
TNFR2 Signaling Regulates the Immunomodulatory Function of Oligodendrocyte Precursor Cells

 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Primary Oligodendrocyte Cultures and Cytokine Stimulation
2.3. Immunocytochemistry
2.4. Flow Cytometry
2.5. Next Generation RNAseq
2.6. Ligand–Receptor Interaction Analysis
2.7. Real-Time RT-PCR
2.8. Western Blot
2.9. Cytokine Array
2.10. Statistical Analysis
3. Results
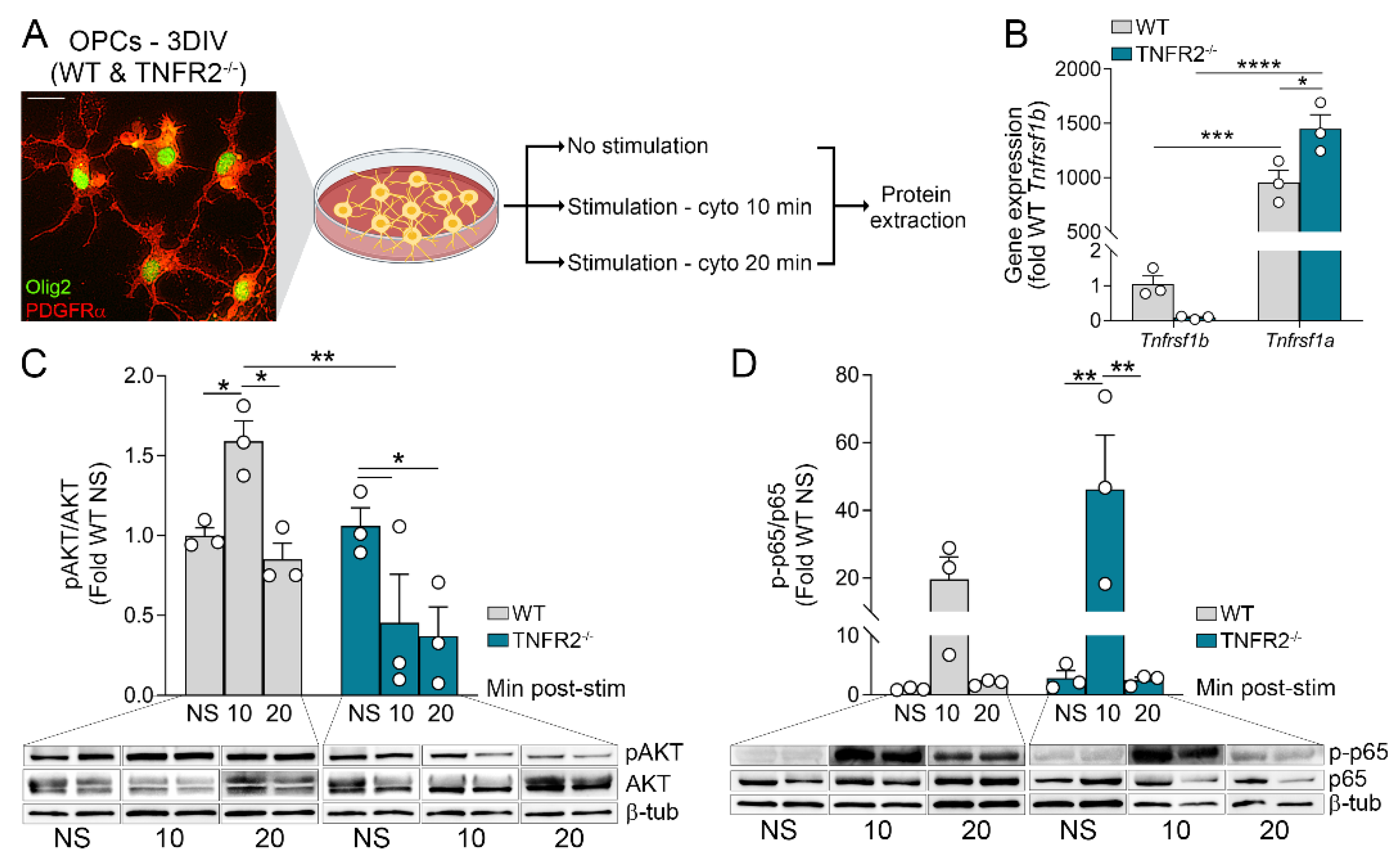
3.1. OPCs Upregulate and Activate TNF Receptors Following Inflammatory Stimulation
3.2. TNFR2 Participates in the Response of OPCs to Inflammatory Conditions
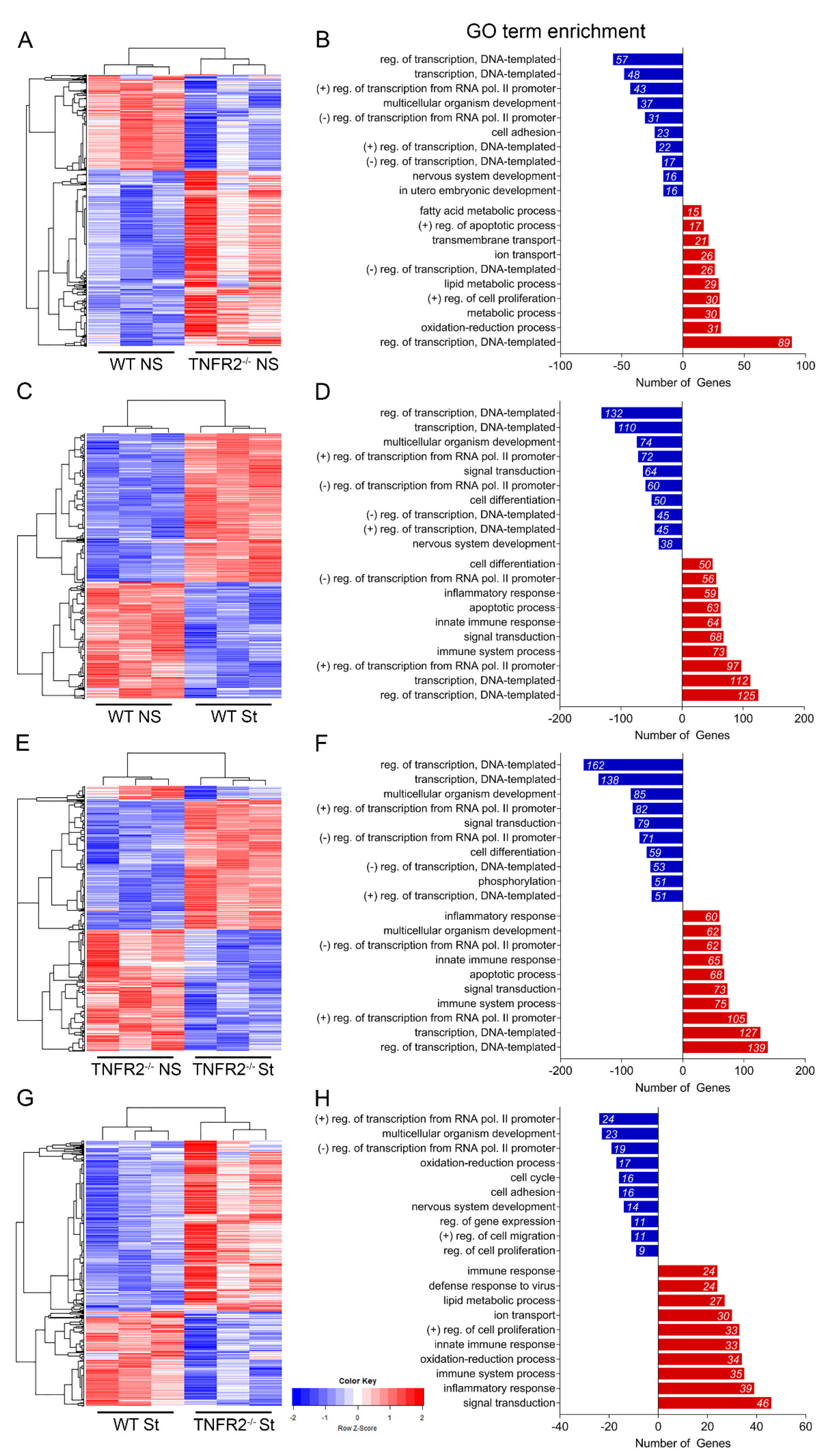
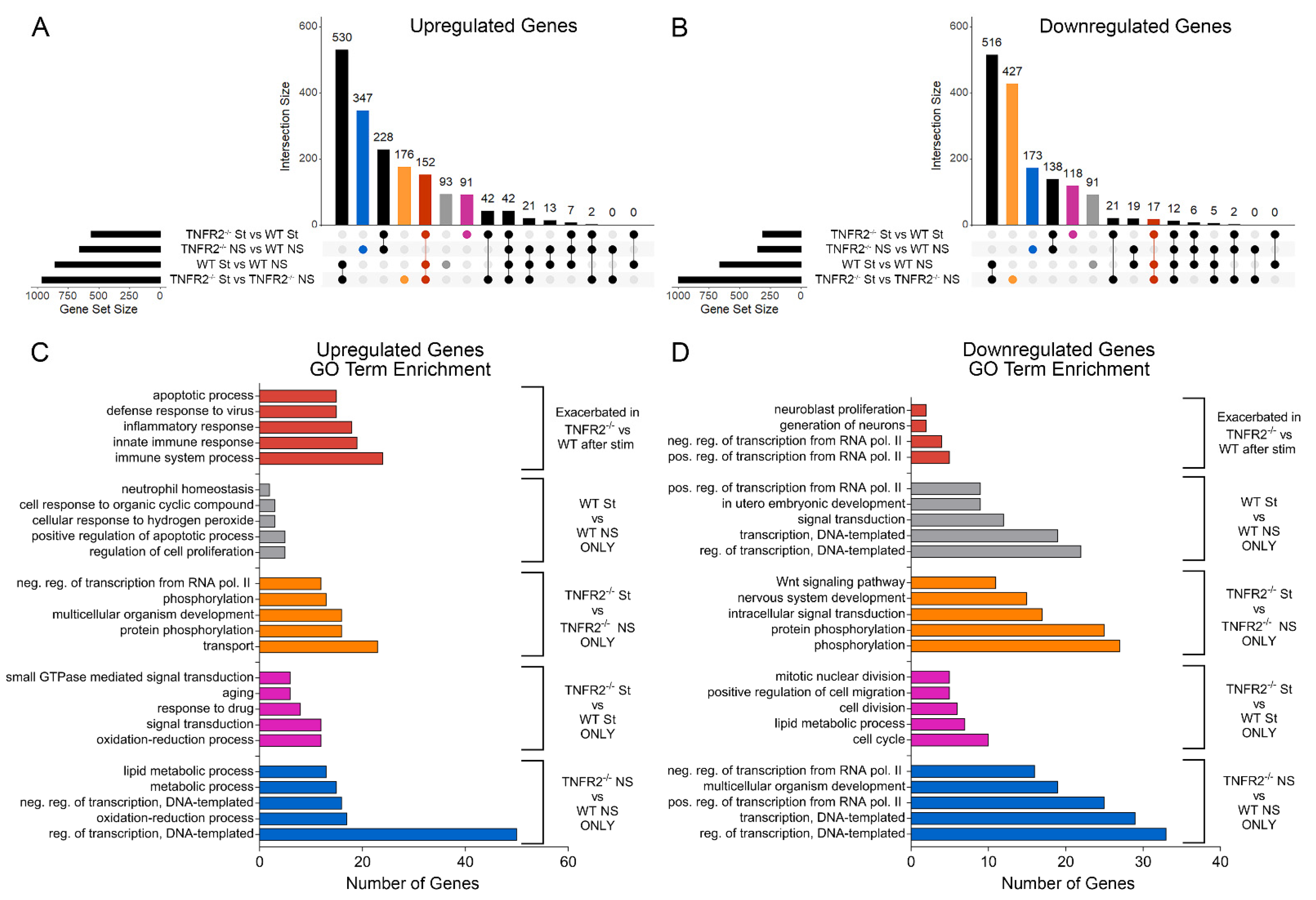
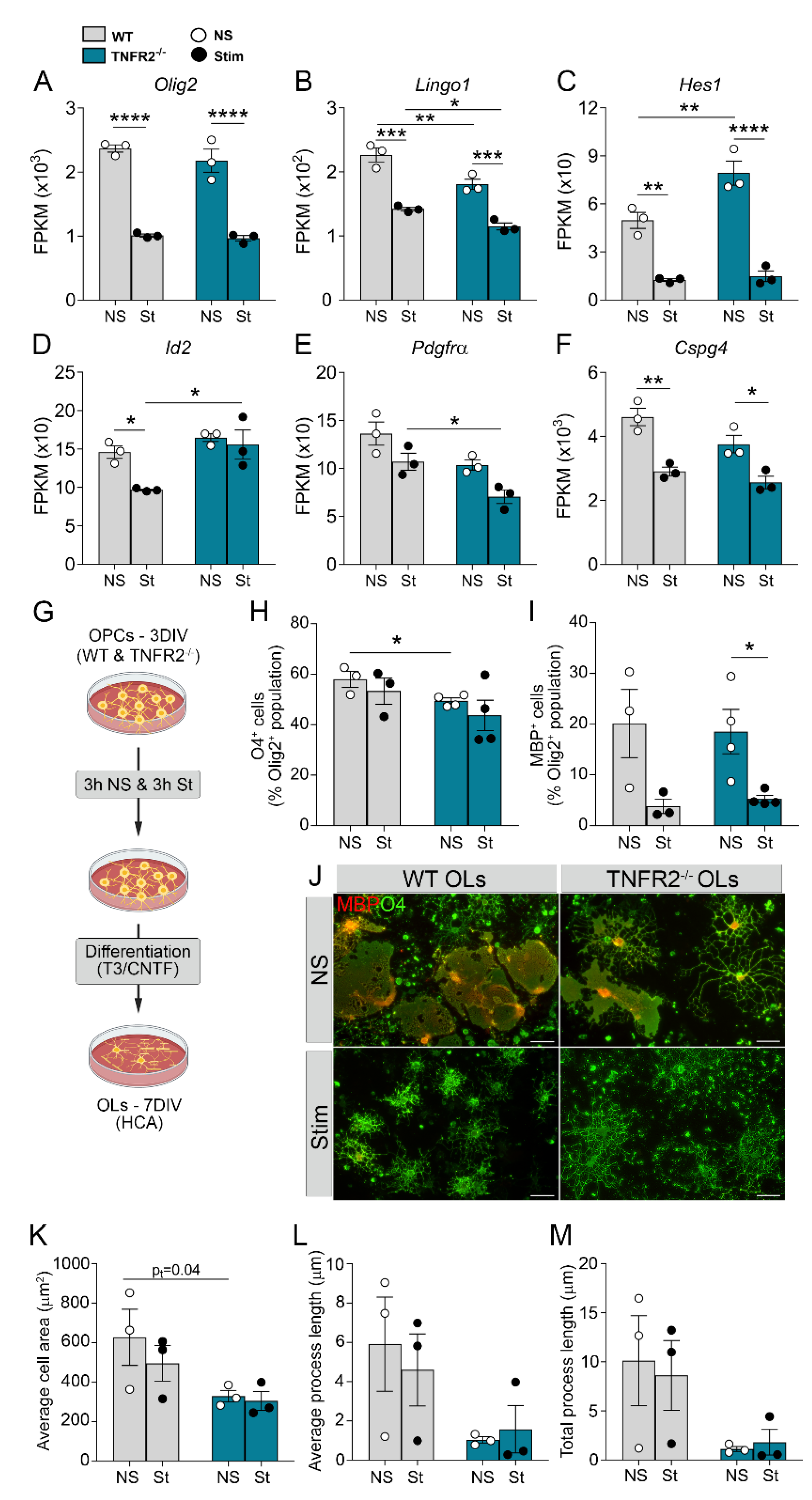
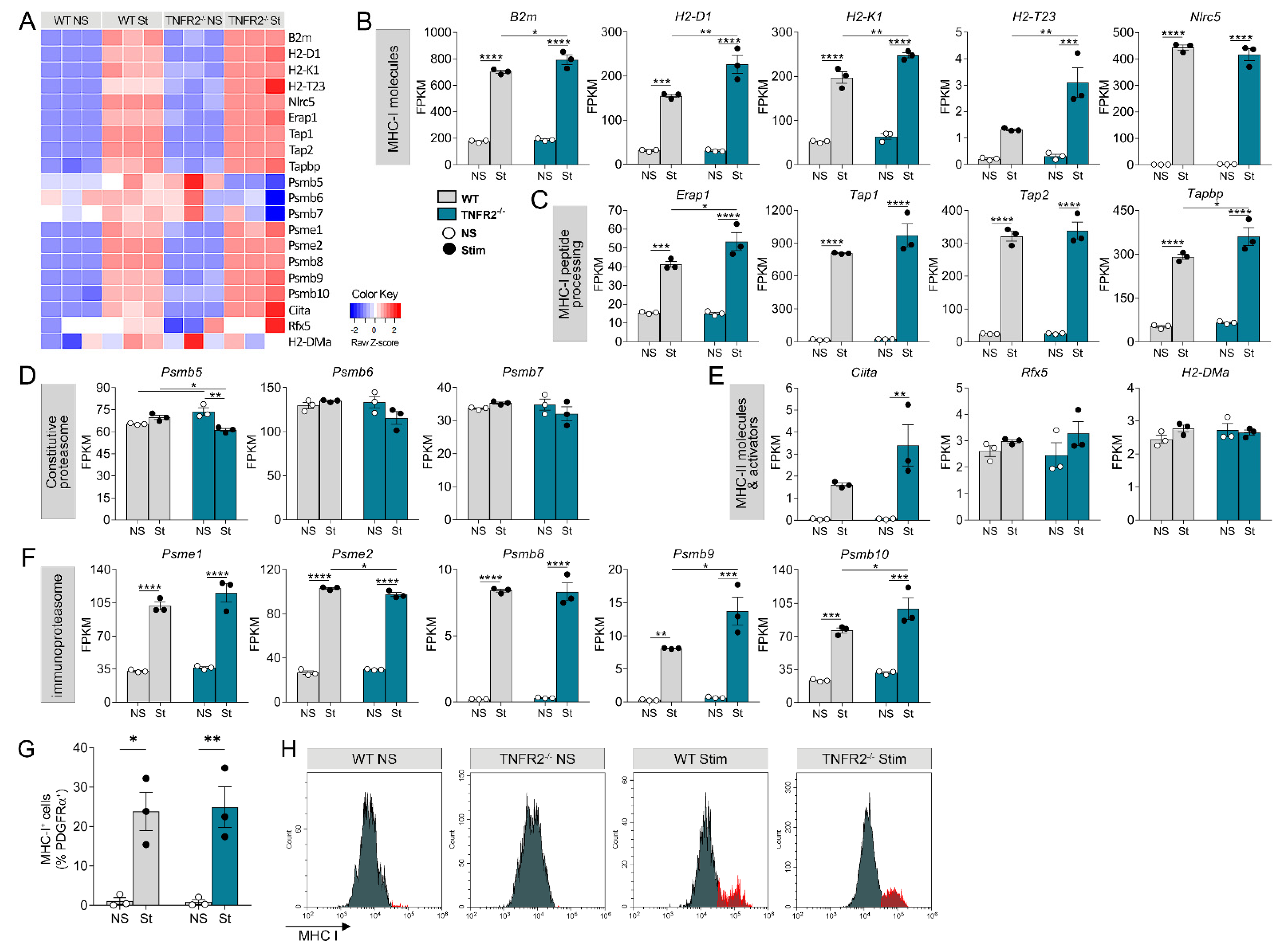
3.3. TNFR2 Ablation Exacerbates Inflammation-Induced Dysregulation of the Cellular Machinery That Sustains OPC Proliferation and Differentiation
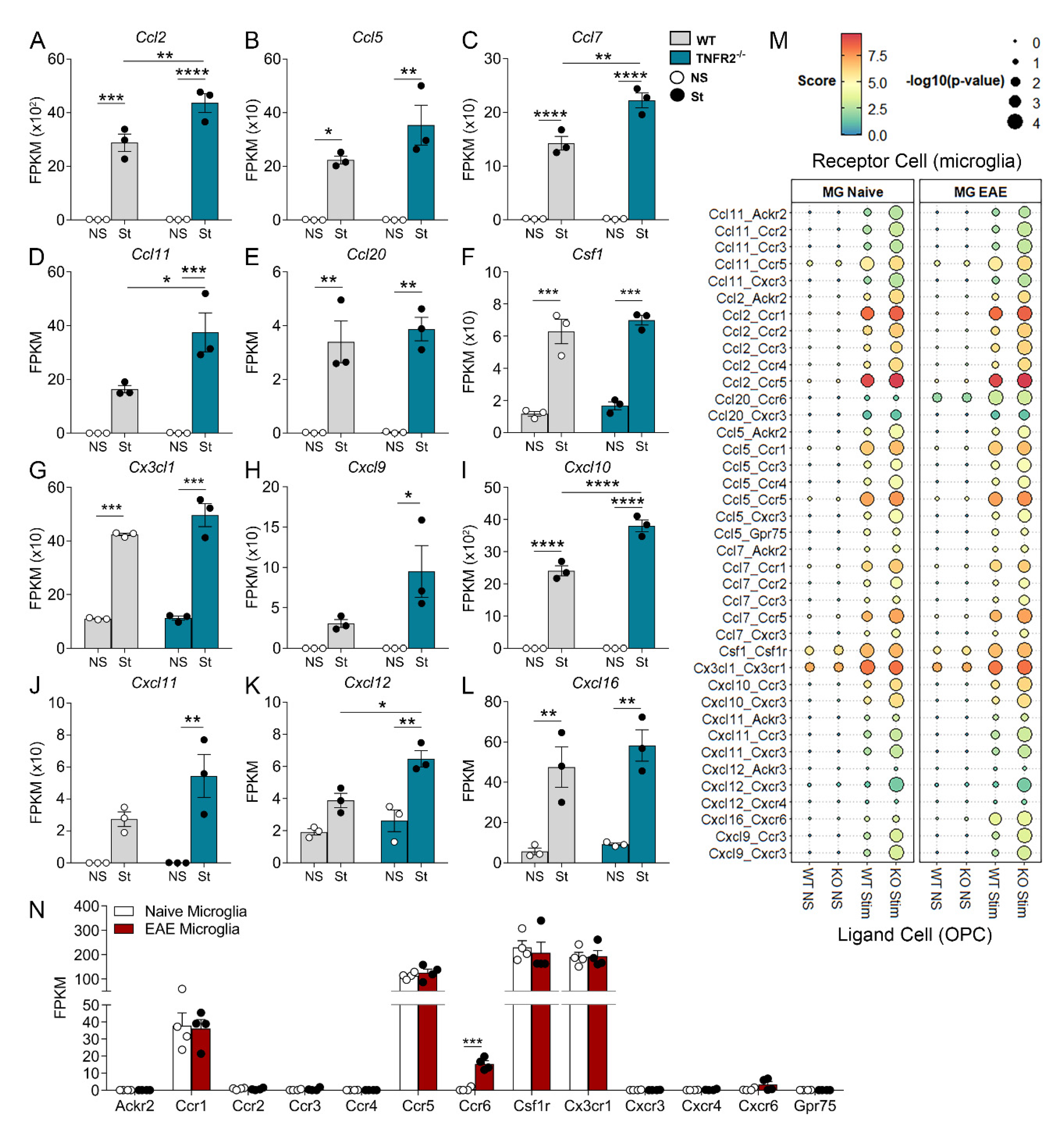
3.4. TNFR2 Ablation Enhances the Immunomodulatory and Inflammatory Function of OPCs in Response to Inflammatory Stimulation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Thompson, A.J.; E Baranzini, S.; Geurts, J.; Hemmer, B.; Ciccarelli, O. Multiple sclerosis. Lancet 2018, 391, 1622–1636. [Google Scholar] [CrossRef]
- Kuhlmann, T.; Ludwin, S.; Prat, A.; Antel, J.; Brück, W.; Lassmann, H. An updated histological classification system for multiple sclerosis lesions. Acta Neuropathol. 2017, 133, 13–24. [Google Scholar] [CrossRef]
- Sørensen, T.L.; Tani, M.; Jensen, J.; Pierce, V.; Lucchinetti, C.; Folcik, V.A.; Qin, S.; Rottman, J.; Sellebjerg, F.; Strieter, R.M.; et al. Expression of specific chemokines and chemokine receptors in the central nervous system of multiple sclerosis patients. J. Clin. Investig. 1999, 103, 807–815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, L.-Y.; Chu, S.-F.; Chen, N.-H. The role of chemokines and chemokine receptors in multiple sclerosis. Int. Immunopharmacol. 2020, 83, 106314. [Google Scholar] [CrossRef]
