
Cell–Cell Fusion and the Roads to Novel Properties of Tumor Hybrid Cells


Abstract
:1. Introduction
2. In Vitro and In Vivo Data Supporting Cell–Cell Fusion in Cancer
3. Human In Vivo Data Supporting the Cell–cell Fusion in Cancer Hypothesis
4. How Do Cancer Cells Fuse?
5. Post-Fusion Processes
6. Novel Properties of Tumor Hybrids
7. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ADAMs | a disintegrin and metalloproteases |
| AKE | autocatalytic karyotypic evolution |
| BMDCs | bone marrow-derived cells |
| BMT | bone marrow transplantation |
| CS/IC | cancer stem/initiating cell |
| E/M | epithelial/mesenchymal |
| EMT | epithelial-mesenchymal transition |
| EpCAM | epithelial cell adhesion molecule |
| FCM | fetal cell microchimerism |
| HST | heterokaryon-to-synkaryon transition |
| HUVECs | human umbilical vein endothelial cells |
| MET | mesenchymal epithelial transition |
| MMP9 | matrix metalloproteinase 9 |
| MSCs | mesenchymal stem/stromal cells |
| NOD/SCID | non-obese diabetic/severe combined immunodeficiency |
| OSCC | oral squamous cell carcinoma |
| PDACs | pancreatic ductal adeno carcinoma |
| PEG | polyethylene glycol |
| PHSP | post-hybrid selection process |
| PR | ploidy reduction |
| TGF-β | transforming growth factor-β |
| TNF-α | tumor necrosis factor-α |
References
- Aguilar, P.S.; Baylies, M.K.; Fleissner, A.; Helming, L.; Inoue, N.; Podbilewicz, B.; Wang, H.; Wong, M. Genetic basis of cell-cell fusion mechanisms. Trends Genet. 2013, 29, 427–437. [Google Scholar] [CrossRef] [Green Version]
- Dittmar, T.; Zänker, K.S. Cell Fusion in Health and Disease: Volume I; Springer: Dordrecht, The Netherlands, 2011; Volume 1. [Google Scholar]
- Dittmar, T.; Zanker, K.S. Cell fusion in Health and Disease: Volume II; Springer: Dordrecht, The Netherlands, 2011; Volume 2. [Google Scholar]
- Hernandez, J.M.; Podbilewicz, B. The hallmarks of cell-cell fusion. Development 2017, 144, 4481–4495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pawelek, J.M.; Chakraborty, A.K. Fusion of tumour cells with bone marrow-derived cells: A unifying explanation for metastasis. Nat. Rev. Cancer 2008, 8, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Platt, J.L. Molecular and cellular mechanisms of Mammalian cell fusion. Adv. Exp. Med. Biol. 2011, 713, 33–64. [Google Scholar] [CrossRef] [PubMed]
- Helming, L.; Gordon, S. Molecular mediators of macrophage fusion. Trends Cell Biol. 2009, 19, 514–522. [Google Scholar] [CrossRef] [PubMed]
- Willkomm, L.; Bloch, W. State of the art in cell-cell fusion. Methods Mol. Biol. 2015, 1313, 1–19. [Google Scholar] [CrossRef]
- Brukman, N.G.; Uygur, B.; Podbilewicz, B.; Chernomordik, L.V. How cells fuse. J. Cell Biol. 2019, 218, 1436–1451. [Google Scholar] [CrossRef] [Green Version]
- Duelli, D.; Lazebnik, Y. Cell fusion: A hidden enemy? Cancer Cell 2003, 3, 445–448. [Google Scholar] [CrossRef] [Green Version]
- Lu, X.; Kang, Y. Cell fusion as a hidden force in tumor progression. Cancer Res. 2009, 69, 8536–8539. [Google Scholar] [CrossRef] [Green Version]
- Manjunath, Y.; Porciani, D.; Mitchem, J.B.; Suvilesh, K.N.; Avella, D.M.; Kimchi, E.T.; Staveley-O’Carroll, K.F.; Burke, D.H.; Li, G.; Kaifi, J.T. Tumor-Cell-Macrophage Fusion Cells as Liquid Biomarkers and Tumor Enhancers in Cancer. Int. J. Mol. Sci. 2020, 21, 1872. [Google Scholar] [CrossRef] [Green Version]
- Weiler, J.; Dittmar, T. Cell Fusion in Human Cancer: The Dark Matter Hypothesis. Cells 2019, 8, 132. [Google Scholar] [CrossRef] [Green Version]
- Laberge, G.S.; Duvall, E.; Haedicke, K.; Pawelek, J. Leukocyte(-)Cancer Cell Fusion-Genesis of a Deadly Journey. Cells 2019, 8, 170. [Google Scholar] [CrossRef] [Green Version]
- Aichel, O. Über Zellverschmelzung mit quantitativ abnormer Chromosomenverteilung als Ursache der Geschwulstbildung. In Vorträge und Aufsätze über Entwicklungsmechanik der Organismen; Roux, W., Ed.; Wilhelm Engelmann: Leipzig, Germany, 1911; pp. 1–115. [Google Scholar]
- Miroshnychenko, D.; Baratchart, E.; Ferrall-Fairbanks, M.C.; Velde, R.V.; Laurie, M.A.; Bui, M.M.; Tan, A.C.; Altrock, P.M.; Basanta, D.; Marusyk, A. Spontaneous cell fusions as a mechanism of parasexual recombination in tumour cell populations. Nat. Ecol. Evol. 2021, 5, 379–391. [Google Scholar] [CrossRef]
