1. Introduction
Kidney fibrosis is a second-line healing program that only occurs if normal kidney repair is impaired or consistently suppressed by ongoing tissue injury and inflammation [
1]. Immediately after kidney injury, the innate immune system is activated and orchestrates the recruitment of inflammatory cells from the circulation. Local, resident immune cells, including macrophages, produce chemoattractant that enhance inflammatory responses by recruiting more leukocytes. During tissue injury, changing tissue environments shape the macrophages phenotype towards the anti-inflammatory to provide them with additional functional properties that meet the tissue’s need to address the danger. At the later stage after injury, macrophages contribute to the removal of fibrous tissue, mediating the resolution phase of healing that includes collagen remodeling. However, little is known about how a particular population of macrophages terminates the repair response and becomes pro or anti fibrotic [
2]. The knowledge of the agent modulating the phenotype of these macrophages or ex-vivo production of such cells possesses great therapeutic potential.
Nishida et al. [
3,
4] already suggested the existence of a subpopulation of infiltrating macrophages with an anti-fibrotic role in the recovery phase of obstructive nephropathy in which the angiotensin II type 1 receptor affected macrophage function. Specifically, the group indicates that the number of macrophages and their phagocytic activity is the determinant factor that defines the anti-fibrotic role of macrophages. Zhang et al. [
5] revealed that the decrease phagocytosis of pro-fibrotic molecules by macrophages resulted in kidney fibrosis in late-stage UUO. Other studies suggest that macrophages are beneficial in different settings when they may help to remove dead cells and debris through their phagocytic ability [
6]. Therefore, it seems clear that macrophages contribute to the removal of fibrous tissue and this fact points to the fundamental role of phagocytosis.
Phagocytosis is an inherent function of tissue macrophages for the removal of apoptotic cells and cellular debris during acute and chronic injury [
4]; however, the implications of this event on macrophage function during tissue remodeling and fibrosis are not known.
Overall, we think that despite the macrophage phenotype, the phagocytosis function of macrophages as effector cells able to phagocytize the extracellular matrix and apoptotic cells contributes to fibrosis development. If macrophages are unable to remove cell debris from the extracellular matrix, or are slow to remove it, extracellular matrix accumulation takes place.
Previous studies of our group have evidenced a role of macrophage carnitine palmitoyl transferase (CPT1a) expression in phagocytosis [
7]. Furthermore, strategies of CPT1a genetic modification confirmed these findings. In CPT1a knockdown macrophages, we found up-regulated iNOS expression leading to an inflammatory state where phagocytosis was found to be impaired. In accordance, CPT1a overexpression by Crispr activation maintained an improved phagocytosis respect Control Crispr plasmid. Our results revealed that macrophage phagocytosis and inflammatory phenotype are dependent on intracellular lipid accumulation and CPT1a expression. Other authors have found a role of CPT1a expression in kidney fibrosis and recent studies confirm its protective role in epithelial cells [
8], but no studies are known about the role of CPT1a on macrophage phagocytosis in kidney fibrosis.
The objective of this work is to know if any population of macrophages during the progression of kidney fibrosis have a reduced phagocytic ability and if increasing the population of phagocytic macrophages in fibrotic kidneys by a new macrophage therapy with highly phagocytic ability could reduce fibrosis development in the experimental model of UUO in mice. For this purpose, we have identified the different macrophage populations during the development of fibrosis and have characterized the phagocytic ability and the expression of CPT1a. Additionally, we have evaluated the capability of a cell therapy with macrophages overexpressing CPT1a, with high phagocytic ability, to ameliorate fibrosis.
2. Materials and Methods
2.1. Animals
Male CD-1 mice (Crl:CD1 (ICR); Charles River Laboratories Italy Srl, Calco, Italy) of 10–12 weeks old were housed in the Unitat d’Experimentació Animal Facilities of the University of Barcelona at the School of Medicine, with food and water ad libitum in a 12:12 h light/dark cycle. All procedures were previously approved by de Comité Ètic d’Experimentació Animal of the University of Barcelona (CEEA-UB).
2.2. Surgical Technique and Experimental Design
Animals were placed in an induction chamber with 5% isoflurane in 1–2 L/min oxygen followed by a maintenance dose at 1.5% with 0.9 L/min oxygen. After confirming a surgical anesthetic plane, ophthalmic ointment was applied, and the surgical field was prepared by removing the hair and disinfecting the skin with a 2% chlorhexidine solution. The animal was then transferred to a sterile field and a suprapelvic transversal incision of 0.5–1 cm was performed. The surrounding external tissue was covered with sterile gauze and the bladder exposed to reveal the left ureter which was ligated with 5/0 silk. Afterwards, the bladder was laid back in place and the incision closed with 4/0 silk. Buprenorphine 0.1 mg/Kg was administered during anesthetic induction and every 6–12 h for a minimum of 72 h. The group of control mice (sham) were operated in the same conditions but without ureter ligation. For the UUO kinetics study, tissues were harvested on day 3, 5 and 7 days (n = 4). For the cells therapy groups, one million of macrophages RAW 264.7 (MØ), macrophages RAW 264.7 transfected with adenovirus CPT1AM (AdCPT1AM) and transfected with GPF (AdGPF1), were administered to the mice intravenously through the tail vein on day 3 of UUO and the tissue was then harvested on day 5 after UUO (48 h post-treatment).
