
Myelopoiesis during Solid Cancers and Strategies for Immunotherapy


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Role of Effector Myeloid Cells in the Tumor Microenvironment
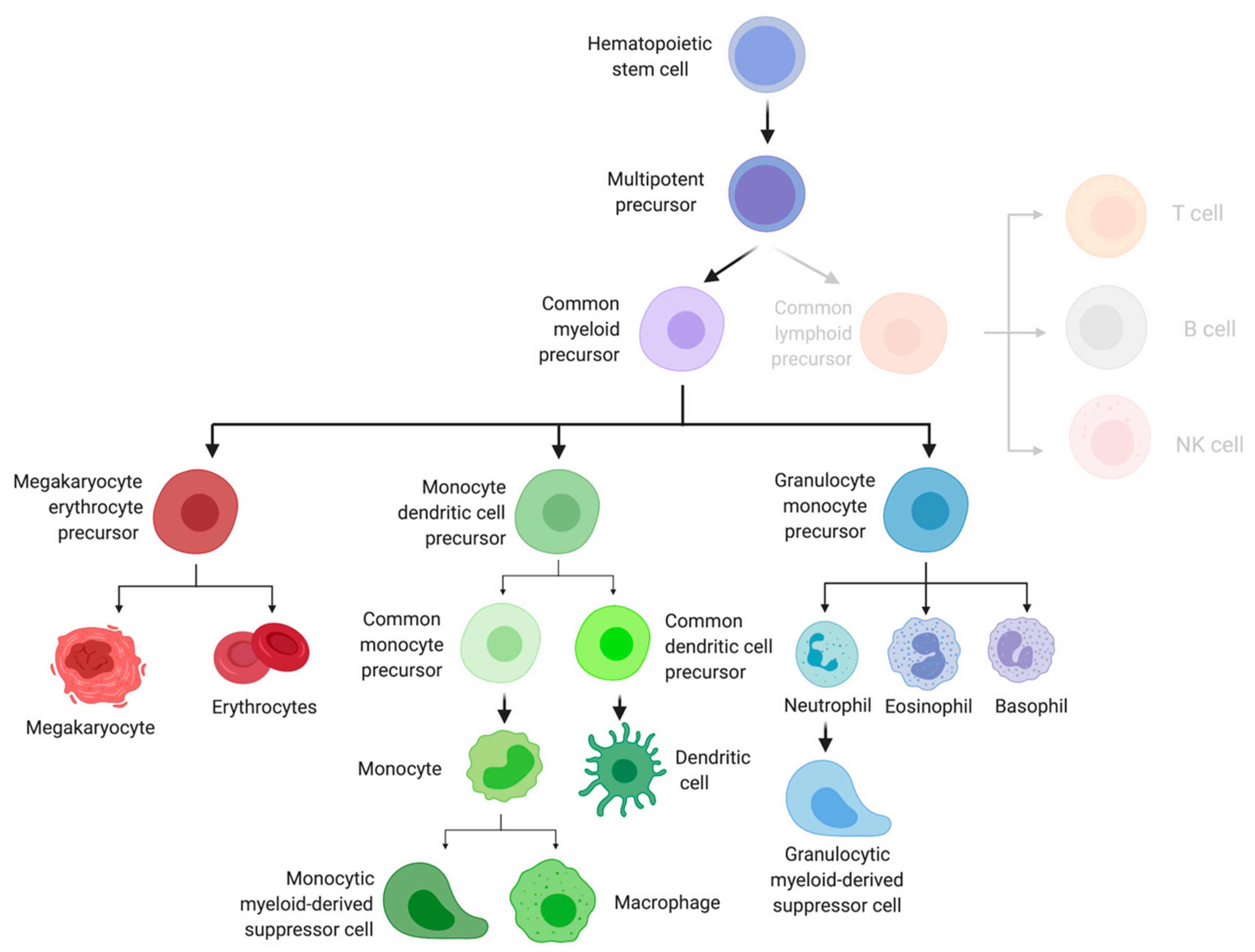
3. Early Myeloid Cell Development
4. Impact of Solid Cancers on Dendritic Cell Differentiation
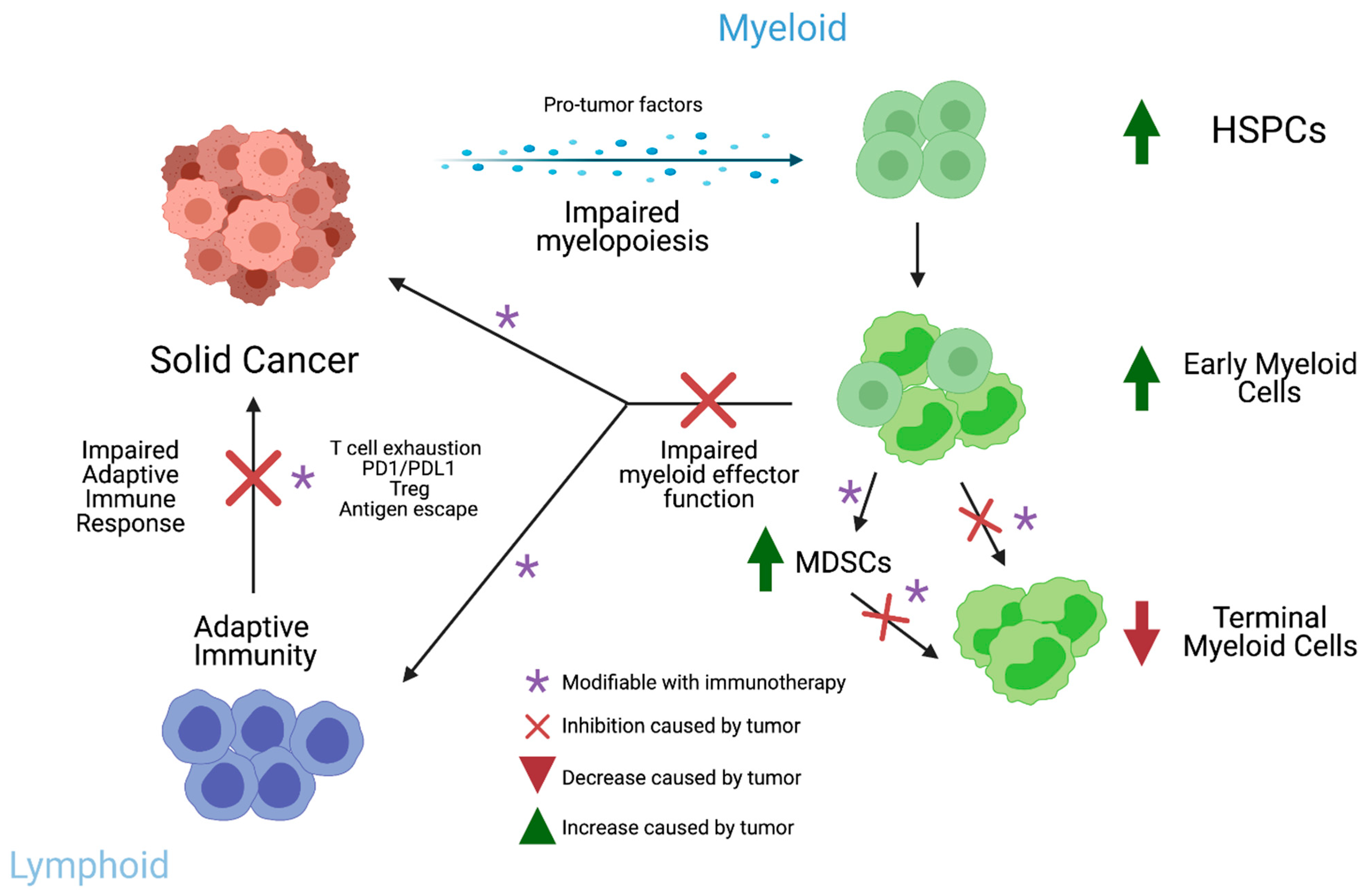
5. Impact of Solid Cancers on Myeloid Progenitors
6. Extramedullary Hematopoiesis
7. Impact of Early Myeloid Cells on Cancer Progression and Metastasis
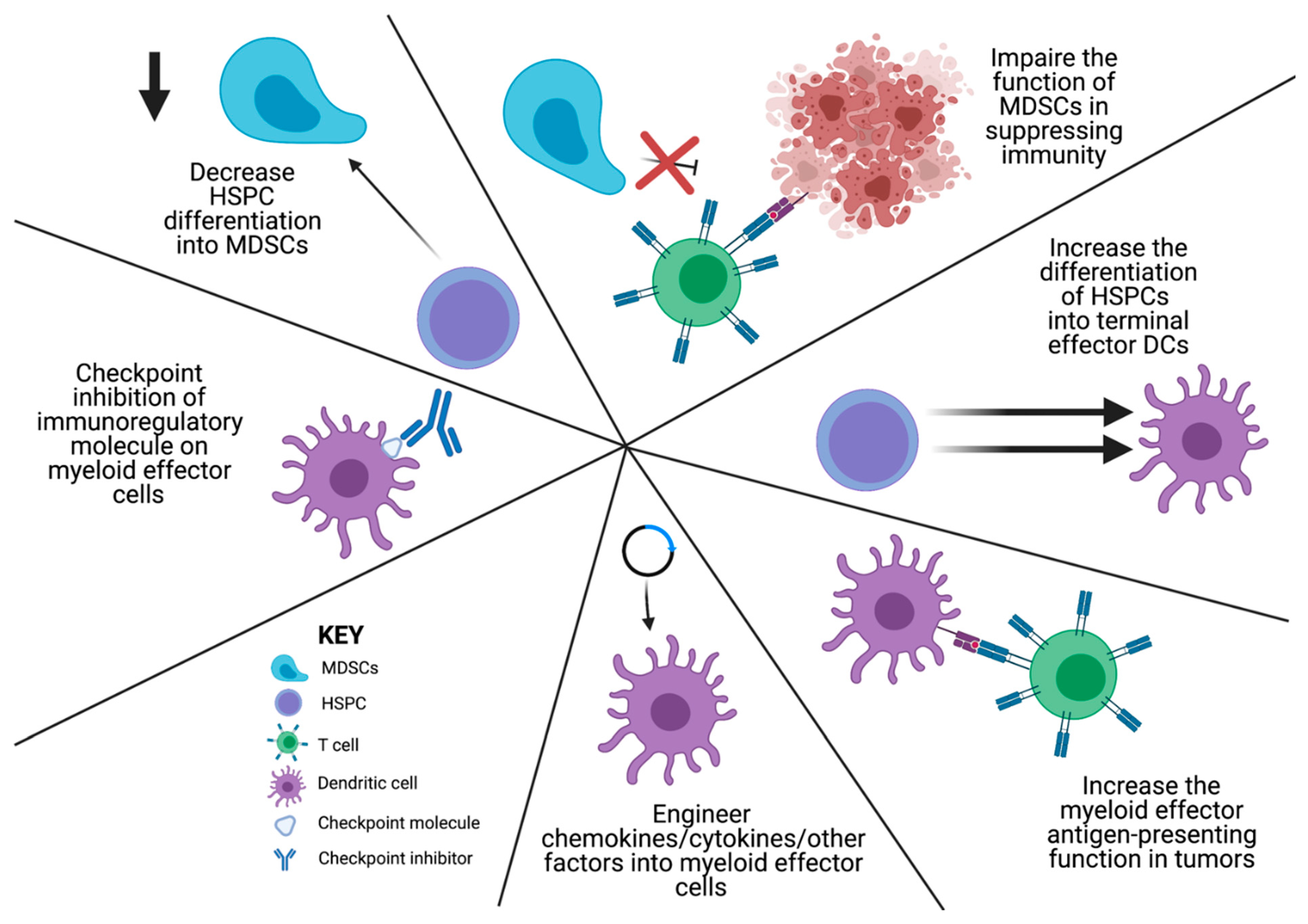
8. Early Hematopoietic Cells as Immunotherapy
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Rosenberg, S.A. Raising the bar: The curative potential of human cancer immunotherapy. Sci. Transl. Med. 2012, 4, 127ps8–128ps8. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A. Cell transfer immunotherapy for metastatic solid cancer--what clinicians need to know. Nat. Rev. Clin. Oncol. 2011, 8, 577–585. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Restifo, N.P. Adoptive cell transfer as personalized immunotherapy for human cancer. Science 2015, 348, 62–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenberg, S.A.; Restifo, N.P.; Yang, J.C.; Morgan, R.A.; Dudley, M.E. Adoptive cell transfer: A clinical path to effective cancer immunotherapy. Nat. Rev. Cancer 2008, 8, 299–308. [Google Scholar] [CrossRef]
- Togashi, Y.; Shitara, K.; Nishikawa, H. Regulatory T cells in cancer immunosuppression—Implications for anticancer therapy. Nat. Rev. Clin. Oncol. 2019, 16, 356–371. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Ostrand-Rosenberg, S.; Bronte, V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012, 12, 253–268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talmadge, J.E.; Gabrilovich, D.I. History of myeloid-derived suppressor cells. Nat. Rev. Cancer 2013, 13, 739–752. [Google Scholar] [CrossRef]
- Ahmadzadeh, M.; Johnson, L.A.; Heemskerk, B.; Wunderlich, J.R.; Dudley, M.E.; White, D.E.; Rosenberg, S.A. Tumor antigen–specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood 2009, 114, 1537–1544. [Google Scholar] [CrossRef]
- Garon, E.B.; Rizvi, N.A.; Hui, R.; Leighl, N.; Balmanoukian, A.S.; Eder, J.P.; Patnaik, A.; Aggarwal, C.; Gubens, M.; Horn, L.; et al. Pembrolizumab for the Treatment of Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 372, 2018–2028. [Google Scholar] [CrossRef]
- Salmon, H.; Idoyaga, J.; Rahman, A.; Leboeuf, M.; Remark, R.; Jordan, S.; Casanova-Acebes, M.; Khudoynazarova, M.; Agudo, J.; Tung, N.; et al. Expansion and Activation of CD103(+) Dendritic Cell Progenitors at the Tumor Site Enhances Tumor Responses to Therapeutic PD-L1 and BRAF Inhibition. Immunity 2016, 44, 924–938. [Google Scholar] [CrossRef] [Green Version]
- Spranger, S.; Dai, D.; Horton, B.; Gajewski, T.F. Tumor-Residing Batf3 Dendritic Cells Are Required for Effector T Cell Trafficking and Adoptive T Cell Therapy. Cancer Cell 2017, 31, 711–723.e714. [Google Scholar] [CrossRef] [Green Version]
