
Implant Fibrosis and the Underappreciated Role of Myofibroblasts in the Foreign Body Reaction


Laboratory of Tissue Repair and Regeneration, Faculty of Dentistry, University of Toronto, Toronto, ON M5G 1G6, Canada
*
Authors to whom correspondence should be addressed.
Cells 2021, 10(7), 1794; https://doi.org/10.3390/cells10071794
Submission received: 31 May 2021
/
Revised: 8 July 2021
/
Accepted: 13 July 2021
/
Published: 15 July 2021
(This article belongs to the Special Issue Biomechanical Signaling and Fibrosis)
Abstract
:Body implants and implantable medical devices have dramatically improved and prolonged the life of countless patients. However, our body repair mechanisms have evolved to isolate, reject, or destroy any object that is recognized as foreign to the organism and inevitably mounts a foreign body reaction (FBR). Depending on its severity and chronicity, the FBR can impair implant performance or create severe clinical complications that will require surgical removal and/or replacement of the faulty device. The number of review articles discussing the FBR seems to be proportional to the number of different implant materials and clinical applications and one wonders, what else is there to tell? We will here take the position of a fibrosis researcher (which, coincidentally, we are) to elaborate similarities and differences between the FBR, normal wound healing, and chronic healing conditions that result in the development of peri-implant fibrosis. After giving credit to macrophages in the inflammatory phase of the FBR, we will mainly focus on the activation of fibroblastic cells into matrix-producing and highly contractile myofibroblasts. While fibrosis has been discussed to be a consequence of the disturbed and chronic inflammatory milieu in the FBR, direct activation of myofibroblasts at the implant surface is less commonly considered. Thus, we will provide a perspective how physical properties of the implant surface control myofibroblast actions and accumulation of stiff scar tissue. Because formation of scar tissue at the surface and around implant materials is a major reason for device failure and extraction surgeries, providing implant surfaces with myofibroblast-suppressing features is a first step to enhance implant acceptance and functional lifetime. Alternative therapeutic targets are elements of the myofibroblast mechanotransduction and contractile machinery and we will end with a brief overview on such targets that are considered for the treatment of other organ fibroses.
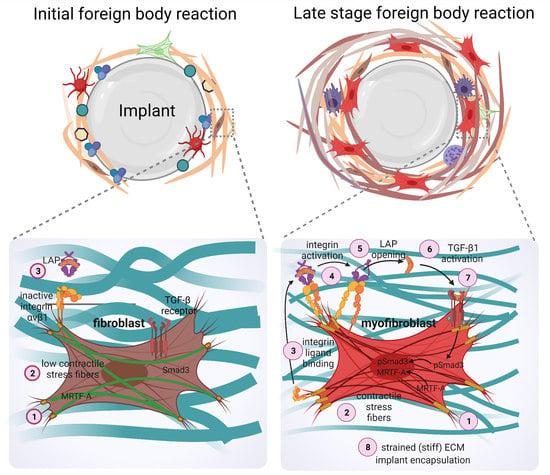
1. Introduction
Implantable medical devices and body replacement parts have revolutionized modern medicine in supporting and/or replacing malfunctioning human organs and tissues. Clinical applications of implants range from tissue repair and reconstruction, prostheses, neural interfacing, biosensors, controlled drug release and electronic pacing. Despite their undisputable benefits for patients, prolonged presence of objects that are not normally part of the organism often provokes tissue repair responses that are collectively known as the foreign body reaction (FBR). The FBR often starts similar to a physiological healing response to lost tissue homeostasis following implantation surgery [1,2] and can be divided into overlapping stages, not unlike the phases that define normal wound healing: (1) protein adsorption and formation of provisional ECM, (2) acute inflammation, and (3) chronic inflammation which in the context of the FBR refers to the infiltration of leukocytes and monocytes 2–5 weeks post-implantation [3].
In this review, we will mainly discuss two subsequent FBR stages that differ from the normal wound healing process and occur at implant surfaces: (4) chronic macrophage activation and foreign body giant cell (FBGC) formation, and (5) fibroblastic cell activation into myofibroblasts that form peri-implant scar tissue—a process called implant fibrosis [4]. We will further concentrate our discussion on larger (i.e., exceeding particle sizes that do not allow cellular uptake) and ‘impenetrable’ implants that, in contrast to porous scaffold materials, do not allow cell invasion. Such solid medical devices include glucose sensors for diabetics, various drug delivery systems, artificial heart valves and pacemakers to alleviate cardiac malfunctions, orthopedic prostheses, and breast implants for plastic and reconstructive surgery [1,5,6]. For excellent overviews and commentaries on other aspects of the FBR, notably the inflammatory cell and growth factor environment, and role of vascularization, the reader is referred to these excellent recent review articles [1,2,4,6,7,8,9,10,11,12].
2. Clinical Complications with Implants
Implant material allergies, bacterial infections, association with cancer and tissue necrosis are some of the most frequent clinical complications associated with the FBR. For instance, exposure to metal-based materials such as those used for orthopedic, dental, or cardiac (pacemaker leads) implant devices can induce material-induced allergies [13], and cause side effects such as joint failure, pain, localized swelling, and cutaneous reactions [14]. Dental implant rejections due to poor bone quality, unresolved carries, and poor dental hygiene are most common [15]. Dental implant failures, if left untreated, can lead to other health issues, such as gum inflammation, infection, and damage to surrounding tissue and teeth [16]. In addition to implant surfaces directly causing FBR as discussed further below, mechanical overloading and implant-wear complications, for instance caused by poor implant positioning, often result in implant failures, [17]. Moreover, biological complications can be caused by the improper placing of dental implants under aseptic measures and infections caused by bacterial plaques [18].
Similarly, mechanical overloading of stainless steel-based orthopedic implants can cause clinical issues by implant loosening, ultimately leading to adverse cell responses and inflammation [19]. However, the main problems with mechanically challenged orthopedic implants such as contemporary knee and hip replacements are production of wear debris and implant corrosion accumulating over time. When joint replacements malfunction, metal and polyethylene particles can wear off the implant and trigger cell responses in the adjacent periprosthetic tissue but also spread systemically to different organs [20].
With our aging population, the demand for implantable devices is also rising to support cardiac and vascular function, such as artificial heart valves, pacemakers, catheters, stents, defibrillators, and prostheses. Cardiac implants can cause local pocket infections and ensuing FBRs that often require device extraction. Pacemakers for instance must be surgically replaced every 10–15 years to function properly and an estimated 10,000–15,000 pacemaker and defibrillators leads are extracted annually in the world [21,22,23,24]. Extraction surgery can face severe clinical complications, such as hemopericardium, major vascular injuries, and subsequent need for open cardiac surgery. As implants remain in the tissue for extended time, the formation of fibrotic tissue encapsulating the implants promotes their adhesion to major veins and cardiac structures [25]. The presence of fibrotic collagenous tissue around defective pacemaker leads increases the risk of lead extraction procedures often resulting in structural damages to vessel walls and heart tissue [21,26,27,28].
The complications associated with FBR are not limited to implantable devices used for medical or therapeutics purposes, but also in cosmetic or reconstructive surgeries. Placement of reconstructive or esthetic breast implants is one of the most common procedures performed in plastic surgery [29,30], with >330,000 implant surgeries performed in the US alone in 2020 [31]. Several complications have been reported with such procedures, including hematoma, seroma, asymmetry, scarring, swelling, and rupture [32]. However, development of a fibrotic capsule and capsular contracture remains the most common complication following breast implant surgery. While formation of an initial collagenous capsule helps positioning the implant and is usually soft and slightly firm, thicker and denser fibrotic capsules develop in >10% of the cases [33,34]. The associated capsular contraction can cause chronic pain and unwanted position changes of the implant which requires reoperation. Over 58,000 breast implant reoperations were performed in 2020 in the US due to capsular contractures [2,35]. Bacterial infections are the next most frequent complication that often require surgical revision [32]. Bacterial infection is suggested to be involved in the fibrotic reaction to implants as well as in the development of anaplastic large cell lymphoma. Breast implant-associated anaplastic large cell lymphoma is often diagnosed as a localized peri-implant seroma containing cancer cells in one breast and to the lesser degree a tumor mass attached to the surface of the fibrotic capsule. In 2011, the FDA reported that women with high-texture high-surface area implants—intended to improve tissue integration—are at the highest risk of contracting anaplastic large cell lymphoma cancer [36,37]. The main treatment option for patients is a complete surgical removal of breast implants, dissection of adjacent diseased tissue and fibrotic capsule [38]. Chemotherapy is often recommended only in patients with advanced disease stage [39]. Another complication of breast reconstruction is mastectomy-related skin flap necrosis that increases morbidity rates as it can lead to impaired wound healing, tissue scarring, infection, and ultimately implant removal and replacement [40,41]. Many of the clinical complication listed above are variations of failing normal tissue and repair processes and it seems important to briefly discuss similarities between normal wound healing and the FBR (Figure 1).
3. Commonalities between the FBR and Normal Wound Healing
Most if not all animal model and clinical implant placings involve surgical damage to the host tissue which inevitably creates a normal wound healing response, often starting with damage to vascularized connective tissue. Plasma proteins released form damaged vessels include albumin, complement factors, fibrinogen, fibronectin, and vitronectin. Together, these constituents form a fibrin-dominated provisional extracellular matrix (ECM), called thrombus or wound clot that stops bleeding [42]. In the presence of an implant, part of this neo-ECM adsorbs to the material surface as a 2–5 nm thick layer of provisional ECM [43,44,45] (Figure 1). In the FBR and during normal wound healing, the provisional ECM provides a structural cell-populated scaffold that stores and continuously releases various mitogens, chemo-attractants, cytokines, and growth factors, all influencing the outcome of the FBR [2,46]. In both cases, hemostasis is followed by and overlaps with a phase of acute inflammation that ranges from hours to few days and usually resolves within one week [7].
Within hours post implantation, neutrophils are among the first immune cells that appear at the tissue- implant interface and their primary task is to clear bacteria and other debris [47]. Degranulating mast cells release histamine and serotonin, causing vasodilatation which allows easier access to other inflammatory cells migrating into the wound site [48,49]. Release of factors such as interleukin (IL)-4 and IL-13 by mast cells also plays an integral role in recruiting monocytes to the site of implanted material where they differentiate into macrophages [50,51,52]. Macrophages eventually become the primary cell population around the implant and at the implant surface [53]. Even in a normally healing wound and around resolving implants, macrophages are present for multiple days to phagocytose dead cells, damaged tissue as well as possible degradation products of the implant. The failure of macrophages to digest implants whose sizes and material properties withstanding phagocytosis is one of the main causes that turn acute into chronic (not resolved) inflammation at the implant site as discussed below.
Macrophages, platelets, and other implant-adjacent cells of wounded tissue, such as keratinocytes, endothelial cells, and adipocytes secrete various profibrotic and angiogenic growth factors, including platelet-derived growth factors (PDGF), vascular endothelial growth factors (VEGF), and transforming growth factor-beta (TGF-β) which among other functions attract and activate injury-adjacent fibroblastic cells [54]. During normal wound healing, fibroblastic cells replace provisional and weak fibrin with stronger collagenous ECM to restore the lost tissue architecture and mechanical integrity. Collagen fibers produced during the normal wound healing phase of the FBR show anisotropic orientations not unlike that of healthy connective tissues such as skin dermis [55]. Overlapping with ECM production, fibroblastic cells develop contractile features that allow first migration into the provisional ECM and later organization of the neo-collagen ECM into mechanically resistant scar. The transition from a quiescent to migratory (proto-myofibroblast) to a contractile phenotype is termed myofibroblast activation [56,57] (Figure 1) and will be discussed further below in the context of implant fibrosis. Myofibroblast are main contributors to the formation of scar tissue that characterizes peri-implant fibrosis and together with their fibroblast cousins become the major cell population in the ECM around implants within 4 weeks [5]. In severe cases, the dense fibrotic capsule entirely isolates the implanted material from the local tissue environment, possibly serving as our body’s last defense mechanism against the foreign object. In contrast, remodelling and contraction of normal wound healing blends into the resolution phase, when myofibroblasts and fibroblasts undergo apoptosis, neo-vasculature decreases, and damaged tissue is repaired [58].
4. What Is Different between Normal Wound Healing and the FBR? The Implant
Although the onset of the FBR shares several features with normal wound healing, the nature of the implanted material has a profound impact on the progression of acute immune and repair reactions into chronic conditions [59]. Depending on their purpose, desired durability and mechanical requirements, implants come in various shapes and sizes. Our body appears to be able to repair without scarring when wounds are small (~1 cm diameter) and cell-free gaps of provisional ECM can be covered by cell migration. Implanted solid or cell-free materials increase the distance that cells must cover to populate wound granulation tissue [60]. Consequently, wounds with sizes greater than 1 cm develop scar features if treated with a biomaterial ECM, but damages up to 30 cm in depth or length can be treated without adverse fibrotic response if the biomaterial is pre-populated with cells from the wound tissue [61]. Such physical conditions may underly different FBR-susceptibilities of materials with identical molecular chemistry but different dimensionality. For instance, alginate spheres with different diameters ranging from 0.3–0.5 mm, 0.7 mm, 1 mm, 1.5 mm implanted into the peritoneal cavity of mice for 2 weeks generate stronger FBRs with decreasing sphere diameters [62]. This effect, possibly generated by the higher curvature of smaller particles, is independent from the implant material since the same results are recapitulated with spheres made of different materials, including solid glass, polycaprolactone, polystyrene, and stainless steel.
In addition to having different dimensions, implants are produced of materials with very different surface chemistries [6]. All these properties influence their biointegration potential by affecting host tissue and plasma protein adsorption—a process called Vroman effect [63]. For example, mass spectrometry analysis of proteins adsorbed at the surface of polyethylene glycol (PEG)-based hydrogels identified more than 200 different adsorbed proteins most of which are associated with wound healing and acute inflammation [64]. Protein coverage and cell interactions with the surface both determine the ability of cells to attach and spread on implants [46,64]. Therefore, modulating implant surface properties is a common strategy to enhance tissue integration while simultaneously reducing the incidence and severity of implant FBR and fibrosis. In addition to or combined with altering implant surface chemistry [65], treatments include modulation of physical parameters such as surface hydrophilicity or wettability [66], porosity [67,68], stiffness (elastic modulus) [69,70], anisotropic ‘roughness’ [71,72], and regular topographies [73,74] (Figure 2).
Surface charge has been shown to alter the efficacy of host protein adsorption to the implant. For example, the plasma proteins fibronectin and vitronectin possess a higher affinity for positively or negatively charged hydrophobic implant surfaces than to uncharged hydrophobic surfaces [75,76,77]. Moreover, it has been suggested that macrophage rather adhere to hydrophobic and cationic implant surfaces than to hydrophilic and anionic surfaces [3]. Likewise, providing implant materials with hydrophilic surfaces, for instance by oxygen plasma treatment, generally improves cell adhesion and spreading, partly by facilitating the initial absorption of ECM proteins. As a result, basic cell functions such as proliferation, survival, and differentiation are enhanced [78,79,80]. Conversely, reducing surface wettability of polyurethane, a material frequently used for medical devices, by using zwitterionic and anionic chemistry decreases macrophage spreading areas which regulates their secretion of proinflammatory cytokines and ability to fuse into FBGCs [81]. Ultra-low-fouling zwitterionic hydrogels are also to prevent peri-implant fibrosis, and promote wound healing and neovascularization in the adjacent tissue when subcutaneously implanted in a mouse model [43]. In addition to coating with zwitterions, non-fouling hydrophilic PEG is another material that reduces protein adsorption to implant surfaces [82,83]. Although one of the possible drawbacks of PEG is its susceptibility to oxidation damage, there are currently no indications of such adverse reactions in vivo [84].
Surface wettability can be changed by physico-chemical treatments and/or by modulating material porosity, which is often used to enhance the stability of implants in the target tissue [85,86]. Laser-generated micropores of ~90 µm enhance the roughness and wettability of titanium surfaces used for dental or bone implants [87], with rougher surfaces promoting spreading of osteoprogenitor cells and improving osseointegration [88,89]. Surface roughness typically refers to irregular topographies with feature sizes that can span from hundreds of millimeters down to the sub-micron level. Whether cell responses are exclusively due to the enhanced wettability and/or caused by increased surface roughness or the created topographies is unclear and combinatorial effects are likely. For instance, silicone implant materials such as those used for breast reconstructive and esthetic surgery are produced with smooth and textured surfaces, both having similar wettability properties [90,91]. Nevertheless, silicone implants with textured ‘rough’ surfaces in a micron-range show protection against implant fibrosis whereas smooth surface implants promote formation of FBGC and become encapsulated by scar tissue [3,92,93,94,95,96,97]. Similarly, matching the molecular topography of silicone implants with that of normal breast connective tissue suppresses FBR and implant fibrosis [98]. Despite their fibrosis-suppressing effect, the safety of textured breast implants has been reviewed by the FDA, as there are studies suggesting that patients with textured breast implants and surface roughness of 300 µm are at a higher risk of developing breast implant-associated anaplastic large-cells lymphoma compared to patients with smooth implants [99,100]. To mitigate such reactions, micro- and nanotextured implant surfaces have been developed with combined smooth and textured surfaces and a surface roughness of 1–100 µm [74,101]. In the range of these dimensions, surface roughness features around 4 µm were recently shown to cause minimal inflammation and FBR in different animal models and humans [74]. Another interesting approach to counteract host reactions is to provide the implant material with a stealth coat of ‘body-own’ ECM [102,103]. For example, subcutaneously implanted tissue expanders coated with decellularized ECM into non-human primates displayed minimal fibrotic encapsulation. Likewise, brain microelectrodes coated with decellularized primary astrocyte-derived ECM suppressed macrophage activation and decreased astrogliosis compared to non-coated microelectrodes [104]. However, while implant coating with human ECM is an interesting approach to mitigate the FBR, it is difficult to achieve for large implants given limited availability of autologous material, and expensive. Furthermore, although such implant surface treatments are experimentally shown to reduce fibrotic reactions but suitability in clinical applications and regulatory approvals are largely pending.