- Su, Y.; Subedee, A.; Bloushtain-Qimron, N.; Savova, V.; Krzystanek, M.; Li, L.; Marusyk, A.; Tabassum, D.P.; Zak, A.; Flacker, M.J.; et al. Somatic Cell Fusions Reveal Extensive Heterogeneity in Basal-like Breast Cancer. Cell Rep. 2015, 11, 1549–1563. [Google Scholar] [CrossRef] [Green Version]
- Wakeling, W.F.; Greetham, J.; Bennett, D.C. Efficient spontaneous fusion between some co-cultured cells, especially murine melanoma cells. Cell Biol. Int. 1994, 18, 207–210. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Kang, Y. Efficient acquisition of dual metastasis organotropism to bone and lung through stable spontaneous fusion between MDA-MB-231 variants. Proc. Natl. Acad. Sci. USA 2009, 106, 9385–9390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clawson, G.A.; Matters, G.L.; Xin, P.; Imamura-Kawasawa, Y.; Du, Z.; Thiboutot, D.M.; Helm, K.F.; Neves, R.I.; Abraham, T. Macrophage-Tumor Cell Fusions from Peripheral Blood of Melanoma Patients. PLoS ONE 2015, 10, e0134320. [Google Scholar] [CrossRef] [Green Version]
- Dittmar, T.; Schwitalla, S.; Seidel, J.; Haverkampf, S.; Reith, G.; Meyer-Staeckling, S.; Brandt, B.H.; Niggemann, B.; Zanker, K.S. Characterization of hybrid cells derived from spontaneous fusion events between breast epithelial cells exhibiting stem-like characteristics and breast cancer cells. Clin. Exp. Metastasis 2011, 28, 75–90. [Google Scholar] [CrossRef]
- Gast, C.E.; Silk, A.D.; Zarour, L.; Riegler, L.; Burkhart, J.G.; Gustafson, K.T.; Parappilly, M.S.; Roh-Johnson, M.; Goodman, J.R.; Olson, B.; et al. Cell fusion potentiates tumor heterogeneity and reveals circulating hybrid cells that correlate with stage and survival. Sci. Adv. 2018, 4, eaat7828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hass, R.; von der Ohe, J.; Ungefroren, H. Potential Role of MSC/Cancer Cell Fusion and EMT for Breast Cancer Stem Cell Formation. Cancers 2019, 11, 1432. [Google Scholar] [CrossRef] [Green Version]
- Powell, A.E.; Anderson, E.C.; Davies, P.S.; Silk, A.D.; Pelz, C.; Impey, S.; Wong, M.H. Fusion between Intestinal epithelial cells and macrophages in a cancer context results in nuclear reprogramming. Cancer Res. 2011, 71, 1497–1505. [Google Scholar] [CrossRef] [Green Version]
- Ramakrishnan, M.; Mathur, S.R.; Mukhopadhyay, A. Fusion derived epithelial cancer cells express hematopoietic markers and contribute to stem cell and migratory phenotype in ovarian carcinoma. Cancer Res. 2013, 73, 5360–5370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shabo, I.; Midtbo, K.; Andersson, H.; Akerlund, E.; Olsson, H.; Wegman, P.; Gunnarsson, C.; Lindstrom, A. Macrophage traits in cancer cells are induced by macrophage-cancer cell fusion and cannot be explained by cellular interaction. BMC Cancer 2015, 15, 922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.N.; Kong, C.F.; Zhao, D.; Cong, X.L.; Wang, S.S.; Ma, L.; Huang, Y.H. Fusion with mesenchymal stem cells differentially affects tumorigenic and metastatic abilities of lung cancer cells. J. Cell. Physiol. 2018, 234, 3570–3582. [Google Scholar] [CrossRef]
- Zhang, L.N.; Zhang, D.D.; Yang, L.; Gu, Y.X.; Zuo, Q.P.; Wang, H.Y.; Xu, J.; Liu, D.X. Roles of cell fusion between mesenchymal stromal/stem cells and malignant cells in tumor growth and metastasis. FEBS J. 2020, 288, 1447–1456. [Google Scholar] [CrossRef]
- Kemeny, L.V.; Kurgyis, Z.; Buknicz, T.; Groma, G.; Jakab, A.; Zanker, K.; Dittmar, T.; Kemeny, L.; Nemeth, I.B. Melanoma Cells Can Adopt the Phenotype of Stromal Fibroblasts and Macrophages by Spontaneous Cell Fusion in Vitro. Int. J. Mol. Sci. 2016, 17, 826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melzer, C.; Ohe, J.V.; Hass, R. Altered Tumor Plasticity after Different Cancer Cell Fusions with MSC. Int. J. Mol. Sci. 2020, 21, 8347. [Google Scholar] [CrossRef] [PubMed]