2.3. Cell Culture
Murine RAW264.7 macrophages, kindly given by Dr. Laura Herrero (Department of Biochemistry and Physiology, University of Barcelona, Spain), were cultured in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin–streptomycin mixture (Gibco, ThermoFisher Scientific, Madrid, Spain). Cells were maintained in a humidified incubator at 37 °C under 5% CO2 and passaged every 2 days when reaching 80% confluence by cell scraping with a slip ratio of 1:3 to 1:6. Cells from passages 9 to 11 were cultured in a 12-well plate at a density of 70,000 cells/well and allowed to grow for 72 h before transduction.
2.4. Adenovirus Transduction
We used an adenovirus vector carrying a permanent active mutant form of CPT1a (AdCPT1AM) which is insensitive to its inhibitor’s malonyl-CoA [
9] and an adenovirus carrying the green fluorescent protein (AdGFP) as a control. RAW 264.7 macrophages were transduced with AdCPT1AM and AdGFP at a multiplicity of infection (MOI) of 100 for 2 h in serum-free DMEM, and after, replaced with complete medium for an additional 72 h. Adenoviruses aliquots were kindly provided by Dr. Laura Herrero under MTA agreement.
2.5. Isolation of Murine Kidney Mononuclear Cells
Kidney mononuclear cells were isolated by enzymatic digestion following a density gradient. Briefly, kidney tissue was decapsulated, mechanically disaggregated into 1–2 mm pieces and enzymatically digested with Collagenase type I (1 mg/mL) for 30 min at 37 °C (Sigma-Aldrich, Madrid, Spain). The cell suspension was then filtered through a 70 µm cell strainer, resuspended