- McGinley, M.P.; Goldschmidt, C.H.; Rae-Grant, A.D. Diagnosis and Treatment of Multiple Sclerosis. JAMA 2021, 325, 765–779. [Google Scholar] [CrossRef]
- Absinta, M.; Lassmann, H.; Trapp, B.D. Mechanisms underlying progression in multiple sclerosis. Curr. Opin. Neurol. 2020, 33, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Kuhlmann, T.; Miron, V.; Cuo, Q.; Wegner, C.; Antel, J.; Bruck, W. Differentiation block of oligodendroglial progenitor cells as a cause for remyelination failure in chronic multiple sclerosis. Brain 2008, 131, 1749–1758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Probert, L. TNF and its receptors in the CNS: The essential, the desirable and the deleterious effects. Neuroscience 2015, 302, 2–22. [Google Scholar] [CrossRef] [Green Version]
- Maimone, D.; Gregory, S.; Arnason, B.G.; Reder, A.T. Cytokine levels in the cerebrospinal fluid and serum of patients with multiple sclerosis. J. Neuroimmunol. 1991, 32, 67–74. [Google Scholar] [CrossRef]
- Sharief, M.M.; Hentges, R. Association between Tumor Necrosis Factor-α and Disease Progression in Patients with Multiple Sclerosis. N. Engl. J. Med. 1991, 325, 467–472. [Google Scholar] [CrossRef] [PubMed]
- Hofman, F.M.; Hinton, D.R.; Johnson, K.; E Merrill, J. Tumor necrosis factor identified in multiple sclerosis brain. J. Exp. Med. 1989, 170, 607–612. [Google Scholar] [CrossRef]
- Selmaj, K.; Raine, C.S.; Cannella, B.; Brosnan, C.F. Identification of lymphotoxin and tumor necrosis factor in multiple sclerosis lesions. J. Clin. Investig. 1991, 87, 949–954. [Google Scholar] [CrossRef] [Green Version]
- Kriegler, M.; Perez, C.; Defay, K.; Albert, I.; Lu, S. A novel form of TNF/cachectin is a cell surface cytotoxic transmembrane protein: Ramifications for the complex physiology of TNF. Cell 1988, 53, 45–53. [Google Scholar] [CrossRef]
