
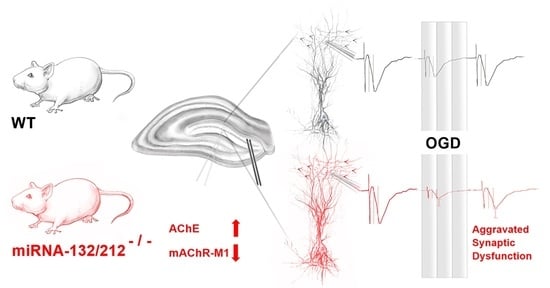
miRNA-132/212 Gene-Deletion Aggravates the Effect of Oxygen-Glucose Deprivation on Synaptic Functions in the Female Mouse Hippocampus

 ,
, 

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Electrophysiology
2.2.1. Hippocampal Slices Preparation
2.2.2. Extracellular Recordings
2.2.3. Oxygen Glucose Deprivation Protocol
2.3. Immunoblotting
2.4. Statistical Analysis
3. Results
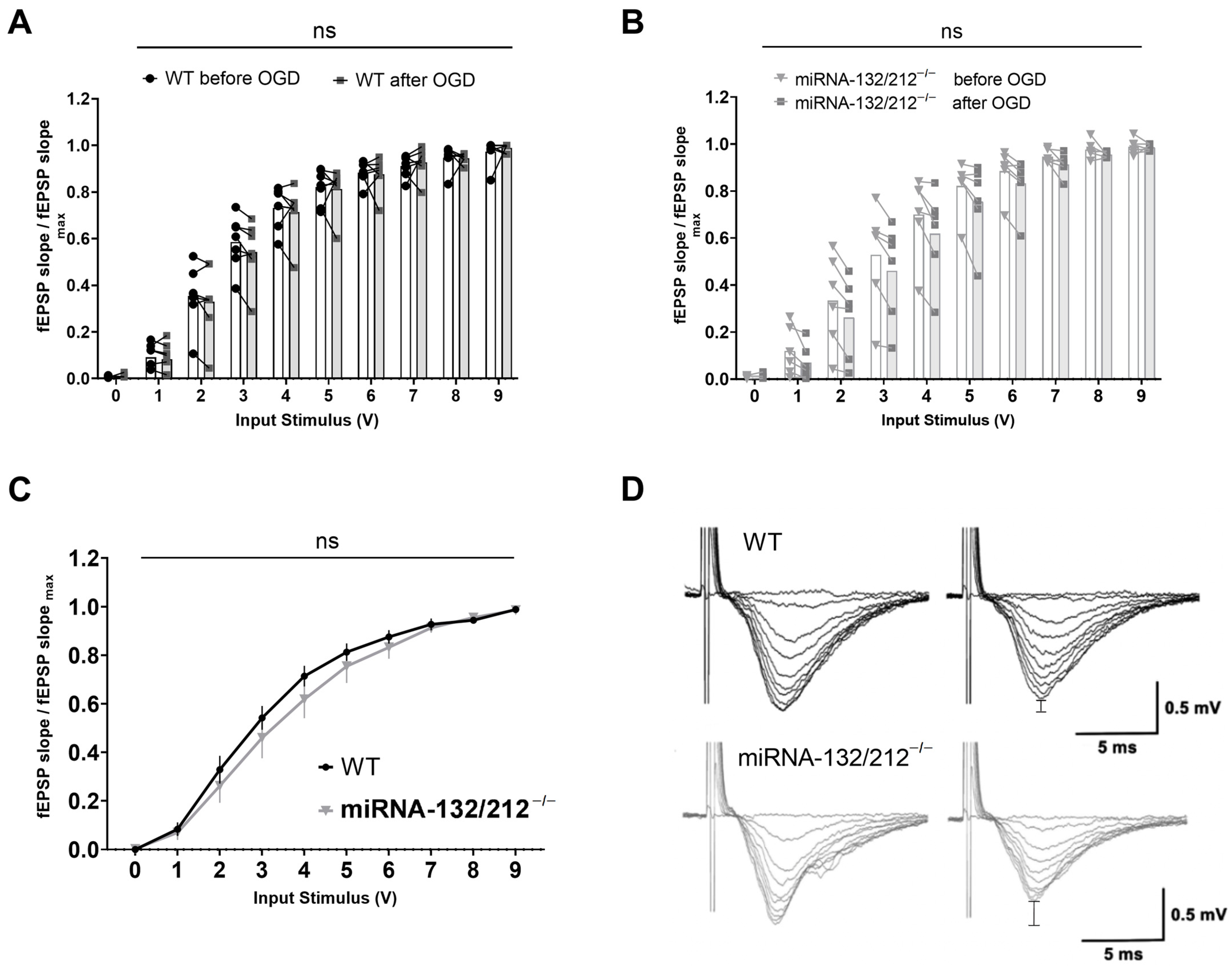
3.1. Dentate Gyri Basal Synaptic Transmission and Short-Term Plasticity Are Unaffected by miRNA 132/212 Gene Deletion
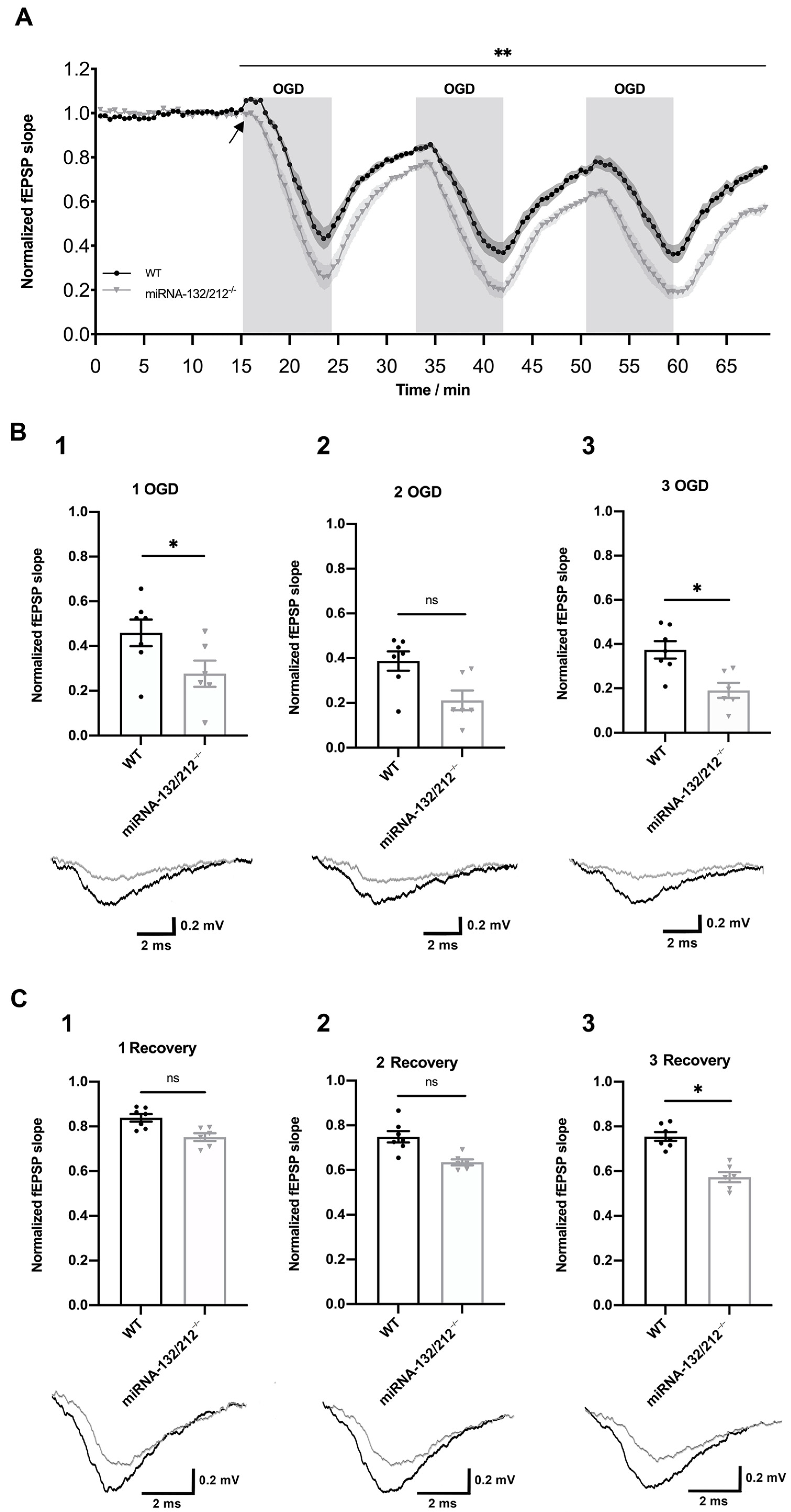
3.2. Dentate Gyri Synaptic Depression Following OGD Is Aggravated in miRNA 132/212−/− Mice
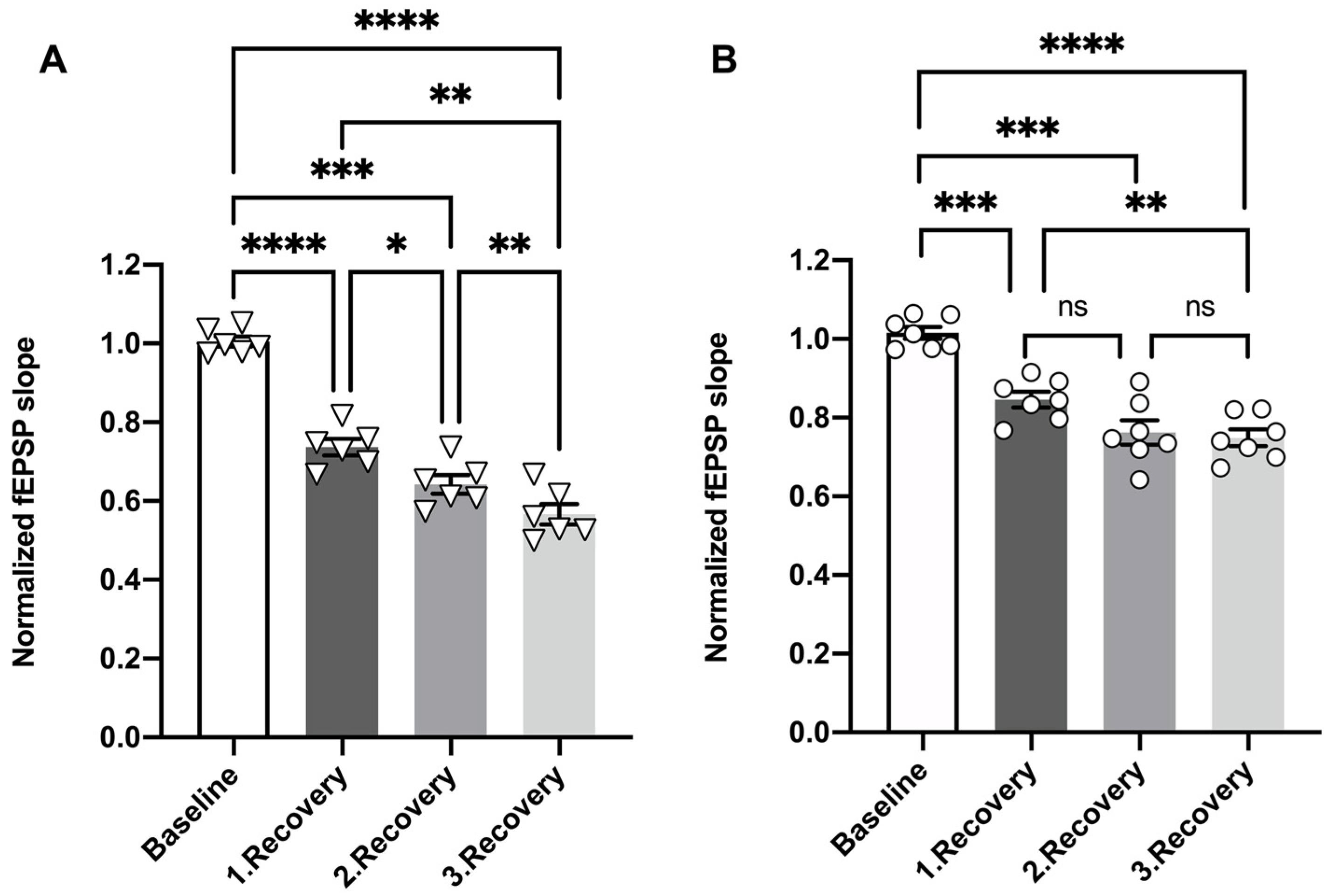
3.3. Episodic OGD Impairs the Recovery of Dentate Gyri Synaptic Transmission in miRNA 132/212−/−
3.4. miRNA 132/212 Gene Deletion Promotes the Vulnerability of Short-Term Synaptic Depression to Deleterious Effects of OGD
3.5. Basal Synaptic Transmission Remains Unaffected in the miRNA 132/212−/− Hippocampus after Episodic OGD Insults
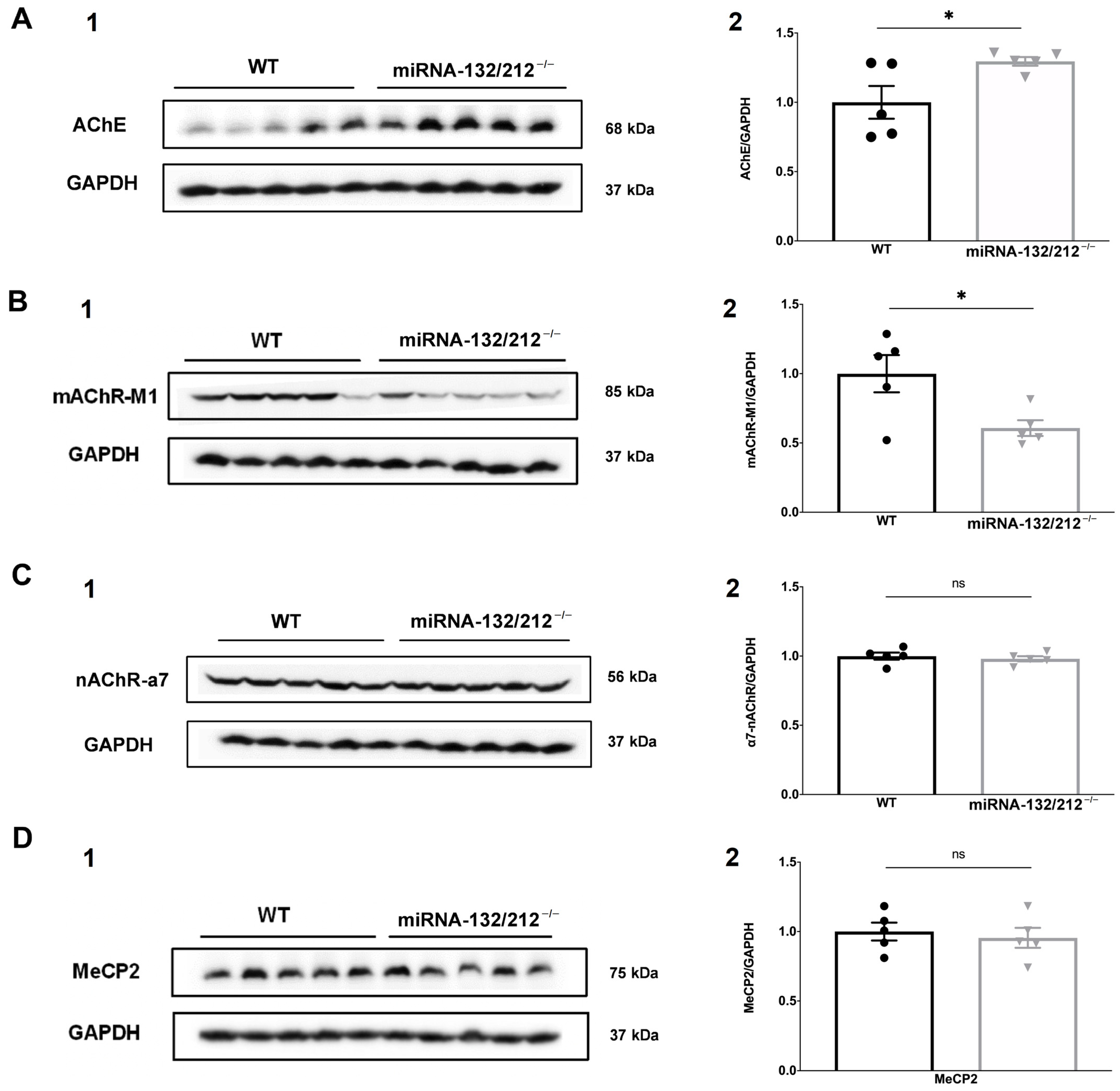
3.6. miRNAs 132/212 Deletion Alters Protein Levels of AChE and mAChR-M1 in the Female Mouse Hippocampus
4. Discussion
4.1. Modeling Ischemia Reperfusion Injuries Ex Vivo to Study the Effects of OGD on Synaptic Transmission
4.2. The miRNA 132/212 Cluster: A Putative Link between Cholinergic Signaling, Hippocampal Synaptic Transmissions and the Resilience to Ischemia Induced Functional Impairment
4.3. Limitations and Perspectives
5. Final Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Campbell, B.C.; De Silva, D.A.; Macleod, M.R.; Coutts, S.B.; Schwamm, L.H.; Davis, S.M.; Donnan, G.A. Ischaemic stroke. Nat. Rev. Dis. Primers 2019, 5, 70. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.O.; Nguyen, M.; Roth, G.A.; Nichols, E.; Alam, T.; Abate, D.; Abd-Allah, F.; Abdelalim, A.; Abraha, H.N.; Abu-Rmeileh, N.M.; et al. Global, regional, and national burden of stroke, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 439–458. [Google Scholar] [CrossRef] [Green Version]
- Powers, W.J.; Rabinstein, A.A.; Ackerson, T.; Adeoye, O.M.; Bambakidis, N.C.; Becker, K.; Biller, J.; Brown, M.; Demaerschalk, B.M.; Hoh, B.; et al. Guidelines for the Early Management of Patients With Acute Ischemic Stroke: 2019 Update to the 2018 Guidelines for the Early Management of Acute Ischemic Stroke: A Guideline for Healthcare Professionals From the American Heart Association/American Stroke Association. Stroke 2019, 50, e344–e418. [Google Scholar] [CrossRef]
- George, P.M.; Steinberg, G.K. Novel Stroke Therapeutics: Unraveling Stroke Pathophysiology and Its Impact on Clinical Treatments. Neuron 2015, 87, 297–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, P.; Zuo, X.; Ji, A. Stroke-induced microRNAs: The potential therapeutic role for stroke. Exp. Ther. Med. 2012, 3, 571–576. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Sandhu, H.K.; Zhi, F.; Hua, F.; Wu, M.; Xia, Y. Effects of hypoxia and ischemia on microRNAs in the brain. Curr. Med. Chem. 2015, 22, 1292–1301. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Fang, M.; Liu, X.Y. Role of microRNAs in stroke and poststroke depression. Sci. World J. 2013, 2013, 459692. [Google Scholar] [CrossRef] [PubMed]
- Vasudeva, K.; Munshi, A. miRNA dysregulation in ischaemic stroke: Focus on diagnosis, prognosis, therapeutic and protective biomarkers. Eur. J. Neurosci. 2020, 52, 3610–3627. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, Y.; Yang, G.-Y. MicroRNAs in Cerebral Ischemia. Stroke Res. Treat. 2013, 2013, 276540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ambros, V. microRNAs: Tiny regulators with great potential. Cell 2001, 107, 823–826. [Google Scholar] [CrossRef] [Green Version]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [Green Version]
