
Changes in Cerebrospinal Fluid Balance of TNF and TNF Receptors in Naïve Multiple Sclerosis Patients: Early Involvement in Compartmentalised Intrathecal Inflammation

 , , ,
, , ,  ,
,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. MS Patient Cohorts
2.2. Control Population
2.3. MRI Acquisition Protocol and Analysis
2.3.1. WM and CL Lesion Detection and Lesion Load Assessment
2.3.2. Cortical Thickness Evaluation and T1 Parcellation
2.4. Immunoassay CSF Protein Analysis
2.5. Bioinformatic Investigation and Pathway Analysis
2.6. Statistics
3. Results
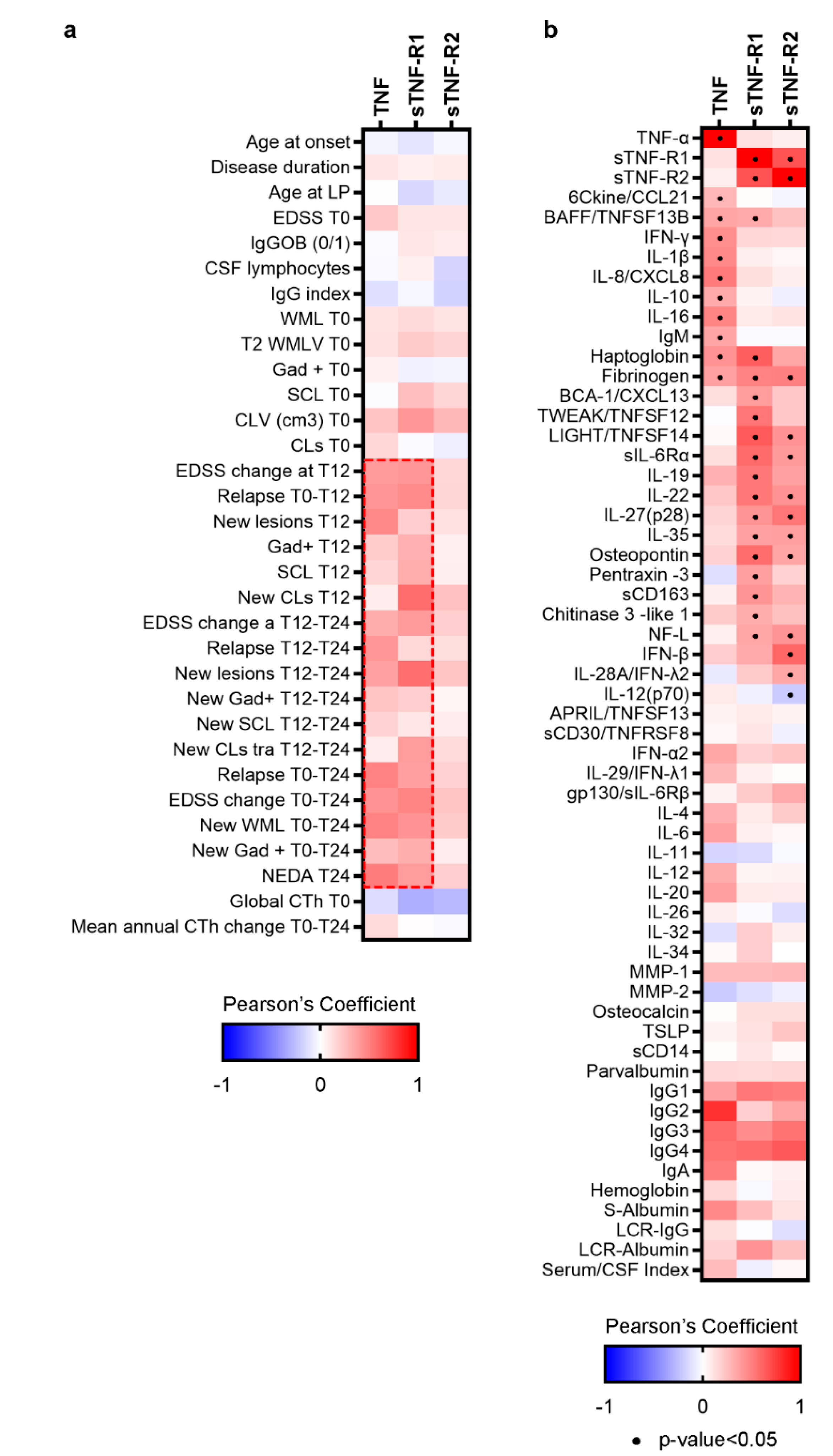
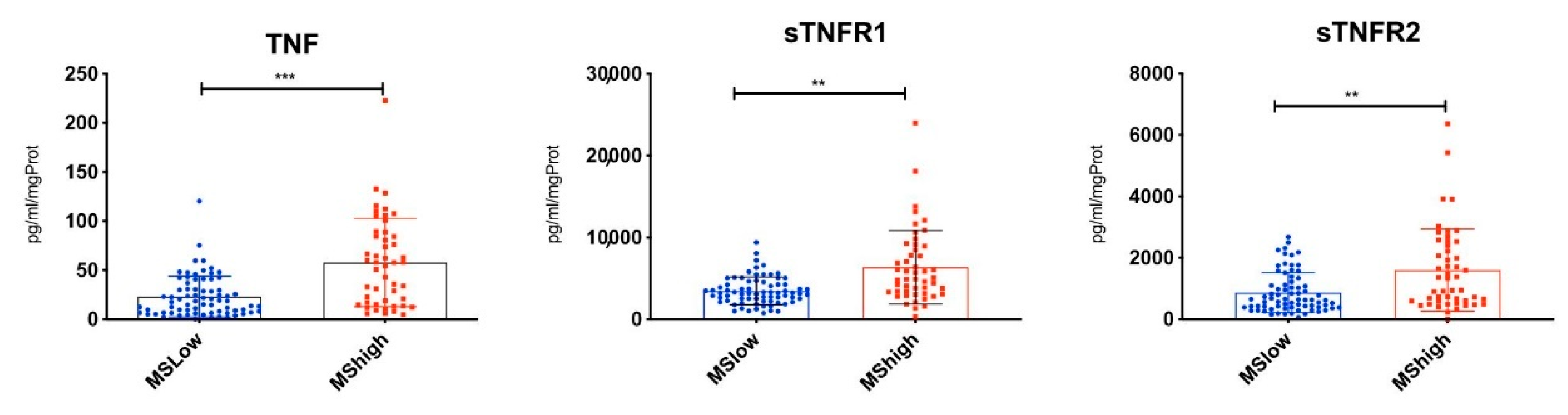
3.1. Differential TNF/TNFRs Protein Expression at Time of Diagnosis
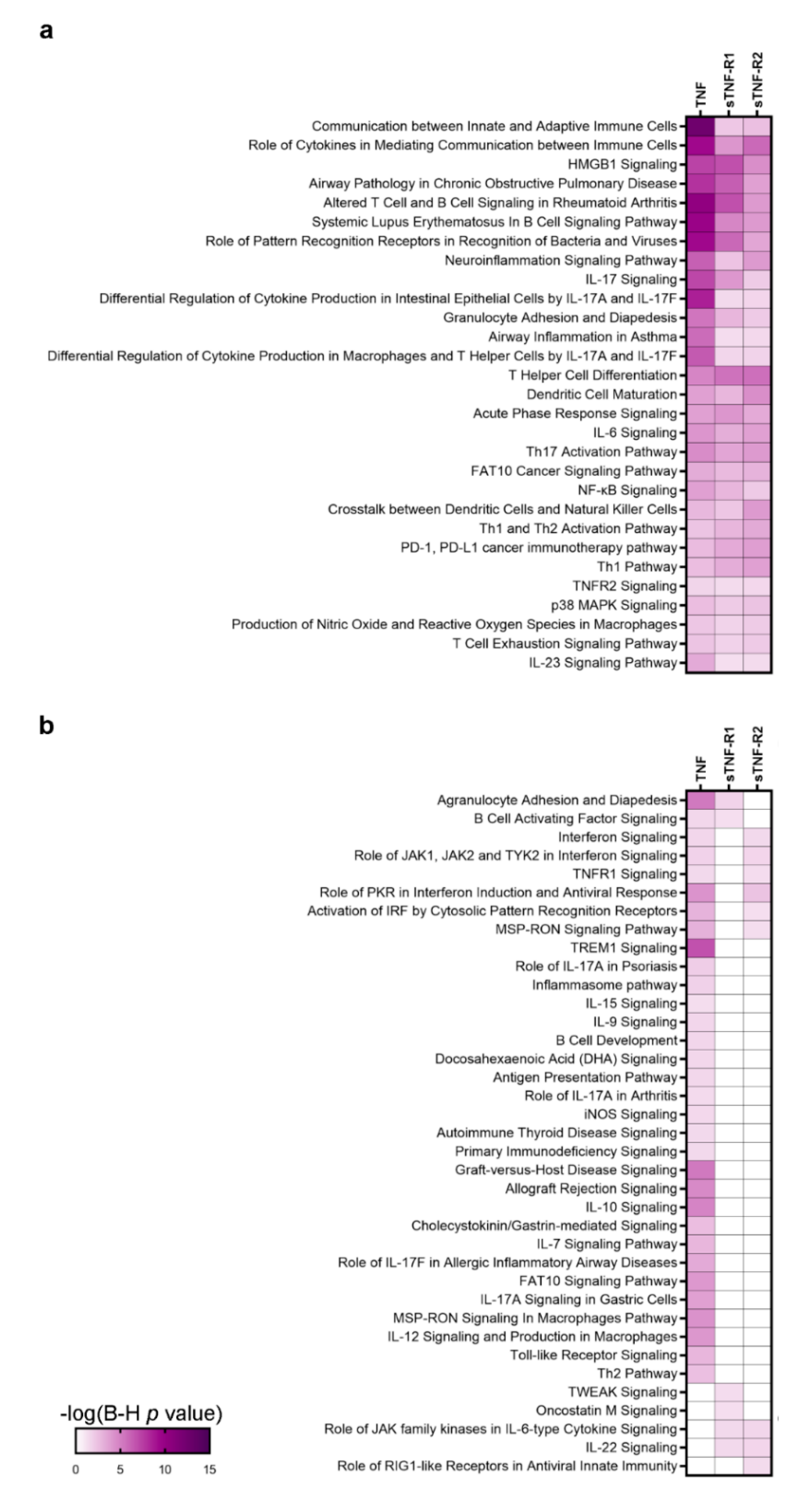
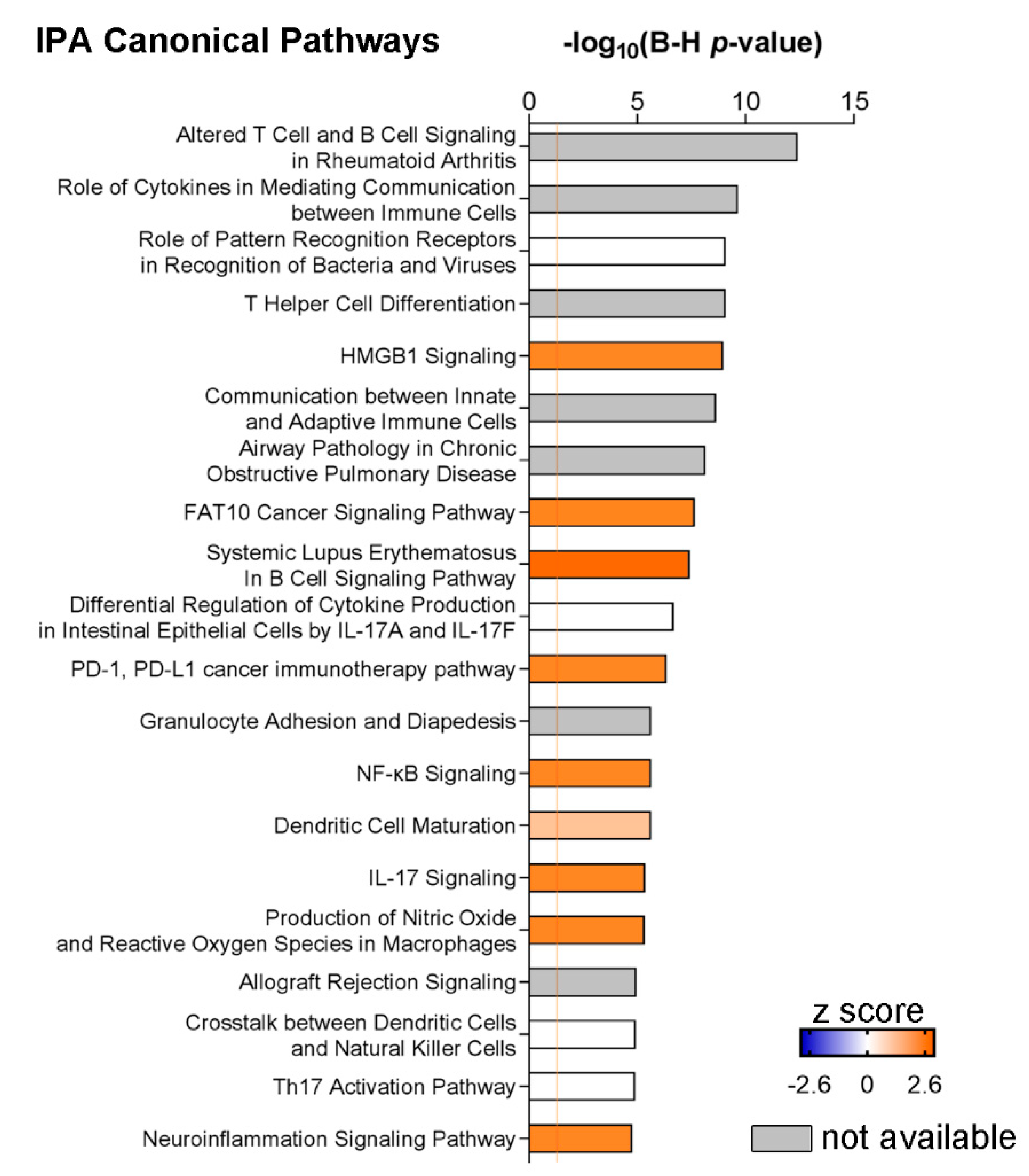
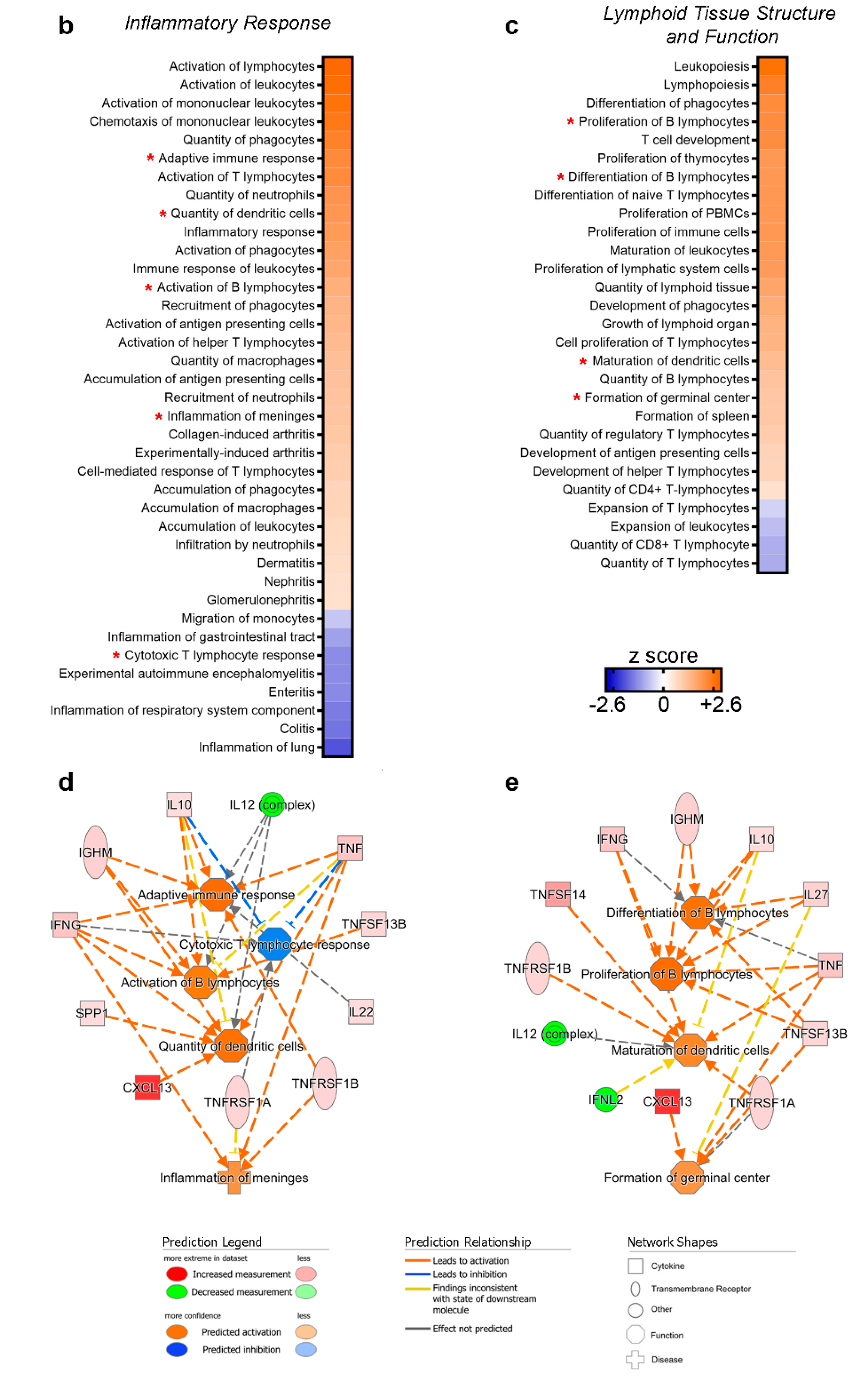
3.2. A Specific Pattern Related to Chronic Inflammation, Acute Phase Response, Dendritic Cells and Neuroinflammation Is Associated with CSF Over-Expression of TNF and sTNFRs
3.3. Highest CSF Levels of TNF, sTNFR1 and sTNFR2 Were Present in MS Patients with Increased Cortical Lesion Loads at the Time of Diagnosis
3.4. CSF Expression of TNF, sTNFR1 and sTNFR2 May Represent Surrogate Markers of Meningeal Inflammation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rossol, M.; Meusch, U.; Pierer, M.; Kaltenhäuser, S.; Häntzschel, H.; Hauschildt, S.; Wagner, U. Interaction between Transmembrane TNF and TNFR1/2 Mediates the Activation of Monocytes by Contact with T Cells. J. Immunol. 2007, 179, 4239–4248. [Google Scholar] [CrossRef] [Green Version]
- Robinson, W.H.; Genovese, M.C.; Moreland, L.W. Demyelinating and neurologic events reported in association with tumor necrosis factor α antagonism: By what mechanisms could tumor necrosis factor α antagonists improve rheumatoid arthritis but exacerbate multiple sclerosis? Arthritis Rheum. 2001, 44, 1977–1983. [Google Scholar] [CrossRef]
