
Cystatin C Deficiency Increases LPS-Induced Sepsis and NLRP3 Inflammasome Activation in Mice

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. General Reagents
2.2. Mice
2.3. Systemic LPS Challenge In Vivo
2.4. Preparation of Macrophages
2.5. qPCR
2.6. Cell Lysate Preparation and Western Blot Analysis
2.7. Cathepsin Activity Measurements
2.8. Cytokine Analysis
2.9. Cytotoxicity, Lactate Dehydrogenase Release
2.10. Flow Cytometry
2.11. Analysis of OCR and ECAR with Seahorse XF Analyser
2.12. Statistical Analysis
3. Results
3.1. CstC KO Mice Are More Sensitive to LPS-Induced Lethal Endotoxemia and Produce Higher Levels of IL-18 in Spleen Post-LPS Administration
3.2. Activity of Lysosomal Cathepsins Is Increased in CstC KO BMDMs upon NLRP3 Inflammasome Activation
3.3. Increased Activation of NLPR3 Inflammasome in CstC KO BMDMs Is Independent of Cysteine Cathepsin Activity
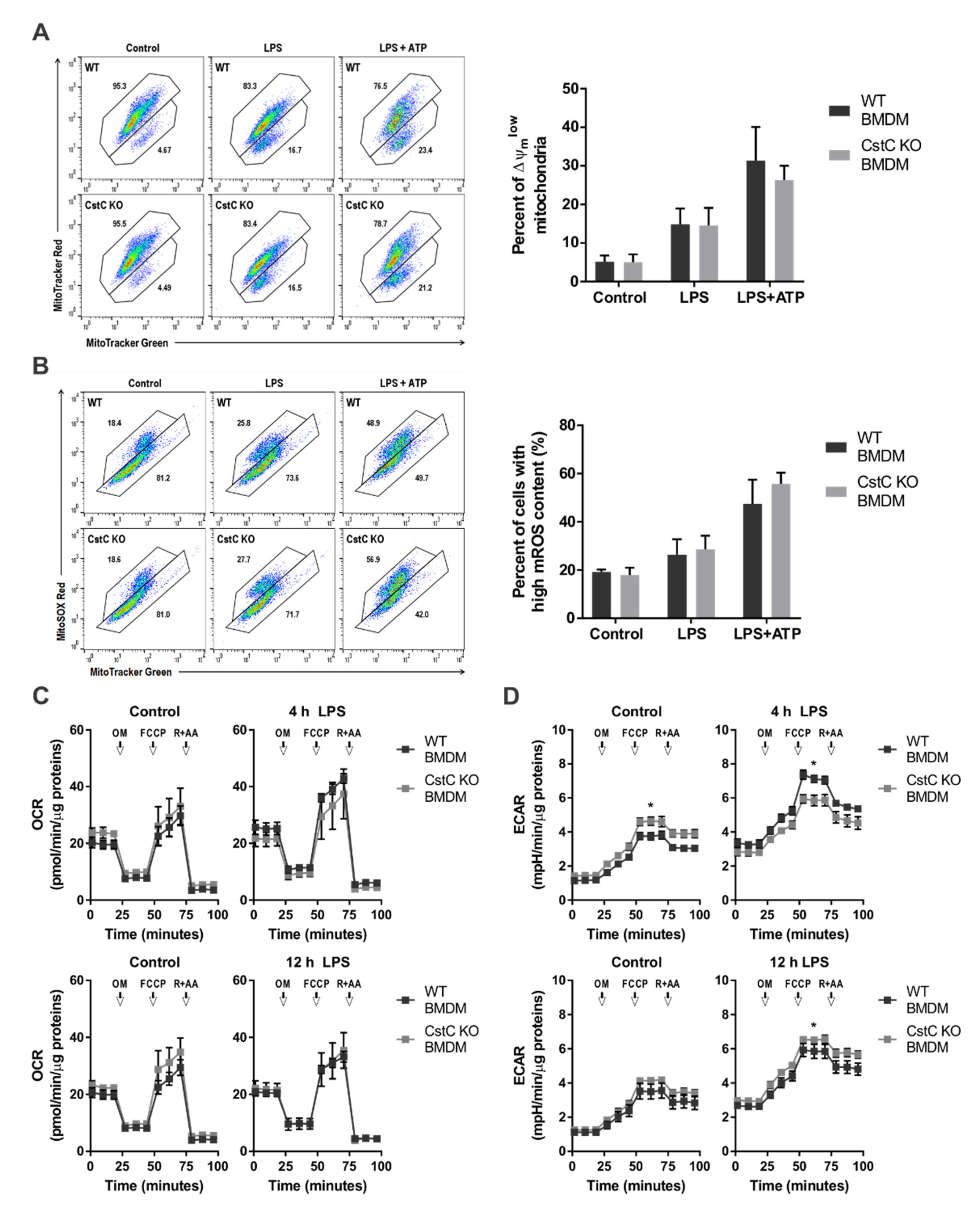
3.4. Cystatin C Deficiency Does Not Affect Mitochondrial Function or ROS Production
3.5. Cystatin C Deficiency in BMDMs upon LPS Stimulation Does Not Affect MAPK Signalling Pathways, Anti-Inflammatory IL-10 Production and Nitric Oxide Production
3.6. CstC Ablation in BMDMs Decreases Autophagy Induction
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Esposito, S.; De Simone, G.; Boccia, G.; De Caro, F.; Pagliano, P. Sepsis and septic shock: New definitions, new diagnostic and therapeutic approaches. J. Glob. Antimicrob. Resist. 2017, 10, 204–212. [Google Scholar] [CrossRef]
- Erridge, C.; Bennett-Guerrero, E.; Poxton, I.R. Structure and function of lipopolysaccharides. Microbes Infect. 2002, 4, 837–851. [Google Scholar] [CrossRef]
- Rosadini, C.V.; Kagan, J.C. Early innate immune responses to bacterial LPS. Curr. Opin. Immunol. 2017, 44, 14–19. [Google Scholar] [CrossRef] [Green Version]
- Martinon, F.; Tschopp, J. Inflammatory caspases and inflammasomes: Master switches of inflammation. Cell Death Differ. 2007, 14, 10–22. [Google Scholar] [CrossRef]
- Liao, P.C.; Chao, L.K.; Chou, J.C.; Dong, W.C.; Lin, C.N.; Lin, C.Y.; Chen, A.; Ka, S.M.; Ho, C.L.; Hua, K.F. Lipopolysaccharide/adenosine triphosphate-mediated signal transduction in the regulation of NLRP3 protein expression and caspase-1-mediated interleukin-1β secretion. Inflamm. Res. 2013, 62, 89–96. [Google Scholar] [CrossRef]
- Swanson, K.V.; Deng, M.; Ting, J.P.Y. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef] [PubMed]