- Lai, E.C. Micro RNAs are complementary to 3′ UTR sequence motifs that mediate negative post-transcriptional regulation. Nat. Genet. 2002, 30, 363–364. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yun, Z.; Gong, L.; Qu, H.; Duan, X.; Jiang, Y.; Zhu, H. Comparison of miRNA Evolution and Function in Plants and Animals. Microrna 2018, 7, 4–10. [Google Scholar] [CrossRef]
- Schaefer, A.; O’Carroll, D.; Tan, C.L.; Hillman, D.; Sugimori, M.; Llinas, R.; Greengard, P. Cerebellar neurodegeneration in the absence of microRNAs. J. Exp. Med. 2007, 204, 1553–1558. [Google Scholar] [CrossRef] [Green Version]
- Vo, N.; Klein, M.E.; Varlamova, O.; Keller, D.M.; Yamamoto, T.; Goodman, R.H.; Impey, S. A cAMP-response element binding protein-induced microRNA regulates neuronal morphogenesis. Proc. Natl. Acad. Sci. USA 2005, 102, 16426–16431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wanet, A.; Tacheny, A.; Arnould, T.; Renard, P. miR-212/132 expression and functions: Within and beyond the neuronal compartment. Nucleic Acids Res. 2012, 40, 4742–4753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giraldez, A.J.; Cinalli, R.M.; Glasner, M.E.; Enright, A.J.; Thomson, J.M.; Baskerville, S.; Hammond, S.M.; Bartel, D.P.; Schier, A.F. MicroRNAs regulate brain morphogenesis in zebrafish. Science 2005, 308, 833–838. [Google Scholar] [CrossRef] [Green Version]
- Cheng, L.C.; Pastrana, E.; Tavazoie, M.; Doetsch, F. miR-124 regulates adult neurogenesis in the subventricular zone stem cell niche. Nat. Neurosci. 2009, 12, 399–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baby, N.; Alagappan, N.; Dheen, S.T.; Sajikumar, S. MicroRNA-134-5p inhibition rescues long-term plasticity and synaptic tagging/capture in an Aβ(1-42)-induced model of Alzheimer’s disease. Aging Cell 2020, 19, e13046. [Google Scholar] [CrossRef] [Green Version]
- Berentsen, B.; Patil, S.; Rønnestad, K.; Goff, K.M.; Pajak, M.; Simpson, T.I.; Wibrand, K.; Bramham, C.R. MicroRNA-34a Acutely Regulates Synaptic Efficacy in the Adult Dentate Gyrus In Vivo. Mol Neurobiol. 2020, 57, 1432–1445. [Google Scholar] [CrossRef] [PubMed]
- Remenyi, J.; van den Bosch, M.W.; Palygin, O.; Mistry, R.B.; McKenzie, C.; Macdonald, A.; Hutvagner, G.; Arthur, J.S.; Frenguelli, B.G.; Pankratov, Y. miR-132/212 knockout mice reveal roles for these miRNAs in regulating cortical synaptic transmission and plasticity. PLoS ONE 2013, 8, e62509. [Google Scholar] [CrossRef]
- Stojanovic, T.; Benes, H.; Awad, A.; Bormann, D.; Monje, F.J. Nicotine abolishes memory-related synaptic strengthening and promotes synaptic depression in the neurogenic dentate gyrus of miR-132/212 knockout mice. Addict. Biol. 2020, 26, e12905. [Google Scholar] [CrossRef] [Green Version]
- Ye, Y.; Xu, H.; Su, X.; He, X. Role of MicroRNA in Governing Synaptic Plasticity. Neural. Plast. 2016, 2016, 4959523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Guo, J.; Wang, Q.; Chen, Y. MicroRNA-132 silencing decreases the spontaneous recurrent seizures. Int. J. Clin. Exp. Med. 2014, 7, 1639–1649. [Google Scholar]