- Gajewski, T.F.; Schreiber, H.; Fu, Y.-X. Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol. 2013, 14, 1014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broz, M.L.; Binnewies, M.; Boldajipour, B.; Nelson, A.E.; Pollack, J.L.; Erle, D.J.; Barczak, A.; Rosenblum, M.D.; Daud, A.; Barber, D.L.; et al. Dissecting the tumor myeloid compartment reveals rare activating antigen-presenting cells critical for T cell immunity. Cancer Cell 2014, 26, 638–652. [Google Scholar] [CrossRef] [Green Version]
- Maier, B.; Leader, A.M.; Chen, S.T.; Tung, N.; Chang, C.; LeBerichel, J.; Chudnovskiy, A.; Maskey, S.; Walker, L.; Finnigan, J.P.; et al. A conserved dendritic-cell regulatory program limits antitumour immunity. Nature 2020, 580, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Ginhoux, F.; Liu, K.; Helft, J.; Bogunovic, M.; Greter, M.; Hashimoto, D.; Price, J.; Yin, N.; Bromberg, J.; Lira, S.A.; et al. The origin and development of nonlymphoid tissue CD103+ DCs. J. Exp. Med. 2009, 206, 3115–3130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veglia, F.; Gabrilovich, D.I. Dendritic cells in cancer: The role revisited. Curr. Opin. Immunol. 2017, 45, 43–51. [Google Scholar] [CrossRef] [Green Version]
- Wculek, S.K.; Cueto, F.J.; Mujal, A.M.; Melero, I.; Krummel, M.F.; Sancho, D. Dendritic cells in cancer immunology and immunotherapy. Nat. Rev. Immunol. 2020, 20, 7–24. [Google Scholar] [CrossRef] [PubMed]
- Bronte, V.; Brandau, S.; Chen, S.-H.; Colombo, M.P.; Frey, A.B.; Greten, T.F.; Mandruzzato, S.; Murray, P.J.; Ochoa, A.; Ostrand-Rosenberg, S.; et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat. Commun. 2016, 7, 12150. [Google Scholar] [CrossRef] [Green Version]
- Kumar, V.; Patel, S.; Tcyganov, E.; Gabrilovich, D.I. The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Trends Immunol. 2016, 37, 208–220. [Google Scholar] [CrossRef] [Green Version]
- Kumar, V.; Cheng, P.; Condamine, T.; Mony, S.; Languino, L.R.; McCaffrey, J.C.; Hockstein, N.; Guarino, M.; Masters, G.; Penman, E.; et al. CD45 Phosphatase Inhibits STAT3 Transcription Factor Activity in Myeloid Cells and Promotes Tumor-Associated Macrophage Differentiation. Immunity 2016, 44, 303–315. [Google Scholar] [CrossRef] [Green Version]
- King, K.Y.; Goodell, M.A. Inflammatory modulation of HSCs: Viewing the HSC as a foundation for the immune response. Nat. Rev. Immunol. 2011, 11, 685–692. [Google Scholar] [CrossRef]
- Wildes, T.J.; Flores, C.T.; Mitchell, D.A. Concise Review: Modulating Cancer Immunity with Hematopoietic Stem and Progenitor Cells. Stem Cells 2019, 37, 166–175. [Google Scholar] [CrossRef] [Green Version]
- Weiskopf, K.; Schnorr, P.J.; Pang, W.W.; Chao, M.P.; Chhabra, A.; Seita, J.; Feng, M.; Weissman, I.L. Myeloid Cell Origins, Differentiation, and Clinical Implications. Microbiol. Spectr. 2016, 4. [Google Scholar] [CrossRef] [Green Version]
