al. Phosphorylation of tyrosine 1070 at the GluN2B subunit is regulated by synaptic activity and critical for surface expression of N-methyl-D-aspartate (NMDA) receptors. J. Biol. Chem. 2015, 290, 22945–22954. [Google Scholar] [CrossRef]
- Zhang, G.; Neubert, T.A.; Jordan, B.A. RNA binding proteins accumulate at the postsynaptic density with synaptic activity. J. Neurosci. 2012, 32, 599–609. [Google Scholar] [CrossRef]
- Royo, M.; Gutiérrez, Y.; Fernández-Monreal, M.; Gutiérrez-Eisman, S.; Jiménez, R.; Jurado, S.; Esteban, J.A. A retention–release mechanism based on RAB11FIP2 for AMPA receptor synaptic delivery during long-term potentiation. J. Cell Sci. 2019, 132, jcs234237. [Google Scholar] [CrossRef] [PubMed]
- Borczyk, M.; Śliwińska, M.A.; Caly, A.; Bernas, T.; Radwanska, K. Neuronal plasticity affects correlation between the size of dendritic spine and its postsynaptic density. Sci. Rep. 2019, 9, 1693. [Google Scholar] [CrossRef]
- Lei, W.; Myers, K.R.; Rui, Y.; Hladyshau, S.; Tsygankov, D.; Zheng, J.Q. Phosphoinositide-dependent enrichment of actin monomers in dendritic spines regulates synapse development and plasticity. J. Cell Biol. 2017, 216, 2551–2564. [Google Scholar] [CrossRef] [PubMed]
- Rui, Y.; Gu, J.; Yu, K.; Hartzell, H.C.; Zheng, J.Q. Inhibition of AMPA receptor trafficking at hippocampal synapses by -amyloid oligomers: The mitochondrial contribution. Mol. Brain 2010, 3, 10. [Google Scholar] [CrossRef]
- Trotter, J.H.; Lussier, A.L.; Psilos, K.E.; Mahoney, H.L.; Sponaugle, A.E.; Hoe, H.S.; Rebeck, G.W.; Weeber, E.J. Extracellular proteolysis of reelin by tissue plasminogen activator following synaptic potentiation. Neuroscience 2014, 274, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Studer, D.; Chai, X.; Graber, W.; Brose, N.; Nestel, S.; Young, C.; Rodriguez, E.P.; Saetzler, K.; Frotscher, M. Structural plasticity of hippocampal mossy fiber synapses as revealed by high-pressure freezing. J. Comp. Neurol. 2012, 53, 4–8. [Google Scholar] [CrossRef]
- Fukazawa, Y.; Saitoh, Y.; Ozawa, F.; Ohta, Y.; Mizuno, K.; Inokuchi, K. Hippocampal LTP is accompanied by enhanced F-actin content within the dendritic spine that is essential for late LTP maintenance in vivo. Neuron 2003, 38, 447–460. [Google Scholar] [CrossRef]
- Harvey, C.D.; Svoboda, K. Locally dynamic synaptic learning rules in pyramidal neuron dendrites. Nature 2007, 450, 1195–1200. [Google Scholar] [CrossRef]
- Matsuzaki, M.; Ellis-Davies, G.C.R.; Nemoto, T.; Miyashita, Y.; Iino, M.; Kasai, H. Dendritic spine geometry is critical for AMPA receptor expression in hippocampal CA1 pyramidal neurons. Nat. Neurosci. 2001, 4, 1086–1092. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wang, X.B.; Frerking, M.; Zhou, Q. Delivery of AMPA receptors to perisynaptic sites precedes the full expression of long-term potentiation. Proc. Natl. Acad. Sci. USA 2008, 105, 11388–11393. [Google Scholar] [CrossRef] [PubMed]
- Tocco, G.; Maren, S.; Shors, T.J.; Baudry, M.; Thompson, R.F. Long-term potentiation is associated with increased [3H]AMPA binding in rat hippocampus. Brain Res. 1992, 573, 228–234. [Google Scholar] [CrossRef]
- Standley, S.; Tocco, G.; Wagle, N.; Baudry, M. High- and low-affinity α-[3H] amino-3-hydroxy-5-methylisoxazole-4-propionic acid ([3H]AMPA) binding sites. Represent immature and mature forms of AMPA receptors and are composed of differentially glycosylated subunits. J. Neurochem. 1998, 70, 2434–2445. [Google Scholar] [CrossRef] [PubMed]
- Behuet, S.; Cremer, J.N.; Cremer, M.; Palomero-Gallagher, N.; Zilles, K.; Amunts, K. Developmental changes of glutamate and GABA receptor densities in Wistar rats. Front. Neuroanat. 2019, 13, 100. [Google Scholar] [CrossRef]
- Hoffman, K.B.; Kessler, M.; Lynch, G. Sialic acid residues indirectly modulate the binding properties of AMPA-type glutamate receptors. Brain Res. 1997, 753, 309–314. [Google Scholar] [CrossRef]