- Black, R.A.; Rauch, C.T.; Kozlosky, C.J.; Peschon, J.J.; Slack, J.L.; Wolfson, M.F.; Castner, B.J.; Stocking, K.L.; Reddy, P.; Srinivasan, S.; et al. A metalloproteinase disintegrin that releases tumour-necrosis factor-α from cells. Nature 1997, 385, 729–733. [Google Scholar] [CrossRef] [PubMed]
- Moss, M.L.; Jin, S.-L.C.; Milla, M.E.; Burkhart, W.; Carter, H.L.; Chen, W.-J.; Clay, W.C.; Didsbury, J.R.; Hassler, D.; Hoffman, C.R.; et al. Cloning of a disintegrin metalloproteinase that processes precursor tumour-necrosis factor-α. Nature 1997, 385, 733–736. [Google Scholar] [CrossRef]
- Grell, M.; Douni, E.; Wajant, H.; Löhden, M.; Clauss, M.; Maxeiner, B.; Georgopoulos, S.; Lesslauer, W.; Kollias, G.; Pfizenmaier, K.; et al. The transmembrane form of tumor necrosis factor is the prime activating ligand of the 80 kDa tumor necrosis factor receptor. Cell 1995, 83, 793–802. [Google Scholar] [CrossRef] [Green Version]
- Holtmann, M.H.; Neurath, M.H.H.A.M.F. Differential TNF-Signaling in Chronic Inflammatory Disorders. Curr. Mol. Med. 2004, 4, 439–444. [Google Scholar] [CrossRef]
- Dostert, C.; Grusdat, M.; Letellier, E.; Brenner, D. The TNF Family of Ligands and Receptors: Communication Modules in the Immune System and Beyond. Physiol. Rev. 2019, 99, 115–160. [Google Scholar] [CrossRef]
- Arnett, H.A.; Mason, J.T.; Marino, M.W.; Suzuki, K.; Matsushima, G.K.; Ting, J.P.-Y. TNFα promotes proliferation of oligodendrocyte progenitors and remyelination. Nat. Neurosci. 2001, 4, 1116–1122. [Google Scholar] [CrossRef]
- Madsen, P.; Desu, H.L.; Vaccari, J.P.D.R.; Florimon, Y.; Ellman, D.G.; Keane, R.W.; Clausen, B.H.; Lambertsen, K.L.; Brambilla, R. Oligodendrocytes modulate the immune-inflammatory response in EAE via TNFR2 signaling. Brain Behav. Immun. 2020, 84, 132–146. [Google Scholar] [CrossRef]