Instead of providing implant materials with a rough irregular surface, different patterning approaches have been developed to elicit defined cell responses by controlling cell attachment. Lithography methods can be applied to polymer implant materials that are shaped during the polymerization process such as polyurethanes, silicones, and various hydrogels. Numerous studies demonstrated cell responses to topographies, typically involving cell alignment and shape confinement on top of elevated structures or in the pits of the designed landscapes [73]. When arranged in highly regular geometries, topographical features can prevent force-dependent alignment of cell-ECM focal adhesions and reduce cell adhesion by preventing elongation of the spreading cells [105]. Likewise, laser-generated complex but regular surface abrasions with nanometer-deep and micrometer-wide topographical features guide the formation of focal adhesions, control cell spreading area, and direct basic cell functions of human mesenchymal stromal cells towards osseointegration of titanium materials [106]. Similar topographical patterning approaches have been used to provide innovative bulk metallic glass materials with brush-like arrays of rods with cross-sectional diameters ranging from 55 nm to 200 nm and heights of ~0.5 µm [107]. In addition to controlling cell spreading by providing different rod surface areas for focal adhesion formation, the different cross-diameters result in different rod bending stiffnesses and thus resistances to cell traction forces, not unlike the silicone micropillar substrates produced in seminal works of the Chen lab [108]. Because different cell types exhibit different pattern preferences, controlled patterning can be used to enhance implant integration by differentially regulating adhesion of distinct cell types. In the example of bulk metallic glass ‘brush’ surfaces, rods with diameters <150 nm prevent macrophage spreading and <100 nm diameter rods prevent endothelial cell spreading. Fibroblasts adhere to all arrays but exert different forces that increase with increasing rod diameters [107]. Similar effects are obtained by etching stiff titanium materials to produce pliable lamellar nanostructures that are being perceived as “soft” by seeded macrophages [109].
These and other studies indicate that perceived stiffness and ‘true’ modulus of the implant surface can be used to control cell responses. Medical implants are carefully designed and fabricated from materials that are compatible with surgical handling but can be orders of magnitude stiffer than the host tissue. Physiologically relevant elastic moduli falling into the mechanosensing range of cells are related to the stiffness of their normal tissue environments. Several studies have used atomic force microscopy to measure the Young’s elastic modulus (stress over strain, in Pa) of various organs and tissues that typically receive implants, such as skin (0.1–10 kPa) [110], brain (0.1–0.5 kPa) [111], fat tissue (1–3 kPa) [112], and liver (1–2 kPa) [113]. Muscle tissues were measured to be moderately stiffer (10–15 kPa) [114] whereas teeth and bone are considered as stiff organs (1–4 GPa) [115]. The range of stiffnesses that cells can discriminate is proportional to their contractile force and the strain they can induce in a material, which is comparably low in macrophages (≤5 kPa) and higher in fibroblastic cells (≤100 kPa) [116,117].
As a result of the mechanical mismatch between implants and host tissue, movement of the tissue results in shear stress and strain at the interface, leading to mechanical activation of inflammatory and fibroblastic cells [7,118,119]. Even breast implants that appear macroscopically soft due to their malleable content are produced with a resistant outer shell that is thousand times stiffer than the surrounding connective host tissue. Our own research has demonstrated that silicone-based soft coatings with an elastic modulus resembling that of skin dermis (~2 kPa) effectively reduce detrimental fibrotic encapsulation of stiffer (~2 MPa) silicone materials used in breast reconstruction by inhibiting activation of fibroblasts in a mouse implant model [69] (Figure 3). Considering that metal-based implants are typically used in tissues that undergo high mechanical load and wear similar to bone and teeth, coting with soft layers is not practicable in these applications. However, implants that are designed to release diffusible compounds (e.g., insulin pumps), to measure local tissue environments (e.g., glucose sensors), or to stimulate tissue (e.g., deep brain stimulation electrodes) have been provided with soft hydrogels that generally reduce FBRs and fibrotic encapsulation in rodent models [120,121,122,123,124]. In the upcoming sections, we will revisit how modulation of implant surface properties specifically affects the activity of macrophages and fibroblasts in engineering strategies to reduce FBR and implant fibrosis.
5. Implant Surfaces from the Perspective of a Macrophage
Macrophages are early responders to surgically placed implants and accumulate at the implantation site for multiple days to phagocytose cell and tissue debris and micron-sized implant abrasion products [125]. The presence of macrophages results in the upregulation of pro-inflammatory and pro-fibrotic cytokines such as IL-1, IL-8, monocyte chemotactic protein-1 (MCP-1), chemokine (C-X-C motif) ligand 13 (CXCL13), and macrophage inflammatory protein (MIP) [2]. These and other factors promote further macrophage recruitment and changes in macrophage activity states during the FBR [67,102]. The inability of macrophages to completely engulf large implants leads to frustrated phagocytosis and chronic inflammatory actions [7], including formation of FBGC [126], and trophic actions on vascular cells [127,128], adaptive immune cells [9,129,130], and fibroblasts [4,131]. All these actions contribute to the FBR and often to implant fibrosis. Consequently, abolishing macrophage recruitment in animal implant studies or using animals with varying degrees of immune perpetuations (i.e., genetic knockouts, chemical and/or antibody guided immune cell depletions) exhibit reduced FBR in the absence of macrophages. For instance, clodronate liposome-induced macrophage deletion blocks monocyte infiltration, FBGC formation, neovascularization, and fibrosis [125,127,132]. Moreover, targeting macrophage receptors such as colony stimulating factor (CSF)-1 receptor, which is significantly upregulated following implantation of materials, completely suppresses development of implant fibrosis [125]. In the following sections, we will focus on strategies that aim in suppressing macrophage activation by physically altering implant surface properties.
The goals of implant surface modifications targeting macrophages are at least two-fold: (1) suppressing the formation of detrimental macrophage phenotypes while stimulating regenerating and resolving activation states [133,134,135], and (2) preventing the formation of FBGCs which typically follows the chronic inflammatory phase of the FBR [126]. Pro-inflammatory macrophages are often called ‘M1’, referring to activation states that can be produced in vitro by classical activation with lipopolysaccharides (LPS) and/or interferon-γ (IFN-γ). M1-like macrophages are the most predominant polarization type present at the surface of implantable materials [67,127] and drive early FBRs, for instance by producing PDGF, tumor necrosis factor (TNF-α), IL-6, granulocyte-CSF (G-CSF), and granulocyte macrophage CSF (GM-CSF) [2,136]. In contrast, pro-regeneration (but also pro-fibrotic) ‘M2’ are generated in culture by stimulation with IL-13 and/or IL-4. It becomes amply clear that M1 and M2 are only extremes of a spectrum of different macrophage activation states and/or subtypes, including M1/M2 hybrid ‘scaffold associated macrophages’ [134,137]. IL-4 and IL-13 are also critically involved in driving macrophage fusion into FBGCs [138] suggesting that the M2-like phenotype is a precondition for the formation of multinucleated FBGCs. FBGCs are formed at implant surfaces by the fusion of macrophages in a frustrated attempt to phagocytose exceedingly large objects, accompanied by production of reactive oxygen species and ECM-degrading proteases that are detrimental to the implant and surrounding tissue structure [2,9,126,139,140].
Numerous studies investigated how macrophage recruitment, adhesion, spreading, activation, and fusion depend on implant surface chemistry and topography [11]. Macrophages primarily use integrins to attach to RGD domains in host proteins that adsorb to the surface of implants, of which αMβ2 integrin (aka Mac-1 receptor or CD11b/CD18) was one of the first receptor described in this context [141,142]. In addition, macrophages also directly attach to the body-foreign material and sense surface characteristics using Toll-like receptors (TLR) and scavenger receptors [143]. Consequently, modulating receptor engagement by modulating implant surfaces affects the ability of macrophages to attach and spread on surfaces [144] and to fuse into FBGCs [138,139,145]. In addition to integrins and RhoA/ROCK signaling [146], transient receptor potential vanilloid 4 (TRPV4) channels appear to be critical in mediating macrophage responses and polarization on differently stiff substrates [147] and macrophage fusion in a Rac1-dependent manner [148].
Spreading ability and shape directly control macrophage polarization states, i.e., their secretory and remodeling activities in addition to the expression of characteristic polarization markers [134,149]. For instance, engagement of integrins and downstream focal adhesion kinase (FAK) and Wnt signaling differentially regulate macrophage phenotypes at titanium implant materials with different degrees of surface roughness in vitro and in vivo [150]. This exemplary study adds to a large body of literature on primary and lineage macrophages and showing that providing implants with defined surface roughness and nano-/micro-topographies allows control over the transition of M1-like into M2-like macrophages by determining their spreading area and shape. The range of micro-roughness feature sizes that promote M2-like phenotypes appears to be remarkably narrow between 0.51–1.36 μm [151]. Likewise, sub-micron to single digit micron-sized surface topographies are forcing macrophage alignment, generate M2-like macrophages, and stimulate macrophage fusion into FBGCs, whereas irregular topographies with similar dimensions or larger and smaller features produce circularly spreading macrophages with M1 characteristics [152,153,154,155,156]. Another narrow window of macrophage phenotype control appears to exist at the nanometer perception level. When seeded onto bulk metallic glass implant materials with 55 nm surface patterns, M1 and M2 pre-polarized macrophages show reduced cell spreading areas, reduced pro-inflammatory profiles, higher phagocytic activities, and higher fusion rates compared to the same cells on non-patterned surfaces or surfaces with 100–200 nm features [157,158,159].
In addition to surface topographies and adhesive patterns, the stiffness of surfaces has been shown to be critical for the responses of human and mouse lineage and primary macrophages to implant materials. Stimulating cultured macrophages with LPS and IFN-γ on stiff (elastic modulus ≥47 kPa) but not on softer (≤27 kPa) polyacrylamide hydrogels elicits pro-inflammatory response in a process depending on TLR4, NF-kB, and p65 signal transduction [160]. In general, growth on ECM-functionalized soft materials results in reduced spreading areas and suppresses pro-inflammatory macrophages phenotypes compared to macrophages grown on stiffer substrates [70,161,162,163,164]. Furthermore, growth on soft culture materials reduces the ability of mouse lineage and human alveolar macrophages to phagocytose micron-sized particles [165]. Likewise, the fusion of macrophages into FBGCs is enhanced by stiffer, more adhesive and 3D versus 2D implant materials [166]. In rodent implantation models, macrophage-driven inflammation is reduced around soft versus stiffer materials or when providing the implant with a soft layer, for instance using kPa-soft PEG hydrogels [70]. Although the definition of soft and stiff can largely vary between studies, elastic moduli around and below 1–5 kPa seem to be perceived as soft by macrophages in most studies. This mechanosensation threshold is higher for fibroblastic cells which drive the formation of scar tissue around implanted materials.
6. A Fibroblast View on the Implant: The Origins of Implant Fibrosis
Inflammatory environment, such reactive oxygen species, phagocytosis, and/or proteolysis alone can severely impact certain implant materials that are degradable such as replacement scaffolds or depend in their function on physiological environment, such as glucose sensors. Formation of fibrotic capsules around implantable devices can additionally lead to the physical isolation of the device from the tissue which can disrupt device sensing functions, cause pain and disconnect cell-seeded implants from nutrient support [5]. Implant surface properties affect macrophage activities which in turn affects fibrogenesis in the implant environment. Materials that skew macrophages into M2-like phenotypes and/or promote fusion into FBGCs seem generally more prone to become encapsulated by fibroblast-derived scar-like ECM [4]. However, it is beginning to be appreciated that implant surface properties also directly contribute to fibrogenesis. Above, we have discussed how material adhesivity/wettability, stiffness, and topography generally affect the engagement of fibroblasts with implant surfaces surface. Next, we will focus on how implant surface properties affect implant fibrosis by exerting control over activation of fibroblastic cells into myofibroblasts.
The formation of fibrotic capsules around implants was originally attributed to fibroblasts producing a highly fibrous and acellular ECM which contains collagen I/III, fibronectin, and proteoglycans [167]. However, it is now acknowledged that peri-implant tissue can additionally comprise multiple layers of contractile myofibroblasts that secrete and organize various ECM components—a hallmark of fibrosis [168,169]. ‘Myofibroblast’ denominates a pathophysiologically relevant fibroblast activation state that is characterized by neo-formation of stress fibers in tissues [56,57] (Figure 3). Stress fiber-mediated contraction confers to myofibroblasts the ability to repair tissues by organizing collagen ECM into mechanically resistant scar tissue. It is the excessive production, contraction, and crosslinking of collagen by transglutaminases [170,171,172], lysyl hydroxylases [173], lysyl oxidases [174], and lysyl oxidase-like enzymes [175] that result in fibrosis [176,177,178,179]. In vitro, all fibroblastic cells form stress fibers with stiff plastic or glass culture surfaces in the presence of serum and should thus be considered functional myofibroblasts [180]. Neo-expression of the smooth muscle actin (SMA) isoform α-SMA in stress fibers has become the most widely used molecular marker of myofibroblasts in culture and in vivo as it further enhances the contractile force of fibroblastic cells [181]. Recruitment of α-SMA into stress fibers is also an excellent indicator of acute intracellular and extracellular stress on different surfaces. During spreading on stiff culture surfaces, cytosolic α-SMA localizes to pre-formed β-cytoplasmic actin stress fibers only when spreading area is maximal, circular spreading symmetry is broken and myofibroblasts start to polarize into multipolar shapes by forming large focal adhesions [182]. Restriction of focal adhesion sizes by micropatterning stiff surfaces with adhesion sized fibronectin islets limits myofibroblast spreading, intracellular stress and α-SMA stress fiber incorporation [183]. The same acute effect is achieved by limiting cell spreading area using patterns of larger fibronectin islands [183,184] or culturing myofibroblasts on soft (<15 kPa) silicone substrates [181,183] (Figure 4).
All these principles are being applied to reduce myofibroblast activation at the surface of implant materials in culture systems and/or animal models of implant fibrosis [185]. In our own studies, we were able to suppress mechanical activation of myofibroblasts and implant fibrosis by covering MPa-stiff implant silicone surfaces with small focal adhesion micropatterns or soft (2 kPa) layers in rat and mouse models of subcutaneous implantation [69,186]. Others inhibited formation of large focal adhesions and fibroblast-to-myofibroblast activation using symmetrical arrays of hexagonal pits with micron-sized dimensions in hydrogels [105] or hierarchical micro/nano-topographical features in silicone materials [187]. A common theme of all these studies is that both, too little or much adhesion will be detrimental to implant integration and function and that both macrophage and fibroblasts have their sweet spots for implant acceptance. Surfaces that allow insufficient cell adhesion are perceived as body-foreign and elicit a strong inflammatory response. Surfaces allowing ‘unlimited’ cell adhesion and spreading signify large objects that activate hungry FBGCs and/or mechanically stimulate fibroblast transition onto fibrogenic myofibroblasts.
7. Outlook: Lessons to Learn from Anti-Fibrosis Strategies against Cell Mechanosensing?
Organ fibrosis comprises a group of heterogeneous connective tissue diseases that often lead to disruption of soft organ architecture and ultimately organ failure. Fibrosis is characterized by the excessive accumulation of disorganized and stiff ECM that often develops due to impaired wound healing as a result of the repetitive or chronic tissue injury [188,189,190]. Various factors, such as persistent infection, environmental exposure to toxin agents or smoke, genetic predisposition, and chronic inflammation contribute to development of tissue fibrosis [191]. Because fibroblast-driven encapsulation of implants by scar-like ECM shares features with organ fibrosis, anti-fibrotic therapies are being tested to preserve the function of implants that are threatened by capsular contractions [192], such as the lung fibrosis-approved drug pirfenidone [193] or using pan-αv integrin inhibitors [69,194]. Other studies targeted collagen type I synthesis using small interfering RNA which resulted in significant decrease the fibrous capsule thickness in a mouse implant model [195] (Table 1). We and others have already amply reviewed current strategies and therapies to counteract fibrosis in different organs [56,190,196,197,198]. For this review, we will limit our summary to strategies that aim to suppress myofibroblast actions but have not yet been considered or tested for implant fibrosis. The focus will be on mechanisms and molecules that are involved in the mechanical activation of myofibroblasts, which we identified as one major mechanism acting at the surface of stiff implants [69,186].
Aberrant ECM mechanosensing by fibroblastic cells and myofibroblast activation are associated with fibroproliferative disease conditions that involve changes in tissue environment and ECM composition [209,210,211]. Cells feel different mechanical stimuli through different cell surface receptors such as integrins, G-protein coupled receptors (GPCRs), or stretch-activated ion channels which activate mechanotransduction downstream signaling pathway [212]. One of the critical steps in the fibrotic phase of the FBR is fibroblast integrin engagement either with host proteins adsorbing to the implant, or to the provisional ECM developing around the object [213]. Integrin-based adhesion complexes mediate transmembrane signaling and intracellular responses to the environmental stimuli such as substrate stiffness [214,215,216,217,218] and thus represent prime anti-fibrosis targets [219,220,221]. In addition to the adhesion patterning approaches already discussed above, functionalization of implant materials with integrin-binding moieties, possibly including growth factor binding sites, has been proposed to guide a normal healing response and suppress the FBR and fibrosis [222,223].
Beyond their classical roles in direct mechanosensing, specific αv integrins are also involved in the activation of latent TGF-β1, a key factor in driving myofibroblast activation [196,224,225,226,227] (Figure 3). Fibroblasts and other cells secrete latent TGF-β1 in complex with its non-covalently bound latency-associated peptide (LAP), which in turn binds to the fibrillin protein family member latent TGF-β1-binding protein 1 (LTBP-1) in the ECM [228]. Integrins αvβ1, αvβ3, αvβ5, and αvβ6 have been demonstrated to physically interact with an RGD domain present in the LAP peptide and mechanically activate latent TGF-β1 by transmitting cytoskeletal forces to the LAP-TGF-β1 complex [229,230,231,232]. If the latent TGF-β1/LTBP-1 complex is integrated into a mechanically resisting matrix, cytoskeletal force exertion leads to a conformation change in the LAP-TGF-β1 ‘straitjacket’ configuration, resulting in activation and secretion of active TGF-β1 [226,227,233,234,235,236]. Our own recent study has shown that blocking αv integrins in a mouse model of implant fibrosis reduces TGF-β1 signaling at the implants surface and almost completely abolishes the formation of a fibrotic peri-implant tissue [69]. This study was inspired by seminal studies using similar approaches successfully in different experimental models of organ fibrosis [194,237].