- Hérault, A.; Binnewies, M.; Leong, S.; Calero-Nieto, F.J.; Zhang, S.Y.; Kang, Y.A.; Wang, X.; Pietras, E.M.; Chu, S.H.; Barry-Holson, K.; et al. Myeloid progenitor cluster formation drives emergency and leukaemic myelopoiesis. Nature 2017, 544, 53–58. [Google Scholar] [CrossRef]
- Sun, J.; Ramos, A.; Chapman, B.; Johnnidis, J.B.; Le, L.; Ho, Y.J.; Klein, A.; Hofmann, O.; Camargo, F.D. Clonal dynamics of native haematopoiesis. Nature 2014, 514, 322–327. [Google Scholar] [CrossRef]
- Grinenko, T.; Eugster, A.; Thielecke, L.; Ramasz, B.; Krüger, A.; Dietz, S.; Glauche, I.; Gerbaulet, A.; von Bonin, M.; Basak, O.; et al. Hematopoietic stem cells can differentiate into restricted myeloid progenitors before cell division in mice. Nat. Commun. 2018, 9, 1898. [Google Scholar] [CrossRef] [PubMed]
- Ji, H.; Ehrlich, L.I.; Seita, J.; Murakami, P.; Doi, A.; Lindau, P.; Lee, H.; Aryee, M.J.; Irizarry, R.A.; Kim, K.; et al. Comprehensive methylome map of lineage commitment from haematopoietic progenitors. Nature 2010, 467, 338–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yáñez, A.; Coetzee, S.G.; Olsson, A.; Muench, D.E.; Berman, B.P.; Hazelett, D.J.; Salomonis, N.; Grimes, H.L.; Goodridge, H.S. Granulocyte-Monocyte Progenitors and Monocyte-Dendritic Cell Progenitors Independently Produce Functionally Distinct Monocytes. Immunity 2017, 47, 890–902. [Google Scholar] [CrossRef] [Green Version]
- Liu, K.; Victora, G.D.; Schwickert, T.A.; Guermonprez, P.; Meredith, M.M.; Yao, K.; Chu, F.F.; Randolph, G.J.; Rudensky, A.Y.; Nussenzweig, M. In vivo analysis of dendritic cell development and homeostasis. Science 2009, 324, 392–397. [Google Scholar] [CrossRef] [Green Version]
- Jaillon, S.; Ponzetta, A.; Di Mitri, D.; Santoni, A.; Bonecchi, R.; Mantovani, A. Neutrophil diversity and plasticity in tumour progression and therapy. Nat. Rev. Cancer 2020, 20, 485–503. [Google Scholar] [CrossRef]
- Méndez-Ferrer, S.; Bonnet, D.; Steensma, D.P.; Hasserjian, R.P.; Ghobrial, I.M.; Gribben, J.G.; Andreeff , M.; Krause, D.S. Bone marrow niches in haematological malignancies. Nat. Rev. Cancer 2020, 20, 285–298. [Google Scholar] [CrossRef]
- Pang, W.W.; Price, E.A.; Sahoo, D.; Beerman, I.; Maloney, W.J.; Rossi, D.J.; Schrier, S.L.; Weissman, I.L. Human bone marrow hematopoietic stem cells are increased in frequency and myeloid-biased with age. Proc. Natl. Acad. Sci. USA 2011, 108, 20012–20017. [Google Scholar] [CrossRef] [Green Version]
- Burda, P.; Laslo, P.; Stopka, T. The role of PU.1 and GATA-1 transcription factors during normal and leukemogenic hematopoiesis. Leukemia 2010, 24, 1249–1257. [Google Scholar] [CrossRef] [Green Version]
- Gabrilovich, D.I.; Chen, H.L.; Girgis, K.R.; Cunningham, H.T.; Meny, G.M.; Nadaf, S.; Kavanaugh, D.; Carbone, D.P. Production of vascular endothelial growth factor by human tumors inhibits the functional maturation of dendritic cells. Nat. Med. 1996, 2, 1096–1103. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Nadaf, S.; Corak, J.; Berzofsky, J.A.; Carbone, D.P. Dendritic cells in antitumor immune responses. II. Dendritic cells grown from bone marrow precursors, but not mature DC from tumor-bearing mice, are effective antigen carriers in the therapy of established tumors. Cell. Immunol. 1996, 170, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Ishida, T.; Oyama, T.; Carbone, D.P.; Gabrilovich, D.I. Defective function of Langerhans cells in tumor-bearing animals is the result of defective maturation from hemopoietic progenitors. J. Immunol. 1998, 161, 4842–4851. [Google Scholar]