- Probert, L. TNF and its receptors in the CNS: The essential, the desirable and the deleterious effects. Neuroscience 2015, 302, 2–22. [Google Scholar] [CrossRef] [Green Version]
- Dopp, J.M.; Sarafian, T.A.; Spinella, F.M.; Kahn, M.A.; Shau, H.; de Vellis, J. Expression of the p75 TNF receptor is linked to TNF-induced NFkappaB translocation and oxyradical neutralization in glial cells. Neurochem. Res. 2002, 27, 1535–1542. [Google Scholar] [CrossRef]
- Pegoretti, V.; Baron, W.; Laman, J.D.; Eisel, U.L.M. Selective Modulation of TNF–TNFRs Signaling: Insights for Multiple Sclerosis Treatment. Front. Immunol. 2018, 9, 925. [Google Scholar] [CrossRef] [PubMed]
- Magliozzi, R.; Howell, O.W.; Nicholas, R.; Cruciani, C.; Castellaro, M.; Romualdi, C.; Rossi, S.; Pitteri, M.; Benedetti, M.D.; Gajofatto, A.; et al. Inflammatory intrathecal profiles and cortical damage in multiple sclerosis. Ann. Neurol. 2018, 83, 739–755. [Google Scholar] [CrossRef]
- Lepennetier, G.; Hracsko, Z.; Unger, M.; Van Griensven, M.; Grummel, V.; Krumbholz, M.; Berthele, A.; Hemmer, B.; Kowarik, M.C. Cytokine and immune cell profiling in the cerebrospinal fluid of patients with neuro-inflammatory diseases. J. Neuroinflammation 2019, 16, 219. [Google Scholar] [CrossRef] [PubMed]
- Magliozzi, R.; Howell, O.W.; Durrenberger, P.; Aricò, E.; James, R.; Cruciani, C.; Reeves, C.; Roncaroli, F.; Nicholas, R.; Reynolds, R. Meningeal inflammation changes the balance of TNF signalling in cortical grey matter in multiple sclerosis. J. Neuroinflammation 2019, 16, 259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- James, R.E.; Schalks, R.; Browne, E.; Eleftheriadou, I.; Munoz, C.P.; Mazarakis, N.D.; Reynolds, R. Persistent elevation of intrathecal pro-inflammatory cytokines leads to multiple sclerosis-like cortical demyelination and neurodegeneration. Acta Neuropathol. Commun. 2020, 8, 66. [Google Scholar] [CrossRef]