- Jimenez-Mateos, E.M.; Engel, T.; Merino-Serrais, P.; McKiernan, R.C.; Tanaka, K.; Mouri, G.; Sano, T.; O’Tuathaigh, C.; Waddington, J.L.; Prenter, S.; et al. Silencing microRNA-134 produces neuroprotective and prolonged seizure-suppressive effects. Nat. Med. 2012, 18, 1087–1094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, L.; Ren, Y.; Cai, H.; Zhao, W.; Song, Y. MicroRNA-132 aggravates epileptiform discharges via suppression of BDNF/TrkB signaling in cultured hippocampal neurons. Brain Res. 2015, 1622, 484–495. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Huang, H.; Zhou, X.; Liu, X.; Ou, S.; Xu, T.; Li, R.; Ma, L.; Chen, Y. MicroRNA-132 Interact with p250GAP/Cdc42 Pathway in the Hippocampal Neuronal Culture Model of Acquired Epilepsy and Associated with Epileptogenesis Process. Neural Plast. 2016, 2016, 5108489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Fatimy, R.; Li, S.; Chen, Z.; Mushannen, T.; Gongala, S.; Wei, Z.; Balu, D.T.; Rabinovsky, R.; Cantlon, A.; Elkhal, A.; et al. MicroRNA-132 provides neuroprotection for tauopathies via multiple signaling pathways. Acta Neuropathol. 2018, 136, 537–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herrera-Espejo, S.; Santos-Zorrozua, B.; Álvarez-González, P.; Lopez-Lopez, E.; Garcia-Orad, Á. A Systematic Review of MicroRNA Expression as Biomarker of Late-Onset Alzheimer’s Disease. Mol. Neurobiol. 2019, 56, 8376–8391. [Google Scholar] [CrossRef]
- Song, Y.; Hu, M.; Zhang, J.; Teng, Z.Q.; Chen, C. A novel mechanism of synaptic and cognitive impairments mediated via microRNA-30b in Alzheimer’s disease. EBioMedicine 2019, 39, 409–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salta, E.; De Strooper, B. microRNA-132: A key noncoding RNA operating in the cellular phase of Alzheimer’s disease. FASEB J. 2017, 31, 424–433. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Mateos, E.M. Role of MicroRNAs in innate neuroprotection mechanisms due to preconditioning of the brain. Front. Neurosci. 2015, 9, 118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, K.F.; Sakamoto, K.; Aten, S.; Snider, K.H.; Loeser, J.; Hesse, A.M.; Page, C.E.; Pelz, C.; Arthur, J.S.; Impey, S.; et al. Targeted deletion of miR-132/-212 impairs memory and alters the hippocampal transcriptome. Learn. Mem. 2016, 23, 61–71. [Google Scholar] [CrossRef] [Green Version]
- Hong, S.; Lee, J.; Seo, H.H.; Lee, C.Y.; Yoo, K.J.; Kim, S.M.; Lee, S.; Hwang, K.C.; Choi, E. Na(+)-Ca(2+) exchanger targeting miR-132 prevents apoptosis of cardiomyocytes under hypoxic condition by suppressing Ca(2+) overload. Biochem. Biophys. Res. Commun. 2015, 460, 931–937. [Google Scholar] [CrossRef]
- Keasey, M.P.; Scott, H.L.; Bantounas, I.; Uney, J.B.; Kelly, S. MiR-132 Is Upregulated by Ischemic Preconditioning of Cultured Hippocampal Neurons and Protects them from Subsequent OGD Toxicity. J. Mol. Neurosci. 2016, 59, 404–410. [Google Scholar] [CrossRef]
- Sun, Z.Z.; Lv, Z.Y.; Tian, W.J.; Yang, Y. MicroRNA-132 protects hippocampal neurons against oxygen-glucose deprivation-induced apoptosis. Int. J. Immunopathol. Pharmacol. 2017, 30, 253–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lusardi, T.A.; Farr, C.D.; Faulkner, C.L.; Pignataro, G.; Yang, T.; Lan, J.; Simon, R.P.; Saugstad, J.A. Ischemic preconditioning regulates expression of microRNAs and a predicted target, MeCP2, in mouse cortex. J. Cereb. Blood Flow Metab. 2010, 30, 744–756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt-Kastner, R.; Freund, T.F. Selective vulnerability of the hippocampus in brain ischemia. Neuroscience 1991, 40, 599–636. [Google Scholar] [CrossRef]
- Cervós-Navarro, J.; Diemer, N.H. Selective vulnerability in brain hypoxia. Crit. Rev. Neurobiol. 1991, 6, 149–182. [Google Scholar] [PubMed]
- Kirino, T.; Tamura, A.; Sano, K. Selective Vulnerability of the Hippocampus to Ischemia—Reversible and Irreversible Types of Ischemic Cell Damage. In Progress in Brain Research; Kogure, K., Hossmann, K.-A., Siesjö, B.K., Welsh, F.A., Eds.; Elsevier: Amsterdam, The Netherlands, 1985; pp. 39–58. [Google Scholar]
- Anderson, C.A.; Arciniegas, D.B. Cognitive sequelae of hypoxic-ischemic brain injury: A review. NeuroRehabilitation 2010, 26, 47–63. [Google Scholar] [CrossRef]