- Lindstrom, A.; Midtbo, K.; Arnesson, L.G.; Garvin, S.; Shabo, I. Fusion between M2-macrophages and cancer cells results in a subpopulation of radioresistant cells with enhanced DNA-repair capacity. Oncotarget 2017, 8, 51370–51386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lizier, M.; Anselmo, A.; Mantero, S.; Ficara, F.; Paulis, M.; Vezzoni, P.; Lucchini, F.; Pacchiana, G. Fusion between cancer cells and macrophages occurs in a murine model of spontaneous neu+ breast cancer without increasing its metastatic potential. Oncotarget 2016, 7, 60793–60806. [Google Scholar] [CrossRef] [Green Version]
- Rachkovsky, M.; Sodi, S.; Chakraborty, A.; Avissar, Y.; Bolognia, J.; McNiff, J.M.; Platt, J.; Bermudes, D.; Pawelek, J. Melanoma x macrophage hybrids with enhanced metastatic potential. Clin. Exp. Metastasis 1998, 16, 299–312. [Google Scholar] [CrossRef]
- Noubissi, F.K.; Harkness, T.; Alexander, C.M.; Ogle, B.M. Apoptosis-induced cancer cell fusion: A mechanism of breast cancer metastasis. FASEB J. 2015, 29, 4036–4045. [Google Scholar] [CrossRef]
- Zeng, C.; Zhang, Y.; Park, S.C.; Eun, J.R.; Nguyen, N.T.; Tschudy-Seney, B.; Jung, Y.J.; Theise, N.D.; Zern, M.A.; Duan, Y. CD34 Liver Cancer Stem Cells Were Formed by Fusion of Hepatobiliary Stem/Progenitor Cells with Hematopoietic Precursor-Derived Myeloid Intermediates. Stem Cells Dev. 2015, 24, 2467–2478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, F.; Liu, T.; Wang, J.; Li, J.; Ma, P.; Ding, H.; Feng, G.; Lin, D.; Xu, Y.; Yang, K. Bone marrow mesenchymal stem cells participate in prostate carcinogenesis and promote growth of prostate cancer by cell fusion in vivo. Oncotarget 2016, 7, 30924–30934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobsen, B.M.; Harrell, J.C.; Jedlicka, P.; Borges, V.F.; Varella-Garcia, M.; Horwitz, K.B. Spontaneous fusion with, and transformation of mouse stroma by, malignant human breast cancer epithelium. Cancer Res. 2006, 66, 8274–8279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldenberg, D.M.; Pavia, R.A.; Tsao, M.C. In vivo hybridisation of human tumour and normal hamster cells. Nature 1974, 250, 649–651. [Google Scholar] [CrossRef]
- Sottile, F.; Aulicino, F.; Theka, I.; Cosma, M.P. Mesenchymal stem cells generate distinct functional hybrids in vitro via cell fusion or entosis. Sci. Rep. 2016, 6, 36863. [Google Scholar] [CrossRef] [Green Version]
- Dornen, J.; Myklebost, O.; Dittmar, T. Cell Fusion of Mesenchymal Stem/Stromal Cells and Breast Cancer Cells Leads to the Formation of Hybrid Cells Exhibiting Diverse and Individual (Stem Cell) Characteristics. Int. J. Mol. Sci. 2020, 21, 9636. [Google Scholar] [CrossRef]
- Chakraborty, A.; Lazova, R.; Davies, S.; Backvall, H.; Ponten, F.; Brash, D.; Pawelek, J. Donor DNA in a renal cell carcinoma metastasis from a bone marrow transplant recipient. Bone Marrow Transplant. 2004, 34, 183–186. [Google Scholar] [CrossRef] [PubMed]
- LaBerge, G.S.; Duvall, E.; Grasmick, Z.; Haedicke, K.; Pawelek, J. A Melanoma Lymph Node Metastasis with a Donor-Patient Hybrid Genome following Bone Marrow Transplantation: A Second Case of Leucocyte-Tumor Cell Hybridization in Cancer Metastasis. PLoS ONE 2017, 12, e0168581. [Google Scholar] [CrossRef]
- Lazova, R.; Laberge, G.S.; Duvall, E.; Spoelstra, N.; Klump, V.; Sznol, M.; Cooper, D.; Spritz, R.A.; Chang, J.T.; Pawelek, J.M. A Melanoma Brain Metastasis with a Donor-Patient Hybrid Genome following Bone Marrow Transplantation: First Evidence for Fusion in Human Cancer. PLoS ONE 2013, 8, e66731. [Google Scholar] [CrossRef]
- Yilmaz, Y.; Lazova, R.; Qumsiyeh, M.; Cooper, D.; Pawelek, J. Donor Y chromosome in renal carcinoma cells of a female BMT recipient: Visualization of putative BMT-tumor hybrids by FISH. Bone Marrow Transplant. 2005, 35, 1021–1024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clawson, G.A.; Kimchi, E.; Patrick, S.D.; Xin, P.; Harouaka, R.; Zheng, S.; Berg, A.; Schell, T.; Staveley-O’Carroll, K.F.; Neves, R.I.; et al. Circulating tumor cells in melanoma patients. PLoS ONE 2012, 7, e41052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manjunath, Y.; Mitchem, J.B.; Suvilesh, K.N.; Avella, D.M.; Kimchi, E.T.; Staveley-O’Carroll, K.F.; Deroche, C.B.; Pantel, K.; Li, G.; Kaifi, J.T. Circulating giant tumor-macrophage fusion cells are independent prognosticators in non-small cell lung cancer patients. J. Thorac. Oncol. 2020, 5, 1460–1471. [Google Scholar] [CrossRef] [PubMed]