- Madsen, P.; Motti, D.; Karmally, S.; Szymkowski, D.E.; Lambertsen, K.L.; Bethea, J.R.; Brambilla, R. Oligodendroglial TNFR2 Mediates Membrane TNF-Dependent Repair in Experimental Autoimmune Encephalomyelitis by Promoting Oligodendrocyte Differentiation and Remyelination. J. Neurosci. 2016, 36, 5128–5143. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010, 11, R25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, H.; Danzi, M.C.; Choi, C.S.; Taherian, M.; Hansen, C.D.; Ellman, D.G.; Madsen, P.; Bixby, J.L.; Lemmon, V.P.; Lambertsen, K.L.; et al. Opposing Functions of Microglial and Macrophagic TNFR2 in the Pathogenesis of Experimental Autoimmune Encephalomyelitis. Cell Rep. 2017, 18, 198–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Efremova, M.; Vento-Tormo, M.; Teichmann, S.A.; Vento-Tormo, R. CellPhoneDB: Inferring cell–cell communication from combined expression of multi-subunit ligand–receptor complexes. Nat. Protoc. 2020, 15, 1484–1506. [Google Scholar] [CrossRef]
- Ramilowski, J.A.; Goldberg, T.; Harshbarger, J.; Kloppmann, E.; Lizio, M.; Satagopam, V.P.; Itoh, M.; Kawaji, H.; Carninci, P.; Rost, B.; et al. Correction: Corrigendum: A draft network of ligand-receptor-mediated multicellular signalling in human. Nat. Commun. 2016, 7, 10706. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Brambilla, R. The contribution of astrocytes to the neuroinflammatory response in multiple sclerosis and experimental autoimmune encephalomyelitis. Acta Neuropathol. 2019, 137, 757–783. [Google Scholar] [CrossRef]
- Plastini, M.J.; Desu, H.L.; Brambilla, R. Dynamic Responses of Microglia in Animal Models of Multiple Sclerosis. Front. Cell. Neurosci. 2020, 14, 269. [Google Scholar] [CrossRef]
- Marchetti, L.; Klein, M.; Schlett, K.; Pfizenmaier, K.; Eisel, U. Tumor Necrosis Factor (TNF)-mediated Neuroprotection against Glutamate-induced Excitotoxicity Is Enhanced by N-Methyl-D-aspartate Receptor Activation. J. Biol. Chem. 2004, 279, 32869–32881. [Google Scholar] [CrossRef] [Green Version]