- Sun, J.H.; Tan, L.; Yu, J.T. Post-stroke cognitive impairment: Epidemiology, mechanisms and management. Ann. Transl. Med. 2014, 2, 80. [Google Scholar] [PubMed]
- Jokinen, H.; Melkas, S.; Ylikoski, R.; Pohjasvaara, T.; Kaste, M.; Erkinjuntti, T.; Hietanen, M. Post-stroke cognitive impairment is common even after successful clinical recovery. Eur. J. Neurol. 2015, 22, 1288–1294. [Google Scholar] [CrossRef]
- Jacquin, A.; Binquet, C.; Rouaud, O.; Graule-Petot, A.; Daubail, B.; Osseby, G.V.; Bonithon-Kopp, C.; Giroud, M.; Béjot, Y. Post-stroke cognitive impairment: High prevalence and determining factors in a cohort of mild stroke. J. Alzheimers Dis. 2014, 40, 1029–1038. [Google Scholar] [CrossRef]
- Zhao, X.; Bai, F.; Zhang, E.; Zhou, D.; Jiang, T.; Zhou, H.; Wang, Q. Electroacupuncture Improves Neurobehavioral Function Through Targeting of SOX2-Mediated Axonal Regeneration by MicroRNA-132 After Ischemic Stroke. Front. Mol. Neurosci. 2018, 11, 471. [Google Scholar] [CrossRef] [Green Version]
- Tasca, C.I.; Dal-Cim, T.; Cimarosti, H. In vitro oxygen-glucose deprivation to study ischemic cell death. Methods Mol. Biol. 2015, 1254, 197–210. [Google Scholar] [PubMed]
- Leuner, B.; Gould, E. Structural plasticity and hippocampal function. Annu. Rev. Psychol. 2010, 61, 111–140. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Song, N. Molecular Mechanism of Adult Neurogenesis and its Association with Human Brain Diseases. J. Cent. Nerv. Syst. Dis. 2016, 8, 5–11. [Google Scholar] [CrossRef]
- Beery, A.K.; Zucker, I. Sex bias in neuroscience and biomedical research. Neurosci. Biobehav. Rev. 2011, 35, 565–572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cordonnier, C.; Sprigg, N.; Sandset, E.C.; Pavlovic, A.; Sunnerhagen, K.S.; Caso, V.; Christensen, H. Women Initiative for Stroke in Europe (WISE) group. Stroke in women—from evidence to inequalities. Nat. Rev. Neurol. 2017, 13, 521–532. [Google Scholar] [CrossRef] [Green Version]
- Cicvaric, A.; Bulat, T.; Bormann, D.; Yang, J.; Auer, B.; Milenkovic, I.; Cabatic, M.; Milicevic, R.; Monje, F.J. Sustained consumption of cocoa-based dark chocolate enhances seizure-like events in the mouse hippocampus. Food Funct. 2018, 9, 1532–1544. [Google Scholar] [CrossRef]
- Cicvaric, A.; Yang, J.; Bulat, T.; Zambon, A.; Dominguez-Rodriguez, M.; Kühn, R.; Sadowicz, M.G.; Siwert, A.; Egea, J.; Pollak, D.D.; et al. Enhanced synaptic plasticity and spatial memory in female but not male FLRT2-haplodeficient mice. Sci. Rep. 2018, 8, 3703. [Google Scholar] [CrossRef]
- Cicvaric, A.; Yang, J.; Krieger, S.; Khan, D.; Kim, E.J.; Dominguez-Rodriguez, M.; Cabatic, M.; Molz, B.; Acevedo Aguilar, J.P.; Milicevic, R.; et al. The brain-tumor related protein podoplanin regulates synaptic plasticity and hippocampus-dependent learning and memory. Ann. Med. 2016, 48, 652–668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massa, F.; Koehl, M.; Wiesner, T.; Grosjean, N.; Revest, J.M.; Piazza, P.V.; Abrous, D.N.; Oliet, S.H. Conditional reduction of adult neurogenesis impairs bidirectional hippocampal synaptic plasticity. Proc. Natl. Acad. Sci. USA 2011, 108, 6644–6649. [Google Scholar] [CrossRef] [Green Version]
- Furling, D.; Ghribi, O.; Lahsaini, A.; Mirault, M.E.; Massicotte, G. Impairment of synaptic transmission by transient hypoxia in hippocampal slices: Improved recovery in glutathione peroxidase transgenic mice. Proc. Natl. Acad. Sci. USA 2000, 97, 4351–4356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hájos, N.; Ellender, T.J.; Zemankovics, R.; Mann, E.O.; Exley, R.; Cragg, S.J.; Freund, T.F.; Paulsen, O. Maintaining network activity in submerged hippocampal slices: Importance of oxygen supply. Eur. J. Neurosci. 2009, 29, 319–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bekenstein, J.W.; Lothman, E.W. Electrophysiological characterization of associational pathway terminating on dentate gyrus granule cells in the rat. Hippocampus 1991, 1, 399–404. [Google Scholar] [CrossRef] [PubMed]