- Bjerkvig, R.; Tysnes, B.B.; Aboody, K.S.; Najbauer, J.; Terzis, A.J. Opinion: The origin of the cancer stem cell: Current controversies and new insights. Nat. Rev. Cancer 2005, 5, 899–904. [Google Scholar] [CrossRef] [PubMed]
- Duncan, A.W.; Hickey, R.D.; Paulk, N.K.; Culberson, A.J.; Olson, S.B.; Finegold, M.J.; Grompe, M. Ploidy reductions in murine fusion-derived hepatocytes. PLoS Genet. 2009, 5, e1000385. [Google Scholar] [CrossRef] [Green Version]
- Duncan, A.W.; Taylor, M.H.; Hickey, R.D.; Hanlon Newell, A.E.; Lenzi, M.L.; Olson, S.B.; Finegold, M.J.; Grompe, M. The ploidy conveyor of mature hepatocytes as a source of genetic variation. Nature 2010, 467, 707–710. [Google Scholar] [CrossRef] [Green Version]
- Hass, R.; von der Ohe, J.; Ungefroren, H. Impact of the Tumor Microenvironment on Tumor Heterogeneity and Consequences for Cancer Cell Plasticity and Stemness. Cancers 2020, 12, 3716. [Google Scholar] [CrossRef]
- Li, R.; Sonik, A.; Stindl, R.; Rasnick, D.; Duesberg, P. Aneuploidy versus gene mutation hypothesis of cancer: Recent study claims mutation, but is found to support aneuploidy. Proc. Natl. Acad. Sci. USA 2000, 97, 3236–3241. [Google Scholar] [CrossRef]
- He, X.; Li, B.; Shao, Y.; Zhao, N.; Hsu, Y.; Zhang, Z.; Zhu, L. Cell fusion between gastric epithelial cells and mesenchymal stem cells results in epithelial-to-mesenchymal transition and malignant transformation. BMC Cancer 2015, 15, 24. [Google Scholar] [CrossRef] [Green Version]
- Soe, K. Osteoclast Fusion: Physiological Regulation of Multinucleation through Heterogeneity-Potential Implications for Drug Sensitivity. Int. J. Mol. Sci. 2020, 21, 7717. [Google Scholar] [CrossRef]
- Vignery, A. Osteoclasts and giant cells: Macrophage-macrophage fusion mechanism. Int. J. Exp. Pathol. 2000, 81, 291–304. [Google Scholar] [CrossRef]
- Alvarez-Dolado, M.; Martinez-Losa, M. Cell fusion and tissue regeneration. Adv. Exp. Med. Biol. 2011, 713, 161–175. [Google Scholar] [CrossRef]
- Eisenberg, L.M.; Eisenberg, C.A. Stem cell plasticity, cell fusion, and transdifferentiation. Birth Defects Res. Part C Embryo Today 2003, 69, 209–218. [Google Scholar] [CrossRef]
- Vassilopoulos, G.; Wang, P.R.; Russell, D.W. Transplanted bone marrow regenerates liver by cell fusion. Nature 2003, 422, 901–904. [Google Scholar] [CrossRef]
- Wurmser, A.E.; Gage, F.H. Stem cells: Cell fusion causes confusion. Nature 2002, 416, 485–487. [Google Scholar] [CrossRef]
- Shabo, I.; Stal, O.; Olsson, H.; Dore, S.; Svanvik, J. Breast cancer expression of CD163, a macrophage scavenger receptor, is related to early distant recurrence and reduced patient survival. Int. J. Cancer 2008, 123, 780–786. [Google Scholar] [CrossRef] [PubMed]
- Melzer, C.; von der Ohe, J.; Hass, R. In vitro fusion of normal and neoplastic breast epithelial cells with human mesenchymal stroma/stem cells (MSC) partially involves TNF receptor signaling. Stem Cells 2018, 36, 977–989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melzer, C.; von der Ohe, J.; Hass, R. In Vivo Cell Fusion between Mesenchymal Stroma/Stem-Like Cells and Breast Cancer Cells. Cancers 2019, 11, 185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gauck, D.; Keil, S.; Niggemann, B.; Zanker, K.S.; Dittmar, T. Hybrid clone cells derived from human breast epithelial cells and human breast cancer cells exhibit properties of cancer stem/initiating cells. BMC Cancer 2017, 17, 515. [Google Scholar] [CrossRef] [Green Version]
- Nagler, C.; Hardt, C.; Zänker, K.S.; Dittmar, T. Co-cultivation of murine BMDCs with 67NR mouse mammary carcinoma cells give rise to highly drug resistant hybrid cells. Cancer Cell Int. 2011, 11, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dittmar, T.; Nagler, C.; Schwitalla, S.; Reith, G.; Niggemann, B.; Zanker, K.S. Recurrence cancer stem cells-made by cell fusion? Med. Hypotheses 2009, 73, 542–547. [Google Scholar] [CrossRef]