in 4 mL DMEM + 20% FBS and centrifugated at 1500 rpm for 5 min at 4 °C. The subsequent pellet was re-suspended in 40% percoll, overlapped on 80% percoll and centrifugated at 400 g for 30 min at room temperature. The obtained fraction was then washed twice and resuspended in facs buffer. Cell counting was preformed using the trypan blue exclusion on a hemocytometer.
2.6. Flow Cytometry
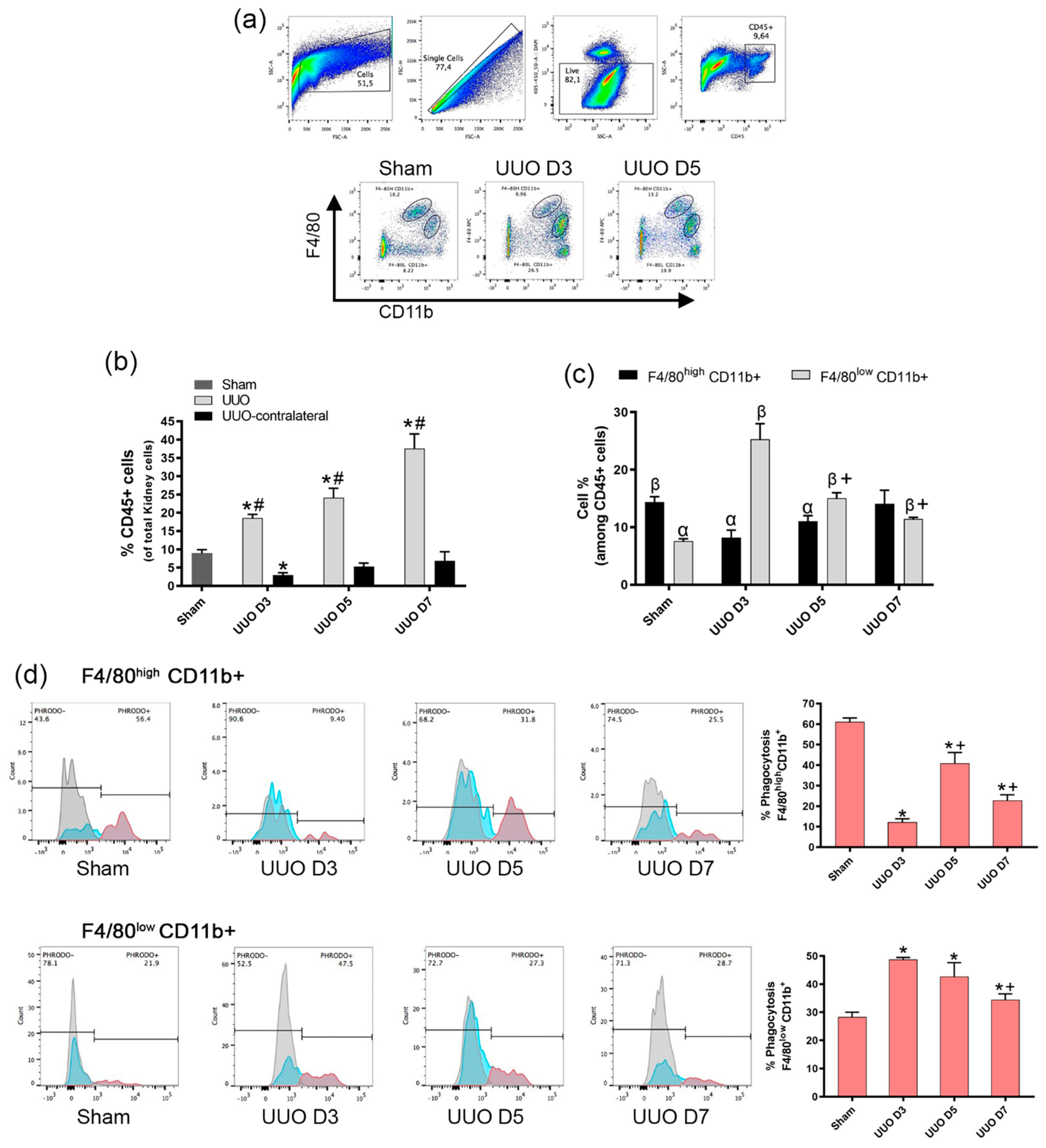
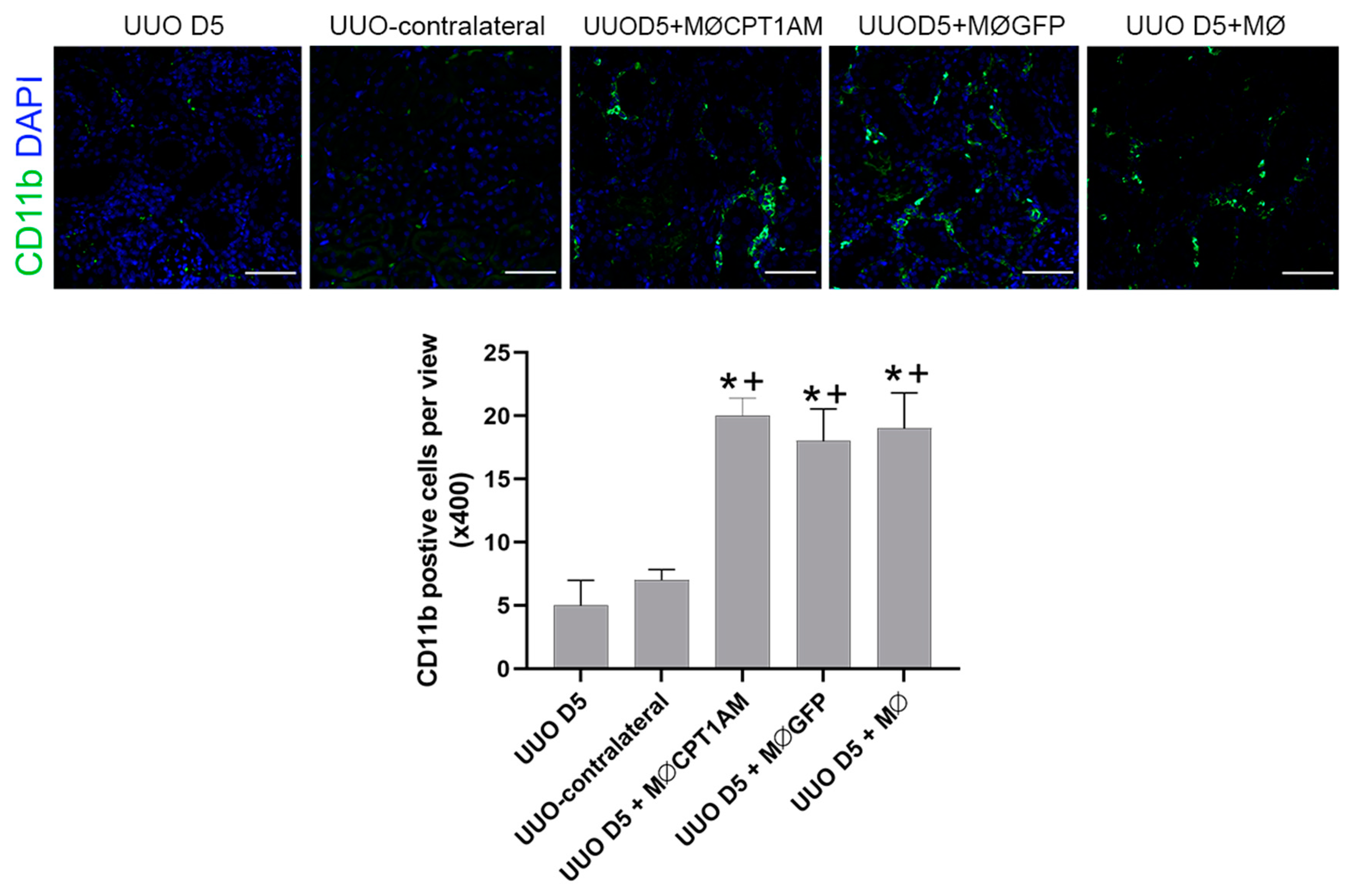
Isolated mice kidney cells were pre-incubated with the anti-mouse CD16/32 antibody (Biolegend, San Diego, CA, USA. Cat#:101320) at 1.0 µg/1.06 cells in 100 µL volume for 5 min on ice prior to immunostaining. After, the cells were stained with fluorescence-conjugated surface antibodies for 30 min at 4 °C, washed and analyzed using Becton-Dickinson (BD) FACSCanto II (BD Bioscience, San Jose, CA, USA). Antibodies used for the analysis were: anti-CD45 (Clone: 30-F11) PerCP-Cy5.5 at dilution 1:100 (Biolegend, San Diego, CA, USA. Cat#:103132), anti-F4/80 (Clone: BM8) APC at dilution 1:150 (Biolegend, San Diego, CA, USA. Cat#:123116), CD11b (Clone: M1/70) PE at dilution 1:50 (Biolegend, San Diego, CA, USA. Cat#:101207). Cell sorting was performed with BD FACSAria II (BD Bioscience, San Jose, CA, USA). Facs-sorted macrophages cells (F4/80 high CD11b+ and F4/80low CD11b+) were acquired from a pool of 4 mice per each group (sham and UUO day 5) in order to obtain a pure RNA isolation for qPCR analysis. Data were analyzed using the FlowJo v10 for Windows (FlowJo LLC, Ashland, OR, USA).