- Almand, B.; Resser, J.R.; Lindman, B.; Nadaf, S.; Clark, J.I.; Kwon, E.D.; Carbone, D.P.; Gabrilovich, D.I. Clinical significance of defective dendritic cell differentiation in cancer. Clin. Cancer Res. 2000, 6, 1755–1766. [Google Scholar] [PubMed]
- Almand, B.; Clark, J.I.; Nikitina, E.; van Beynen, J.; English, N.R.; Knight, S.C.; Carbone, D.P.; Gabrilovich, D.I. Increased Production of Immature Myeloid Cells in Cancer Patients: A Mechanism of Immunosuppression in Cancer. J. Immunol. 2001, 166, 678–689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garrity, T.; Pandit, R.; Wright, M.A.; Benefield, J.; Keni, S.; Young, M.R. Increased presence of CD34+ cells in the peripheral blood of head and neck cancer patients and their differentiation into dendritic cells. Int. J. Cancer 1997, 73, 663–669. [Google Scholar] [CrossRef]
- Bronte, V.; Chappell, D.B.; Apolloni, E.; Cabrelle, A.; Wang, M.; Hwu, P.; Restifo, N.P. Unopposed production of granulocyte-macrophage colony-stimulating factor by tumors inhibits CD8+ T cell responses by dysregulating antigen-presenting cell maturation. J. Immunol. 1999, 162, 5728–5737. [Google Scholar] [PubMed]
- Wang, T.; Niu, G.; Kortylewski, M.; Burdelya, L.; Shain, K.; Zhang, S.; Bhattacharya, R.; Gabrilovich, D.; Heller, R.; Coppola, D.; et al. Regulation of the innate and adaptive immune responses by Stat-3 signaling in tumor cells. Nat. Med. 2004, 10, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Nefedova, Y.; Huang, M.; Kusmartsev, S.; Bhattacharya, R.; Cheng, P.; Salup, R.; Jove, R.; Gabrilovich, D. Hyperactivation of STAT3 Is Involved in Abnormal Differentiation of Dendritic Cells in Cancer. J. Immunol. 2004, 172, 464–474. [Google Scholar] [CrossRef] [PubMed]
- Strauss, L.; Sangaletti, S.; Consonni, F.M.; Szebeni, G.; Morlacchi, S.; Totaro, M.G.; Porta, C.; Anselmo, A.; Tartari, S.; Doni, A.; et al. RORC1 Regulates Tumor-Promoting “Emergency” Granulo-Monocytopoiesis. Cancer Cell 2015, 28, 253–269. [Google Scholar] [CrossRef] [Green Version]
- Bayne, L.J.; Beatty, G.L.; Jhala, N.; Clark, C.E.; Rhim, A.D.; Stanger, B.Z.; Vonderheide, R.H. Tumor-Derived Granulocyte-Macrophage Colony-Stimulating Factor Regulates Myeloid Inflammation and T Cell Immunity in Pancreatic Cancer. Cancer Cell 2012, 21, 822–835. [Google Scholar] [CrossRef] [Green Version]
- Mandruzzato, S.; Solito, S.; Falisi, E.; Francescato, S.; Chiarion-Sileni, V.; Mocellin, S.; Zanon, A.; Rossi, C.R.; Nitti, D.; Bronte, V. IL4Rα+ myeloid-derived suppressor cell expansion in cancer patients. J. Immunol. 2009, 182, 6562–6568. [Google Scholar] [CrossRef] [Green Version]