- Melzer, C.; von der Ohe, J.; Hass, R. Enhanced metastatic capacity of breast cancer cells after interaction and hybrid formation with mesenchymal stroma/stem cells (MSC). Cell Commun. Signal. 2018, 16, 2. [Google Scholar] [CrossRef] [Green Version]
- Fais, S.; Overholtzer, M. Cell-in-cell phenomena in cancer. Nat. Rev. Cancer 2018, 18, 758–766. [Google Scholar] [CrossRef] [PubMed]
- Lens, S.M.A.; Medema, R.H. Cytokinesis defects and cancer. Nat. Rev. Cancer 2019, 19, 32–45. [Google Scholar] [CrossRef]
- Shabo, I.; Olsson, H.; Sun, X.F.; Svanvik, J. Expression of the macrophage antigen CD163 in rectal cancer cells is associated with early local recurrence and reduced survival time. Int. J. Cancer 2009, 125, 1826–1831. [Google Scholar] [CrossRef] [PubMed]
- Shabo, I.; Olsson, H.; Stal, O.; Svanvik, J. Breast cancer expression of DAP12 is associated with skeletal and liver metastases and poor survival. Clin. Breast Cancer 2013, 13, 371–377. [Google Scholar] [CrossRef]
- Andersen, T.L.; Boissy, P.; Sondergaard, T.E.; Kupisiewicz, K.; Plesner, T.; Rasmussen, T.; Haaber, J.; Kolvraa, S.; Delaisse, J.M. Osteoclast nuclei of myeloma patients show chromosome translocations specific for the myeloma cell clone: A new type of cancer-host partnership? J. Pathol. 2007, 211, 10–17. [Google Scholar] [CrossRef]
- Andersen, T.L.; Soe, K.; Sondergaard, T.E.; Plesner, T.; Delaisse, J.M. Myeloma cell-induced disruption of bone remodelling compartments leads to osteolytic lesions and generation of osteoclast-myeloma hybrid cells. Br. J. Haematol. 2010, 148, 551–561. [Google Scholar] [CrossRef]
- Kurgyis, Z.; Kemeny, L.V.; Buknicz, T.; Groma, G.; Olah, J.; Jakab, A.; Polyanka, H.; Zanker, K.; Dittmar, T.; Kemeny, L.; et al. Melanoma-Derived BRAF(V600E) Mutation in Peritumoral Stromal Cells: Implications for in Vivo Cell Fusion. Int. J. Mol. Sci. 2016, 17, 980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shabo, I.; Svanvik, J. Expression of macrophage antigens by tumor cells. Adv. Exp. Med. Biol. 2011, 714, 141–150. [Google Scholar] [CrossRef]
- Silvestris, F.; Ciavarella, S.; Strippoli, S.; Dammacco, F. Cell fusion and hyperactive osteoclastogenesis in multiple myeloma. Adv. Exp. Med. Biol. 2011, 714, 113–128. [Google Scholar] [CrossRef] [PubMed]
- Clawson, G.A.; Matters, G.L.; Xin, P.; McGovern, C.; Wafula, E.; dePamphilis, C.; Meckley, M.; Wong, J.; Stewart, L.; D’Jamoos, C.; et al. “Stealth dissemination” of macrophage-tumor cell fusions cultured from blood of patients with pancreatic ductal adenocarcinoma. PLoS ONE 2017, 12, e0184451. [Google Scholar] [CrossRef] [Green Version]
- Cirello, V.; Fugazzola, L. Novel insights into the link between fetal cell microchimerism and maternal cancers. J. Cancer Res. Clin. Oncol. 2016, 142, 1697–1704. [Google Scholar] [CrossRef] [PubMed]
- Dornen, J.; Sieler, M.; Weiler, J.; Keil, S.; Dittmar, T. Cell Fusion-Mediated Tissue Regeneration as an Inducer of Polyploidy and Aneuploidy. Int. J. Mol. Sci. 2020, 21, 1811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huppertz, B.; Bartz, C.; Kokozidou, M. Trophoblast fusion: Fusogenic proteins, syncytins and ADAMs, and other prerequisites for syncytial fusion. Micron 2006, 37, 509–517. [Google Scholar] [CrossRef]
- Mi, S.; Lee, X.; Li, X.; Veldman, G.M.; Finnerty, H.; Racie, L.; LaVallie, E.; Tang, X.Y.; Edouard, P.; Howes, S.; et al. Syncytin is a captive retroviral envelope protein involved in human placental morphogenesis. Nature 2000, 403, 785–789. [Google Scholar] [CrossRef]
- Muir, A.; Lever, A.M.; Moffett, A. Human endogenous retrovirus-W envelope (syncytin) is expressed in both villous and extravillous trophoblast populations. J. Gen. Virol. 2006, 87, 2067–2071. [Google Scholar] [CrossRef] [Green Version]
- Millay, D.P.; O’Rourke, J.R.; Sutherland, L.B.; Bezprozvannaya, S.; Shelton, J.M.; Bassel-Duby, R.; Olson, E.N. Myomaker is a membrane activator of myoblast fusion and muscle formation. Nature 2013, 499, 301–305. [Google Scholar] [CrossRef] [Green Version]
- Leikina, E.; Gamage, D.G.; Prasad, V.; Goykhberg, J.; Crowe, M.; Diao, J.; Kozlov, M.M.; Chernomordik, L.V.; Millay, D.P. Myomaker and Myomerger Work Independently to Control Distinct Steps of Membrane Remodeling during Myoblast Fusion. Dev. Cell 2018, 46, 767–780.e767. [Google Scholar] [CrossRef] [Green Version]