- Picon, C.; Jayaraman, A.; James, R.; Beck, C.; Gallego, P.; Witte, M.E.; van Horssen, J.; Mazarakis, N.D.; Reynolds, R. Neuron-specific activation of necroptosis signaling in multiple sclerosis cortical grey matter. Acta Neuropathol. 2021, 141, 585–604. [Google Scholar] [CrossRef] [PubMed]
- Gardner, C.; Magliozzi, R.; Durrenberger, P.F.; Howell, O.W.; Rundle, J.; Reynolds, R. Cortical grey matter demyelination can be induced by elevated pro-inflammatory cytokines in the subarachnoid space of MOG-immunized rats. Brain 2013, 136, 3596–3608. [Google Scholar] [CrossRef] [Green Version]
- Magliozzi, R.; Scalfari, A.; Pisani, A.I.; Ziccardi, S.; Marastoni, D.; Pizzini, F.B.; Bajrami, A.; Tamanti, A.; Guandalini, M.; Bonomi, S.; et al. The CSF Profile Linked to Cortical Damage Predicts Multiple Sclerosis Activity. Ann. Neurol. 2020, 88, 562–573. [Google Scholar] [CrossRef] [PubMed]
- Polman, C.H.; Reingold, S.C.; Banwell, B.; Clanet, M.; Cohen, J.A.; Filippi, M.; Fujihara, K.; Havrdova, E.; Hutchinson, M.; Kappos, L.; et al. Diagnostic criteria for multiple sclerosis: 2010 Revisions to the McDonald criteria. Ann. Neurol. 2011, 69, 292–302. [Google Scholar] [CrossRef] [Green Version]
- Calabrese, M.; Castellaro, M. Cortical Gray Matter MR Imaging in Multiple Sclerosis. Neuroimaging Clin. N. Am. 2017, 27, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Geurts, J.J.G.G.; Roosendaal, S.D.; Calabrese, M.; Ciccarelli, O.; Agosta, F.; Chard, D.T.; Gass, A.; Huerga, E.; Moraal, B.; Pareto, D.; et al. Consensus recommendations for MS cortical lesion scoring using double inversion recovery MRI. Neurology 2011, 76, 418–424. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, M.; Battaglini, M.; Giorgio, A.; Atzori, M.; Bernardi, V.; Mattisi, I.; Gallo, P.; De Stefano, N. Imaging distribution and frequency of cortical lesions in patients with multiple sclerosis. Neurology 2010, 75, 1234–1240. [Google Scholar] [CrossRef]
- Roura, E.; Oliver, A.; Cabezas, M.; Valverde, S.; Pareto, D.; Vilanova, J.C.; Ramió-Torrentà, L.; Rovira, À.; Lladó, X. A toolbox for multiple sclerosis lesion segmentation. Neuroradiology 2015, 57, 1031–1043. [Google Scholar] [CrossRef]