- Zea, A.H.; Rodriguez, P.C.; Atkins, M.B.; Hernandez, C.; Signoretti, S.; Zabaleta, J.; McDermott, D.; Quiceno, D.; Youmans, A.; O’Neill, A.; et al. Arginase-producing myeloid suppressor cells in renal cell carcinoma patients: A mechanism of tumor evasion. Cancer Res. 2005, 65, 3044–3048. [Google Scholar] [CrossRef] [Green Version]
- Gallina, G.; Dolcetti, L.; Serafini, P.; De Santo, C.; Marigo, I.; Colombo, M.P.; Basso, G.; Brombacher, F.; Borrello, I.; Zanovello, P.; et al. Tumors induce a subset of inflammatory monocytes with immunosuppressive activity on CD8+ T cells. J. Clin. Investig. 2006, 116, 2777–2790. [Google Scholar] [CrossRef]
- Filipazzi, P.; Valenti, R.; Huber, V.; Pilla, L.; Canese, P.; Iero, M.; Castelli, C.; Mariani, L.; Parmiani, G.; Rivoltini, L. Identification of a new subset of myeloid suppressor cells in peripheral blood of melanoma patients with modulation by a granulocyte-macrophage colony-stimulation factor-based antitumor vaccine. J. Clin. Oncol. 2007, 25, 2546–2553. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.C.; Sun, H.W.; Chen, H.T.; Liang, J.; Yu, X.J.; Wu, C.; Wang, Z.; Zheng, L. Circulating hematopoietic stem and progenitor cells are myeloid-biased in cancer patients. Proc. Natl. Acad. Sci. USA 2014, 111, 4221–4226. [Google Scholar] [CrossRef] [Green Version]
- Sun, H.-W.; Wu, W.-C.; Chen, H.-T.; Xu, Y.-T.; Yang, Y.-Y.; Chen, J.; Yu, X.-J.; Wang, Z.; Shuang, Z.-Y.; Zheng, L. Glutamine Deprivation Promotes the Generation and Mobilization of MDSCs by Enhancing Expression of G-CSF and GM-CSF. Front. Immunol. 2021, 11, 616367. [Google Scholar] [CrossRef]
- Sio, A.; Chehal, M.K.; Tsai, K.; Fan, X.; Roberts, M.E.; Nelson, B.H.; Grembecka, J.; Cierpicki, T.; Krebs, D.L.; Harder, K.W. Dysregulated hematopoiesis caused by mammary cancer is associated with epigenetic changes and hox gene expression in hematopoietic cells. Cancer Res. 2013, 73, 5892–5904. [Google Scholar] [CrossRef] [Green Version]
- Casbon, A.J.; Reynaud, D.; Park, C.; Khuc, E.; Gan, D.D.; Schepers, K.; Passegue, E.; Werb, Z. Invasive breast cancer reprograms early myeloid differentiation in the bone marrow to generate immunosuppressive neutrophils. Proc. Natl. Acad. Sci. USA 2015, 112, E566–E575. [Google Scholar] [CrossRef] [Green Version]
- Fernández-García, V.; González-Ramos, S.; Martín-Sanz, P.; Castrillo, A.; Boscá, L. Contribution of Extramedullary Hematopoiesis to Atherosclerosis. The Spleen as a Neglected Hub of Inflammatory Cells. Front. Immunol. 2020, 11, 586527. [Google Scholar] [CrossRef]