- Kato, K.; Satouh, Y.; Nishimasu, H.; Kurabayashi, A.; Morita, J.; Fujihara, Y.; Oji, A.; Ishitani, R.; Ikawa, M.; Nureki, O. Structural and functional insights into IZUMO1 recognition by JUNO in mammalian fertilization. Nat. Commun. 2016, 7, 12198. [Google Scholar] [CrossRef] [Green Version]
- Inoue, N.; Ikawa, M.; Isotani, A.; Okabe, M. The immunoglobulin superfamily protein Izumo is required for sperm to fuse with eggs. Nature 2005, 434, 234–238. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, E.; Wright, G.J. Cross-species fertilization: The hamster egg receptor, Juno, binds the human sperm ligand, Izumo1. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2015, 370, 20140101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bjerregaard, B.; Holck, S.; Christensen, I.J.; Larsson, L.I. Syncytin is involved in breast cancer-endothelial cell fusions. Cell. Mol. Life Sci. 2006, 63, 1906–1911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strick, R.; Ackermann, S.; Langbein, M.; Swiatek, J.; Schubert, S.W.; Hashemolhosseini, S.; Koscheck, T.; Fasching, P.A.; Schild, R.L.; Beckmann, M.W.; et al. Proliferation and cell-cell fusion of endometrial carcinoma are induced by the human endogenous retroviral Syncytin-1 and regulated by TGF-beta. J. Mol. Med. 2007, 85, 23–38. [Google Scholar] [CrossRef]
- Yan, T.L.; Wang, M.; Xu, Z.; Huang, C.M.; Zhou, X.C.; Jiang, E.H.; Zhao, X.P.; Song, Y.; Song, K.; Shao, Z.; et al. Up-regulation of syncytin-1 contributes to TNF-alpha-enhanced fusion between OSCC and HUVECs partly via Wnt/beta-catenin-dependent pathway. Sci. Rep. 2017, 7, 40983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, H.; Liu, T.; Zhao, Z.; Chen, Y.; Zeng, J.; Liu, S.; Zhu, F. Mutations in 3′-long terminal repeat of HERV-W family in chromosome 7 upregulate syncytin-1 expression in urothelial cell carcinoma of the bladder through interacting with c-Myb. Oncogene 2014, 33, 3947–3958. [Google Scholar] [CrossRef]
- Larsson, L.I.; Holck, S.; Christensen, I.J. Prognostic role of syncytin expression in breast cancer. Hum. Pathol. 2007, 38, 726–731. [Google Scholar] [CrossRef]
- Davies, P.S.; Powell, A.E.; Swain, J.R.; Wong, M.H. Inflammation and proliferation act together to mediate intestinal cell fusion. PLoS ONE 2009, 4, e6530. [Google Scholar] [CrossRef] [PubMed]
- Johansson, C.B.; Youssef, S.; Koleckar, K.; Holbrook, C.; Doyonnas, R.; Corbel, S.Y.; Steinman, L.; Rossi, F.M.; Blau, H.M. Extensive fusion of haematopoietic cells with Purkinje neurons in response to chronic inflammation. Nat. Cell Biol. 2008, 10, 575–583. [Google Scholar] [CrossRef] [Green Version]
- McNally, A.K.; Anderson, J.M. Macrophage fusion and multinucleated giant cells of inflammation. Adv. Exp. Med. Biol. 2011, 713, 97–111. [Google Scholar] [CrossRef] [PubMed]
- Weiler, J.; Dittmar, T. Minocycline impairs TNF-alpha-induced cell fusion of M13SV1-Cre cells with MDA-MB-435-pFDR1 cells by suppressing NF-kappaB transcriptional activity and its induction of target-gene expression of fusion-relevant factors. Cell Commun. Signal. 2019, 17, 71. [Google Scholar] [CrossRef] [Green Version]
- Weiler, J.; Mohr, M.; Zanker, K.S.; Dittmar, T. Matrix metalloproteinase-9 (MMP9) is involved in the TNF-alpha-induced fusion of human M13SV1-Cre breast epithelial cells and human MDA-MB-435-pFDR1 cancer cells. Cell Commun. Signal. 2018, 16, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skokos, E.A.; Charokopos, A.; Khan, K.; Wanjala, J.; Kyriakides, T.R. Lack of TNF-alpha-induced MMP-9 production and abnormal E-cadherin redistribution associated with compromised fusion in MCP-1-null macrophages. Am. J. Pathol. 2011, 178, 2311–2321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hotokezaka, H.; Sakai, E.; Ohara, N.; Hotokezaka, Y.; Gonzales, C.; Matsuo, K.; Fujimura, Y.; Yoshida, N.; Nakayama, K. Molecular analysis of RANKL-independent cell fusion of osteoclast-like cells induced by TNF-alpha, lipopolysaccharide, or peptidoglycan. J. Cell. Biochem. 2007, 101, 122–134. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Bigler, D.; Ito, Y.; White, J.M. Sequence-specific interaction between the disintegrin domain of mouse ADAM 3 and murine eggs: Role of beta1 integrin-associated proteins CD9, CD81, and CD98. Mol. Biol. Cell 2001, 12, 809–820. [Google Scholar] [CrossRef]