- Bronzino, J.D.; Blaise, J.H.; Morgane, P.J. The paired-pulse index: A measure of hippocampal dentate granule cell modulation. Ann. Biomed. Eng. 1997, 25, 870–873. [Google Scholar] [CrossRef] [PubMed]
- Naylor, D.E.; Wasterlain, C.G. GABA synapses and the rapid loss of inhibition to dentate gyrus granule cells after brief perforant-path stimulation. Epilepsia 2005, 46 (Suppl. 5), 142–147. [Google Scholar] [CrossRef] [Green Version]
- Fusco, I.; Cherchi, F.; Catarzi, D.; Colotta, V.; Varano, F.; Pedata, F.; Pugliese, A.M.; Coppi, E. Functional characterization of a novel adenosine A2B receptor agonist on short-term plasticity and synaptic inhibition during oxygen and glucose deprivation in the rat CA1 hippocampus. Brain Res. Bull. 2019, 151, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Fusco, I.; Ugolini, F.; Lana, D.; Coppi, E.; Dettori, I.; Gaviano, L.; Nosi, D.; Cherchi, F.; Pedata, F.; Giovannini, M.G.; et al. The Selective Antagonism of Adenosine A2B Receptors Reduces the Synaptic Failure and Neuronal Death Induced by Oxygen and Glucose Deprivation in Rat CA1 Hippocampus in Vitro. Front. Pharmacol. 2018, 9, 399. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.E.; Ko, I.G.; Kim, C.J.; Chung, J.Y.; Yi, J.W.; Choi, J.H.; Jang, M.S.; Han, J.H. Dexmedetomidine promotes the recovery of the field excitatory postsynaptic potentials (fEPSPs) in rat hippocampal slices exposed to oxygen-glucose deprivation. Neurosci. Lett. 2016, 631, 91–96. [Google Scholar] [CrossRef]
- Levin, S.G.; Godukhin, O.V. Anti-inflammatory cytokines, TGF-beta1 and IL-10, exert anti-hypoxic action and abolish posthypoxic hyperexcitability in hippocampal slice neurons: Comparative aspects. Exp. Neurol. 2011, 232, 329–332. [Google Scholar] [CrossRef]
- Pugliese, A.M.; Traini, C.; Cipriani, S.; Gianfriddo, M.; Mello, T.; Giovannini, M.G.; Galli, A.; Pedata, F. The adenosine A2A receptor antagonist ZM241385 enhances neuronal survival after oxygen-glucose deprivation in rat CA1 hippocampal slices. Br. J. Pharmacol. 2009, 157, 818–830. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, R.; Hirota, K.; Roth, S.H.; Yamazaki, M. Anoxic depolarization of rat hippocampal slices is prevented by thiopental but not by propofol or isoflurane. Br. J. Anaesth. 2005, 94, 486–491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, M.; Reuter, J.; Gerich, F.J.; Hildebrandt, B.; Hägele, S.; Katschinski, D.; Müller, M. Enhanced hypoxia susceptibility in hippocampal slices from a mouse model of rett syndrome. J. Neurophysiol. 2009, 101, 1016–1032. [Google Scholar] [CrossRef]
- Pugliese, A.M.; Latini, S.; Corradetti, R.; Pedata, F. Brief, repeated, oxygen-glucose deprivation episodes protect neurotransmission from a longer ischemic episode in the in vitro hippocampus: Role of adenosine receptors. Br. J. Pharmacol. 2003, 140, 305–314. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, E.; Yamamoto, S.; Kudo, Y.; Mihara, S.; Higashi, H. Mechanisms underlying the rapid depolarization produced by deprivation of oxygen and glucose in rat hippocampal CA1 neurons in vitro. J. Neurophysiol. 1997, 78, 891–902. [Google Scholar] [CrossRef] [Green Version]
- Wall, A.M.; Corcoran, A.E.; O’Halloran, K.D.; O’Connor, J.J. Effects of prolyl-hydroxylase inhibition and chronic intermittent hypoxia on synaptic transmission and plasticity in the rat CA1 and dentate gyrus. Neurobiol. Dis. 2014, 62, 8–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Youssef, F.F.; Addae, J.I.; McRae, A.; Stone, T.W. Long-term potentiation protects rat hippocampal slices from the effects of acute hypoxia. Brain Res. 2001, 907, 144–150. [Google Scholar] [CrossRef]