2.7. Phagocytosis Assay
One vial of pHrodo Green E. coli BioParticles conjugate (Invitrogen, Waltham, MA, USA. Cat#:P35366) was suspended in 2 mL of Live Cell Imaging Solution (LCIS) (Invitrogen, Waltham, MA, USA. Cat#:A14291DJ) at 1 mg/mL, thoroughly vortexed and sonicated following the manufacturer’s instructions. Bioparticles were previously tittered, and the final concentration of 120 μg/mL was our optimal concentration. After the isolation of murine kidney cells, the cells were incubated with pHrodo Green E. coli Bio-Particles diluted in LCIS at 120 μg/mL for 90 min at 37 °C. As control, cells were incubated with the same dose of phrodo but at 4 °C. Phagocytosis assay was performed in FACSCanto II. For the phagocytosis assay of the transduced cell, RAW 264.7 transduced cells were incubated with pHrodo Red E. coli Bio-Particles (Invitrogen, Waltham, MA, USA. Cat#:P35361) diluted in LCIS at 55 μg/mL for 90 min at 37 °C and the phagocytosis evaluated through fluorescence microscopy and quantify by spectrofluorometer at excitation 560 nm and emission 585 nm.
2.8. Histopathological Examination
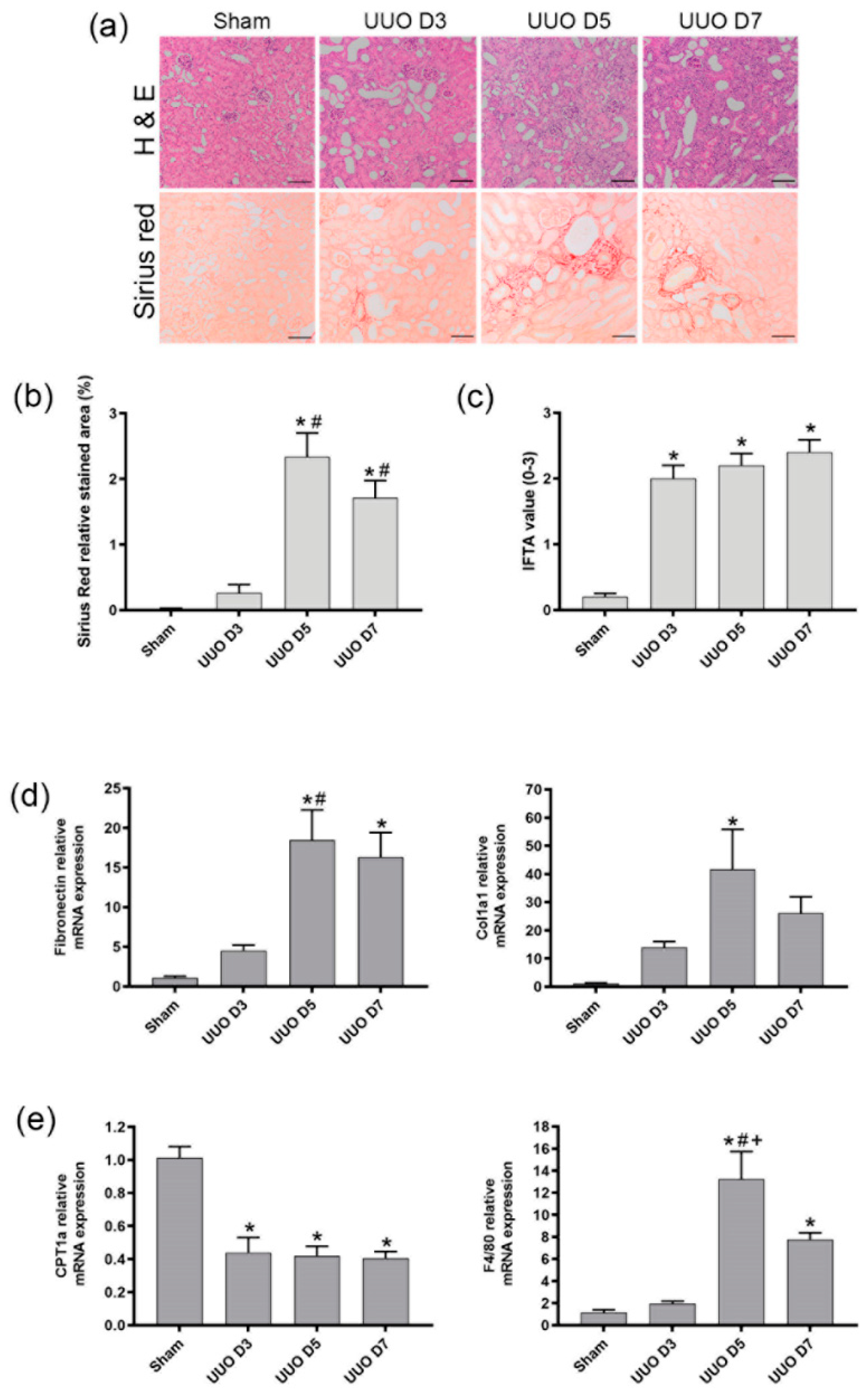
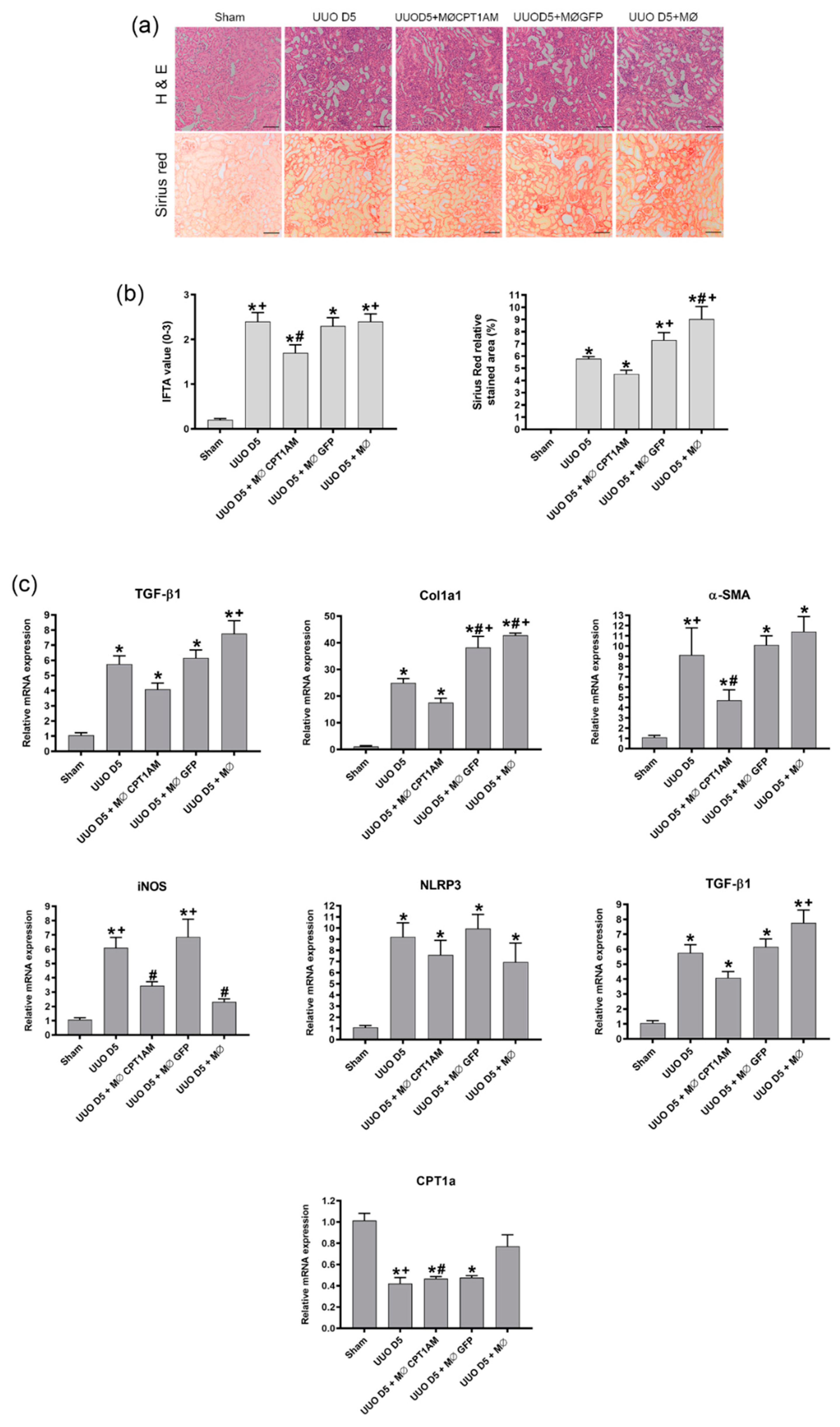
Transversal cross sections of mice kidney were fixed in 4% paraformaldehyde and embedded in paraffin for histological analysis by hematoxylin-eosin and Sirius red stains. Slides were analyzed by optical microscopy, in a Zeiss Axiophot microscope, to assess fibrosis and were quantified from each non-overlapping cortical field from the cortical region using ImageJ software. For histological analysis, 7 fields per kidney section were evaluated per animal. Pathological evaluation of interstitial fibrosis (IFTA) was assessed following Banff criteria and graded as follows: 0, no damage; 1, Mild interstitial fibrosis and tubular atrophy (<25% of cortical area); 2, Moderate interstitial fibrosis and tubular atrophy (26–50% of cortical area); and 3, Severe interstitial fibrosis and tubular atrophy/loss (>50% of cortical area). Sirius red values were obtained as relative stained area (%) of each field.