- Moldes-Anaya, A.S.; Fonnum, F.; Eriksen, G.S.; Rundberget, T.; Walaas, S.I.; Wigestrand, M.B. In vitro neuropharmacological evaluation of penitrem-induced tremorgenic syndromes: Importance of the GABAergic system. Neurochem. Int. 2011, 59, 1074–1081. [Google Scholar] [CrossRef]
- Henley, J.M. Subcellular localization and molecular pharmacology of distinct populations of [3H]-AMPA binding sites in rat hippocampus. Br. J. Pharmacol. 1995, 115, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Wright, V.L.; Georgiou, P.; Bailey, A.; Heal, D.J.; Bailey, C.P.; Wonnacott, S. Inhibition of alpha7 nicotinic receptors in the ventral hippocampus selectively attenuates reinstatement of morphine-conditioned place preference and associated changes in AMPA receptor binding. Addict. Biol. 2019, 24, 590–603. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, A.; Tajima, Y.; Kunugi, A.; Kimura, H. Electrophysiological characterization of a novel AMPA receptor potentiator, TAK-137, in rat hippocampal neurons. Neurosci. Lett. 2019, 712, 134488. [Google Scholar] [CrossRef] [PubMed]
- Dai, W.; Gao, X.; Xiao, D.; Li, Y.L.; Zhou, X.B.; Yong, Z.; Su, R. Bin The impact and mechanism of a novel allosteric AMPA receptor modulator LCX001 on protection against respiratory depression in rodents. Front. Pharmacol. 2019, 10, 105. [Google Scholar] [CrossRef]
- Kunugi, A.; Tanaka, M.; Suzuki, A.; Tajima, Y.; Suzuki, N.; Suzuki, M.; Nakamura, S.; Kuno, H.; Yokota, A.; Sogabe, S.; et al. TAK-137, an AMPA-R potentiator with little agonistic effect, has a wide therapeutic window. Neuropsychopharmacology 2019, 44, 961–970. [Google Scholar] [CrossRef]
- Szepesi, Z.; Bijata, M.; Ruszczycki, B.; Kaczmarek, L.; Wlodarczyk, J. Matrix metalloproteinases regulate the formation of dendritic spine head protrusions during chemically induced long-term potentiation. PLoS ONE 2013, 8, e63314. [Google Scholar] [CrossRef] [PubMed]
- Fu, A.K.; Ip, N.Y. Regulation of postsynaptic signaling in structural synaptic plasticity. Curr. Opin. Neurobiol. 2017, 45, 148–155. [Google Scholar] [CrossRef]
- Honkura, N.; Matsuzaki, M.; Noguchi, J.; Ellis-Davies, G.C.R.; Kasai, H. The subspine organization of actin fibers regulates the structure and plasticity of dendritic spines. Neuron 2008, 57, 719–729. [Google Scholar] [CrossRef]
- Tolias, K.F.; Duman, J.G.; Um, K. Control of synapse development and plasticity by Rho GTPase regulatory proteins. Prog. Neurobiol. 2011, 94, 133–148. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, B.W.; Bamburg, J.R. Cycling of actin assembly in synaptosomes and neurotransmitter release. Neuron 1989, 3, 257–265. [Google Scholar] [CrossRef]
- Wolf, M.; Zimmermann, A.M.; Görlich, A.; Gurniak, C.B.; Sassoè-Pognetto, M.; Friauf, E.; Witke, W.; Rust, M.B. ADF/Cofilin controls synaptic actin dynamics and regulates synaptic vesicle mobilization and exocytosis. Cereb. Cortex 2015, 25, 2863–2875. [Google Scholar] [CrossRef]
- Pelucchi, S.; Stringhi, R.; Marcello, E. Dendritic spines in Alzheimer’s disease: How the actin cytoskeleton contributes to synaptic failure. Int. J. Mol. Sci. 2020, 21, 908. [Google Scholar] [CrossRef] [PubMed]
- Latif-Hernandez, A.; Losada, P.M.; Yang, T.; Tran, K.C.; Liu, H.; Lehallier, B.; Massa, S.M.; Longo, F.M. Elucidating emerging therapeutics: P75 receptor modulation reverts tauopathy associated alterations in synapse-relevant gene expression signatures. Alzheimers Dement. 2020, 16, e045764. [Google Scholar] [CrossRef]
- El Fatimy, R.; Li, S.; Chen, Z.; Mushannen, T.; Gongala, S.; Wei, Z.; Balu, D.T.; Rabinovsky, R.; Cantlon, A.; Elkhal, A.; et al. MicroRNA-132 provides neuroprotection for tauopathies via multiple signaling pathways. Acta Neuropathol. 2018, 136, 537–555. [Google Scholar] [CrossRef] [PubMed]
- Baglietto-Vargas, D.; Prieto, G.A.; Limon, A.; Forner, S.; Rodriguez-Ortiz, C.J.; Ikemura, K.; Ager, R.R.; Medeiros, R.; Trujillo-Estrada, L.; Martini, A.C.; et al. Impaired AMPA signaling and cytoskeletal alterations induce early synaptic dysfunction in a mouse model of Alzheimer’s disease. Aging Cell 2018, 17, e12791. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).