- Webster, J.D.; Vucic, D. The Balance of TNF Mediated Pathways Regulates Inflammatory Cell Death Signaling in Healthy and Diseased Tissues. Front. Cell Dev. Biol. 2020, 8, 365. [Google Scholar] [CrossRef]
- Falcão, A.M.; Van Bruggen, D.; Marques, S.; Meijer, M.; Jäkel, S.; Agirre, E.; Samudyata; Floriddia, E.M.; Vanichkina, D.P.; Ffrench-Constant, C.; et al. Disease-specific oligodendrocyte lineage cells arise in multiple sclerosis. Nat. Med. 2018, 24, 1837–1844. [Google Scholar] [CrossRef] [PubMed]
- Chew, L.-J.; King, W.C.; Kennedy, A.; Gallo, V. Interferon-γ inhibits cell cycle exit in differentiating oligodendrocyte progenitor cells. Glia 2005, 52, 127–143. [Google Scholar] [CrossRef]
- Balabanov, R.; Strand, K.; Goswami, R.; McMahon, E.; Begolka, W.; Miller, S.D.; Popko, B. Interferon- -Oligodendrocyte Interactions in the Regulation of Experimental Autoimmune Encephalomyelitis. J. Neurosci. 2007, 27, 2013–2024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonora, M.; De Marchi, E.; Patergnani, S.; Suski, J.M.; Celsi, F.; Bononi, A.; Giorgi, C.; Marchi, S.; Rimessi, A.; Duszynski, J.; et al. Tumor necrosis factor-α impairs oligodendroglial differentiation through a mitochondria-dependent process. Cell Death Differ. 2014, 21, 1198–1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Zhang, C.-J.; Martin, B.N.; Bulek, K.; Kang, Z.; Zhao, J.; Bian, G.; Carman, J.A.; Gao, J.; Dongre, A.; et al. IL-17 induced NOTCH1 activation in oligodendrocyte progenitor cells enhances proliferation and inflammatory gene expression. Nat. Commun. 2017, 8, 15508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirby, L.; Jin, J.; Cardona, J.G.; Smith, M.D.; Martin, K.A.; Wang, J.; Strasburger, H.; Herbst, L.; Alexis, M.; Karnell, J.; et al. Oligodendrocyte precursor cells present antigen and are cytotoxic targets in inflammatory demyelination. Nat. Commun. 2019, 10, 1–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pantoja, I.E.M.; Smith, M.; Rajbhandari, L.; Cheng, L.; Gao, Y.; Mahairaki, V.; Venkatesan, A.; Calabresi, P.A.; Fitzgerald, K.C.; Whartenby, K.A. iPSCs from people with MS can differentiate into oligodendrocytes in a homeostatic but not an inflammatory milieu. PLoS ONE 2020, 15, e0233980. [Google Scholar] [CrossRef]