2.9. Quantitative Real-time Polymerase Chain Reaction (qPCR)
Total RNA was isolated using the RNeasy Mini kit (Qiagen. Hilden, Gerrmany). A total amount of 1μg RNA was reverse transcribed into cDNA using the iScript cDNA synthesis Kit (Bio-Rad, Hercules, CA, USA). qPCR assay was performed on a Bio-Rad CFX96 Touch Real Time PCR detection system with SooAdvanced Universal SYBR Green Supermix (Bio-Rad, Hercules, CA, USA). Levels of mRNA were normalized to those of reference genes GAPDH and 18s rRNA and expressed as fold change respect control or sham. Sequences of specific primers are listed in
Table 1.
2.10. Statistical Analysis
Data were reported as mean ± SEM of at least three independent experiments. Group means were compared with Student’s t test for two groups or one-way ANOVA followed by Tukey᾿s post-hoc test for multiple comparisons. Values were considered statistically significant if p value ≤ 0.05. The analyses were performed in GraphPad Prism version 9.1.0 for Windows (GraphPad Software, San Diego, CA, USA).
4. Discussion
Among the main macrophage phenotypes, it seems that M1 (inflammatory macrophages) and M2 (regenerative macrophages) are involved in the fibrotic process, but the involvement is controversial and, in some studies, depends on the fibrosis stage after injury. It has been found that elimination of M2 macrophages at the advanced stage of fibrosis in UUO animals mitigated the formation of renal fibrosis, but elimination of M1 macrophages in the early UUO stage did not modify the fibrotic features [
6]. In the same sense, other authors indicated that it was M2 macrophage depletion, rather than M1, that specifically inhibited the mesenchymal epithelial transition and, therefore, renal fibrosis [
10]. On the contrary, others found that the increase in the proportion of M2 macrophages plays an important beneficial role in the resolution of renal fibrosis, being the most characteristic feature of the early recovery phase after UUO [
11]. Recent studies in our group indicate that intravenous administration of a stable population of M2 macrophage reduces the impact of the fibrosis [
12].
Macrophage dynamics during the different phases of CKD progression are not fully known, and assessment of the predominant macrophage phenotype may be relevant in terms of defining the type of therapy [
13].
Anders and Ryu [
14] have proposed to classify tissue macrophages according to their predominant roles in different phases of kidney disease: pro-inflammatory, anti-inflammatory, profibrotic and fibrolytic macrophages. However, there is a lack of information regarding macrophage types and their dynamics, plasticity, and function in human CKD. Macrophages represent a spectrum of activated phenotypes rather than discrete stable subpopulations. Therefore, better strategies, based in the macrophage function and phenotype, should be studied to induce truly regenerative and reparative macrophages therapies.
Belliere et al. described a switch of macrophage phenotype from M1 to M2 during disease progression in a mouse model of rhabdomyolysis-induced AKI [
15], whereby F4/80
lowCD11b
high M1 kidney macrophages were dominant 2 days after the induction of rhabdomyolysis, whereas F4/80
highCD11b+ M2 cells were dominant by day 8. Importantly, analysis at the single cell level indicated the existence of considerable phenotypic heterogeneity, with some macrophages expressing both M1 and M2 markers. Likewise, an M1 to M2 transition was also evident in a UUO model of renal fibrosis, which was promoted by the actions of a tissue type plasminogen activator [
16].
Several publications strongly suggest that the expression of F4/80+CD11b+ cells in the obstructed kidneys starts going up after at least 4 days of UUO [
17,
18,
19]. Our results are in line with these results, indicating that under control conditions (sham mice), the F4/80
highCD11b+ fraction was significantly larger than the F4/80
lowCD11b+ fraction (
Figure 4b), but the latter was increased at day 3 after UUO, while the former was reduced on day 3. Nonetheless, during the UUO progress, F4/80
highCD11b+ increases as other authors suggest, while the F4/80
lowCD11b+ decreases during the progression of fibrosis.