- Wang, H.; Yushkevich, P.A. Multi-atlas segmentation with joint label fusion and corrective learning—An open source implementation. Front. Neuroinform. 2013, 7, 27. [Google Scholar] [CrossRef] [Green Version]
- Teunissen, C.E.; Petzold, A.; Bennett, J.L.; Berven, F.S.; Brundin, L.; Comabella, M.; Franciotta, D.; Frederiksen, J.L.; Fleming, J.O.; Furlan, R.; et al. A consensus protocol for the standardization of cerebrospinal fluid collection and biobanking. Neurology 2009, 73, 1914–1922. [Google Scholar] [CrossRef] [Green Version]
- Magliozzi, R.; Hametner, S.; Facchiano, F.; Marastoni, D.; Rossi, S.; Castellaro, M.; Poli, A.; Lattanzi, F.; Visconti, A.; Nicholas, R.; et al. Iron homeostasis, complement, and coagulation cascade as CSF signature of cortical lesions in early multiple sclerosis. Ann. Clin. Transl. Neurol. 2019, 6, 2150–2163. [Google Scholar] [CrossRef] [Green Version]
- Pezzini, F.; Bianchi, M.; Benfatto, S.; Griggio, F.; Doccini, S.; Carrozzo, R.; Dapkunas, A.; Delledonne, M.; Santorelli, F.M.; Lalowski, M.M.; et al. The Networks of Genes Encoding Palmitoylated Proteins in Axonal and Synaptic Compartments Are Affected in PPT1 Overexpressing Neuronal-Like Cells. Front. Mol. Neurosci. 2017, 10, 266. [Google Scholar] [CrossRef]
- Brambilla, R.; Ashbaugh, J.J.; Magliozzi, R.; Dellarole, A.; Karmally, S.; Szymkowski, D.E.; Bethea, J.R. Inhibition of soluble tumour necrosis factor is therapeutic in experimental autoimmune encephalomyelitis and promotes axon preservation and remyelination. Brain 2011, 134, 2736–2754. [Google Scholar] [CrossRef] [PubMed]
- Taoufik, E.; Tseveleki, V.; Chu, S.Y.; Tselios, T.; Karin, M.; Lassmann, H.; Szymkowski, D.E.; Probert, L. Transmembrane tumour necrosis factor is neuroprotective and regulates experimental autoimmune encephalomyelitis via neuronal nuclear factor-κB. Brain 2011, 134, 2722–2735. [Google Scholar] [CrossRef] [PubMed]