- McMahon, E.J.; Cook, D.N.; Suzuki, K.; Matsushima, G.K. Absence of Macrophage-Inflammatory Protein-1α Delays Central Nervous System Demyelination in the Presence of an Intact Blood-Brain Barrier. J. Immunol. 2001, 167, 2964–2971. [Google Scholar] [CrossRef] [Green Version]
- Murphy, C.A.; Hoek, R.M.; Wiekowski, M.T.; Lira, S.A.; Sedgwick, J.D. Interactions Between Hemopoietically Derived TNF and Central Nervous System-Resident Glial Chemokines Underlie Initiation of Autoimmune Inflammation in the Brain. J. Immunol. 2002, 169, 7054–7062. [Google Scholar] [CrossRef] [Green Version]
- Bartosik-Psujek, H.; Stelmasiak, Z. The levels of chemokines CXCL8, CCL2 and CCL5 in multiple sclerosis patients are linked to the activity of the disease. Eur. J. Neurol. 2005, 12, 49–54. [Google Scholar] [CrossRef]
- Balashov, K.E.; Rottman, J.B.; Weiner, H.L.; Hancock, W.W. CCR5+ and CXCR3+ T cells are increased in multiple sclerosis and their ligands MIP-1 and IP-10 are expressed in demyelinating brain lesions. Proc. Natl. Acad. Sci. USA 1999, 96, 6873–6878. [Google Scholar] [CrossRef] [Green Version]
- Trebst, C.; Sørensen, T.L.; Kivisäkk, P.; Cathcart, M.K.; Hesselgesser, J.; Horuk, R.; Sellebjerg, F.; Lassmann, H.; Ransohoff, R.M. CCR1+/CCR5+ Mononuclear Phagocytes Accumulate in the Central Nervous System of Patients with Multiple Sclerosis. Am. J. Pathol. 2001, 159, 1701–1710. [Google Scholar] [CrossRef] [Green Version]
- Jalonen, T.O.; Pulkkinen, K.; Ukkonen, M.; Saarela, M.; Elovaara, I. Differential intracellular expression of CCR5 and chemokines in multiple sclerosis subtypes. J. Neurol. 2002, 249, 576–583. [Google Scholar] [CrossRef]