In addition to driving pro-fibrotic cell programs thorough ‘canonical’ Smad signaling pathways, TGF-β1 also acts through the PI3K/Akt pathway, mitogen activated protein kinases (MAPKs) including Jun N-terminal kinases (JNKs), p38 MAPK (aka MAPK14), extracellular signal-regulated kinases (ERKs), and the Rho/Rho-associated protein kinase (ROCK) pathway [196,238,239]. Rho/ROCK signaling is a key regulatory pathway of actin-myosin contractility and central in controlling myofibroblast contraction [177,240,241], downstream of the engagement of the TGF-β1 receptor, and GPCR receptors such as proteinase-activated receptor 1 (PAR-1) or lysophosphatic acid receptor 1 (LPAR1) in a stressed environment [242,243,244,245]. Consequently, these receptor-signaling pathways are considered as anti-fibrosis targets [246]. Rho/ROCK signaling further regulates mechanical and receptor-mediated activation of Yes-associated protein 1 (YAP) and transcriptional coactivator with PDZ-binding motif (TAZ) [247,248]. YAP and TAZ are downstream co-activators of the Hippo signaling pathway, consisting of a cascade of mammalian sterile 20-like kinase 1/2 (MTS1/2) and large tumor suppressor kinase 1/2 (LATS1/2) [249,250]. YAP and TAZ are regulated by the activated and phosphorylated serine/threonine kinases LATS1/2, with YAP/TAZ phosphorylation resulting in cytoplasmic sequestration or proteasomal degradation [251]. Conversely, and relevant for fibrosis, MST1/2 activity is decreased by F-actin polymerization leading to inactivation of the Hippo complex. In this scenario and in direct dependence on force [252], non-phosphorylated YAP and TAZ translocate to the cell nucleus where they predominantly associate with transcription factors, such as TEA-domain family member (TEAD) but also Smad, p73, and Runt (RunX), to mediate transcription of fibrosis-related genes, including CCN2 and microRNA-21 [253,254,255,256]. YAP and TAZ are involved in myofibroblast activation and the onset of fibrosis in several organs, including lung, kidney, and liver [257,258,259,260,261]. Inhibition of selective GPCRs upstream of YAP/Taz signaling was shown to be effective in blocking myofibroblast activation and halt animal fibrosis [262].
Myocardin-related transcription factor (MRTF-A) is another mechanosensitive transcriptional effector that has been implicated in coordinating profibrotic signaling in myofibroblasts [209]. Upon mechanical stimulation and/or increased actin polymerization into stress fibers, MRTF-A becomes liberated from its inhibitory complex with G-actin in the cytoplasm, which then allows translocation of MRTF into the nucleus. In complex with serum response factor (SRF), nuclear MRTF-A regulate transcription of several cytoskeletal and ECM proteins that are associated with myofibroblast activation and fibrosis, including CCN2 and α-SMA [263,264,265,266]. Taken together, different mechanotransduction signaling pathways play distinct roles in myofibroblast activation and contraction in conditions of organ fibrosis. Targeting these pathways in the context of implanted solid materials that are almost always perceived as being stiff has great potential to alleviate peri-implant fibrosis.
8. Conclusions and Open Questions
Macrophage activation and fusion into FBGCs are hallmarks of the FBR and have been studied extensively because of their detrimental effect on medical device functions and surrounding tissue integrity. However, the consequences of chronic inflammation on fibroblastic cell populations in the vicinity of a—usually stiff—foreign body is less well documented. We and others have previously reviewed how macrophage-fibroblast interactions contribute to normal tissue repair and how dysregulation results in fibrosis; similar cross-talk is expected to take place in the environment of an implant [4,131]. Not unlike the sequalae taking place during organ fibrosis, chronic persistence of macrophages promotes activation of fibroblasts and expression of pro-fibrotic genes through paracrine signaling [267,268]. For instance, secretion and/or presentation of TGF-β1 by macrophages is a major contributor to the activation of fibroblasts and deposition of ECM in both, fibrosis and implant scarring [177,269] in a very localized niche environment [69,270]. In addition to secretory factors, the ECM itself plays a major role in mediating cellular behavior and intracellular communication. ECM guidance such as stiffness gradients and fibroblast induced contraction events in the ECM modulate macrophage-fibroblast communication [271,272,273,274]. However, not all fibrotic reactions to implanted materials appear to depend on macrophage actions. For example, macrophage depletion in a transgenic macrophage Fas-induced apoptosis (MaFIA) mouse model suppresses infiltration of inflammatory cells into hexamethylenediisocyanate-crosslinked dermal sheep collagen scaffolds which leads to an increased fibrotic capsule size around implants [275]. Furthermore, depletion of other immune cells including T-cells, NK cells, and mast cells depletion does not suppress the formation of the FBGC and fibrotic encapsulation of implants [276,277,278].
Stiff materials placed in provisional ECM create boundary conditions that are sensed over long distances by fibroblastic cells [279,280,281,282]. It is tempting to speculate that implants attract migrating fibroblasts, just by presenting an obstacle where lines of tension converge in provisional ECM. A review on the FBR from the perspective of a fibrosis researcher with a knack for cell biomechanics necessarily falls short in reporting on other exciting aspects of the FBR, such as the role of adaptive immune cells and design principles for of immunomodulatory biomaterials [130,135,283]. We also purposely excluded porous scaffold materials and degradation properties from our discussion which are intriguing both from a chemical composition and from a mechanical point of view—We hope we can be forgiven. Even in our core area of expertise, we did not even tempt to discuss a current hot topic in the fibroblast field: fibroblast origin and progeny [284]. The publication rate of single RNA sequencing studies classifying fibroblastic cells in various fibrotic organs and conditions has reached impressive if not overwhelming levels. However, we were not aware of a similar assessment of fibroblasts in peri-implant tissue—it will likely be different next week and then become topic of another review.
Funding
The research of B.H. is supported by a foundation grant from the Canadian Institutes of Health Research (grant #375597) and support from the John Evans Leadership funds (grants #36050 and #38861) and innovation funds (‘Fibrosis Network, grant #36349′) from the Canada Foundation for Innovation (CFI) and the Ontario Research Fund (ORF). P.P. has been funded through a fellowship through Mathematics of Information Technology and Complex Systems (Mitacs Accelerate, grant# IT13587).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
We thank Ronen Schuster (Hinz lab) for assisting with figure design. Schemes were produced with Biorender (biorender.com) under paid license.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Ratner, B.D. Biomaterials: Been There, Done That, and Evolving into the Future. Annu. Rev. Biomed. Eng. 2019, 21, 171–191. [Google Scholar] [CrossRef]
- Anderson, J.M.; Rodriguez, A.; Chang, D.T. Foreign body reaction to biomaterials. Semin. Immunol. 2008, 20, 86–100. [Google Scholar] [CrossRef] [Green Version]
- Anderson, J.M. Exploiting the inflammatory response on biomaterials research and development. J. Mater. Sci. Mater. Electron. 2015, 26, 121. [Google Scholar] [CrossRef]
- Witherel, C.E.; Abebayehu, D.; Barker, T.H.; Spiller, K.L. Macrophage and Fibroblast Interactions in Biomaterial-Mediated Fibrosis. Adv. Health Mater. 2019, 8, e1801451. [Google Scholar] [CrossRef]
- Veiseh, O.; Vegas, A.J. Domesticating the foreign body response: Recent advances and applications. Adv. Drug Deliv. Rev. 2019, 144, 148–161. [Google Scholar] [CrossRef]
- Williams, D.F. Biocompatibility Pathways: Biomaterials-Induced Sterile Inflammation, Mechanotransduction, and Principles of Biocompatibility Control. ACS Biomater. Sci. Eng. 2017, 3, 2–35. [Google Scholar] [CrossRef]
- Klopfleisch, R.; Jung, F. The pathology of the foreign body reaction against biomaterials. J. Biomed. Mater. Res. Part A 2017, 105, 927–940. [Google Scholar] [CrossRef]
- Grainger, D.W. All charged up about implanted biomaterials. Nat. Biotechnol. 2013, 31, 507–509. [Google Scholar] [CrossRef] [PubMed]
- Adusei, K.M.; Ngo, T.B.; Sadtler, K. T lymphocytes as critical mediators in tissue regeneration, fibrosis, and the foreign body response. Acta Biomater. 2021. [Google Scholar] [CrossRef] [PubMed]
- Greiner, A.M.; Sales, A.; Chen, H.; Biela, S.A.; Kaufmann, D.; Kemkemer, R. Nano- and microstructured materials for in vitro studies of the physiology of vascular cells. Beilstein J. Nanotechnol. 2016, 7, 1620–1641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abaricia, J.O.; Farzad, N.; Heath, T.J.; Simmons, J.; Morandini, L.; Olivares-Navarrete, R. Control of innate immune response by biomaterial surface topography, energy, and stiffness. Acta Biomater. 2021. [Google Scholar] [CrossRef]
- Mariani, E.; Lisignoli, G.; Borzì, R.M.; Pulsatelli, L. Biomaterials: Foreign Bodies or Tuners for the Immune Response? Int. J. Mol. Sci. 2019, 20, 636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosner, G.A.; Fonacier, L.S. Hypersensitivity to biomedical implants: Prevention and diagnosis. Allergy Asthma Proc. 2017, 38, 177–183. [Google Scholar] [CrossRef]
- Schalock, P.C.; Thyssen, J.P. Patch Testers’ Opinions Regarding Diagnostic Criteria for Metal Hypersensitivity Reactions to Metallic Implants. Dermatitis 2013, 24, 183–185. [Google Scholar] [CrossRef] [PubMed]
- Porter, J.A.; Von Fraunhofer, J.A. Success or failure of dental implants? A literature review with treatment considerations. Gen. Dent. 2005, 53, 423. [Google Scholar] [PubMed]
- Liaw, K.; Delfini, R.H.; Abrahams, J.J. Dental Implant Complications. Semin. Ultrasound CT MR 2015, 36, 427–433. [Google Scholar] [CrossRef]
- Tolman, D.E.; Laney, W.R.; Tolman, D.E.; Laney, W.R. Tissue-integrated prosthesis complications. Int. J. Oral Maxillofac. Implant. 1992, 7, 477–484. [Google Scholar] [CrossRef]
- Quirynen, M.; De Soete, M.; Van Steenberghe, D. Infectious risks for oral implants: A review of the literature. Clin. Oral Implant. Res. 2002, 13, 1–19. [Google Scholar] [CrossRef]
- Gibon, E.; Amanatullah, D.F.; Loi, F.; Pajarinen, J.; Nabeshima, A.; Yao, Z.; Hamadouche, M.; Goodman, S.B. The biological response to orthopaedic implants for joint replacement: Part I: Metals. J. Biomed. Mater. Res. Part B Appl. Biomater. 2017, 105, 2162–2173. [Google Scholar] [CrossRef]
- Urban, R.M.; Jacobs, J.; Tomlinson, M.J.; Gavrilovic, J.; Black, J.; Peoc’H, M. Dissemination of Wear Particles to the Liver, Spleen, and Abdominal Lymph Nodes of Patients with Hip or Knee Replacement. J. Bone Jt. Surg. Am. Vol. 2000, 82, 457–477. [Google Scholar] [CrossRef]
- Hauser, R.G.; Katsiyiannis, W.T.; Gornick, C.C.; Almquist, A.K.; Kallinen, L.M. Deaths and cardiovascular injuries due to device-assisted implantable cardioverter-defibrillator and pacemaker lead extraction. Europace 2009, 12, 395–401. [Google Scholar] [CrossRef]
- Nof, E.; Epstein, L. Complications of cardiac implants: Handling device infections. Eur. Hear. J. 2013, 34, 229–236. [Google Scholar] [CrossRef] [Green Version]
- Tarakji, K.G.; Chan, E.J.; Cantillon, D.J.; Doonan, A.L.; Hu, T.; Schmitt, S.; Fraser, T.G.; Kim, A.; Gordon, S.M.; Wilkoff, B. Cardiac implantable electronic device infections: Presentation, management, and patient outcomes. Hear. Rhythm. 2010, 7, 1043–1047. [Google Scholar] [CrossRef]
- Baman, T.S.; Gupta, S.K.; Valle, J.A.; Yamada, E. Risk Factors for Mortality in Patients with Cardiac Device-Related Infection. Circ. Arrhythmia Electrophysiol. 2009, 2, 129–134. [Google Scholar] [CrossRef] [Green Version]
- Keiler, J.; Schulze, M.; Sombetzki, M.; Heller, T.; Tischer, T.; Grabow, N.; Wree, A.; Bänsch, D. Neointimal fibrotic lead encapsulation—Clinical challenges and demands for implantable cardiac electronic devices. J. Cardiol. 2017, 70, 7–17. [Google Scholar] [CrossRef] [Green Version]
- Epstein, L.M.; Love, C.J.; Wilkoff, B.; Chung, M.K.; Hackler, J.W.; Bongiorni, M.G.; Segreti, L.; Carrillo, R.G.; Baltodano, P.; Fischer, A.; et al. Superior Vena Cava Defibrillator Coils Make Transvenous Lead Extraction More Challenging and Riskier. J. Am. Coll. Cardiol. 2013, 61, 987–989. [Google Scholar] [CrossRef] [Green Version]
- Smith, M.C.; Love, C.J. Extraction of Transvenous Pacing and ICD Leads. Pacing Clin. Electrophysiol. 2008, 31, 736–752. [Google Scholar] [CrossRef] [PubMed]
- Yakish, S.J.; Narula, A.; Foley, R.; Kohut, A.; Kutalek, S. Superior Vena Cava Echocardiography as a Screening Tool to Predict Cardiovascular Implantable Electronic Device Lead Fibrosis. J. Cardiovasc. Ultrasound 2015, 23, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Borenstein, A.; Friedman, O. Combined Breast Implant Explantation and Multilevel Mastopexy Technique. Plast. Reconstr. Surg. Glob. Open 2019, 7, e2429. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, A.; Maxwell, G.P. The Evolution of Breast Implants. Clin. Plast. Surg. 2015, 42, 399–404. [Google Scholar] [CrossRef] [PubMed]
- American_Society_of_Plastic_Surgeons. Cosmetic Surgery Procedures. National Plastic Surgery Statistics. 2020. Available online: https://www.plasticsurgery.org/documents/News/Statistics/2020/plastic-surgery-statistics-report-2020.pdf (accessed on 14 July 2021).