- Williams, S.K.; Maier, O.; Fischer, R.; Fairless, R.; Hochmeister, S.; Stojic, A.; Pick, L.; Haar, D.; Musiol, S.; Storch, M.K.; et al. Antibody-Mediated Inhibition of TNFR1 Attenuates Disease in a Mouse Model of Multiple Sclerosis. PLoS ONE 2014, 9, e90117. [Google Scholar] [CrossRef] [PubMed]
- Madsen, P.M.; Motti, D.; Karmally, S.; Szymkowski, D.E.; Lambertsen, K.L.; Bethea, J.R.; Brambilla, R. Oligodendroglial TNFR2 mediates membrane TNF-dependent repair in experimental autoimmune encephalomyelitis by promoting oligodendrocyte differentiation and remyelination. J. Neurosci. 2016, 36, 5128–5143. [Google Scholar] [CrossRef] [Green Version]
- Rossi, S.; Motta, C.; Studer, V.; Barbieri, F.; Buttari, F.; Bergami, A.; Sancesario, G.; Bernardini, S.; De Angelis, G.; Martino, G.; et al. Tumor necrosis factor is elevated in progressive multiple sclerosis and causes excitotoxic neurodegeneration. Mult. Scler. J. 2014, 20, 304–312. [Google Scholar] [CrossRef]
- Donninelli, G.; Studer, V.; Brambilla, L.; Zecca, C.; Peluso, D.; Laroni, A.; Michelis, D.; Mantegazza, R.; Confalonieri, P.; Volpe, E. Immune Soluble Factors in the Cerebrospinal Fluid of Progressive Multiple Sclerosis Patients Segregate Into Two Groups. Front. Immunol. 2021, 12, 428. [Google Scholar] [CrossRef]
- Bai, Z.; Chen, D.; Wang, L.; Zhao, Y.; Liu, T.; Yu, Y.; Yan, T.; Cheng, Y. Cerebrospinal Fluid and Blood Cytokines as Biomarkers for Multiple Sclerosis: A Systematic Review and Meta-Analysis of 226 Studies With 13,526 Multiple Sclerosis Patients. Front. Neurosci. 2019, 13, 1026. [Google Scholar] [CrossRef]
- Rincón-Arévalo, H.; Sanchez-Parra, C.C.; Castaño, D.; Yassin, L.; Vásquez, G. Regulatory B Cells and Mechanisms. Int. Rev. Immunol. 2016, 35, 156–176. [Google Scholar] [CrossRef]
- Shen, P.; Roch, T.; Lampropoulou, V.; O’Connor, R.A.; Stervbo, U.; Hilgenberg, E.; Ries, S.; Dang, V.D.; Jaimes, Y.; Daridon, C.; et al. IL-35-producing B cells are critical regulators of immunity during autoimmune and infectious diseases. Nature 2014, 507, 366–370. [Google Scholar] [CrossRef] [Green Version]