- Sharma, D.; Kanneganti, T.D. The cell biology of inflammasomes: Mechanisms of inflammasome activation and regulation. J. Cell Biol. 2016, 213, 617–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoseini, Z.; Sepahvand, F.; Rashidi, B.; Sahebkar, A.; Masoudifar, A.; Mirzaei, H. NLRP3 inflammasome: Its regulation and involvement in atherosclerosis. J. Cell. Physiol. 2018, 233, 2116–2132. [Google Scholar] [CrossRef]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Kayagaki, N.; Warming, S.; Lamkanfi, M.; Walle, L.V.; Louie, S.; Dong, J.; Newton, K.; Qu, Y.; Liu, J.; Heldens, S.; et al. Non-canonical inflammasome activation targets caspase-11. Nature 2011, 479, 117–121. [Google Scholar] [CrossRef]
- Kayagaki, N.; Wong, M.T.; Stowe, I.B.; Ramani, S.R.; Gonzalez, L.C.; Akashi-Takamura, S.; Miyake, K.; Zhang, J.; Lee, W.P.; Muszynśki, A.; et al. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science 2013, 341, 1246–1249. [Google Scholar] [CrossRef]
- Hagar, J.A.; Powell, D.A.; Aachoui, Y.; Ernst, R.K.; Miao, E.A. Cytoplasmic LPS activates caspase-11: Implications in TLR4-independent endotoxic shock. Science 2013, 341, 1250–1253. [Google Scholar] [CrossRef] [Green Version]
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T.; et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 2015, 526, 666–671. [Google Scholar] [CrossRef] [PubMed]
- Viganò, E.; Mortellaro, A. Caspase-11: The driving factor for noncanonical inflammasomes. Eur. J. Immunol. 2013, 43, 2240–2245. [Google Scholar] [CrossRef]
- Yu, L.; Chen, Y.; Tooze, S.A. Autophagy pathway: Cellular and molecular mechanisms. Autophagy 2018, 14, 207–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saitoh, T.; Fujita, N.; Jang, M.H.; Uematsu, S.; Yang, B.G.; Satoh, T.; Omori, H.; Noda, T.; Yamamoto, N.; Komatsu, M.; et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1β production. Nature 2008, 456, 264–268. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, T.; Akira, S. Regulation of inflammasomes by autophagy. J. Allergy Clin. Immunol. 2016, 138, 28–36. [Google Scholar] [CrossRef] [Green Version]
- Sun, Q.; Fan, J.; Billiar, T.R.; Scott, M.J. Inflammasome and autophagy regulation: A two-way street. Mol. Med. 2017, 23, 188–195. [Google Scholar] [CrossRef]
- Biasizzo, M.; Kopitar-Jerala, N. Interplay Between NLRP3 Inflammasome and Autophagy. Front. Immunol. 2020, 11, 591803. [Google Scholar] [CrossRef] [PubMed]
- Turk, V.; Stoka, V.; Vasiljeva, O.; Renko, M.; Sun, T.; Turk, B.; Turk, D. Cysteine cathepsins: From structure, function and regulation to new frontiers. Biochim. Biophys. Acta Proteins Proteomics 2012, 1824, 68–88. [Google Scholar] [CrossRef] [Green Version]