- Deva, A.K.; Adams, W.P., Jr.; Vickery, K. The Role of Bacterial Biofilms in Device-Associated Infection. Plast. Reconstr. Surg. 2013, 132, 1319–1328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coroneos, C.J.; Selber, J.C.; Offodile, A.C., 2nd; Butler, C.E.; Clemens, M.W. US FDA Breast Implant Postapproval Studies: Long-term Outcomes in 99,993 Patients. Ann Surg. 2019, 269, 30–36. [Google Scholar] [CrossRef]
- Headon, H.; Kasem, A.; Mokbel, K. Capsular Contracture after Breast Augmentation: An Update for Clinical Practice. Arch. Plast. Surg. 2015, 42, 532–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steiert, A.; Sorg, H.; Boyce, M. Capsular contracture by silicone breast implants: Possible causes, biocompatibility, and prophylactic strategies. Med. Devices Évid. Res. 2013, 6, 211–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collett, D.J.; Rakhorst, H.; Lennox, P.; Magnusson, M.; Cooter, R.; Deva, A. Current Risk Estimate of Breast Implant–Associated Anaplastic Large Cell Lymphoma in Textured Breast Implants. Plast. Reconstr. Surg. 2019, 143, 30S–40S. [Google Scholar] [CrossRef]
- Stack, A.; Ali, N.; Khan, N. Breast Implant-associated Anaplastic Large Cell Lymphoma: A Review with Emphasis on the Role of Brentuximab Vedotin. J. Cell Immunol. 2020, 2, 80–89. [Google Scholar] [PubMed]
- Clemens, M.W.; Medeiros, L.J.; Butler, C.E.; Hunt, K.K.; Fanale, M.A.; Horwitz, S.; Weisenburger, D.D.; Liu, J.; Morgan, E.; Kanagal-Shamanna, R.; et al. Complete Surgical Excision Is Essential for the Management of Patients with Breast Implant–Associated Anaplastic Large-Cell Lymphoma. J. Clin. Oncol. 2016, 34, 160–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clemens, M.W.; Brody, G.S.; Mahabir, R.C.; Miranda, R.N. How to Diagnose and Treat Breast Implant-Associated Anaplastic Large Cell Lymphoma. Plast Reconstr. Surg. 2018, 141, 586e–599e. [Google Scholar] [CrossRef]
- Sue, G.R.; Long, C.; Lee, G.K. Management of Mastectomy Skin Necrosis in Implant Based Breast Reconstruction. Ann. Plast. Surg. 2017, 78, S208–S211. [Google Scholar] [CrossRef]
- Hultman, C.S.; Daiza, S. Skin-Sparing Mastectomy Flap Complications After Breast Reconstruction: Review of Incidence, Management, and Outcome. Ann. Plast. Surg. 2003, 50, 249–255. [Google Scholar] [CrossRef]
- Rivera-Chacon, D.M.; Alvarado-Velez, M.; Acevedo-Morantes, C.; Singh, S.P.; Gultepe, E.; Nagesha, D.; Sridhar, S.; Ramirez-Vick, J. Fibronectin and Vitronectin Promote Human Fetal Osteoblast Cell Attachment and Proliferation on Nanoporous Titanium Surfaces. J. Biomed. Nanotechnol. 2013, 9, 1092–1097. [Google Scholar] [CrossRef]
- Zhang, L.; Cao, Z.; Bai, T.; Carr, L.R.; Ella-Menye, J.-R.; Irvin, C.; Ratner, B.D.; Jiang, S. Zwitterionic hydrogels implanted in mice resist the foreign-body reaction. Nat. Biotechnol. 2013, 31, 553–556. [Google Scholar] [CrossRef]
- Gorbet, M.B.; Sefton, M.V. Biomaterial-associated thrombosis: Roles of coagulation factors, complement, platelets and leukocytes. Biomaterials 2004, 25, 5681–5703. [Google Scholar] [CrossRef]
- Jenney, C.R.; Anderson, J.M. Adsorbed serum proteins responsible for surface dependent human macrophage behavior. J. Biomed. Mater. Res. 1999, 49, 435–447. [Google Scholar] [CrossRef]
- Wells, L.A.; Guo, H.; Emili, A.; Sefton, M.V. The profile of adsorbed plasma and serum proteins on methacrylic acid copolymer beads: Effect on complement activation. Biomaterials 2017, 118, 74–83. [Google Scholar] [CrossRef]
- Zdolsek, J.; Eaton, J.W.; Tang, L. Histamine release and fibrinogen adsorption mediate acute inflammatory responses to biomaterial implants in humans. J. Transl. Med. 2007, 5, 31. [Google Scholar] [CrossRef] [Green Version]
- Wulff, B.C.; Wilgus, T.A. Mast cell activity in the healing wound: More than meets the eye? Exp. Dermatol. 2013, 22, 507–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komi, D.E.A.; Khomtchouk, K.; Maria, P.L.S. A Review of the Contribution of Mast Cells in Wound Healing: Involved Molecular and Cellular Mechanisms. Clin. Rev. Allergy Immunol. 2020, 58, 298–312. [Google Scholar] [CrossRef]
- Janeway, C.A., Jr.; Medzhitov, R. Innate immune recognition. Annu. Rev. Immunol. 2002, 20, 197–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, C.; Liu, L.; Rung, S.; Wang, Y.; Ma, Y.; Hu, C.; Zhao, X.; Man, Y.; Qu, Y. Modulation of foreign body reaction and macrophage phenotypes concerning microenvironment. J. Biomed. Mater. Res. Part A 2020, 108, 127–135. [Google Scholar] [CrossRef]
- Van Dyken, S.J.; Locksley, R.M. Interleukin-4- and interleukin-13-mediated alternatively activated macrophages: Roles in homeostasis and disease. Annu. Rev. Immunol. 2013, 31, 317–343. [Google Scholar] [CrossRef] [Green Version]
- Kenneth Ward, W. A review of the foreign-body response to subcutaneously-implanted devices: The role of macrophages and cytokines in biofouling and fibrosis. J. Diabetes Sci. Technol. 2008, 2, 768–777. [Google Scholar] [CrossRef] [Green Version]
- Hinz, B. The role of myofibroblasts in wound healing. Curr. Res. Transl. Med. 2016, 64, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Safferling, K.; Sütterlin, T.; Westphal, K.; Ernst, C.; Breuhahn, K.; James, M.; Jäger, D.; Halama, N.; Grabe, N. Wound healing revised: A novel reepithelialization mechanism revealed by in vitro and in silico models. J. Cell Biol. 2013, 203, 691–709. [Google Scholar] [CrossRef] [Green Version]
- Pakshir, P.; Noskovicova, N.; Lodyga, M.; Son, D.O.; Schuster, R.; Goodwin, A.; Karvonen, H.; Hinz, B. The myofibroblast at a glance. J. Cell Sci. 2020, 133, jcs227900. [Google Scholar] [CrossRef] [PubMed]
- Schuster, R.; Rockel, J.S.; Kapoor, M.; Hinz, B. The inflammatory speech of fibroblasts. Immunol. Rev. 2021. [Google Scholar] [CrossRef]
- Hinz, B.; Lagares, D. Evasion of apoptosis by myofibroblasts: A hallmark of fibrotic diseases. Nat. Rev. Rheumatol. 2020, 16, 11–31. [Google Scholar] [CrossRef] [PubMed]
- Luttikhuizen, D.T.; Harmsen, M.C.; Van Luyn, M.J. Cellular and Molecular Dynamics in the Foreign Body Reaction. Tissue Eng. 2006, 12, 1955–1970. [Google Scholar] [CrossRef] [PubMed]
- Dorin, R.P.; Pohl, H.G.; De Filippo, R.E.; Yoo, J.J.; Atala, A. Tubularized urethral replacement with unseeded matrices: What is the maximum distance for normal tissue regeneration? World J. Urol. 2008, 26, 323–326. [Google Scholar] [CrossRef]
- Atala, A. Engineering organs. Curr. Opin. Biotechnol. 2009, 20, 575–592. [Google Scholar] [CrossRef] [PubMed]
- Veiseh, O.; Doloff, J.; Ma, M.; Vegas, A.F.; Tam, H.H.; Bader, A.R.; Li, J.; Langan, E.; Wyckoff, J.; Loo, W.; et al. Size- and shape-dependent foreign body immune response to materials implanted in rodents and non-human primates. Nat. Mater. 2015, 14, 643–651. [Google Scholar] [CrossRef] [Green Version]
- Vroman, L.; Adams, A.L.; Fischer, G.C.; Munoz, P.C. Interaction of high molecular weight kininogen, factor XII, and fibrinogen in plasma at interfaces. Blood 1980, 55, 156–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swartzlander, M.D.; Barnes, C.A.; Blakney, A.K.; Kaar, J.L.; Kyriakides, T.R.; Bryant, S.J. Linking the foreign body response and protein adsorption to PEG-based hydrogels using proteomics. Biomaterials 2015, 41, 26–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thang, L.; Thevenot, P.; Hu, W. Surface Chemistry Influences Implant Biocompatibility. Curr. Top. Med. Chem. 2008, 8, 270–280. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.; Loppnow, H.; Groth, T. A macrophage/fibroblast co-culture system using a cell migration chamber to study inflammatory effects of biomaterials. Acta Biomater. 2015, 26, 54–63. [Google Scholar] [CrossRef]
- Sussman, E.; Halpin, M.C.; Muster, J.; Moon, R.; Ratner, B.D. Porous Implants Modulate Healing and Induce Shifts in Local Macrophage Polarization in the Foreign Body Reaction. Ann. Biomed. Eng. 2014, 42, 1508–1516. [Google Scholar] [CrossRef] [PubMed]
- Garg, K.; Pullen, N.; Oskeritzian, C.A.; Ryan, J.J.; Bowlin, G.L. Macrophage functional polarization (M1/M2) in response to varying fiber and pore dimensions of electrospun scaffolds. Biomaterials 2013, 34, 4439–4451. [Google Scholar] [CrossRef] [Green Version]
- Noskovicova, N.; Schuster, R.; van Putten, S.; Ezzo, M.; Koehler, A.; Boo, S.; Coelho, N.M.; Griggs, D.; Ruminski, P.; McCulloch, C.A.; et al. Suppression of the fibrotic encapsulation of silicone implants by inhibiting the mechanical activation of pro-fibrotic TGF-beta. Nat. Biomed. Eng. 2021. [Google Scholar] [CrossRef] [PubMed]
- Blakney, A.K.; Swartzlander, M.D.; Bryant, S.J. The effects of substrate stiffness on the in vitro activation of macrophages and in vivo host response to poly(ethylene glycol)-based hydrogels. J. Biomed. Mater. Res. A 2012, 100, 1375–1386. [Google Scholar] [CrossRef] [Green Version]
- Prasad, B.R.; Brook, M.A.; Smith, T.; Zhao, S.; Chen, Y.; Sheardown, H.; D’Souza, R.; Rochev, Y. Controlling cellular activity by manipulating silicone surface roughness. Colloids Surf. B Biointerfaces 2010, 78, 237–242. [Google Scholar] [CrossRef]
- Ananth, H.; Kundapur, V.; Mohammed, H.S.; Anand, M.; Amarnath, G.S.; Mankar, S. A Review on Biomaterials in Dental Implantology. Int. J. Biomed. Sci. IJBS 2015, 11, 113–120. [Google Scholar] [PubMed]
- Ermis, M.; Antmen, E.; Hasirci, V. Micro and Nanofabrication methods to control cell-substrate interactions and cell behavior: A review from the tissue engineering perspective. Bioact. Mater. 2018, 3, 355–369. [Google Scholar] [CrossRef] [PubMed]
- Doloff, J.C.; Veiseh, O.; de Mezerville, R.; Sforza, M.; Perry, T.A.; Haupt, J.; Jamiel, M.; Chambers, C.; Nash, A.; Aghlara-Fotovat, S.; et al. The surface topography of silicone breast implants mediates the foreign body response in mice, rabbits and humans. Nat. Biomed. Eng. 2021. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.J.; Clegg, R.; Leavesley, D.; Pearcy, M. Mediation of Biomaterial–Cell Interactions by Adsorbed Proteins: A Review. Tissue Eng. 2005, 11, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Liamas, E.; Kubiak-Ossowska, K.; Black, R.A.; Thomas, O.R.; Zhang, Z.J.; Mulheran, P.A. Adsorption of Fibronectin Fragment on Surfaces Using Fully Atomistic Molecular Dynamics Simulations. Int. J. Mol. Sci. 2018, 19, 3321. [Google Scholar] [CrossRef] [Green Version]
- Felgueiras, H.P.; Evans, M.D.M.; Migonney, V. Contribution of fibronectin and vitronectin to the adhesion and morphology of MC3T3-E1 osteoblastic cells to poly(NaSS) grafted Ti6Al4V. Acta Biomater. 2015, 28, 225–233. [Google Scholar] [CrossRef]
- Xu, L.C.; Siedlecki, C.A. Effects of surface wettability and contact time on protein adhesion to biomaterial surfaces. Biomaterials 2007, 28, 3273–3283. [Google Scholar] [CrossRef] [Green Version]
- Wei, J.; Igarashi, T.; Okumori, N.; Igarashi, T.; Maetani, T.; Liu, B.; Yoshinari, M. Influence of surface wettability on competitive protein adsorption and initial attachment of osteoblasts. Biomed. Mater. 2009, 4, 045002. [Google Scholar] [CrossRef]
- Tan, F.; Al-Rubeai, M. Customizable Implant-specific and Tissue-Specific Extracellular Matrix Protein Coatings Fabricated Using Atmospheric Plasma. Front. Bioeng. Biotechnol. 2019, 7, 247. [Google Scholar] [CrossRef] [PubMed]
- Khandwekar, A.; Rho, C.K. Modulation of cellular responses on engineered polyurethane implants. J. Biomed. Mater. Res. Part A 2012, 100, 2211–2222. [Google Scholar] [CrossRef]
- He, Y.; Hower, J.; Chen, S.; Bernards, M.T.; Chang, Y.; Jiang, S. Molecular Simulation Studies of Protein Interactions with Zwitterionic Phosphorylcholine Self-Assembled Monolayers in the Presence of Water. Langmuir 2008, 24, 10358–10364. [Google Scholar] [CrossRef]
- Damodaran, V.B.; Murthy, N.S. Bio-inspired strategies for designing antifouling biomaterials. Biomater. Res. 2016, 20, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandor, M.; Singh, D.; Silverman, R.P.; Xu, H.; De Deyne, P.G. Comparative Host Response of 2 Human Acellular Dermal Matrices in a Primate Implant Model. Eplasty 2014, 14, e7. [Google Scholar] [PubMed]
- Mathieu, V.; Vayron, R.; Richard, G.; Lambert, G.; Naili, S.; Meningaud, J.-P.; Haiat, G. Biomechanical determinants of the stability of dental implants: Influence of the bone–implant interface properties. J. Biomech. 2014, 47, 3–13. [Google Scholar] [CrossRef]
- Vagaská, B.; Bacáková, L.; Filová, E.; Balík, K. Osteogenic cells on bio-inspired materials for bone tissue engineering. Physiol. Res. 2009, 59, 309–322. [Google Scholar]
- Tang, D.; Yang, L.-Y.; Ou, K.-L.; Oreffo, R. Repositioning Titanium: An In Vitro Evaluation of Laser-Generated Microporous, Microrough Titanium Templates as a Potential Bridging Interface for Enhanced Osseointegration and Durability of Implants. Front. Bioeng. Biotechnol. 2017, 5, 77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, J.Y.; Hansen, J.C.; Siedlecki, C.A.; Runt, J.; Donahue, H.J. Human foetal osteoblastic cell response to polymer-demixed nanotopographic interfaces. J. R. Soc. Interface 2005, 2, 97–108. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Chen, S.E.; Shao, J.; Beucken, J.J.J.P.V.D. Combinatorial Surface Roughness Effects on Osteoclastogenesis and Osteogenesis. ACS Appl. Mater. Interfaces 2018, 10, 36652–36663. [Google Scholar] [CrossRef] [Green Version]
- Harvey, A.G.; Hill, E.; Bayat, A. Designing implant surface topography for improved biocompatibility. Expert Rev. Med. Devices 2013, 10, 257–267. [Google Scholar] [CrossRef]
- Wixtrom, R.N.; Garadi, V.; Leopold, J.; Canady, J.W. Device-Specific Findings of Imprinted-Texture Breast Implants: Characteristics, Risks, and Benefits. Aesthetic Surg. J. 2019, 40, 167–173. [Google Scholar] [CrossRef]
- Wong, C.-H.; Samuel, M.; Tan, B.-K.; Song, C. Capsular Contracture in Subglandular Breast Augmentation with Textured versus Smooth Breast Implants: A Systematic Review. Plast. Reconstr. Surg. 2006, 118, 1224–1236. [Google Scholar] [CrossRef]
- Liu, X.; Zhou, L.; Pan, F.; Gao, Y.; Yuan, X.; Fan, D. Comparison of the Postoperative Incidence Rate of Capsular Contracture among Different Breast Implants: A Cumulative Meta-Analysis. PLoS ONE 2015, 10, e0116071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, B.H.; Kim, B.H.; Kim, S.; Lee, K.; Bin Choy, Y.; Heo, C.Y. Silicone breast implant modification review: Overcoming capsular contracture. Biomater. Res. 2018, 22, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnsley, G.P.; Sigurdson, L.J.; Barnsley, S.E. Textured Surface Breast Implants in the Prevention of Capsular Contracture among Breast Augmentation Patients: A Meta-Analysis of Randomized Controlled Trials. Plast. Reconstr. Surg. 2006, 117, 2182–2190. [Google Scholar] [CrossRef] [PubMed]
- Stevens, W.G.; Nahabedian, M.Y.; Calobrace, M.B.; Harrington, J.L.; Capizzi, P.J.; Cohen, R.; d’Incelli, R.C.; Beckstrand, M. Risk factor analysis for capsular contracture: A 5-year Sientra study analysis using round, smooth, and textured implants for breast augmentation. Plast Reconstr Surg. 2013, 132, 1115–1123. [Google Scholar] [CrossRef] [PubMed]
- Derby, B.M.; Codner, M.A. Textured silicone breast implant use in primary augmentation: Core data update and review. Plast. Reconstr. Surg. 2015, 135, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Barr, S.; Hill, E.; Bayat, A. Development, fabrication and evaluation of a novel biomimetic human breast tissue derived breast implant surface. Acta Biomater. 2017, 49, 260–271. [Google Scholar] [CrossRef] [Green Version]