- van Oosten, B.W.; Barkhof, F.; Truyen, L.; Boringa, J.B.; Bertelsmann, F.W.; von Blomberg, B.M.E.; Woody, J.N.; Hartung, H.-P.; Polman, C.H. Increased MRI activity and immune activation in two multiple sclerosis patients treated with the monoclonal anti-tumor necrosis factor antibody cA2. Neurology 1996, 47, 1531–1534. [Google Scholar] [CrossRef] [PubMed]
- Arnason, B.G.W. TNF neutralization in MS: Results of a randomized, placebo-controlled multicenter study. Neurology 1999, 53, 457. [Google Scholar] [CrossRef] [Green Version]
- Seror, R.; Richez, C.; Sordet, C.; Rist, S.; Gossec, L.; Direz, G.; Houvenagel, E.; Berthelot, J.-M.; Pagnoux, C.; Dernis, E.; et al. Pattern of demyelination occurring during anti-TNF-α therapy: A French national survey. Rheumatology 2013, 52, 868–874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ronin, E.; Pouchy, C.; Khosravi, M.; Hilaire, M.; Grégoire, S.; Casrouge, A.; Kassem, S.; Sleurs, D.; Martin, G.H.; Chanson, N.; et al. Tissue-restricted control of established central nervous system autoimmunity by TNF receptor 2–expressing Treg cells. Proc. Natl. Acad. Sci. USA 2021, 118, e2014043118. [Google Scholar] [CrossRef]
- Suvannavejh, G.C.; Lee, H.O.; Padilla, J.; Dal Canto, M.C.; Barrett, T.A.; Miller, S.D. Divergent roles for p55 and p75 tumor necrosis factor receptors in the pathogenesis of MOG35-55-induced experimental autoimmune encephalomyelitis. Cell. Immunol. 2000, 205, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Arnett, H.A.; Mason, J.; Marino, M.; Suzuki, K.; Matsushima, G.K.; Ting, J.P.Y. TNFα promotes proliferation of oligodendrocyte progenitors and remyelination. Nat. Neurosci. 2001, 4, 1116–1122. [Google Scholar] [CrossRef]