Our study has identified two different types of macrophages which exhibit a differential phenotype, being the F4/80
lowCD11b+ population with high phagocytosis predominant at the initial phases of injury, while the F4/80
highCD11b+ with low phagocytosis was dominant at the later phases of UUO development. Moreover, there is a net decrease in phagocytosis, since the population with higher phagocytic ability decreases with respect to day 3 following ureter obstruction, while the population with low phagocytic ability increased as shown in
Figure 2. Further, when cells were isolated from fibrotic kidneys, the F4/80
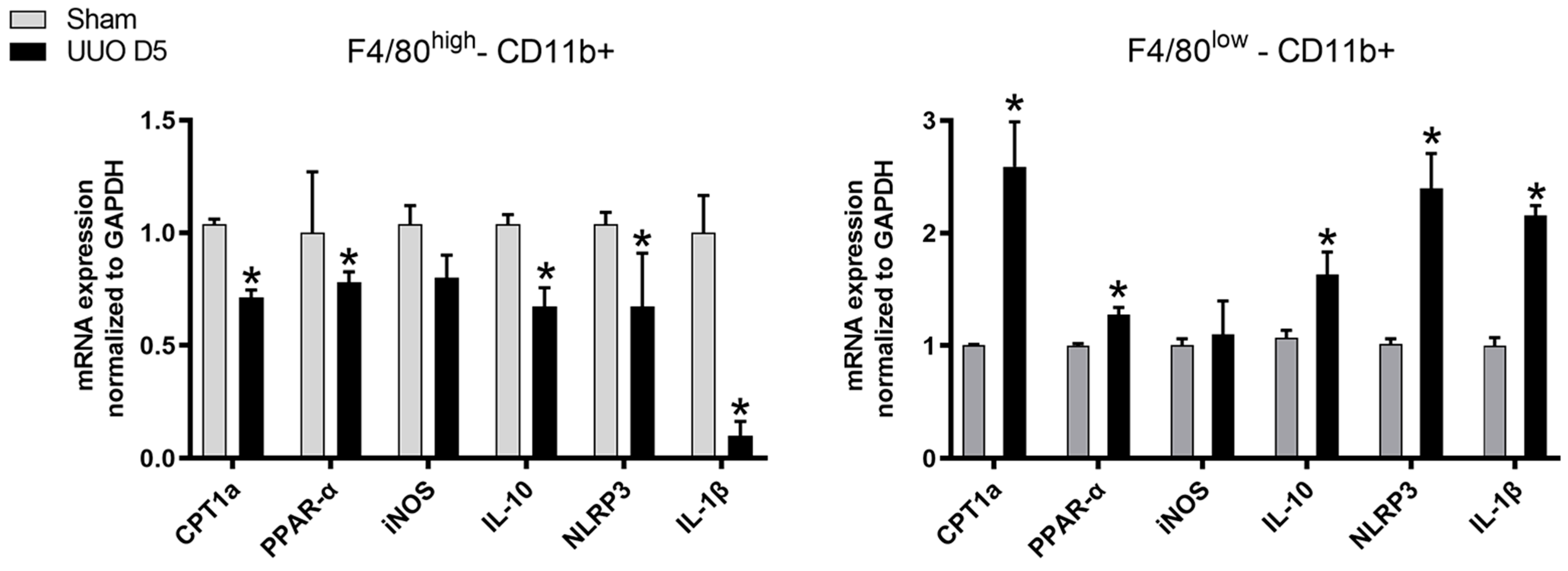
high population, characterized by low phagocytosis, expressed a decrease of PPAR-α expression and its target gene CPT1a as well as IL-10, NLRP3, IL-1β compared to the F4/80
low population of macrophages which presented elevated mRNA levels.
Some authors previously [
17] indicated that the CD11b+F4/80
low has shown a phenotype profile of M1-type activation, while CD11b+F4/80
high shows a phenotype indicative of M2-type activation.
Our results indicate that the phenotype of CD11b+F4/80low could be like M1 since an increase in NLRP3 and IL-1β could be observed, but the fact that IL-10 also increased indicates a mixed phenotype. In contrast, CD11b+F4/80high has a trend towards M2 phenotype, since NLRP3 and IL-1β are decreased, but the diminished levels of IL-10 point to a mixed phenotype again. However, our intention was to determine whether these two populations differ in CPT1a expression, which has been confirmed in the present study.
Other studies [
20] previously confirmed marked changes in the proportion and gene expression of at least 12 myeloid cell subsets involved in kidney injury and repair in a reversible UUO model. In addition, they described a novel Mmp121 macrophage subset that acts during repair. The Mmp121 cluster–expressed genes are implicated in efferocytosis and lipid transport, suggesting that they may be involved in the clearance of apoptotic cells, thus pointing again to a potential role of macrophage phagocytosis in kidney repair.
The association of defective phagocytic ability of mononuclear cells and exaggerated inflammatory and fibrotic responses has been proposed earlier [
21] and the F4/80
high population, which has been characterized as embryo-derived macrophages, are known to exhibit a higher phagocytic ability than F4/80
low bone marrow-derived macrophages (BMDM) [
22]. Our results agree with these authors, since in sham conditions, the F4/80
low CD11b+ cells are more phagocytic that F4/80
high CD11+, but in fibrotic conditions, there is a shift between populations as F4/80
low CD11b+ become less phagocytic than the F4/80
high CD11b+.
One potential explanation could be that after UUO, the F4/80
high CD11b+ population exhibits a decrease in the expression of CPT1a that leads to a decrease in phagocytosis since a direct relationship was previously observed between CPT1a as inductor of phagocytosis [
7]. One could speculate that in fibrotic conditions, the higher phagocytic population, the F4/80
high CD11b+population, is able to phagocytose detritus and to be converted in foam cells, thus leading to a downregulation of CPT1a that in turns reduces the phagocytosis of this population during fibrosis. On the other hand, the F4/80
high CD11b+ cells have been previously described as embryo derived macrophages with high glycolysis [
22] that could also potentially result in a decrease in CPT1a expression as observed in our experiments. Nevertheless, the present study has not addressed these issues.
These observations support the importance of macrophage dynamics during fibrosis development. and suggest a potential therapeutic approach to reduce fibrosis in the UUO model by enrichment of the kidney resident macrophage population with a higher proportion of exogenous phagocytic macrophages.