- Aghababaei, M.; Hogg, K.; Perdu, S.; Robinson, W.P.; Beristain, A.G. ADAM12-directed ectodomain shedding of E-cadherin potentiates trophoblast fusion. Cell Death Differ. 2015, 22, 1970–1984. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, D.; Morrison, C.J.; Overall, C.M. Matrix metalloproteinases: What do they not do? New substrates and biological roles identified by murine models and proteomics. Biochim. Biophys. Acta 2010, 1803, 39–54. [Google Scholar] [CrossRef] [Green Version]
- Balkwill, F.; Mantovani, A. Inflammation and cancer: Back to Virchow? Lancet 2001, 357, 539–545. [Google Scholar] [CrossRef]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef]
- Dvorak, H.F. Tumors: Wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Engl. J. Med. 1986, 315, 1650–1659. [Google Scholar]
- Fortuna, M.B.; Dewey, M.J.; Furmanski, P. Cell fusion in tumor development and progression: Occurrence of cell fusion in primary methylcholanthrene-induced tumorigenesis. Int. J. Cancer 1989, 44, 731–737. [Google Scholar] [CrossRef]
- Yan, B.; Wang, J.; Liu, L. Chemotherapy promotes tumour cell hybridization in vivo. Tumour Biol. 2015, 37, 5025–5030. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, A.K.; Sodi, S.; Rachkovsky, M.; Kolesnikova, N.; Platt, J.T.; Bolognia, J.L.; Pawelek, J.M. A spontaneous murine melanoma lung metastasis comprised of host x tumor hybrids. Cancer Res. 2000, 60, 2512–2519. [Google Scholar]
- Sun, W.; Kang, K.S.; Morita, I.; Trosko, J.E.; Chang, C.C. High susceptibility of a human breast epithelial cell type with stem cell characteristics to telomerase activation and immortalization. Cancer Res. 1999, 59, 6118–6123. [Google Scholar] [PubMed]
- Ly, P.; Cleveland, D.W. Rebuilding Chromosomes After Catastrophe: Emerging Mechanisms of Chromothripsis. Trends Cell Biol. 2017, 27, 917–930. [Google Scholar] [CrossRef]
- Rode, A.; Maass, K.K.; Willmund, K.V.; Lichter, P.; Ernst, A. Chromothripsis in cancer cells: An update. Int. J. Cancer 2016, 138, 2322–2333. [Google Scholar] [CrossRef] [PubMed]
- Chunduri, N.K.; Storchova, Z. The diverse consequences of aneuploidy. Nat. Cell Biol. 2019, 21, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Merchak, K.; Lee, W.; Grande, J.P.; Cascalho, M.; Platt, J.L. Cell Fusion Connects Oncogenesis with Tumor Evolution. Am. J. Pathol. 2015, 185, 2049–2060. [Google Scholar] [CrossRef]
- Fahlbusch, S.S.; Keil, S.; Epplen, J.T.; Zanker, K.S.; Dittmar, T. Comparison of hybrid clones derived from human breast epithelial cells and three different cancer cell lines regarding in vitro cancer stem/initiating cell properties. BMC Cancer 2020, 20, 446. [Google Scholar] [CrossRef] [PubMed]
- Delespaul, L.; Merle, C.; Lesluyes, T.; Lagarde, P.; Le Guellec, S.; Perot, G.; Baud, J.; Carlotti, M.; Danet, C.; Fevre, M.; et al. Fusion-mediated chromosomal instability promotes aneuploidy patterns that resemble human tumors. Oncogene 2019, 38, 6083–6094. [Google Scholar] [CrossRef] [PubMed]
- Delespaul, L.; Gelabert, C.; Lesluyes, T.; Le Guellec, S.; Perot, G.; Leroy, L.; Baud, J.; Merle, C.; Lartigue, L.; Chibon, F. Cell-cell fusion of mesenchymal cells with distinct differentiations triggers genomic and transcriptomic remodelling toward tumour aggressiveness. Sci. Rep. 2020, 10, 21634. [Google Scholar] [CrossRef]
- Lartigue, L.; Merle, C.; Lagarde, P.; Delespaul, L.; Lesluyes, T.; Le Guellec, S.; Perot, G.; Leroy, L.; Coindre, J.M.; Chibon, F. Genome remodeling upon mesenchymal tumor cell fusion contributes to tumor progression and metastatic spread. Oncogene 2020, 39, 4198–4211. [Google Scholar] [CrossRef] [PubMed]
- Miller, F.R.; McInerney, D.; Rogers, C.; Miller, B.E. Spontaneous fusion between metastatic mammary tumor subpopulations. J. Cell. Biochem. 1988, 36, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Sun, X.; Wang, C.Y.; Hu, P.; Chu, C.Y.; Liu, S.; Zhau, H.E.; Chung, L.W. Spontaneous cancer-stromal cell fusion as a mechanism of prostate cancer androgen-independent progression. PLoS ONE 2012, 7, e42653. [Google Scholar] [CrossRef] [Green Version]