- Shaltiel, G.; Hanan, M.; Wolf, Y.; Barbash, S.; Kovalev, E.; Shoham, S.; Soreq, H. Hippocampal microRNA-132 mediates stress-inducible cognitive deficits through its acetylcholinesterase target. Brain Struct. Funct. 2013, 218, 59–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanieh, H.; Alzahrani, A. MicroRNA-132 suppresses autoimmune encephalomyelitis by inducing cholinergic anti-inflammation: A new Ahr-based exploration. Eur. J. Immunol. 2013, 43, 2771–2782. [Google Scholar]
- Mishra, N.; Friedson, L.; Hanin, G.; Bekenstein, U.; Volovich, M.; Bennett, E.R.; Greenberg, D.S.; Soreq, H. Antisense miR-132 blockade via the AChE-R splice variant mitigates cortical inflammation. Sci. Rep. 2017, 7, 42755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, F.; Li, Y.; Jiang, R.; Nie, C.; Zeng, Z.; Zhao, N.; Huang, C.; Shao, Q.; Ding, C.; Qing, C.; et al. miR-132 inhibits lipopolysaccharide-induced inflammation in alveolar macrophages by the cholinergic anti-inflammatory pathway. Exp. Lung Res. 2015, 41, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Shaked, I.; Meerson, A.; Wolf, Y.; Avni, R.; Greenberg, D.; Gilboa-Geffen, A.; Soreq, H. MicroRNA-132 potentiates cholinergic anti-inflammatory signaling by targeting acetylcholinesterase. Immunity 2009, 31, 965–973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, M.; Li, N.; Xu, J.; Wu, L.; Li, M.; Tong, H.; Wang, F.; Liu, W.; Feng, Y. Experimental study on the regulation of the cholinergic pathway in renal macrophages by microRNA-132 to alleviate inflammatory response. Open Chem. 2018, 16, 176. [Google Scholar] [CrossRef] [Green Version]
- Yang, F.W.; Wang, H.; Wang, C.; Chi, G.N. Upregulation of acetylcholinesterase caused by downregulation of microRNA-132 is responsible for the development of dementia after ischemic stroke. J. Cell. Biochem. 2020, 121, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Bond, C.E.; Zimmermann, M.; Greenfield, S.A. Upregulation of alpha7 Nicotinic Receptors by Acetylcholinesterase C-Terminal Peptides. PLoS ONE 2009, 4, e4846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Duysen, E.G.; Volpicelli-Daley, L.A.; Levey, A.I.; Lockridge, O. Regulation of muscarinic acetylcholine receptor function in acetylcholinesterase knockout mice. Pharmacol. Biochem. Behav. 2003, 74, 977–986. [Google Scholar] [CrossRef]
- Furukawa, S.; Sameshima, H.; Yang, L.; Ikenoue, T. Acetylcholine receptor agonist reduces brain damage induced by hypoxia-ischemia in newborn rats. Reprod. Sci. 2011, 18, 172–179. [Google Scholar] [CrossRef]
- Hirota, K.; Fukuda, R.; Takabuchi, S.; Kizaka-Kondoh, S.; Adachi, T.; Fukuda, K.; Semenza, G.L. Induction of hypoxia-inducible factor 1 activity by muscarinic acetylcholine receptor signaling. J. Biol. Chem. 2004, 279, 41521–41528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mou, L.; Gates, A.; Mosser, V.A.; Tobin, A.; Jackson, D.A. Transient hypoxia induces sequestration of M1 and M2 muscarinic acetylcholine receptors. J. Neurochem. 2006, 96, 510–519. [Google Scholar] [CrossRef] [PubMed]
- Sapozhnikova, T.A.; Borisevich, S.S.; Kireeva, D.R.; Gabdrakhmanova, S.F.; Khisamutdinova, R.Y.; Makara, N.S.; Gibadullina, N.N.; Khursan, S.L.; Zarudii, F.S. Effects of novel hexahydropyrimidine derivatives as potential ligands of M1 muscarinic acetylcholine receptor on cognitive function, hypoxia-induced lethality, and oxidative stress in rodents. Behav. Brain Res. 2019, 373, 112109. [Google Scholar] [CrossRef] [PubMed]
- Shibata, S.; Koutaroh, K.; Tominaga, K.; Tanaka, T.; Watanabe, S. Effect of muscarinic cholinergic drugs on ischemia-induced decreases in glucose uptake and CA1 field potentials in rat hippocampus slices. Eur. J. Pharmacol. 1992, 221, 113–119. [Google Scholar] [PubMed]