- Trebst, C.; König, F.; Ransohoff, R.; Brück, W.; Stangel, M. CCR5 expression on macrophages/microglia is associated with early remyelination in multiple sclerosis lesions. Mult. Scler. J. 2008, 14, 728–733. [Google Scholar] [CrossRef] [PubMed]
- Howland, K.C.; Ausubel, L.J.; London, C.A.; Abbas, A.K. The Roles of CD28 and CD40 Ligand in T Cell Activation and Tolerance. J. Immunol. 2000, 164, 4465–4470. [Google Scholar] [CrossRef] [Green Version]
- Munroe, M.E.; Bishop, G.A. A Costimulatory Function for T Cell CD40. J. Immunol. 2007, 178, 671–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elgueta, R.; Benson, M.J.; De Vries, V.C.; Wasiuk, A.; Guo, Y.; Noelle, R.J. Molecular mechanism and function of CD40/CD40L engagement in the immune system. Immunol. Rev. 2009, 229, 152–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giustiniani, J.; Marie-Cardine, A.; Bensussan, A. A Soluble Form of the MHC Class I-Specific CD160 Receptor Is Released from Human Activated NK Lymphocytes and Inhibits Cell-Mediated Cytotoxicity. J. Immunol. 2007, 178, 1293–1300. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, K.; Sloan, S.; Bennett, M.L.; Scholze, A.R.; O’Keeffe, S.; Phatnani, H.P.; Guarnieri, P.; Caneda, C.; Ruderisch, N.; et al. An RNA-Sequencing Transcriptome and Splicing Database of Glia, Neurons, and Vascular Cells of the Cerebral Cortex. J. Neurosci. 2014, 34, 11929–11947. [Google Scholar] [CrossRef]
- Harris, J.J.; Jolivet, R.; Attwell, D. Synaptic Energy Use and Supply. Neuron 2012, 75, 762–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liemburg-Apers, D.; Willems, P.H.; Koopman, W.J.H.; Grefte, S. Interactions between mitochondrial reactive oxygen species and cellular glucose metabolism. Arch. Toxicol. 2015, 89, 1209–1226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camandola, S.; Mattson, M.P. Brain metabolism in health, aging, and neurodegeneration. EMBO J. 2017, 36, 1474–1492. [Google Scholar] [CrossRef] [PubMed]