- Yamamoto, K.; Miwa, Y.; Abe-Suzuki, S.; Abe, S.; Kirimura, S.; Onishi, I.; Kitagawa, M.; Kurata, M. Extramedullary hematopoiesis: Elucidating the function of the hematopoietic stem cell niche (Review). Mol. Med. Rep. 2016, 13, 587–591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, C.H. Homeostatic and pathogenic extramedullary hematopoiesis. J. Blood Med. 2010, 1, 13–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.; Hua, Q.; Zheng, L. Generation of Myeloid Cells in Cancer: The Spleen Matters. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Cortez-Retamozo, V.; Etzrodt, M.; Newton, A.; Rauch, P.J.; Chudnovskiy, A.; Berger, C.; Ryan, R.J.; Iwamoto, Y.; Marinelli, B.; Gorbatov, R.; et al. Origins of tumor-associated macrophages and neutrophils. Proc. Natl. Acad. Sci. USA 2012, 109, 2491–2496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.; Ning, H.; Liu, M.; Lin, J.; Luo, S.; Zhu, W.; Xu, J.; Wu, W.-C.; Liang, J.; Shao, C.-K.; et al. Spleen mediates a distinct hematopoietic progenitor response supporting tumor-promoting myelopoiesis. J. Clin. Investig. 2018, 128, 3425–3438. [Google Scholar] [CrossRef] [Green Version]
- Levy, L.; Mishalian, I.; Bayuch, R.; Zolotarov, L.; Michaeli, J.; Fridlender, Z.G. Splenectomy inhibits non-small cell lung cancer growth by modulating anti-tumor adaptive and innate immune response. Oncoimmunology 2015, 4, e998469. [Google Scholar] [CrossRef] [Green Version]
- Kamran, N.; Li, Y.; Sierra, M.; Alghamri, M.S.; Kadiyala, P.; Appelman, H.D.; Edwards, M.; Lowenstein, P.R.; Castro, M.G. Melanoma induced immunosuppression is mediated by hematopoietic dysregulation. Oncoimmunology 2018, 7, e1408750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, B.M.; Hiam, K.J.; Burnett, C.E.; Venida, A.; DeBarge, R.; Tenvooren, I.; Marquez, D.M.; Cho, N.W.; Carmi, Y.; Spitzer, M.H. Systemic dysfunction and plasticity of the immune macroenvironment in cancer models. Nat. Med. 2020, 26, 1125–1134. [Google Scholar] [CrossRef]
- Bao, Y.; Liu, Z.; Guo, M.; Li, B.; Sun, X.; Wang, L. Extramedullary hematopoiesis secondary to malignant solid tumors: A case report and literature review. Cancer Manag. Res. 2018, 10, 1461–1470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Leo, E.K.; Shah, C.P.; Grajo, J.R.; Liu, X.; Parekh, H. Extramedullary Hematopoiesis in Mismatch Repair Deficient Colon Cancer Patient on Adjuvant Chemotherapy. Cureus 2021, 13, e12899. [Google Scholar] [CrossRef] [PubMed]
- Lyden, D.; Hattori, K.; Dias, S.; Costa, C.; Blaikie, P.; Butros, L.; Chadburn, A.; Heissig, B.; Marks, W.; Witte, L.; et al. Impaired recruitment of bone-marrow-derived endothelial and hematopoietic precursor cells blocks tumor angiogenesis and growth. Nat. Med. 2001, 7, 1194–1201. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, R.N.; Riba, R.D.; Zacharoulis, S.; Bramley, A.H.; Vincent, L.; Costa, C.; MacDonald, D.D.; Jin, D.K.; Shido, K.; Kerns, S.A.; et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature 2005, 438, 820–827. [Google Scholar] [CrossRef]
- Giles, A.J.; Reid, C.M.; Evans, J.D.; Murgai, M.; Vicioso, Y.; Highfill, S.L.; Kasai, M.; Vahdat, L.; Mackall, C.L.; Lyden, D.; et al. Activation of Hematopoietic Stem/Progenitor Cells Promotes Immunosuppression Within the Pre-metastatic Niche. Cancer Res. 2016, 76, 1335–1347. [Google Scholar] [CrossRef] [Green Version]