- Tohgi, H.; Utsugisawa, K.; Nagane, Y. Protective effect of nicotine through nicotinic acetylcholine receptor α7 on hypoxia-induced membrane disintegration and DNA fragmentation of cultured PC12 cells. Neurosci. Lett. 2000, 285, 91–94. [Google Scholar] [CrossRef]
- Maraula, G.; Traini, C.; Mello, T.; Coppi, E.; Galli, A.; Pedata, F.; Pugliese, A.M. Effects of oxygen and glucose deprivation on synaptic transmission in rat dentate gyrus: Role of A2A adenosine receptors. Neuropharmacology 2013, 67, 511–520. [Google Scholar] [CrossRef]
- Iadecola, C. The Neurovascular Unit Coming of Age: A Journey through Neurovascular Coupling in Health and Disease. Neuron 2017, 96, 17–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doolette, D.J.; Kerr, D.I.B. Hyperexcitability in CA1 of the rat hippocampal slice following hypoxia or adenosine. Brain Res. 1995, 677, 127–137. [Google Scholar] [CrossRef]
- Godukhin, O.; Savin, A.; Kalemenev, S.; Levin, S. Neuronal hyperexcitability induced by repeated brief episodes of hypoxia in rat hippocampal slices: Involvement of ionotropic glutamate receptors and L-type Ca(2+) channels. Neuropharmacology 2002, 42, 459–466. [Google Scholar] [CrossRef]
- Latini, S.; Bordoni, F.; Corradetti, R.; Pepeu, G.; Pedata, F. Effect of A2A adenosine receptor stimulation and antagonism on synaptic depression induced by in vitro ischaemia in rat hippocampal slices. Br. J. Pharmacol. 1999, 128, 1035–1044. [Google Scholar] [CrossRef] [Green Version]
- Aitken, P.G.; Schiff, S.J. Selective neuronal vulnerability to hypoxia in vitro. Neurosci. Lett. 1986, 67, 92–96. [Google Scholar] [CrossRef]
- Lalonde, C.C.; Mielke, J.G. Selective vulnerability of hippocampal sub-fields to oxygen-glucose deprivation is a function of animal age. Brain Res. 2014, 1543, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, H.; Mizuguchi, A.; Aoki, M. Differential responses between CA1 pyramidal cells and granule cells to ischemic insult in rat hippocampal slices. Neurosci. Lett. 1996, 203, 195–198. [Google Scholar] [CrossRef]
- Dengler, C.G.; Coulter, D.A. Normal and epilepsy-associated pathologic function of the dentate gyrus. Prog. Brain Res. 2016, 226, 155–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naylor, D. Changes in nonlinear signal processing in rat hippocampus associated with loss of paired-pulse inhibition or epileptogenesis. Epilepsia 2002, 43 (Suppl. 5), 188–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiff, S.J.; Somjen, G.G. Hyperexcitability following moderate hypoxia in hippocampal tissue slices. Brain Res. 1985, 337, 337–340. [Google Scholar] [CrossRef]
- Peng, B.W.; Justice, J.A.; He, X.H.; Sanchez, R.M. Decreased A-currents in hippocampal dentate granule cells after seizure-inducing hypoxia in the immature rat. Epilepsia. 2013, 54, 1223–1231. [Google Scholar] [CrossRef] [Green Version]
- Sarecka-Hujar, B.; Kopyta, I. Poststroke epilepsy: Current perspectives on diagnosis and treatment. Neuropsychiatr. Dis. Treat. 2018, 15, 95–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soreq, H.; Seidman, S. Acetylcholinesterase—New roles for an old actor. Nat. Rev. Neurosci. 2001, 2, 294–302. [Google Scholar] [CrossRef] [PubMed]
- Kathner-Schaffert, C.; Karapetow, L.; Günther, M.; Rudolph, M.; Dahab, M.; Baum, E.; Lehmann, T.; Witte, O.W.; Redecker, C.; Schmeer, C.W.; et al. Early Stroke Induces Long-Term Impairment of Adult Neurogenesis Accompanied by Hippocampal-Mediated Cognitive Decline. Cells 2019, 8, 1654. [Google Scholar] [CrossRef] [Green Version]