- Loch-Wilkinson, A.; Beath, K.J.; Knight, R.J.W.; Wessels, W.L.F.; Magnusson, M.; Papadopoulos, T.; Connell, T.; Lofts, J.; Locke, M.; Hopper, I.; et al. Breast Implant-Associated Anaplastic Large Cell Lymphoma in Australia and New Zealand: High-Surface-Area Textured Implants Are Associated with Increased Risk. Plast. Reconstr. Surg. 2017, 140, 645–654. [Google Scholar] [CrossRef]
- Tevis, S.E.; Hunt, K.K.; Miranda, R.N.; Lange, C.; Pinnix, C.C.; Iyer, S.; Butler, C.E.; Clemens, M.W. Breast Implant-Associated Anaplastic Large Cell Lymphoma: A Prospective Series of 52 Patients. Ann. Surg. 2020. [Google Scholar] [CrossRef]
- Sforza, M.; Zaccheddu, R.; Alleruzzo, A.; Seno, A.; Mileto, D.; Paganelli, A.; Sulaiman, H.; Payne, M.; Maurovich-Horvat, L. Preliminary 3-Year Evaluation of Experience with SilkSurface and VelvetSurface Motiva Silicone Breast Implants: A Single-Center Experience With 5813 Consecutive Breast Augmentation Cases. Aesthetic Surg. J. 2018, 38, S62–S73. [Google Scholar] [CrossRef]
- Wolf, M.T.; Dearth, C.L.; Ranallo, C.A.; LoPresti, S.T.; Carey, L.E.; Daly, K.A.; Brown, B.N.; Badylak, S.F. Macrophage polarization in response to ECM coated polypropylene mesh. Biomaterials 2014, 35, 6838–6849. [Google Scholar] [CrossRef] [Green Version]
- Bracaglia, L.G.; Fisher, J.P. Extracellular Matrix-Based Biohybrid Materials for Engineering Compliant, Matrix-Dense Tissues. Adv. Heal. Mater. 2015, 4, 2475–2487. [Google Scholar] [CrossRef] [Green Version]
- Oakes, R.; Polei, M.D.; Skousen, J.L.; Tresco, P.A. An astrocyte derived extracellular matrix coating reduces astrogliosis surrounding chronically implanted microelectrode arrays in rat cortex. Biomaterials 2018, 154, 1–11. [Google Scholar] [CrossRef]
- Robotti, F.; Bottan, S.; Fraschetti, F.; Mallone, A.; Pellegrini, G.; Lindenblatt, N.; Starck, C.; Falk, V.; Poulikakos, D.; Ferrari, A. A micron-scale surface topography design reducing cell adhesion to implanted materials. Sci. Rep. 2018, 8, 10887. [Google Scholar] [CrossRef] [PubMed]
- Klos, A.; Sedao, X.; Itina, T.E.; Helfenstein-Didier, C.; Donnet, C.; Peyroche, S.; Vico, L.; Guignandon, A.; Dumas, V. Ultrafast Laser Processing of Nanostructured Patterns for the Control of Cell Adhesion and Migration on Titanium Alloy. Nanomaterials 2020, 10, 864. [Google Scholar] [CrossRef]
- Padmanabhan, J.; Kinser, E.R.; Stalter, M.A.; Duncan-Lewis, C.; Balestrini, J.L.; Sawyer, A.J.; Schroers, J.; Kyriakides, T.R. Engineering Cellular Response Using Nanopatterned Bulk Metallic Glass. ACS Nano 2014, 8, 4366–4375. [Google Scholar] [CrossRef]
- Tan, J.L.; Tien, J.; Pirone, D.M.; Gray, D.S.; Bhadriraju, K.; Chen, C.S. Cells lying on a bed of microneedles: An approach to isolate mechanical force. Proc. Natl. Acad. Sci. USA 2003, 100, 1484–1489. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Wang, D.; Peng, F.; Qiu, J.; Ouyang, L.; Qiao, Y.; Liu, X. Nanostructural Surfaces with Different Elastic Moduli Regulate the Immune Response by Stretching Macrophages. Nano Lett. 2019, 19, 3480–3489. [Google Scholar] [CrossRef] [PubMed]
- Achterberg, V.F.; Buscemi, L.; Diekmann, H.; Smith-Clerc, J.; Schwengler, H.; Meister, J.-J.; Wenck, H.; Gallinat, S.; Hinz, B. The Nano-Scale Mechanical Properties of the Extracellular Matrix Regulate Dermal Fibroblast Function. J. Investig. Dermatol. 2014, 134, 1862–1872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christ, A.F.; Franze, K.; Gautier, H.O.B.; Moshayedi, P.; Fawcett, J.; Franklin, R.; Karadottir, R.T.; Guck, J. Mechanical difference between white and gray matter in the rat cerebellum measured by scanning force microscopy. J. Biomech. 2010, 43, 2986–2992. [Google Scholar] [CrossRef] [PubMed]
- Butcher, D.T.; Alliston, T.; Weaver, V.M. A tense situation: Forcing tumour progression. Nat. Rev. Cancer 2009, 9, 108–122. [Google Scholar] [CrossRef]
- Georges, P.; Hui, J.-J.; Gombos, Z.; McCormick, M.E.; Wang, A.Y.; Uemura, M.; Mick, R.; Janmey, P.A.; Furth, E.E.; Wells, R.G. Increased stiffness of the rat liver precedes matrix deposition: Implications for fibrosis. Am. J. Physiol. Liver Physiol. 2007, 293, G1147–G1154. [Google Scholar] [CrossRef] [PubMed]
- Majkut, S.F.; Discher, D.E. Cardiomyocytes from late embryos and neonates do optimal work and striate best on substrates with tissue-level elasticity: Metrics and mathematics. Biomech. Model. Mechanobiol. 2012, 11, 1219–1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, S.P.; Marshall, S.J.; Ryder, M.I.; Marshall, G.W. The tooth attachment mechanism defined by structure, chemical composition and mechanical properties of collagen fibers in the periodontium. Biomaterials 2007, 28, 5238–5245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janmey, P.A.; Fletcher, D.A.; Reinhart-King, C.A. Stiffness Sensing by Cells. Physiol. Rev. 2020, 100, 695–724. [Google Scholar] [CrossRef]
- Discher, D.E.; Smith, L.; Cho, S.; Colasurdo, M.; Garcia, A.J.; Safran, S. Matrix Mechanosensing: From Scaling Concepts in Omics Data to Mechanisms in the Nucleus, Regeneration, and Cancer. Annu. Rev. Biophys. 2017, 46, 295–315. [Google Scholar] [CrossRef]
- Franze, K.; Janmey, P.A.; Guck, J. Mechanics in Neuronal Development and Repair. Annu. Rev. Biomed. Eng. 2013, 15, 227–251. [Google Scholar] [CrossRef]
- Cox, T.R.; Erler, J.T. Remodeling and homeostasis of the extracellular matrix: Implications for fibrotic diseases and cancer. Dis. Model. Mech. 2011, 4, 165–178. [Google Scholar] [CrossRef] [Green Version]
- Minev, I.R.; Musienko, P.; Hirsch, A.; Barraud, Q.; Wenger, N.; Moraud, E.M.; Gandar, J.; Capogrosso, M.; Milekovic, T.; Asboth, L.; et al. Electronic dura mater for long-term multimodal neural interfaces. Science 2015, 347, 159–163. [Google Scholar] [CrossRef] [Green Version]
- Moshayedi, P.; Ng, G.; Kwok, J.; Yeo, G.S.H.; Bryant, C.E.; Fawcett, J.; Franze, K.; Guck, J. The relationship between glial cell mechanosensitivity and foreign body reactions in the central nervous system. Biomaterials 2014, 35, 3919–3925. [Google Scholar] [CrossRef] [Green Version]
- Sindhu, K.; Bansode, N.; Rémy, M.; Morel, C.; Bareille, R.; Hagedorn, M.; Hinz, B.; Barthélémy, P.; Chassande, O.; Boiziau, C. New injectable self-assembled hydrogels that promote angiogenesis through a bioactive degradation product. Acta Biomater. 2020, 115, 197–209. [Google Scholar] [CrossRef]
- LeComte, A.; Descamps, E.; Bergaud, C. A review on mechanical considerations for chronically-implanted neural probes. J. Neural. Eng. 2018, 15, 031001. [Google Scholar] [CrossRef]
- Du, Z.J.; Kolarcik, C.L.; Kozai, T.D.; Luebben, S.D.; Sapp, S.A.; Zheng, X.S.; Nabity, J.A.; Cui, X.T. Ultrasoft microwire neural electrodes improve chronic tissue integration. Acta Biomater. 2017, 53, 46–58. [Google Scholar] [CrossRef] [Green Version]
- Doloff, J.; Veiseh, O.; Vegas, A.J.; Tam, H.H.; Farah, S.; Ma, M.; Li, J.; Bader, A.; Chiu, A.; Sadraei, A.; et al. Colony stimulating factor-1 receptor is a central component of the foreign body response to biomaterial implants in rodents and non-human primates. Nat. Mater. 2017, 16, 671–680. [Google Scholar] [CrossRef] [PubMed]
- Miron, R.J.; Bosshardt, D.D. Multinucleated Giant Cells: Good Guys or Bad Guys? Tissue Eng. Part B Rev. 2018, 24, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Dondossola, E.; Holzapfel, B.M.; Alexander, S.; Filippini, S.; Hutmacher, D.W.; Friedl, P. Examination of the foreign body response to biomaterials by nonlinear intravital microscopy. Nat. Biomed. Eng. 2017, 1, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spiller, K.L.; Anfang, R.R.; Spiller, K.J.; Ng, J.; Nakazawa, K.R.; Daulton, J.; Vunjak-Novakovic, G. The role of macrophage phenotype in vascularization of tissue engineering scaffolds. Biomaterials 2014, 35, 4477–4488. [Google Scholar] [CrossRef] [Green Version]
- Anderson, J.M.; McNally, A.K. Biocompatibility of implants: Lymphocyte/macrophage interactions. Semin. Immunopathol. 2011, 33, 221–233. [Google Scholar] [CrossRef]
- Chung, L.; Maestas, D.R., Jr.; Housseau, F.; Elisseeff, J.H. Key players in the immune response to biomaterial scaffolds for regenerative medicine. Adv. Drug Deliv. Rev. 2017, 114, 184–192. [Google Scholar] [CrossRef]
- Pakshir, P.; Hinz, B. The big five in fibrosis: Macrophages, myofibroblasts, matrix, mechanics, and miscommunication. Matrix Biol. 2018, 68, 81–93. [Google Scholar] [CrossRef]
- Mooney, J.E.; Rolfe, B.; Osborne, G.; Sester, D.P.; van Rooijen, N.; Campbell, G.R.; Hume, D.; Campbell, J.H. Cellular Plasticity of Inflammatory Myeloid Cells in the Peritoneal Foreign Body Response. Am. J. Pathol. 2010, 176, 369–380. [Google Scholar] [CrossRef]
- Smith, T.D.; Nagalla, R.R.; Chen, E.Y.; Liu, W.F. Harnessing macrophage plasticity for tissue regeneration. Adv. Drug Deliv. Rev. 2017, 114, 193–205. [Google Scholar] [CrossRef]
- Martin, K.E.; García, A.J. Macrophage phenotypes in tissue repair and the foreign body response: Implications for biomaterial-based regenerative medicine strategies. Acta Biomater. 2021. [Google Scholar] [CrossRef] [PubMed]
- Graney, P.; Lurier, E.B.; Spiller, K.L. Biomaterials and Bioactive Factor Delivery Systems for the Control of Macrophage Activation in Regenerative Medicine. ACS Biomater. Sci. Eng. 2017, 4, 1137–1148. [Google Scholar] [CrossRef] [PubMed]
- Bonner, J.C. Regulation of PDGF and its receptors in fibrotic diseases. Cytokine Growth Factor Rev. 2004, 15, 255–273. [Google Scholar] [CrossRef] [PubMed]
- Sadtler, K.; Wolf, M.T.; Ganguly, S.; Moad, C.A.; Chung, L.; Majumdar, S.; Housseau, F.; Pardoll, D.M.; Elisseeff, J.H. Divergent immune responses to synthetic and biological scaffolds. Biomaterials 2019, 192, 405–415. [Google Scholar] [CrossRef]
- McNally, A.K.; Anderson, J.M. Phenotypic expression in human monocyte-derived interleukin-4-induced foreign body giant cells and macrophages in vitro: Dependence on material surface properties. J. Biomed. Mater. Res. A 2015, 103, 1380–1390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheikh, Z.; Brooks, P.J.; Barzilay, O.; Fine, N.; Glogauer, M. Macrophages, Foreign Body Giant Cells and Their Response to Implantable Biomaterials. Materials 2015, 8, 5671–5701. [Google Scholar] [CrossRef] [Green Version]
- Milde, R.; Ritter, J.; Tennent, G.A.; Loesch, A.; Martinez, F.O.; Gordon, S.; Pepys, M.B.; Verschoor, A.; Helming, L. Multinucleated Giant Cells Are Specialized for Complement-Mediated Phagocytosis and Large Target Destruction. Cell Rep. 2015, 13, 1937–1948. [Google Scholar] [CrossRef] [Green Version]
- Aiyelabegan, H.T.; Sadroddiny, E. Fundamentals of protein and cell interactions in biomaterials. Biomed. Pharmacother. 2017, 88, 956–970. [Google Scholar] [CrossRef]
- Altieri, D.C.; Mannucci, P.M.; Capitanio, A.M. Binding of fibrinogen to human monocytes. J. Clin. Investig. 1986, 78, 968–976. [Google Scholar] [CrossRef] [Green Version]
- Love, R.J.; Jones, K.S. The recognition of biomaterials: Pattern recognition of medical polymers and their adsorbed biomolecules. J. Biomed. Mater. Res. Part A 2013, 101, 2740–2752. [Google Scholar] [CrossRef]
- Lee, T.T.; Garcia, J.R.; Paez, J.I.; Singh, A.; Phelps, E.A.; Weis, S.; Shafiq, Z.; Shekaran, A.; Del Campo, A.; Garcia, A.; et al. Light-triggered in vivo activation of adhesive peptides regulates cell adhesion, inflammation and vascularization of biomaterials. Nat. Mater. 2015, 14, 352–360. [Google Scholar] [CrossRef]
- Helming, L.; Gordon, S. Molecular mediators of macrophage fusion. Trends Cell Biol. 2009, 19, 514–522. [Google Scholar] [CrossRef] [PubMed]
- Sridharan, R.; Cavanagh, B.; Cameron, A.R.; Kelly, D.J.; O’Brien, F.J. Material stiffness influences the polarization state, function and migration mode of macrophages. Acta Biomater. 2019, 89, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Dutta, B.; Goswami, R.; Rahaman, S.O. TRPV4 Plays a Role in Matrix Stiffness-Induced Macrophage Polarization. Front. Immunol. 2020, 11, 3150. [Google Scholar] [CrossRef]
- Arya, R.K.; Goswami, R.; Rahaman, S.O. Mechanotransduction via a TRPV4-Rac1 signaling axis plays a role in multinucleated giant cell formation. J. Biol. Chem. 2021, 296, 100129. [Google Scholar] [CrossRef] [PubMed]
- Boersema, G.S.; Grotenhuis, N.; Bayon, Y.; Lange, J.F.; Bastiaansen-Jenniskens, Y.M. The Effect of Biomaterials Used for Tissue Regeneration Purposes on Polarization of Macrophages. Biores. Open Access 2016, 5, 6–14. [Google Scholar] [CrossRef] [PubMed]
- Abaricia, J.O.; Shah, A.H.; Chaubal, M.; Hotchkiss, K.M.; Olivares-Navarrete, R. Wnt signaling modulates macrophage polarization and is regulated by biomaterial surface properties. Biomaterials 2020, 243, 119920. [Google Scholar] [CrossRef]
- Zhang, Y.; Cheng, X.; Jansen, J.A.; Yang, F.; Beucken, J.J.V.D. Titanium surfaces characteristics modulate macrophage polarization. Mater. Sci. Eng. C 2019, 95, 143–151. [Google Scholar] [CrossRef]
- Wang, T.; Luu, T.U.; Chen, A.; Khine, M.; Liu, W.F. Topographical modulation of macrophage phenotype by shrink-film multi-scale wrinkles. Biomater. Sci. 2016, 4, 948–952. [Google Scholar] [CrossRef]
- Luu, T.; Gott, S.C.; Woo, B.W.K.; Rao, M.; Liu, W.F. Micro- and Nanopatterned Topographical Cues for Regulating Macrophage Cell Shape and Phenotype. ACS Appl. Mater. Interfaces 2015, 7, 28665–28672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McWhorter, F.; Wang, T.; Nguyen, P.; Chung, T.; Liu, W.F. Modulation of macrophage phenotype by cell shape. Proc. Natl. Acad. Sci. USA 2013, 110, 17253–17258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Jones, J.A.; Xu, Y.; Low, H.-Y.; Anderson, J.M.; Leong, K.W. Characterization of topographical effects on macrophage behavior in a foreign body response model. Biomaterials 2010, 31, 3479–3491. [Google Scholar] [CrossRef] [PubMed]
- Moon, H.; Cremmel, C.V.M.; Kulpa, A.; Jaeger, N.A.F.; Kappelhoff, R.; Overall, C.M.; Waterfield, J.D.; Brunette, D.M. Novel grooved substrata stimulate macrophage fusion, CCL2 and MMP-9 secretion. J. Biomed. Mater. Res. Part A 2016, 104, 2243–2254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shayan, M.; Padmanabhan, J.; Morris, A.H.; Cheung, B.; Smith, R.; Schroers, J.; Kyriakides, T.R. Nanopatterned bulk metallic glass-based biomaterials modulate macrophage polarization. Acta Biomater. 2018, 75, 427–438. [Google Scholar] [CrossRef]
- Mohiuddin, M.; Pan, H.-A.; Hung, Y.-C.; Huang, G.S. Control of growth and inflammatory response of macrophages and foam cells with nanotopography. Nanoscale Res. Lett. 2012, 7, 394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Padmanabhan, J.; Augelli, M.J.; Cheung, B.; Kinser, E.R.; Cleary, B.; Kumar, P.; Wang, R.; Sawyer, A.J.; Li, R.; Schwarz, U.; et al. Regulation of cell-cell fusion by nanotopography. Sci. Rep. 2016, 6, 33277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Previtera, M.L.; Sengupta, A. Substrate Stiffness Regulates Proinflammatory Mediator Production through TLR4 Activity in Macrophages. PLoS ONE 2015, 10, e0145813. [Google Scholar] [CrossRef] [PubMed]
- Irwin, E.F.; Saha, K.; Rosenbluth, M.; Gamble, L.J.; Castner, D.G.; Healy, K.E. Modulus-dependent macrophage adhesion and behavior. J. Biomater. Sci. Polym. Ed. 2008, 19, 1363–1382. [Google Scholar] [CrossRef] [Green Version]
- Adlerz, K.M.; Aranda-Espinoza, H.; Hayenga, H.N. Substrate elasticity regulates the behavior of human monocyte-derived macrophages. Eur. Biophys. J. 2016, 45, 301–309. [Google Scholar] [CrossRef]