- Fresegna, D.; Bullitta, S.; Musella, A.; Rizzo, F.R.; De Vito, F.; Guadalupi, L.; Caioli, S.; Balletta, S.; Sanna, K.; Dolcetti, E.; et al. Re-Examining the Role of TNF in MS Pathogenesis and Therapy. Cells 2020, 9, 2290. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mean | Std. Deviation | |
|---|---|---|
| Age at onset | 35.36 | 12.24 |
| Age at LP | 37.78 | 12.71 |
| EDSS T0 | 1.83 | 1.06 |
| T2 WM lesions | 8.29 | 3.76 |
| T2WM lesions volume | 835.10 | 502.70 |
| Gad + lesions T0 | 0.13 | 0.48 |
| Spinal cord lesions T0 | 0.51 | 0.96 |
| CLs volume (cm3) T0 | 215.60 | 297.80 |
| CLs number T0 | 7.72 | 5.90 |
| EDSS change a T12 | 0.22 | 0.38 |
| Relapse T0-T12 | 0.26 | 0.48 |
| New T2 WM lesions T12 | 0.79 | 1.36 |
| New Gad+ lesions T12 | 0.29 | 0.61 |
| New spinal cord lesions T12 | 0.14 | 0.38 |
| New CLs T12 | 0.88 | 1.48 |
| EDSS change T12-T24 | 0.27 | 0.45 |
| Relapse T12-T24 | 0.20 | 0.43 |
| New T2 WM lesions T12-T24 | 0.70 | 1.12 |
| NewGad+ lesions T12-T24 | 0.18 | 0.44 |
| New spinal cord lesions T12-T24 | 0.14 | 0.34 |
| New CLs T12-T24 | 0.82 | 1.31 |
| Relapse T0-T24 | 0.44 | 0.76 |
| EDSS change T0-T24 | 0.45 | 0.70 |
| New T2 WM lesionsT0-T24 | 1.69 | 2.37 |
| New Gad+ lesions T0-T24 | 0.46 | 0.85 |
| Molecule | MS-High | MS-Low | Fold Change | ||
|---|---|---|---|---|---|
| Mean | StDev | Mean | StDev | ||
| TNF-α *** | 57.72 | 25.37 | 23.21 | 20.74 | 2.49 |
| sTNF-R1 ** | 6367.02 | 545.69 | 3465.76 | 1698.50 | 1.84 |
| sTNF-R2 ** | 1601.67 | 357.31 | 866.46 | 652.85 | 1.85 |
| BAFF/ TNFSF13B ** | 10,926.91 | 760.80 | 9049.70 | 6004.98 | 1.21 |
| LIGHT/ TNFSF14 ** | 663.90 | 105.88 | 156.56 | 256.93 | 4.24 |
| IFN-γ ** | 24.77 | 9.33 | 9.85 | 14.12 | 2.51 |
| BCA-1/ CXCL13 *** | 27.77 | 3.81 | 3.17 | 4.87 | 8.75 |
| IL-10 ** | 22.19 | 3.22 | 18.04 | 17.12 | 1.23 |
| IL-22 * | 52.64 | 26.72 | 33.62 | 36.19 | 1.57 |
| IL-27(p28) * | 271.39 | 51.76 | 139.56 | 171.48 | 1.94 |
| Osteopontin/SPP1 ** | 108,631.45 | 10,203.80 | 70,400.60 | 81,511.52 | 1.54 |
| Pentraxin-3 * | 529.91 | 72.04 | 282.65 | 414.24 | 1.87 |