- Qian, B.-Z.; Li, J.; Zhang, H.; Kitamura, T.; Zhang, J.; Campion, L.R.; Kaiser, E.A.; Snyder, L.A.; Pollard, J.W. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 2011, 475, 222–225. [Google Scholar] [CrossRef] [Green Version]
- Si, Y.; Tsou, C.-L.; Croft, K.; Charo, I.F. CCR2 mediates hematopoietic stem and progenitor cell trafficking to sites of inflammation in mice. J. Clin. Investig. 2010, 120, 1192–1203. [Google Scholar] [CrossRef]
- Serbina, N.V.; Pamer, E.G. Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat. Immunol. 2006, 7, 311–317. [Google Scholar] [CrossRef]
- Innamarato, P.; Kodumudi, K.; Asby, S.; Schachner, B.; Hall, M.; Mackay, A.; Wiener, D.; Beatty, M.; Nagle, L.; Creelan, B.C.; et al. Reactive Myelopoiesis Triggered by Lymphodepleting Chemotherapy Limits the Efficacy of Adoptive T Cell Therapy. Mol. Ther. 2020, 28, 2252–2270. [Google Scholar] [CrossRef]
- Diaz-Montero, C.M.; Salem, M.L.; Nishimura, M.I.; Garrett-Mayer, E.; Cole, D.J.; Montero, A.J. Increased circulating myeloid-derived suppressor cells correlate with clinical cancer stage, metastatic tumor burden, and doxorubicin-cyclophosphamide chemotherapy. Cancer Immunol. Immunother. 2009, 58, 49–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veglia, F.; Perego, M.; Gabrilovich, D. Myeloid-derived suppressor cells coming of age. Nat. Immunol. 2018, 19, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Cannarile, M.A.; Weisser, M.; Jacob, W.; Jegg, A.-M.; Ries, C.H.; Rüttinger, D. Colony-stimulating factor 1 receptor (CSF1R) inhibitors in cancer therapy. J. Immunother. Cancer 2017, 5, 53. [Google Scholar] [CrossRef]
- Goswami, S.; Sahai, E.; Wyckoff, J.B.; Cammer, M.; Cox, D.; Pixley, F.J.; Stanley, E.R.; Segall, J.E.; Condeelis, J.S. Macrophages promote the invasion of breast carcinoma cells via a colony-stimulating factor-1/epidermal growth factor paracrine loop. Cancer Res. 2005, 65, 5278–5283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Machiels, J.-P.; Gomez-Roca, C.; Michot, J.-M.; Zamarin, D.; Mitchell, T.; Catala, G.; Eberst, L.; Jacob, W.; Jegg, A.-M.; Cannarile, M.A.; et al. Phase Ib study of anti-CSF-1R antibody emactuzumab in combination with CD40 agonist selicrelumab in advanced solid tumor patients. J. Immunother. Cancer 2020, 8, e001153. [Google Scholar] [CrossRef]
- Akkari, L.; Bowman, R.L.; Tessier, J.; Klemm, F.; Handgraaf, S.M.; de Groot, M.; Quail, D.F.; Tillard, L.; Gadiot, J.; Huse, J.T.; et al. Dynamic changes in glioma macrophage populations after radiotherapy reveal CSF-1R inhibition as a strategy to overcome resistance. Sci. Transl. Med. 2020, 12, eaaw7843. [Google Scholar] [CrossRef]
- Pyonteck, S.M.; Akkari, L.; Schuhmacher, A.J.; Bowman, R.L.; Sevenich, L.; Quail, D.F.; Olson, O.C.; Quick, M.L.; Huse, J.T.; Teijeiro, V.; et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med. 2013, 19, 1264–1272. [Google Scholar] [CrossRef] [Green Version]