- Féréol, S.; Fodil, R.; Labat, B.; Galiacy, S.; Laurent, V.M.; Louis, B.; Isabey, D.; Planus, E. Sensitivity of alveolar macrophages to substrate mechanical and adhesive properties. Cell Motil. Cytoskelet. 2006, 63, 321–340. [Google Scholar] [CrossRef]
- Scott, R.A.; Kiick, K.L.; Akins, R.E. Substrate stiffness directs the phenotype and polarization state of cord blood derived macrophages. Acta Biomater. 2021, 122, 220–235. [Google Scholar] [CrossRef] [PubMed]
- Patel, N.R.; Bole, M.; Chen, C.; Hardin, C.C.; Kho, A.T.; Mih, J.; Deng, L.; Butler, J.; Tschumperlin, D.; Fredberg, J.J.; et al. Cell elasticity determines macrophage function. PLoS ONE 2012, 7, e41024. [Google Scholar] [CrossRef] [Green Version]
- Fang, J.Y.; Yang, Z.; Han, B. Switch of macrophage fusion competency by 3D matrices. Sci. Rep. 2020, 10, 10348. [Google Scholar] [CrossRef]
- Akilbekova, D.; Bratlie, K.M. Quantitative Characterization of Collagen in the Fibrotic Capsule Surrounding Implanted Polymeric Microparticles through Second Harmonic Generation Imaging. PLoS ONE 2015, 10, e0130386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, J.H.; Efendy, J.L.; Campbell, G.R. Novel Vascular Graft Grown Within Recipient’s Own Peritoneal Cavity. Circ. Res. 1999, 85, 1173–1178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duffield, J.S.; Lupher, M.L.; Thannickal, V.J.; Wynn, T.A. Host Responses in Tissue Repair and Fibrosis. Annu. Rev. Pathol. Mech. Dis. 2013, 8, 241–276. [Google Scholar] [CrossRef] [Green Version]
- Schuppan, D.; Ashfaq-Khan, M.; Yang, A.T.; Kim, Y.O. Liver fibrosis: Direct antifibrotic agents and targeted therapies. Matrix Biol. 2018, 68-69, 435–451. [Google Scholar] [CrossRef]
- Fell, S.; Wang, Z.; Blanchard, A.; Nanthakumar, C.; Griffin, M. Transglutaminase 2: A novel therapeutic target for idiopathic pulmonary fibrosis using selective small molecule inhibitors. Amino Acids 2021, 53, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Hauser, S.; Wodtke, R.; Tondera, C.; Wodtke, J.; Neffe, A.T.; Hampe, J.; Lendlein, A.; Löser, R.; Pietzsch, J. Characterization of Tissue Transglutaminase as a Potential Biomarker for Tissue Response toward Biomaterials. ACS Biomater. Sci. Eng. 2018, 5, 5979–5989. [Google Scholar] [CrossRef] [PubMed]
- Piersma, B.; Bank, R.A. Collagen cross-linking mediated by lysyl hydroxylase 2: An enzymatic battlefield to combat fibrosis. Essays Biochem. 2019, 63, 377–387. [Google Scholar] [CrossRef] [PubMed]
- Vallet, S.D.; Ricard-Blum, S. Lysyl oxidases: From enzyme activity to extracellular matrix cross-links. Essays Biochem. 2019, 63, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Li, S.; Li, W. LOX/LOXL in pulmonary fibrosis: Potential therapeutic targets. J. Drug Target. 2018, 27, 790–796. [Google Scholar] [CrossRef]
- Tschumperlin, D.J.; Lagares, D. Mechano-therapeutics: Targeting Mechanical Signaling in Fibrosis and Tumor Stroma. Pharmacol. Ther. 2020, 212, 107575. [Google Scholar] [CrossRef]
- Hinz, B.; McCulloch, C.A.; Coelho, N.M. Mechanical regulation of myofibroblast phenoconversion and collagen contraction. Exp. Cell Res. 2019, 379, 119–128. [Google Scholar] [CrossRef]
- Klingberg, F.; Hinz, B.; White, E.S. The myofibroblast matrix: Implications for tissue repair and fibrosis. J. Pathol. 2013, 229, 298–309. [Google Scholar] [CrossRef] [Green Version]
- Neff, L.S.; Bradshaw, A.D. Cross your heart? Collagen cross-links in cardiac health and disease. Cell. Signal. 2021, 79, 109889. [Google Scholar] [CrossRef] [PubMed]
- Tomasek, J.J.; Gabbiani, G.; Hinz, B.; Chaponnier, C.; Brown, R.A. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat. Rev. Mol. Cell Biol. 2002, 3, 349–363. [Google Scholar] [CrossRef]
- Hinz, B.; Celetta, G.; Tomasek, J.J.; Gabbiani, G.; Chaponnier, C. Alpha-Smooth Muscle Actin Expression Upregulates Fibroblast Contractile Activity. Mol. Biol. Cell 2001, 12, 2730–2741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinz, B.; Dugina, V.; Ballestrem, C.; Wehrle-Haller, B.; Chaponnier, C. Alpha-smooth muscle actin Is crucial for focal adhesion maturation in myofibroblasts. Mol. Biol. Cell. 2003, 14, 2508–2519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goffin, J.M.; Pittet, P.; Csucs, G.; Lussi, J.W.; Meister, J.-J.; Hinz, B. Focal adhesion size controls tension-dependent recruitment of α-smooth muscle actin to stress fibers. J. Cell Biol. 2006, 172, 259–268. [Google Scholar] [CrossRef] [Green Version]
- Godbout, C.; Castella, L.F.; Smith, E.A.; Talele, N.; Chow, M.L.; Garonna, A.; Hinz, B. The Mechanical Environment Modulates Intracellular Calcium Oscillation Activities of Myofibroblasts. PLoS ONE 2013, 8, e64560. [Google Scholar] [CrossRef] [PubMed]
- Smithmyer, M.E.; Sawicki, L.A.; Kloxin, A.M. Hydrogel scaffolds as in vitro models to study fibroblast activation in wound healing and disease. Biomater. Sci. 2014, 2, 634–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Majd, H.; Scherer, S.S.; Boo, S.; Ramondetti, S.; Cambridge, E.; Raffoul, W.; Friedrich, M.; Pittet, B.; Pioletti, D.; Hinz, B.; et al. Novel micropatterns mechanically control fibrotic reactions at the surface of silicone implants. Biomaterials 2015, 54, 136–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kyle, D.J.; Oikonomou, A.; Hill, E.; Bayat, A. Development and functional evaluation of biomimetic silicone surfaces with hierarchical micro/nano-topographical features demonstrates favourable in vitro foreign body response of breast-derived fibroblasts. Biomaterials 2015, 52, 88–102. [Google Scholar] [CrossRef]
- Rockey, D.C.; Bell, P.D.; Hill, J.A. Fibrosis—A Common Pathway to Organ Injury and Failure. N. Engl. J. Med. 2015, 372, 1138–1149. [Google Scholar] [CrossRef]
- Fernández-Colino, A.; Iop, L.; Ferreira, M.S.V.; Mela, P. Fibrosis in tissue engineering and regenerative medicine: Treat or trigger? Adv. Drug Deliv. Rev. 2019, 146, 17–36. [Google Scholar] [CrossRef]
- Henderson, N.C.; Rieder, F.; Wynn, T.A. Fibrosis: From mechanisms to medicines. Nat. Cell Biol. 2020, 587, 555–566. [Google Scholar] [CrossRef]
- Wynn, T.A.; Ramalingam, T.R. Mechanisms of fibrosis: Therapeutic translation for fibrotic disease. Nat. Med. 2012, 18, 1028–1040. [Google Scholar] [CrossRef] [Green Version]
- Love, R.J.; Jones, K.S. Biomaterials, fibrosis, and the use of drug delivery systems in future antifibrotic strategies. Crit. Rev. Biomed. Eng. 2009, 37, 259–281. [Google Scholar] [CrossRef]
- Gancedo, M.; Ruiz-Corro, L.; Salazar-Montes, A.; Rincón, A.R.; Armendáriz-Borunda, J. Pirfenidone Prevents Capsular Contracture After Mammary Implantation. Aesthetic Plast. Surg. 2008, 32, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Henderson, N.C.; Arnold, T.D.; Katamura, Y.; Giacomini, M.M.; Rodriguez, J.D.; McCarty, J.H. Targeting of alphav integrin identifies a core molecular pathway that regulates fibrosis in several organs. Nat. Med. 2013, 19, 1617–1624. [Google Scholar] [CrossRef] [Green Version]
- Rujitanaroj, P.-O.; Jao, B.; Yang, R.; Wang, F.; Anderson, J.M.; Wang, J.; Chew, S.Y. Controlling fibrous capsule formation through long-term down-regulation of collagen type I (COL1A1) expression by nanofiber-mediated siRNA gene silencing. Acta Biomater. 2013, 9, 4513–4524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lodyga, M.; Hinz, B. TGF-beta1—A truly transforming growth factor in fibrosis and immunity. Semin. Cell Dev. Biol. 2020, 101, 123–139. [Google Scholar] [CrossRef] [PubMed]
- Hinz, B. Targeting the myofibroblast to improve wound healing. Volume I: “Therapies and regeneration“. In Wound Healing Biomaterials; Ågren, M.S., Ed.; Woodhead Publishing Limited: Cambridge, UK; Springer Science and Business Media Publishing: Cambridge, UK, 2016; pp. 69–100. [Google Scholar]
- Distler, J.H.W.; Gyorfi, A.H.; Ramanujam, M.; Whitfield, M.L.; Konigshoff, M.; Lafyatis, R. Shared and distinct mechanisms of fibrosis. Nat. Rev. Rheumatol. 2019, 15, 705–730. [Google Scholar] [CrossRef]
- Takahashi, H.; Wang, Y.; Grainger, D.W. Device-based local delivery of siRNA against mammalian target of rapamycin (mTOR) in a murine subcutaneous implant model to inhibit fibrous encapsulation. J. Control Release 2010, 147, 400–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saika, S.; Kawashima, Y.; Miyamoto, T.; Okada, Y.; Ichitanakaa, S.-; Ohmi, S.; Minamide, A.; Yamanaka, O.; Ohnishi, Y.; Ooshima, A.; et al. Immunolocalization of Prolyl 4-Hydroxylase Subunits, α-Smooth Muscle Actin, and Extracellular Matrix Components in Human Lens Capsules with Lens Implants. Exp. Eye Res. 1998, 66, 283–294. [Google Scholar] [CrossRef]
- Zeplin, P.H.; Larena-Avellaneda, A.; Schmidt, K. Surface Modification of Silicone Breast Implants by Binding the Antifibrotic Drug Halofuginone Reduces Capsular Fibrosis. Plast. Reconstr. Surg. 2010, 126, 266–274. [Google Scholar] [CrossRef] [PubMed]
- Li, A.G.; Quinn, M.J.; Siddiqui, Y.; Wood, M.D.; Federiuk, I.F.; Duman, H.M. Elevation of transforming growth factor beta (TGFbeta) and its downstream mediators in subcutaneous foreign body capsule tissue. J. Biomed. Mater. Res. A 2007, 82, 498–508. [Google Scholar] [CrossRef]
- Avula, M.N.; Rao, A.N.; McGill, L.D.; Grainger, D.W.; Solzbacher, F. Modulation of the foreign body response to implanted sensor models through device-based delivery of the tyrosine kinase inhibitor, masitinib. Biomaterials 2013, 34, 9737–9746. [Google Scholar] [CrossRef]
- Ward, W.K.; Li, A.G.; Siddiqui, Y.; Federiuk, I.F.; Wang, X.-J. Increased expression of Interleukin-13 and connective tissue growth factor, and their potential roles during foreign body encapsulation of subcutaneous implants. J. Biomater. Sci. Polym. Ed. 2008, 19, 1065–1072. [Google Scholar] [CrossRef]
- Castro, P.; Marques, S.M.; Viana, C.T.; Campos, P.P.; Ferreira, M.A.; Barcelos, L.S.; Andrade, S.P. Deletion of the chemokine receptor CCR2 attenuates foreign body reaction to implants in mice. Microvasc. Res. 2014, 95, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.A.; McNally, A.K.; Chang, D.T.; Qin, L.A.; Meyerson, H.; Colton, E.; Kwon, I.K.; Matsuda, T.; Anderson, J.M. Matrix metalloproteinases and their inhibitors in the foreign body reaction on biomaterials. J. Biomed. Mater. Res. Part A 2008, 84, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Ouyang, L.; Ci, L.; Chen, B.; Lv, D.; Li, Q.; Sun, Y.; Fei, J.; Bao, S.; Liu, X.; et al. Pravastatin regulates host foreign-body reaction to polyetheretherketone implants via miR-29ab1-mediated SLIT3 upregulation. Biomaterials 2019, 203, 12–22. [Google Scholar] [CrossRef]
- Klueh, U.; Dorsky, D.I.; Kreutzer, D.L. Enhancement of implantable glucose sensor function in vivo using gene transfer-induced neovascularization. Biomaterials 2005, 26, 1155–1163. [Google Scholar] [CrossRef]
- Tschumperlin, D.J.; Ligresti, G.; Hilscher, M.B.; Shah, V.H. Mechanosensing and fibrosis. J. Clin. Investig. 2018, 128, 74–84. [Google Scholar] [CrossRef] [Green Version]
- Santos, A.; Lagares, D. Matrix Stiffness: The Conductor of Organ Fibrosis. Curr. Rheumatol. Rep. 2018, 20, 2. [Google Scholar] [CrossRef]
- Kuehlmann, B.; Bonham, C.A.; Zucal, I.; Prantl, L.; Gurtner, G.C. Mechanotransduction in Wound Healing and Fibrosis. J. Clin. Med. 2020, 9, 1423. [Google Scholar] [CrossRef] [PubMed]
- Martinac, B. The ion channels to cytoskeleton connection as potential mechanism of mechanosensitivity. Biochim. Biophys. Acta (BBA) Biomembr. 2014, 1838, 682–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jo, J.; Nansa, S.A.; Kim, D.-H. Molecular Regulators of Cellular Mechanoadaptation at Cell–Material Interfaces. Front. Bioeng. Biotechnol. 2020, 8, 608569. [Google Scholar] [CrossRef]
- Sun, Z.; Guo, S.S.; Fässler, R. Integrin-mediated mechanotransduction. J. Cell Biol. 2016, 215, 445–456. [Google Scholar] [CrossRef] [PubMed]
- Bachmann, M.; Kukkurainen, S.; Hytonen, V.P.; Wehrle-Haller, B. Cell Adhesion by Integrins. Physiol. Rev. 2019, 99, 1655–1699. [Google Scholar] [CrossRef]
- Jansen, K.A.; Atherton, P.; Ballestrem, C. Mechanotransduction at the cell-matrix interface. Semin. Cell Dev. Biol. 2017, 71, 75–83. [Google Scholar] [CrossRef]
- Revach, O.-Y.; Grosheva, I.; Geiger, B. Biomechanical regulation of focal adhesion and invadopodia formation. J. Cell Sci. 2020, 133. [Google Scholar] [CrossRef] [PubMed]
- Elosegui-Artola, A.; Oria, R.; Chen, Y.; Kosmalska, A.; Pérez-González, C.; Castro, N.; Zhu, C.; Trepat, X.; Roca-Cusachs, P. Mechanical regulation of a molecular clutch defines force transmission and transduction in response to matrix rigidity. Nat. Cell Biol. 2016, 18, 540–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hintermann, E.; Christen, U. The Many Roles of Cell Adhesion Molecules in Hepatic Fibrosis. Cells 2019, 8, 1503. [Google Scholar] [CrossRef] [Green Version]
- Schnittert, J.; Bansal, R.; Storm, G.; Prakash, J. Integrins in wound healing, fibrosis and tumor stroma: High potential targets for therapeutics and drug delivery. Adv. Drug Deliv. Rev. 2018, 129, 37–53. [Google Scholar] [CrossRef]
- Schulz, J.N.; Plomann, M.; Sengle, G.; Gullberg, D.; Krieg, T.; Eckes, B. New developments on skin fibrosis—Essential signals emanating from the extracellular matrix for the control of myofibroblasts. Matrix Biol. 2018, 68, 522–532. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, H.; Hagood, J.S. Cooperative signaling between integrins and growth factor receptors in fibrosis. J. Mol. Med. 2021, 99, 213–224. [Google Scholar] [CrossRef] [PubMed]
- Martino, M.; Briquez, P.S.; Güç, E.; Tortelli, F.; Kilarski, W.W.; Metzger, S.; Rice, J.J.; Kuhn, G.A.; Müller, R.; Swartz, M.A.; et al. Growth Factors Engineered for Super-Affinity to the Extracellular Matrix Enhance Tissue Healing. Science 2014, 343, 885–888. [Google Scholar] [CrossRef]
- Wipff, P.J.; Hinz, B. Integrins and the activation of latent transforming growth factor beta1—An intimate relationship. Eur. J. Cell Biol. 2008, 87, 601–615. [Google Scholar] [CrossRef] [PubMed]
- Hinz, B. It has to be the alphav: Myofibroblast integrins activate latent TGF-beta1. Nat. Med. 2013, 19, 1567–1568. [Google Scholar] [CrossRef] [Green Version]
- Wilson, S.E. TGF beta -1, -2 and -3 in the modulation of fibrosis in the cornea and other organs. Exp. Eye Res. 2021, 207, 108594. [Google Scholar] [CrossRef]
- Kim, K.K.; Sheppard, D.; Chapman, H.A. TGF-beta1 Signaling and Tissue Fibrosis. Cold Spring Harb. Perspect. Biol. 2018, 10, a022293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rifkin, D.B.; Rifkin, W.; Zilberberg, L. LTBPs in biology and medicine: LTBP diseases. Matrix Biol. 2018, 71, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Annes, J.P.; Chen, Y.; Munger, J.S.; Rifkin, D.B. Integrin αVβ6-mediated activation of latent TGF-β requires the latent TGF-β binding protein-1. J. Cell Biol. 2004, 165, 723–734. [Google Scholar] [CrossRef] [PubMed]
- Munger, J.S.; Huang, X.; Kawakatsu, H.; Griffiths, M.J.; Dalton, S.L.; Wu, J.; Pittet, J.F.; Kaminski, N.; Garat, C.; Matthay, M.A.; et al. The integrin alpha v beta 6 binds and activates latent TGF beta 1: A mechanism for regulating pulmonary inflammation and fibrosis. Cell 1999, 96, 319–328. [Google Scholar] [CrossRef] [Green Version]
- Mu, D.; Cambier, S.; Fjellbirkeland, L.; Baron, J.L.; Munger, J.S.; Kawakatsu, H. The integrin alpha(v)beta8 mediates epithelial homeostasis through MT1-MMP-dependent activation of TGF-beta1. J. Cell Biol. 2002, 157, 493–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenkins, G. The role of proteases in transforming growth factor-beta activation. Int. J. Biochem. Cell Biol. 2008, 40, 1068–1078. [Google Scholar] [CrossRef]