- Muthuraju, S.; Maiti, P.; Solanki, P.; Sharma, A.K.; Amitabh; Singh, S.B.; Prasad, D.; Ilavazhagan, G. Acetylcholinesterase inhibitors enhance cognitive functions in rats following hypobaric hypoxia. Behav. Brain Res. 2009, 203, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Muthuraju, S.; Maiti, P.; Pati, S.; Solanki, P.; Sharma, A.K.; Singh, S.B.; Prasad, D.; Ilavazhagan, G. Role of cholinergic markers on memory function of rats exposed to hypobaric hypoxia. Eur. J. Pharmacol. 2011, 672, 96–105. [Google Scholar] [CrossRef]
- Shirahata, M.; Hirasawa, S.; Okumura, M.; Mendoza, J.A.; Okumura, A.; Balbir, A.; Fitzgerald, R.S. Identification of M1 and M2 muscarinic acetylcholine receptors in the cat carotid body chemosensory system. Neuroscience 2004, 128, 635–644. [Google Scholar] [CrossRef]
- Yang, Y.; Ju, J.; Deng, M.; Wang, J.; Liu, H.; Xiong, L.; Zhang, J. Hypoxia Inducible Factor 1α Promotes Endogenous Adaptive Response in Rat Model of Chronic Cerebral Hypoperfusion. Int. J. Mol. Sci. 2017, 18, 3. [Google Scholar] [CrossRef] [Green Version]
- Choudhary, R.; Malairaman, U.; Katyal, A. Inhibition of 12/15 LOX ameliorates cognitive and cholinergic dysfunction in mouse model of hypobaric hypoxia via. attenuation of oxidative/nitrosative stress. Neuroscience 2017, 359, 308–324. [Google Scholar] [CrossRef]
- Aten, S.; Hansen, K.F.; Hoyt, K.R.; Obrietan, K. The miR-132/212 locus: A complex regulator of neuronal plasticity, gene expression and cognition. RNA Dis. 2016, 3, e1375. [Google Scholar] [PubMed]
- Xing, C.; Arai, K.; Lo, E.H.; Hommel, M. Pathophysiologic cascades in ischemic stroke. Int. J. Stroke 2012, 7, 378–385. [Google Scholar] [CrossRef] [PubMed]
- Sohrabji, F.; Selvamani, A. Sex differences in miRNA as therapies for ischemic stroke. Neurochem. Int. 2019, 127, 56–63. [Google Scholar] [CrossRef]
- Kiessling, S.; Ucar, A.; Chowdhury, K.; Oster, H.; Eichele, G. Genetic background-dependent effects of murine micro RNAs on circadian clock function. PLoS ONE 2017, 12, e0176547. [Google Scholar] [CrossRef]
- Lenz, M.; Vlachos, A.; Maggio, N. Ischemic long-term-potentiation (iLTP): Perspectives to set the threshold of neural plasticity toward therapy. Neural. Regen. Res. 2015, 10, 1537–1539. [Google Scholar] [CrossRef] [PubMed]
- Burek, M.; König, A.; Lang, M.; Fiedler, J.; Oerter, S.; Roewer, N.; Bohnert, M.; Thal, S.C.; Blecharz-Lang, K.G.; Woitzik, J.; et al. Hypoxia-Induced MicroRNA-212/132 Alter Blood-Brain Barrier Integrity Through Inhibition of Tight Junction-Associated Proteins in Human and Mouse Brain Microvascular Endothelial Cells. Transl. Stroke Res. 2019, 10, 672–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bormann, D.; Stojanovic, T.; Cicvaric, A.; Schuld, G.J.; Cabatic, M.; Ankersmit, H.J.; Monje, F.J. miRNA-132/212 Gene-Deletion Aggravates the Effect of Oxygen-Glucose Deprivation on Synaptic Functions in the Female Mouse Hippocampus. Cells 2021, 10, 1709. https://doi.org/10.3390/cells10071709
Bormann D, Stojanovic T, Cicvaric A, Schuld GJ, Cabatic M, Ankersmit HJ, Monje FJ. miRNA-132/212 Gene-Deletion Aggravates the Effect of Oxygen-Glucose Deprivation on Synaptic Functions in the Female Mouse Hippocampus. Cells. 2021; 10(7):1709. https://doi.org/10.3390/cells10071709
Chicago/Turabian StyleBormann, Daniel, Tamara Stojanovic, Ana Cicvaric, Gabor J. Schuld, Maureen Cabatic, Hendrik Jan Ankersmit, and Francisco J. Monje. 2021. "miRNA-132/212 Gene-Deletion Aggravates the Effect of Oxygen-Glucose Deprivation on Synaptic Functions in the Female Mouse Hippocampus" Cells 10, no. 7: 1709. https://doi.org/10.3390/cells10071709
APA StyleBormann, D., Stojanovic, T., Cicvaric, A., Schuld, G. J., Cabatic, M., Ankersmit, H. J., & Monje, F. J. (2021). miRNA-132/212 Gene-Deletion Aggravates the Effect of Oxygen-Glucose Deprivation on Synaptic Functions in the Female Mouse Hippocampus. Cells, 10(7), 1709. https://doi.org/10.3390/cells10071709