- Fleury, C.; Mignotte, B.; Vayssière, J.-L. Mitochondrial reactive oxygen species in cell death signaling. Biochimie 2002, 84, 131–141. [Google Scholar] [CrossRef]
- Le Bras, M.; Clément, M.-V.; Pervaiz, S.; Brenner, C. Reactive oxygen species and the mitochondrial signaling pathway of cell death. Histol. Histopathol. 2005, 20, 205–219. [Google Scholar] [PubMed]
- The Lenercept Multiple Sclerosis Study Group and The University of British Columbia MS/MRI Analysis Group. TNF neu-tralization in MS: Results of a randomized, placebo-controlled multicenter study. Neurology 1999, 53, 457–465. [Google Scholar] [CrossRef] [Green Version]
- Magliozzi, R.; Howell, O.; Durrenberger, P.; Aricò, E.; James, R.; Cruciani, C.; Reeves, C.; Roncaroli, F.; Nicholas, R.; Reynolds, R. Meningeal inflammation changes the balance of TNF signalling in cortical grey matter in multiple sclerosis. J. Neuroinflamm. 2019, 16, 1–16. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Desu, H.L.; Illiano, P.; Choi, J.S.; Ascona, M.C.; Gao, H.; Lee, J.K.; Brambilla, R. TNFR2 Signaling Regulates the Immunomodulatory Function of Oligodendrocyte Precursor Cells. Cells 2021, 10, 1785. https://doi.org/10.3390/cells10071785
Desu HL, Illiano P, Choi JS, Ascona MC, Gao H, Lee JK, Brambilla R. TNFR2 Signaling Regulates the Immunomodulatory Function of Oligodendrocyte Precursor Cells. Cells. 2021; 10(7):1785. https://doi.org/10.3390/cells10071785
Chicago/Turabian StyleDesu, Haritha L., Placido Illiano, James S. Choi, Maureen C. Ascona, Han Gao, Jae K. Lee, and Roberta Brambilla. 2021. "TNFR2 Signaling Regulates the Immunomodulatory Function of Oligodendrocyte Precursor Cells" Cells 10, no. 7: 1785. https://doi.org/10.3390/cells10071785
APA StyleDesu, H. L., Illiano, P., Choi, J. S., Ascona, M. C., Gao, H., Lee, J. K., & Brambilla, R. (2021). TNFR2 Signaling Regulates the Immunomodulatory Function of Oligodendrocyte Precursor Cells. Cells, 10(7), 1785. https://doi.org/10.3390/cells10071785