- Shi, M.; Zhu, J.; Wang, R.; Chen, X.; Mi, L.; Walz, T. Latent TGF-beta structure and activation. Nature 2011, 474, 343–349. [Google Scholar] [CrossRef] [Green Version]
- Buscemi, L.; Ramonet, D.; Klingberg, F.; Formey, A.; Smith-Clerc, J.; Meister, J.J. The single-molecule mechanics of the latent TGF-beta1 complex. Curr. Biol. 2011, 21, 2046–2054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klingberg, F.; Chow, M.L.; Koehler, A.; Boo, S.; Buscemi, L.; Quinn, T.M. Prestress in the extracellular matrix sensitizes latent TGF-beta1 for activation. J. Cell Biol. 2014, 207, 283–297. [Google Scholar] [CrossRef] [PubMed]
- Henderson, N.C.; Sheppard, D. Integrin-mediated regulation of TGFbeta in fibrosis. Biochim. Biophys. Acta 2013, 1832, 891–896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reed, N.I.; Jo, H.; Chen, C.; Tsujino, K.; Arnold, T.D.; DeGrado, W.F. The alphavbeta1 integrin plays a critical in vivo role in tissue fibrosis. Sci. Transl. Med. 2015, 7, 288ra79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derynck, R.; Budi, E.H. Specificity, versatility, and control of TGF-beta family signaling. Sci. Signal. 2019, 12, eaav5183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heldin, C.H.; Moustakas, A. Signaling Receptors for TGF-beta Family Members. Cold Spring Harb. Perspect. Biol. 2016, 8, a022053. [Google Scholar] [CrossRef] [Green Version]
- Follonier Castella, L.; Buscemi, L.; Godbout, C.; Meister, J.J.; Hinz, B. A new lock-step mechanism of matrix remodelling based on subcellular contractile events. J. Cell Sci. 2010, 123, 1751–1760. [Google Scholar] [CrossRef] [Green Version]
- Follonier Castella, L.; Gabbiani, G.; McCulloch, C.A.; Hinz, B. Regulation of myofibroblast activities: Calcium pulls some strings behind the scene. Exp. Cell Res. 2010, 316, 2390–2401. [Google Scholar] [CrossRef]
- Sakai, N.; Chun, J.; Duffield, J.S.; Lagares, D.; Wada, T.; Luster, A.D.; Tager, A.M. Lysophosphatidic acid signaling through its receptor initiates profibrotic epithelial cell fibroblast communication mediated by epithelial cell derived connective tissue growth factor. Kidney Int. 2017, 91, 628–641. [Google Scholar] [CrossRef] [Green Version]
- Ungefroren, H.; Gieseler, F.; Kaufmann, R.; Settmacher, U.; Lehnert, H.; Rauch, B.H. Signaling Crosstalk of TGF-beta/ALK5 and PAR2/PAR1: A Complex Regulatory Network Controlling Fibrosis and Cancer. Int. J. Mol. Sci. 2018, 19, 1568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johan, M.Z.; Samuel, M.S. Rho–ROCK signaling regulates tumor-microenvironment interactions. Biochem. Soc. Trans. 2018, 47, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Dooling, L.J.; Discher, D.E. Inhibiting Tumor Fibrosis and Actomyosin through GPCR activation. Trends Cancer 2019, 5, 197–199. [Google Scholar] [CrossRef] [PubMed]
- Haak, A.J.; Ducharme, M.T.; Espinosa, A.M.D.; Tschumperlin, D.J. Targeting GPCR Signaling for Idiopathic Pulmonary Fibrosis Therapies. Trends Pharmacol. Sci. 2020, 41, 172–182. [Google Scholar] [CrossRef] [PubMed]
- Dupont, S.; Morsut, L.; Aragona, M.; Enzo, E.; Giulitti, S.; Cordenonsi, M.; Zanconato, F.; Le Digabel, J.; Forcato, M.; Bicciato, S.; et al. Role of YAP/TAZ in mechanotransduction. Nat. Cell Biol. 2011, 474, 179–183. [Google Scholar] [CrossRef]
- Yu, F.-X.; Zhao, B.; Panupinthu, N.; Jewell, J.L.; Lian, I.; Wang, L.H.; Zhao, J.; Yuan, H.; Tumaneng, K.; Li, H.; et al. Regulation of the Hippo-YAP Pathway by G-Protein-Coupled Receptor Signaling. Cell 2012, 150, 780–791. [Google Scholar] [CrossRef] [Green Version]
- Dasgupta, I.; McCollum, D. Control of cellular responses to mechanical cues through YAP/TAZ regulation. J. Biol. Chem. 2019, 294, 17693–17706. [Google Scholar] [CrossRef] [Green Version]
- Rausch, V.; Hansen, C.G. The Hippo Pathway, YAP/TAZ, and the Plasma Membrane. Trends Cell Biol. 2020, 30, 32–48. [Google Scholar] [CrossRef] [PubMed]
- Totaro, A.; Panciera, T.; Piccolo, S. YAP/TAZ upstream signals and downstream responses. Nat. Cell Biol. 2018, 20, 888–899. [Google Scholar] [CrossRef] [PubMed]
- Elosegui-Artola, A.; Andreu, I.; Beedle, A.; Lezamiz, A.; Uroz, M.; Kosmalska, A.J.; Oria, R.; Kechagia, J.Z.; Rico-Lastres, P.; Le Roux, A.-L.; et al. Force Triggers YAP Nuclear Entry by Regulating Transport across Nuclear Pores. Cell 2017, 171, 1397–1410.e14. [Google Scholar] [CrossRef]
- Kofler, M.; Speight, P.; Little, D.; Di Ciano-Oliveira, C.; Szászi, K.; Kapus, A. Mediated nuclear import and export of TAZ and the underlying molecular requirements. Nat. Commun. 2018, 9, 4966. [Google Scholar] [CrossRef] [Green Version]
- Noguchi, S.; Saito, A.; Nagase, T. YAP/TAZ Signaling as a Molecular Link between Fibrosis and Cancer. Int. J. Mol. Sci. 2018, 19, 3674. [Google Scholar] [CrossRef] [Green Version]
- Piersma, B.; Bank, R.A.; Boersema, M. Signaling in Fibrosis: TGF-beta, WNT, and YAP/TAZ Converge. Front. Med. 2015, 2, 59. [Google Scholar] [CrossRef]
- Pan, D. The Hippo Signaling Pathway in Development and Cancer. Dev. Cell 2010, 19, 491–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szeto, S.G.; Narimatsu, M.; Lu, M.; He, X.; Sidiqi, A.M.; Tolosa, M.F. YAP/TAZ Are Mechanoregulators of TGF-beta-Smad Signaling and Renal Fibrogenesis. J. Am. Soc. Nephrol. 2016, 27, 3117–3128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, K.; Pritchett, J.; Llewellyn, J.; Mullan, A.F.; Athwal, V.S.; Dobie, R.; Harvey, E.; Zeef, L.; Farrow, S.; Streuli, C.; et al. PAK proteins and YAP-1 signalling downstream of integrin beta-1 in myofibroblasts promote liver fibrosis. Nat. Commun. 2016, 7, 12502. [Google Scholar] [CrossRef]
- Piersma, B.; de Rond, S.; Werker, P.M.; Boo, S.; Hinz, B.; van Beuge, M.M.; Bank, R.A. YAP1 Is a Driver of Myofibroblast Differentiation in Normal and Diseased Fibroblasts. Am. J. Pathol. 2015, 185, 3326–3337. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Lagares, D.; Choi, K.M.; Stopfer, L.; Marinković, A.; Vrbanac, V.; Probst, C.K.; Hiemer, S.E.; Sisson, T.H.; Horowitz, J.; et al. Mechanosignaling through YAP and TAZ drives fibroblast activation and fibrosis. Am. J. Physiol. Cell. Mol. Physiol. 2015, 308, L344–L357. [Google Scholar] [CrossRef] [Green Version]
- Talele, N.; Fradette, J.; Davies, J.E.; Kapus, A.; Hinz, B. Expression of α-Smooth Muscle Actin Determines the Fate of Mesenchymal Stromal Cells. Stem Cell Rep. 2015, 4, 1016–1030. [Google Scholar] [CrossRef] [Green Version]
- Haak, A.J.; Kostallari, E.; Sicard, D.; Ligresti, G.; Choi, K.M.; Caporarello, N.; Jones, D.L.; Tan, Q.; Meridew, J.; Espinosa, A.M.D.; et al. Selective YAP/TAZ inhibition in fibroblasts via dopamine receptor D1 agonism reverses fibrosis. Sci. Transl. Med. 2019, 11, eaau6296. [Google Scholar] [CrossRef] [PubMed]
- Gau, D.; Roy, P. SRF’ing and SAP’ing—The role of MRTF proteins in cell migration. J. Cell Sci. 2018, 131, jcs218222. [Google Scholar] [CrossRef] [Green Version]
- Esnault, C.; Stewart, A.; Gualdrini, F.; East, P.; Horswell, S.; Matthews, N.; Treisman, R. Rho-actin signaling to the MRTF coactivators dominates the immediate transcriptional response to serum in fibroblasts. Genes Dev. 2014, 28, 943–958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Small, E.M.; Thatcher, J.E.; Sutherland, L.B.; Kinoshita, H.; Gerard, R.D.; Richardson, J.A. Myocardin-related transcription factor-a controls myofibroblast activation and fibrosis in response to myocardial infarction. Circ. Res. 2010, 107, 294–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bialik, J.F.; Ding, M.; Speight, P.; Dan, Q.; Miranda, M.Z.; Di Ciano-Oliveira, C.; Kofler, M.M.; Rotstein, O.D.; Pedersen, S.F.; Szászi, K.; et al. Profibrotic epithelial phenotype: A central role for MRTF and TAZ. Sci. Rep. 2019, 9, 4323. [Google Scholar] [CrossRef]
- Song, E.; Ouyang, N.; Horbelt, M.; Antus, B.; Wang, M.; Exton, M.S. Influence of alternatively and classically activated macrophages on fibrogenic activities of human fibroblasts. Cell Immunol. 2000, 204, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Ding, J.; Ma, Z.; Iwashina, T.; Tredget, E.E. Alternatively activated macrophages derived from THP-1 cells promote the fibrogenic activities of human dermal fibroblasts. Wound Repair Regen. 2017, 25, 377–388. [Google Scholar] [CrossRef]
- Franz, S.; Rammelt, S.; Scharnweber, D.; Simon, J.C. Immune responses to implants—A review of the implications for the design of immunomodulatory biomaterials. Biomaterials 2011, 32, 6692–6709. [Google Scholar] [CrossRef]
- Lodyga, M.; Cambridge, E.; Karvonen, H.M.; Pakshir, P.; Wu, B.; Boo, S. Cadherin-11-mediated adhesion of macrophages to myofibroblasts establishes a profibrotic niche of active TGF-beta. Sci. Signal. 2019, 12, eaao3469. [Google Scholar] [CrossRef]
- Reinhart-King, C.A.; Dembo, M.; Hammer, D.A. Cell-Cell Mechanical Communication through Compliant Substrates. Biophys. J. 2008, 95, 6044–6051. [Google Scholar] [CrossRef] [Green Version]
- Paszek, M.J.; Zahir, N.; Johnson, K.R.; Lakins, J.N.; Rozenberg, G.I.; Gefen, A.; Reinhart-King, C.A.; Margulies, S.S.; Dembo, M.; Boettiger, D.; et al. Tensional homeostasis and the malignant phenotype. Cancer Cell 2005, 8, 241–254. [Google Scholar] [CrossRef] [Green Version]
- Lo, C.-M.; Wang, H.-B.; Dembo, M.; Wang, Y.-L. Cell Movement Is Guided by the Rigidity of the Substrate. Biophys. J. 2000, 79, 144–152. [Google Scholar] [CrossRef] [Green Version]
- Pakshir, P.; Alizadehgiashi, M.; Wong, B.; Coelho, N.M.; Chen, X.; Gong, Z. Dynamic fibroblast contractions attract remote macrophages in fibrillar collagen matrix. Nat. Commun. 2019, 10, 1850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bank, R.A.; Zandstra, J.; Room, H.; Petersen, A.H.; van Putten, S.M. Biomaterial Encapsulation Is Enhanced in the Early Stages of the Foreign Body Reaction During Conditional Macrophage Depletion in Transgenic Macrophage Fas-Induced Apoptosis Mice. . Tissue Eng. Part A 2017, 23, 1078–1087. [Google Scholar] [CrossRef]
- Rodriguez, A.; Macewanm, S.R.; Meyerson, H.; Kirk, J.T.; Anderson, J.M. The foreign body reaction in T-cell-deficient mice. J. Biomed. Mater. Res. A 2009, 90, 106–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Jao, B.; McNally, A.K.; Anderson, J.M. In vivo quantitative and qualitative assessment of foreign body giant cell formation on biomaterials in mice deficient in natural killer lymphocyte subsets, mast cells, or the interleukin-4 receptorα and in severe combined immunodeficient mice. J. Biomed. Mater. Res. A 2014, 102, 2017–2023. [Google Scholar] [CrossRef]
- Avula, M.N.; Rao, A.N.; McGill, L.D.; Grainger, D.W.; Solzbacher, F. Foreign body response to subcutaneous biomaterial implants in a mast cell-deficient Kit(w-Sh) murine model. Acta Biomater. 2014, 10, 1856–1863. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, H.; Janmey, P.A.; McCulloch, C.A. Lateral boundary mechanosensing by adherent cells in a collagen gel system. Biomaterials 2014, 35, 1138–1149. [Google Scholar] [CrossRef]
- Hall, M.S.; Alisafaei, F.; Ban, E.; Feng, X.; Hui, C.Y.; Shenoy, V.B.; Wu, M. Fibrous nonlinear elasticity enables positive mechanical feedback between cells and ECMs. Proc. Natl. Acad. Sci. USA 2016, 113, 14043–14048. [Google Scholar] [CrossRef] [Green Version]
- Baker, B.; Trappmann, B.; Wang, W.Y.; Sakar, M.S.; Kim, I.L.; Shenoy, V.B.; Burdick, J.A.; Chen, C. Cell-mediated fibre recruitment drives extracellular matrix mechanosensing in engineered fibrillar microenvironments. Nat. Mater. 2015, 14, 1262–1268. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Abhilash, A.; Chen, C.; Wells, R.G.; Shenoy, V.B. Long-Range Force Transmission in Fibrous Matrices Enabled by Tension-Driven Alignment of Fibers. Biophys. J. 2014, 107, 2592–2603. [Google Scholar] [CrossRef] [Green Version]
- Bloise, N.; Rountree, I.; Polucha, C.; Montagna, G.; Visai, L.; Coulombe, K.L.K.; Munarin, F. Engineering Immunomodulatory Biomaterials for Regenerating the Infarcted Myocardium. Front. Bioeng. Biotechnol. 2020, 8, 292. [Google Scholar] [CrossRef] [PubMed]
- Hannan, R.T.; Peirce, S.; Barker, T.H. Fibroblasts: Diverse Cells Critical to Biomaterials Integration. ACS Biomater. Sci. Eng. 2018, 4, 1223–1232. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Comparison between normal wound healing stages and phases of the FBR. (A) Normal wound healing is compromised of the same molecular players as the foreign body reaction (FBR) to implants (B) In fact, the FBR begins as a normal wound healing response, but the persistent presence of the biomaterial results in sustained fibrosis and scar tissue formation. Following the initial blood-biomaterial interaction and provisional ECM formation, acute inflammation, followed by chronic inflammation and fusion of macrophages into foreign body giant cells (FBGCs) occur in a subsequent fashion. Scheme was prepared using Biorender with kind support from Ronen Schuster.
Figure 1.
Comparison between normal wound healing stages and phases of the FBR. (A) Normal wound healing is compromised of the same molecular players as the foreign body reaction (FBR) to implants (B) In fact, the FBR begins as a normal wound healing response, but the persistent presence of the biomaterial results in sustained fibrosis and scar tissue formation. Following the initial blood-biomaterial interaction and provisional ECM formation, acute inflammation, followed by chronic inflammation and fusion of macrophages into foreign body giant cells (FBGCs) occur in a subsequent fashion. Scheme was prepared using Biorender with kind support from Ronen Schuster.
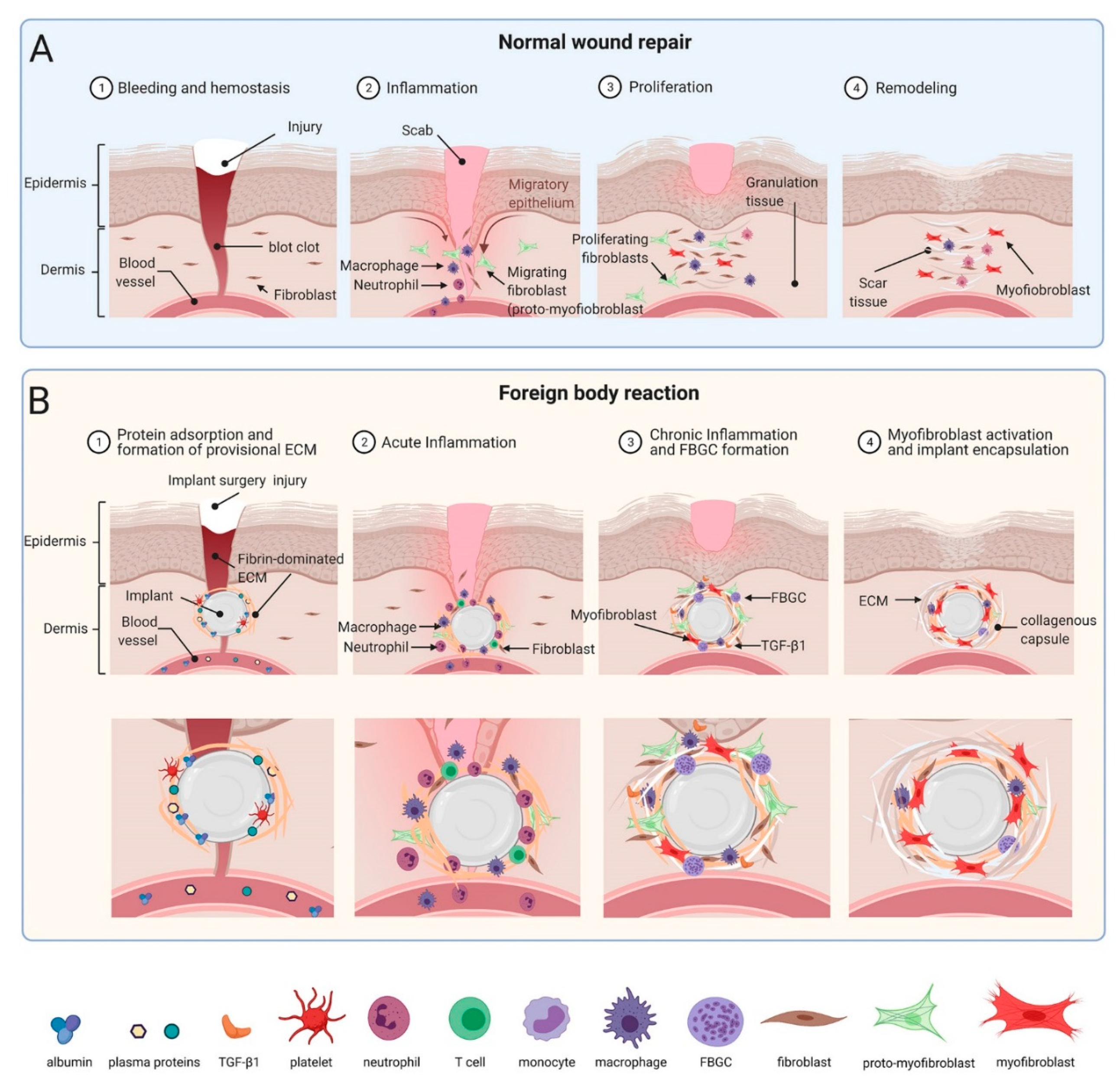
Figure 2.
Modulating implant surface properties is a common strategy to enhance tissue integration while simultaneously reducing the incidence and severity of implant FBR and fibrosis. Protein coverage and cell interactions with the surface both determine the ability of cells to attach and spread on implants. Various physical and chemical modifications to the Implants surface are used to inhibit FBR. (A) Chemical modifications of surface properties include coating with local and slow releasing anti-fibrotic drugs to prevent implant fibrosis. In addition, subcutaneously implanted tissue expanders coated with decellularized ECM into non-human primates display minimal fibrotic. Inhibiting αv-integrin binding, e.g., with the RGD-peptidomimetic inhibitor CWHM-12 attenuate implant encapsulation by preventing mechanical activation of latent TGF-β1 and myofibroblasts. (B) In addition to or combined with altering implant surface chemistry, other treatments include modulation of physical parameters such as surface hydrophilicity or wettability, porosity, stiffness (elastic modulus), anisotropic ‘roughness’, and regular topographies. Perceived stiffness and ‘true’ modulus of the implant surface can be used to control cell responses. Restriction of focal adhesion sizes by micropatterning stiff surfaces with adhesion sized fibronectin islets limits myofibroblast spreading, intracellular stress and α-SMA stress fiber incorporation. Modulating implant geometry (size, thickness, shape, and pore size) or coating with low fouling material such as zwitterions reduces protein adsorption to implant surfaces and reduces implant fibrosis. Scheme was prepared using Biorender.