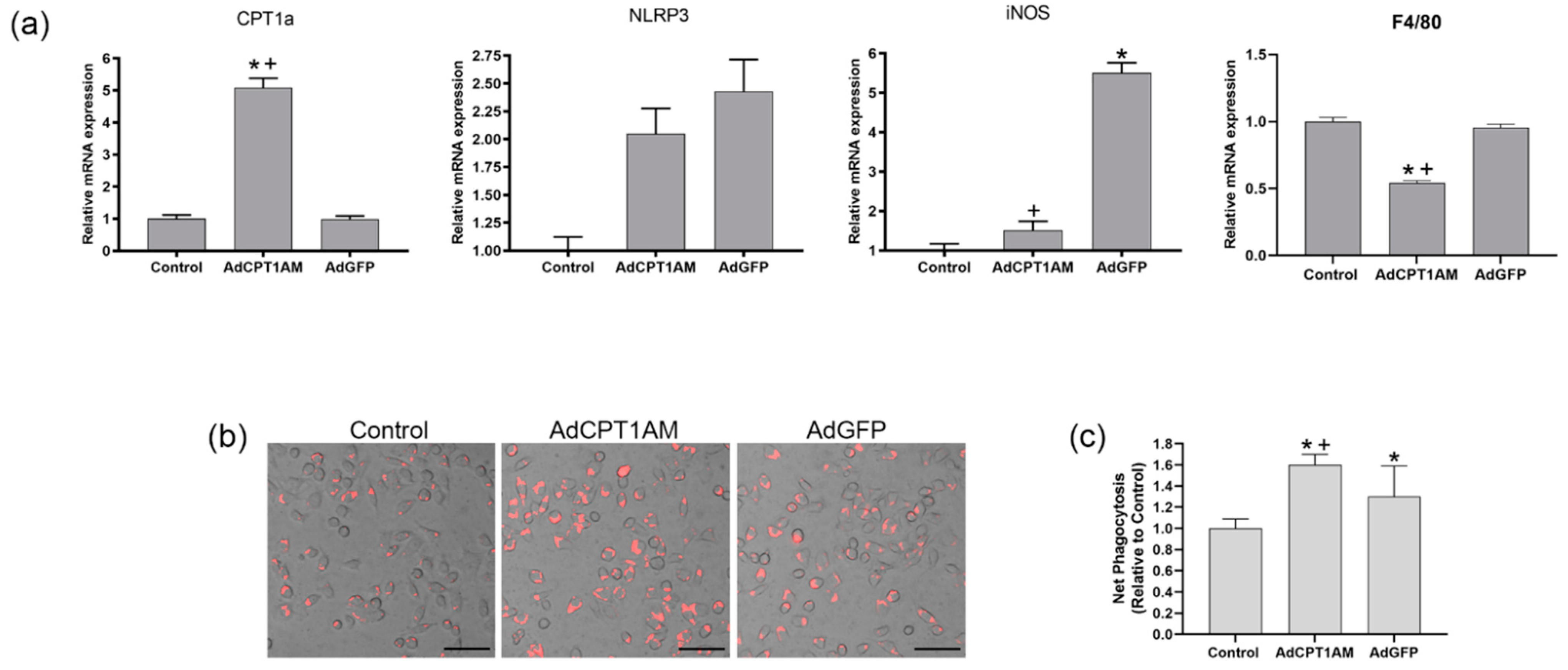
We have tested the effect of a therapy with a stable population of macrophages which exhibit high phagocytosis ability.
As indicated in
Figure 4, macrophages overexpressing CPT1a have a high phagocytic ability. This has been observed in previous studies [
7] that revealed that CPT1a overexpression decreases lipid content and stimulates phagocytosis while CPT1a knockdown promotes an increase in lipid accumulation and in the pro-inflammatory marker iNOS associated with an impaired phagocytosis and down-regulation of NLRP3. Other authors have observed that a NOX4 dependent inactivation of CPT1a leads to a reduction in NLRP3 activation in macrophages [
23]. This study indicates that CPT1a overexpressing macrophages show an increase in NLRP3 and iNOS expression but without statistical significance, suggesting that this level of CPT1a overexpression does not lead to an inflammatory profile. In addition, as observed in
Figure S2, the C75 compound, which is known to inhibit CPT1a activity, induced an increase in the fibrotic markers although a reduction in TGF-β1 was observed. Therefore, it could be said that CPT1a expression is related to the development of fibrosis although TGF-β1 is decreased.
In addition, the phagocytic ability of macrophages overexpressing CPT1a is superior; hence, using these cells as a therapy could reestablish the reduced macrophage phagocytosis observed in kidney fibrosis.
These observations suggest that the expression of CPT1a in macrophages could counteract the increase in the low phagocytic (F4/80high−CD11b) population and that the increase in phagocytic macrophages results in an amelioration of fibrosis.
Our results indicate that cell therapy with macrophages overexpressing CPT1a with high phagocytic ability applied to the UUO model reduces the expression of the fibrotic markers, fibronectin, α-SMA, Col1a1 and TGF-β1, indicating the potential of our therapy to revert fibrosis development. Previous studies in the UUO model have demonstrated that macrophage therapy with a stable population of M2 reduced the impact of fibrosis in front of other macrophage therapies which do not exhibited a stable phenotype [
12]: thus, indicating that if macrophages maintain a stable M2 phenotype, fibrosis could be overcome.


 ,
, 









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}