- Condeelis, J.; Pollard, J.W. Macrophages: Obligate partners for tumor cell migration, invasion, and metastasis. Cell 2006, 124, 263–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacDonald, K.P.; Palmer, J.S.; Cronau, S.; Seppanen, E.; Olver, S.; Raffelt, N.C.; Kuns, R.; Pettit, A.R.; Clouston, A.; Wainwright, B. An antibody against the colony-stimulating factor 1 receptor depletes the resident subset of monocytes and tissue-and tumor-associated macrophages but does not inhibit inflammation. BloodJ. Am. Soc. Hematol. 2010, 116, 3955–3963. [Google Scholar] [CrossRef] [Green Version]
- Mok, S.; Koya, R.C.; Tsui, C.; Xu, J.; Robert, L.; Wu, L.; Graeber, T.G.; West, B.L.; Bollag, G.; Ribas, A. Inhibition of CSF-1 receptor improves the antitumor efficacy of adoptive cell transfer immunotherapy. Cancer Res. 2014, 74, 153–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Knolhoff, B.L.; Meyer, M.A.; Nywening, T.M.; West, B.L.; Luo, J.; Wang-Gillam, A.; Goedegebuure, S.P.; Linehan, D.C.; DeNardo, D.G. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. 2014, 74, 5057–5069. [Google Scholar] [CrossRef] [Green Version]
- Wrzesinski, C.; Paulos, C.M.; Gattinoni, L.; Palmer, D.C.; Kaiser, A.; Yu, Z.; Rosenberg, S.A.; Restifo, N.P. Hematopoietic stem cells promote the expansion and function of adoptively transferred antitumor CD8 T cells. J. Clin. Investig. 2007, 117, 492–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flores, C.; Pham, C.; Snyder, D.; Yang, S.; Sanchez-Perez, L.; Sayour, E.; Cui, X.; Kemeny, H.; Friedman, H.; Bigner, D.D.; et al. Novel role of hematopoietic stem cells in immunologic rejection of malignant gliomas. Oncoimmunology 2015, 4, e994374. [Google Scholar] [CrossRef]
- Flores, C.T.; Wildes, T.J.; Drake, J.A.; Moore, G.L.; Dean, B.D.; Abraham, R.S.; Mitchell, D.A. Lin(-)CCR2(+) hematopoietic stem and progenitor cells overcome resistance to PD-1 blockade. Nat. Commun. 2018, 9, 4313. [Google Scholar] [CrossRef]
- Wildes, T.J.; Grippin, A.; Dyson, K.A.; Wummer, B.M.; Damiani, D.J.; Abraham, R.S.; Flores, C.T.; Mitchell, D.A. Cross-talk between T Cells and Hematopoietic Stem Cells during Adoptive Cellular Therapy for Malignant Glioma. Clin. Cancer Res. 2018, 24, 3955–3966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaczanowska, S.; Beury, D.W.; Gopalan, V.; Tycko, A.K.; Qin, H.; Clements, M.E.; Drake, J.; Nwanze, C.; Murgai, M.; Rae, Z.; et al. Genetically engineered myeloid cells rebalance the core immune suppression program in metastasis. Cell 2021. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wildes, T.J.; DiVita Dean, B.; Flores, C.T. Myelopoiesis during Solid Cancers and Strategies for Immunotherapy. Cells 2021, 10, 968. https://doi.org/10.3390/cells10050968
Wildes TJ, DiVita Dean B, Flores CT. Myelopoiesis during Solid Cancers and Strategies for Immunotherapy. Cells. 2021; 10(5):968. https://doi.org/10.3390/cells10050968
Chicago/Turabian StyleWildes, Tyler J., Bayli DiVita Dean, and Catherine T. Flores. 2021. "Myelopoiesis during Solid Cancers and Strategies for Immunotherapy" Cells 10, no. 5: 968. https://doi.org/10.3390/cells10050968