- Friedl, P.; Alexander, S. Cancer invasion and the microenvironment: Plasticity and reciprocity. Cell 2011, 147, 992–1009. [Google Scholar] [CrossRef] [Green Version]
- Jiang, E.; Yan, T.; Xu, Z.; Shang, Z. Tumor Microenvironment and Cell Fusion. Biomed Res. Int. 2019, 2019, 5013592. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Hoefflin, R.; Lahrmann, B.; Warsow, G.; Hubschmann, D.; Spath, C.; Walter, B.; Chen, X.; Hofer, L.; Macher-Goeppinger, S.; Tolstov, Y.; et al. Spatial niche formation but not malignant progression is a driving force for intratumoural heterogeneity. Nat. Commun. 2016, 7, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Lloyd, M.C.; Cunningham, J.J.; Bui, M.M.; Gillies, R.J.; Brown, J.S.; Gatenby, R.A. Darwinian Dynamics of Intratumoral Heterogeneity: Not Solely Random Mutations but Also Variable Environmental Selection Forces. Cancer Res. 2016, 76, 3136–3144. [Google Scholar] [CrossRef] [Green Version]
- Marusyk, A.; Janiszewska, M.; Polyak, K. Intratumor Heterogeneity: The Rosetta Stone of Therapy Resistance. Cancer Cell 2020, 37, 471–484. [Google Scholar] [CrossRef]
- Li, L.; Neaves, W.B. Normal stem cells and cancer stem cells: The niche matters. Cancer Res. 2006, 66, 4553–4557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Carloni, V.; Mazzocca, A.; Mello, T.; Galli, A.; Capaccioli, S. Cell fusion promotes chemoresistance in metastatic colon carcinoma. Oncogene 2013, 32, 2649–2660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirzayans, R.; Murray, D. Intratumor Heterogeneity and Therapy Resistance: Contributions of Dormancy, Apoptosis Reversal (Anastasis) and Cell Fusion to Disease Recurrence. Int. J. Mol. Sci. 2020, 21, 1308. [Google Scholar] [CrossRef] [Green Version]
- Mohr, M.; Zaenker, K.S.; Dittmar, T. Fusion in cancer: An explanatory model for aneuploidy, metastasis formation, and drug resistance. Methods Mol. Biol. 2015, 1313, 21–40. [Google Scholar] [CrossRef] [PubMed]
- Uygur, B.; Leikina, E.; Melikov, K.; Villasmil, R.; Verma, S.K.; Vary, C.P.H.; Chernomordik, L.V. Interactions with Muscle Cells Boost Fusion, Stemness, and Drug Resistance of Prostate Cancer Cells. Mol. Cancer Res. 2019, 17, 806–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]



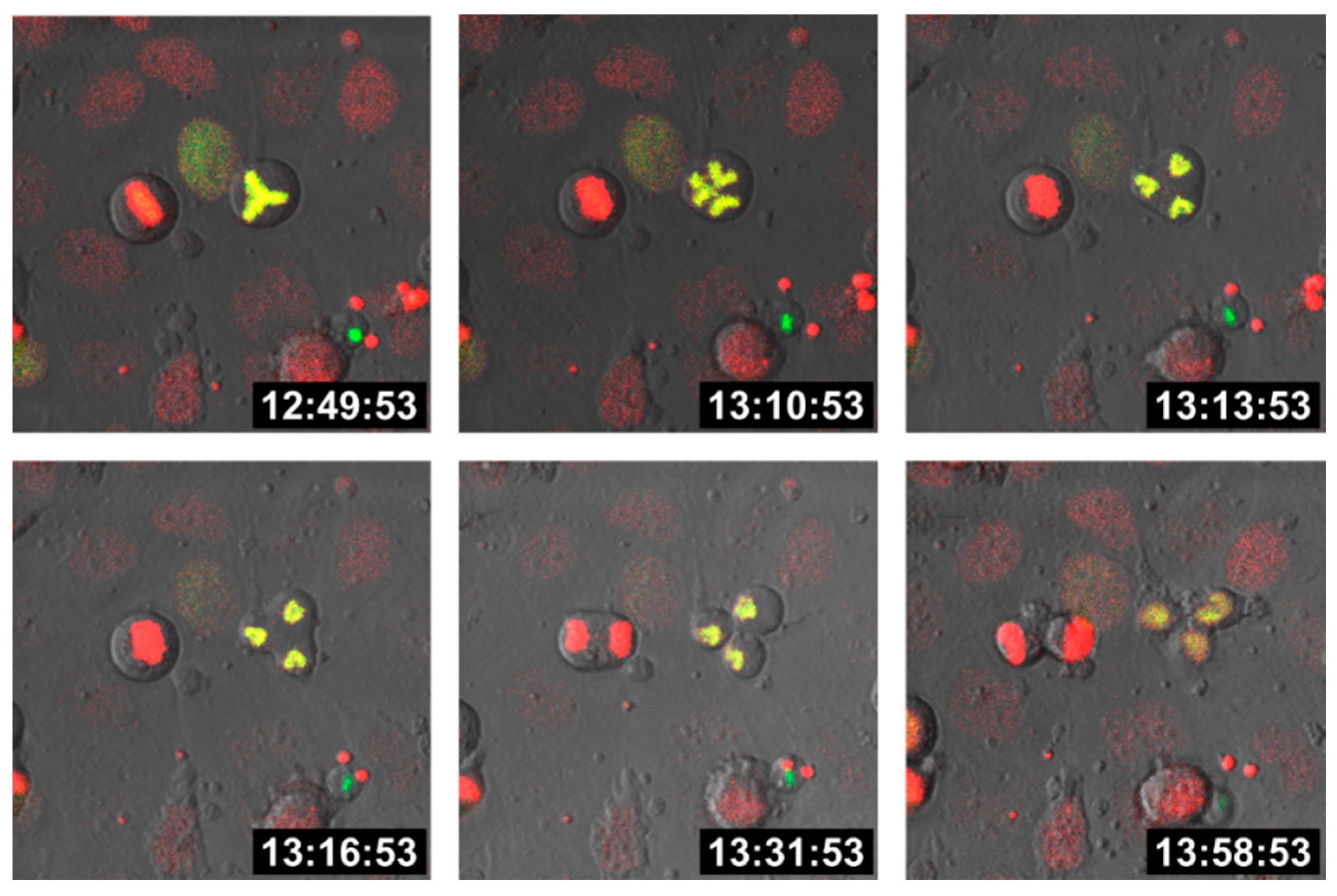{kind=link}
{kind=link}
| Tumor Type | Normal Cells | Marker | Reference |
|---|---|---|---|
| breast cancer | macrophages | CD163, MAC387, DAP12 | [59,69,73] |
| colon/colorectal cancer | macrophages | CD163, MAC387, DAP12 | [68,73] |
| CD14, CD45, cytokeratin | [20] | ||
| epithelial ovarian cancer | BMDCs | CD45, CXCR4 | [25] |
| melanoma | macrophages | CD14, CD45, cytokeratin | [20] |
| stromal cells | BRAF (V600E) mutation | [72] | |
| BMDCs | STR-analysis * | [42,43] | |
| multiple myeloma | osteoclasts | myeloma specific translocations | [70,71,74] |
| non-small cell lung cancer | macrophages | CD14, CD45, cytokeratin, EpCAM | [46] |
| pancreatic ductal adenocarcinoma | macrophages | CD45, cytokeratin | [45,75] |
| BMDCs | CD45, EpCAM, Y chromosome * | [22] | |
| renal cell carcinoma | BMDCs | blood group alleles *, Y-chromosome * | [41,44] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sieler, M.; Weiler, J.; Dittmar, T. Cell–Cell Fusion and the Roads to Novel Properties of Tumor Hybrid Cells. Cells 2021, 10, 1465. https://doi.org/10.3390/cells10061465
Sieler M, Weiler J, Dittmar T. Cell–Cell Fusion and the Roads to Novel Properties of Tumor Hybrid Cells. Cells. 2021; 10(6):1465. https://doi.org/10.3390/cells10061465
Chicago/Turabian StyleSieler, Mareike, Julian Weiler, and Thomas Dittmar. 2021. "Cell–Cell Fusion and the Roads to Novel Properties of Tumor Hybrid Cells" Cells 10, no. 6: 1465. https://doi.org/10.3390/cells10061465
APA StyleSieler, M., Weiler, J., & Dittmar, T. (2021). Cell–Cell Fusion and the Roads to Novel Properties of Tumor Hybrid Cells. Cells, 10(6), 1465. https://doi.org/10.3390/cells10061465