Figure 2.
Modulating implant surface properties is a common strategy to enhance tissue integration while simultaneously reducing the incidence and severity of implant FBR and fibrosis. Protein coverage and cell interactions with the surface both determine the ability of cells to attach and spread on implants. Various physical and chemical modifications to the Implants surface are used to inhibit FBR. (A) Chemical modifications of surface properties include coating with local and slow releasing anti-fibrotic drugs to prevent implant fibrosis. In addition, subcutaneously implanted tissue expanders coated with decellularized ECM into non-human primates display minimal fibrotic. Inhibiting αv-integrin binding, e.g., with the RGD-peptidomimetic inhibitor CWHM-12 attenuate implant encapsulation by preventing mechanical activation of latent TGF-β1 and myofibroblasts. (B) In addition to or combined with altering implant surface chemistry, other treatments include modulation of physical parameters such as surface hydrophilicity or wettability, porosity, stiffness (elastic modulus), anisotropic ‘roughness’, and regular topographies. Perceived stiffness and ‘true’ modulus of the implant surface can be used to control cell responses. Restriction of focal adhesion sizes by micropatterning stiff surfaces with adhesion sized fibronectin islets limits myofibroblast spreading, intracellular stress and α-SMA stress fiber incorporation. Modulating implant geometry (size, thickness, shape, and pore size) or coating with low fouling material such as zwitterions reduces protein adsorption to implant surfaces and reduces implant fibrosis. Scheme was prepared using Biorender.

Figure 3.
Myofibroblast mechanisms of fibrotic encapsulation of implanted stiff materials. (A) During the FBR, inflammatory cells secrete profibrotic cytokines which recruit fibroblasts to the implantation site. The stiff implant surface and surrounding extracellular matrix (ECM) mechanically activate fibroblasts into myofibroblasts at different levels. (1) Formation of focal adhesions with the ECM allows (2) formation of α-SMA-positive stress fibers and development of high contractile forces, evidenced by translocation of myocardin-related transcription factor (MRTF)-A from the cytosol into the nucleus. (3) Following integrin binding to ligands such as the latency associate peptide (LAP), (4) cell contraction and mechanically resisting stiff ECM enhance activation of integrins in αv integrin-containing focal adhesions, including integrin β1. (5) Mechanically activated αvβ1 integrin binds with high affinity to the LAP portion of the ECM-bound large latent TGF-β1 complex (latent TGF-β1 binding protein LTBP not shown). (6) Transmission of cell forces to the stable connection results in unfolding of LAP and release of active TGF-β1. (7) Active TGF-β1 binds to the TGF-β receptor complex, promoting phosphorylation of Smad and translocation to the nucleus. (8) Both, TGF-β1/pSmad3 and MRTF-A signaling drive pro-fibrotic programs that further enhance myofibroblast activation and encapsulation of stiff implants with a stiff ECM capsule. (B) Reducing implant surface stiffness and/or inhibiting αv-integrin binding with the RGD-peptidomimetic inhibitor CWHM-12 at the beginning of this cascade both attenuate implant encapsulation by preventing mechanical activation of β1 integrin, latent TGF-β1, and myofibroblasts. Scheme was prepared using Biorender. Reproduced with permission from [69]. (C) Immunostaining of a cross-section through the peri-implant tissue forming after 7 days around 2 MPa-stiff silicone disks, implanted under the dorsal skin of a mouse. Myofibroblasts (α-SMA, green) accumulate at the implant surface together with CD68-positive macrophages (red) and in deeper layers of fibrotic tissue. Diameter of the implant: 6 mm.
Figure 3.
Myofibroblast mechanisms of fibrotic encapsulation of implanted stiff materials. (A) During the FBR, inflammatory cells secrete profibrotic cytokines which recruit fibroblasts to the implantation site. The stiff implant surface and surrounding extracellular matrix (ECM) mechanically activate fibroblasts into myofibroblasts at different levels. (1) Formation of focal adhesions with the ECM allows (2) formation of α-SMA-positive stress fibers and development of high contractile forces, evidenced by translocation of myocardin-related transcription factor (MRTF)-A from the cytosol into the nucleus. (3) Following integrin binding to ligands such as the latency associate peptide (LAP), (4) cell contraction and mechanically resisting stiff ECM enhance activation of integrins in αv integrin-containing focal adhesions, including integrin β1. (5) Mechanically activated αvβ1 integrin binds with high affinity to the LAP portion of the ECM-bound large latent TGF-β1 complex (latent TGF-β1 binding protein LTBP not shown). (6) Transmission of cell forces to the stable connection results in unfolding of LAP and release of active TGF-β1. (7) Active TGF-β1 binds to the TGF-β receptor complex, promoting phosphorylation of Smad and translocation to the nucleus. (8) Both, TGF-β1/pSmad3 and MRTF-A signaling drive pro-fibrotic programs that further enhance myofibroblast activation and encapsulation of stiff implants with a stiff ECM capsule. (B) Reducing implant surface stiffness and/or inhibiting αv-integrin binding with the RGD-peptidomimetic inhibitor CWHM-12 at the beginning of this cascade both attenuate implant encapsulation by preventing mechanical activation of β1 integrin, latent TGF-β1, and myofibroblasts. Scheme was prepared using Biorender. Reproduced with permission from [69]. (C) Immunostaining of a cross-section through the peri-implant tissue forming after 7 days around 2 MPa-stiff silicone disks, implanted under the dorsal skin of a mouse. Myofibroblasts (α-SMA, green) accumulate at the implant surface together with CD68-positive macrophages (red) and in deeper layers of fibrotic tissue. Diameter of the implant: 6 mm.

Figure 4.
Control of fibroblast-to-myofibroblast activation by adhesion patterns and substrate stiffness. (A) Activation of non-contractile (‘quiescent’) tissue fibroblasts into highly contractile, α-SMA stress fiber forming myofibroblasts passes over consecutive activation stages, one of which is characterized by α-SMA-negative stress fibers. This so-called pro-myofibroblast produces extracellular matrix and is the prevalent fibroblast phenotype in conventional cell culture. Myofibroblast activation stages can be controlled in culture by altering substrate adhesion patterns or stiffness. (B–D) Myofibroblasts from various origins were seeded onto different 2D culture substrates, followed by immunostaining for various proteins, color-coded as indicated. Acute incorporation of the myofibroblast marker α-SMA into stress fibers is increasing as a function of (B) the size of small fibronectin attachment islets that accommodate single focal adhesions, (C) area of large fibronectin adhesive islands that house single cells, and (D) stiffness of fibronectin-coated silicone elastomer substrates (elastic modulus in kPa). All these factors directly affect the ability of myofibroblasts to develop intracellular stress (actomyosin contractility). Scale bars: 20 µm. Scheme produced with Biorender.com.
Figure 4.
Control of fibroblast-to-myofibroblast activation by adhesion patterns and substrate stiffness. (A) Activation of non-contractile (‘quiescent’) tissue fibroblasts into highly contractile, α-SMA stress fiber forming myofibroblasts passes over consecutive activation stages, one of which is characterized by α-SMA-negative stress fibers. This so-called pro-myofibroblast produces extracellular matrix and is the prevalent fibroblast phenotype in conventional cell culture. Myofibroblast activation stages can be controlled in culture by altering substrate adhesion patterns or stiffness. (B–D) Myofibroblasts from various origins were seeded onto different 2D culture substrates, followed by immunostaining for various proteins, color-coded as indicated. Acute incorporation of the myofibroblast marker α-SMA into stress fibers is increasing as a function of (B) the size of small fibronectin attachment islets that accommodate single focal adhesions, (C) area of large fibronectin adhesive islands that house single cells, and (D) stiffness of fibronectin-coated silicone elastomer substrates (elastic modulus in kPa). All these factors directly affect the ability of myofibroblasts to develop intracellular stress (actomyosin contractility). Scale bars: 20 µm. Scheme produced with Biorender.com.
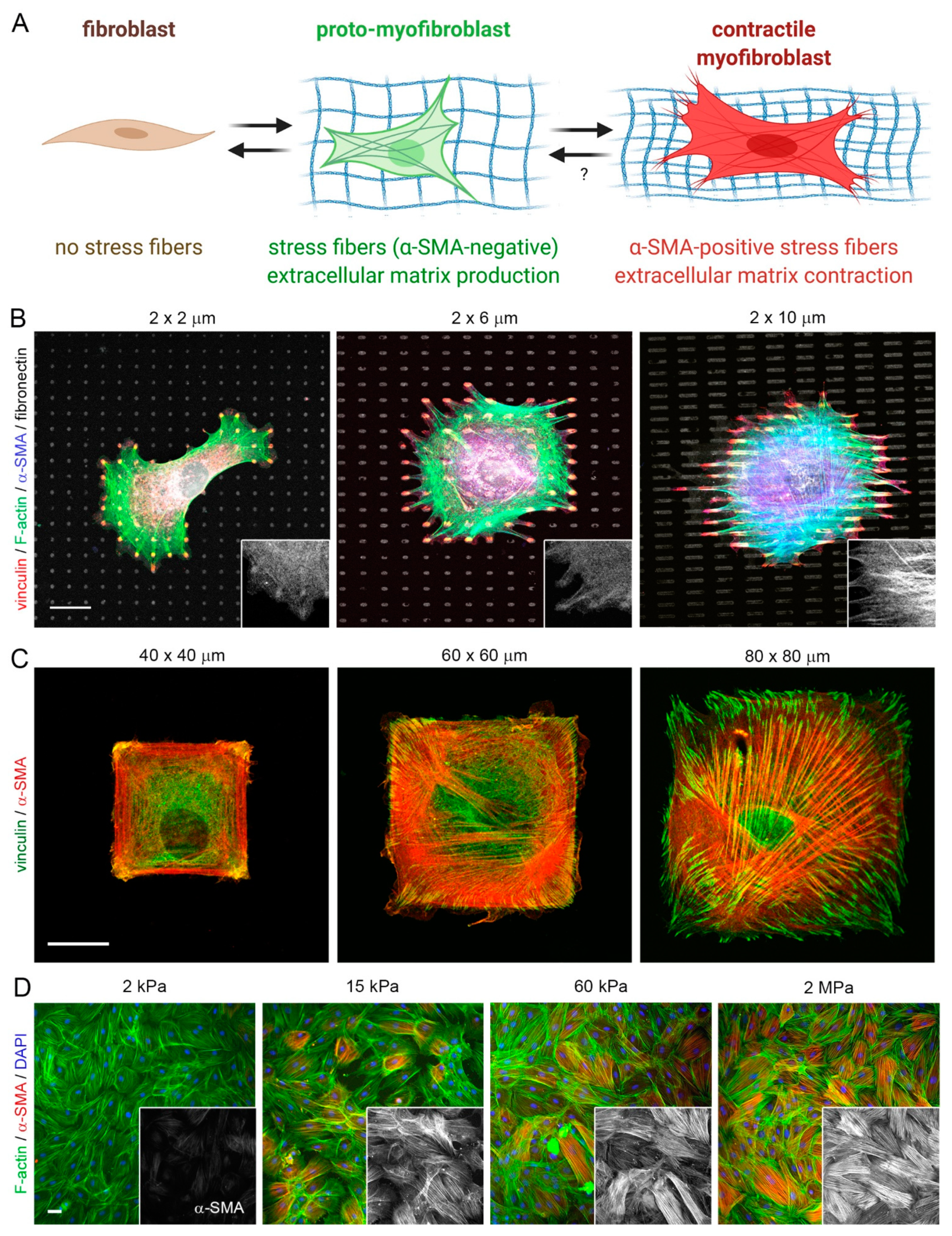

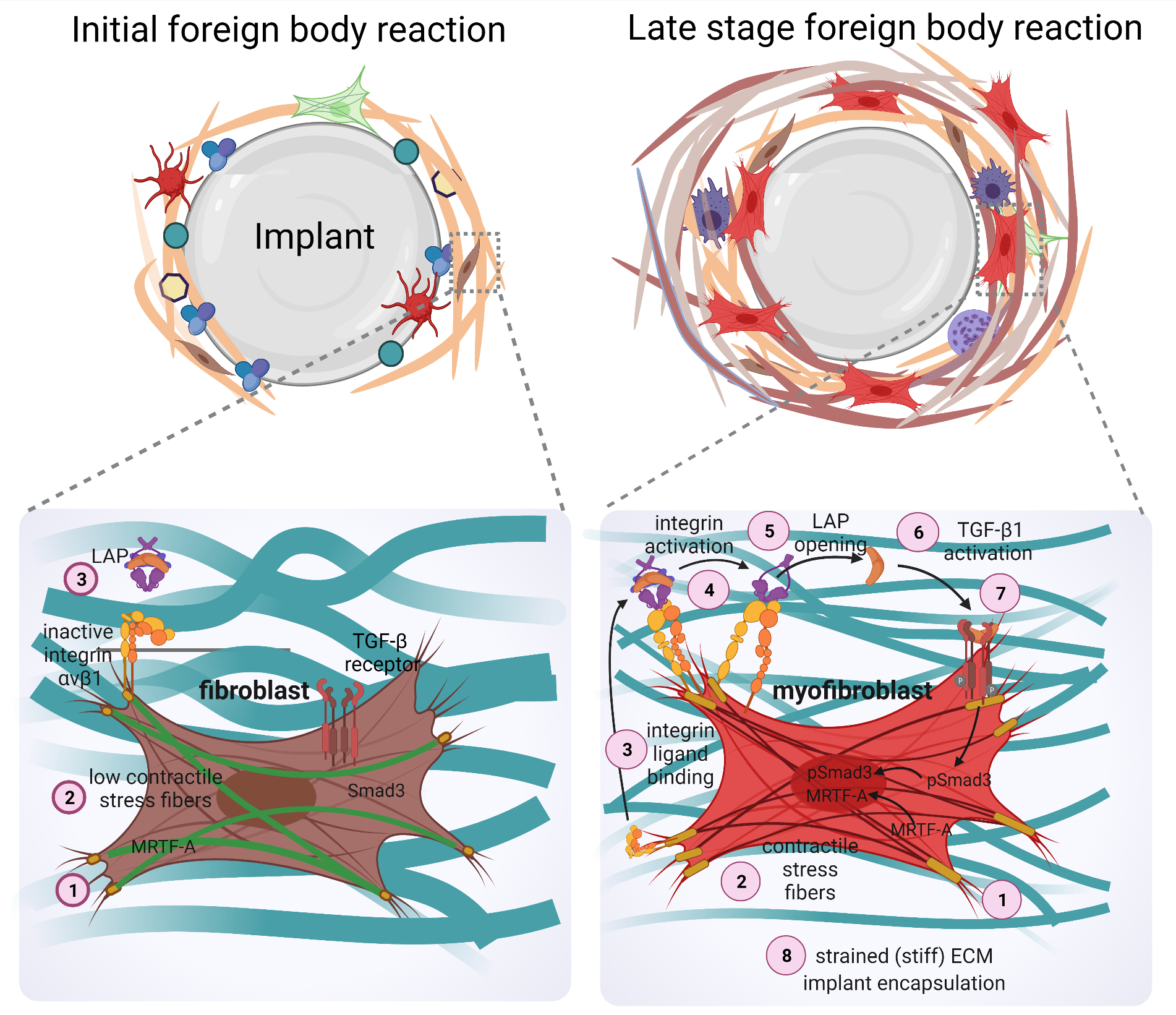{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Current strategies to target the fibrotic phase of the FBR.
| Inhibiting Compound | FBR Pathway Target | Implant Material | Implant Model | Species | Study Length (In Vivo) | References |
|---|---|---|---|---|---|---|
| Prolyl-4-hydroxylase inhibitors | Collagen synthesis | N/A | Intraocular implants | Human | 19 months | [192,193,195,199,200] |
| Col1 siRNA (Nanofiber scaffold-mediated RNA interference) | Collagen synthesis | siRNA–poly(caprolactone-co-ethylethylene phosphate) nanofibers | Posterior dorsal areas | Rat | 4 weeks | [195] |
| Rapamycin (mTOR) siRNA | Type I collagen synthesis | Poly(ethylene glycol) (PEG)-based hydrogel coatings | Subcutaneous siRNA-releasing device implantation | Mice | 2 weeks | [199] |
| Halofuginone | Type I collagen synthesis | Silicone discs | Subcutaneous implantation | Rat | 3 months | [201] |
| Relaxin, BMP-7, hepatocyte growth factor, SMAD7 | TGF-β | Mock biosensors | Subcutaneous implantation | Rat | 55 days | [192,193,202] |
| Pirfenidone | TGF-β and Collagen synthesis | Smooth and textured silicone implants | Submammary implantation | Rat | 8 weeks | [193] |
| Masitinib | Tyrosine-kinase | Polyester fiber model | Subcutaneous implantation | Mice | 4 weeks | [203] |
| Antisense oligonucleotides, cAMP, TNF | CCN2 | polyether-polyurethane sponges | Subcutaneous implantation | Mice | 14 days | [204,205] |
| Monoclonal antibodies specific for MMPs and TIMP-1 | Pharmacological inhibition of MMP-1,-8,-13, and -18 | In vitro human monocyte assay | In vitro | Human | In vitro | [206] |
| Pravastatin | Neovascularization and AMPK/mTOR pathway | Medical-grade Polyetheretherketone (SP) | Subcutaneous implantation | Mice | 4 weeks | [207] |
| VEGF | Neovascularization | Commercial glucose sensors | Subcutaneous implantation | Mice | 10 days | [208] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Noskovicova, N.; Hinz, B.; Pakshir, P. Implant Fibrosis and the Underappreciated Role of Myofibroblasts in the Foreign Body Reaction. Cells 2021, 10, 1794. https://doi.org/10.3390/cells10071794
AMA Style
Noskovicova N, Hinz B, Pakshir P. Implant Fibrosis and the Underappreciated Role of Myofibroblasts in the Foreign Body Reaction. Cells. 2021; 10(7):1794. https://doi.org/10.3390/cells10071794
Chicago/Turabian StyleNoskovicova, Nina, Boris Hinz, and Pardis Pakshir. 2021. "Implant Fibrosis and the Underappreciated Role of Myofibroblasts in the Foreign Body Reaction" Cells 10, no. 7: 1794. https://doi.org/10.3390/cells10071794
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.