| sCD163 ** | 55,404.70 | 2111.60 | 44,373.96 | 35,703.43 | 1.24 |
| Chitinase 3-like 1 ** | 52,758.42 | 4285.75 | 37,362.73 | 30,994.71 | 1.41 |
| IgM * | 707.97 | 61.54 | 346.19 | 338.14 | 2.05 |
| Fibrinogen * | 1483.38 | 184.83 | 941.78 | 6.64 | 1.58 |
| NF-L * | 2.82 | 0.68 | 1.62 | 0.95 | 1.74 |
| IL-28A/IFN-λ2 * | 243.89 | 67.45 | 350.47 | 91.71 | 0.57 |
| IL-12(p70) * | 9.00 | 3.87 | 13.81 | 2.53 | 0.62 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Magliozzi, R.; Pezzini, F.; Pucci, M.; Rossi, S.; Facchiano, F.; Marastoni, D.; Montagnana, M.; Lippi, G.; Reynolds, R.; Calabrese, M. Changes in Cerebrospinal Fluid Balance of TNF and TNF Receptors in Naïve Multiple Sclerosis Patients: Early Involvement in Compartmentalised Intrathecal Inflammation. Cells 2021, 10, 1712. https://doi.org/10.3390/cells10071712
Magliozzi R, Pezzini F, Pucci M, Rossi S, Facchiano F, Marastoni D, Montagnana M, Lippi G, Reynolds R, Calabrese M. Changes in Cerebrospinal Fluid Balance of TNF and TNF Receptors in Naïve Multiple Sclerosis Patients: Early Involvement in Compartmentalised Intrathecal Inflammation. Cells. 2021; 10(7):1712. https://doi.org/10.3390/cells10071712
Chicago/Turabian StyleMagliozzi, Roberta, Francesco Pezzini, Mairi Pucci, Stefania Rossi, Francesco Facchiano, Damiano Marastoni, Martina Montagnana, Giuseppe Lippi, Richard Reynolds, and Massimiliano Calabrese. 2021. "Changes in Cerebrospinal Fluid Balance of TNF and TNF Receptors in Naïve Multiple Sclerosis Patients: Early Involvement in Compartmentalised Intrathecal Inflammation" Cells 10, no. 7: 1712. https://doi.org/10.3390/cells10071712
APA StyleMagliozzi, R., Pezzini, F., Pucci, M., Rossi, S., Facchiano, F., Marastoni, D., Montagnana, M., Lippi, G., Reynolds, R., & Calabrese, M. (2021). Changes in Cerebrospinal Fluid Balance of TNF and TNF Receptors in Naïve Multiple Sclerosis Patients: Early Involvement in Compartmentalised Intrathecal Inflammation. Cells, 10(7), 1712. https://doi.org/10.3390/cells10071712