- Kopitar-Jerala, N. The role of cystatins in cells of the immune system. FEBS Lett. 2006, 580, 6295–6301. [Google Scholar] [CrossRef] [Green Version]
- Vray, B.; Hartmann, S.; Hoebeke, J. Immunomodulatory properties of cystatins. Cell. Mol. Life Sci. 2002, 59, 1503–1512. [Google Scholar] [CrossRef]
- Magister, Š.; Kos, J. Cystatins in immune system. J. Cancer 2013, 4, 45–56. [Google Scholar] [CrossRef] [Green Version]
- Maher, K.; Kokelj, B.J.; Butinar, M.; Mikhaylov, G.; Manček-Keber, M.; Stoka, V.; Vasiljeva, O.; Turk, B.; Grigoryev, S.A.; Kopitar-Jerala, N. A role for stefin B (cystatin B) in inflammation and endotoxemia. J. Biol. Chem. 2014, 289, 31736–31750. [Google Scholar] [CrossRef] [Green Version]
- Maher, K.; Završnik, J.; Jerič-Kokelj, B.; Vasiljeva, O.; Turk, B.; Kopitar-Jerala, N. Decreased IL-10 expression in stefin B-deficient macrophages is regulated by the MAP kinase and STAT-3 signaling pathways. FEBS Lett. 2014, 588, 720–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turk, V.; Stoka, V.; Turk, D. Cystatins: Biochemical and structural properties, and medical relevance. Front. Biosci. 2008, 13, 5406–5420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, B.; Zhou, Y.; Halabisky, B.; Lo, I.; Cho, S.H.; Mueller-Steiner, S.; Devidze, N.; Wang, X.; Grubb, A.; Gan, L. Cystatin C-Cathepsin B Axis Regulates Amyloid Beta Levels and Associated Neuronal Deficits in an Animal Model of Alzheimer’s Disease. Neuron 2008, 60, 247–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaur, G.; Mohan, P.; Pawlik, M.; DeRosa, S.; Fajiculay, J.; Che, S.; Grubb, A.; Ginsberg, S.D.; Nixon, R.A.; Levy, E. Cystatin C rescues degenerating neurons in a cystatin B-knockout mouse model of progressive myoclonus epilepsy. Am. J. Pathol. 2010, 177, 2256–2267. [Google Scholar] [CrossRef]
- Pirttilä, T.J.; Lukasiuk, K.; Håkansson, K.; Grubb, A.; Abrahamson, M.; Pitkänen, A. Cystatin C modulates neurodegeneration and neurogenesis following status epilepticus in mouse. Neurobiol. Dis. 2005, 20, 241–253. [Google Scholar] [CrossRef]
- Tizon, B.; Sahoo, S.; Yu, H.; Gauthier, S.; Kumar, A.R.; Mohan, P.; Figliola, M.; Pawlik, M.; Grubb, A.; Uchiyama, Y.; et al. Induction of Autophagy by Cystatin C: A Mechanism That Protects Murine Primary Cortical Neurons and Neuronal Cell Lines. PLoS ONE 2010, 5, e9819. [Google Scholar] [CrossRef]
- Huh, C.G.; Håkansson, K.; Nathanson, C.M.; Thorgeirsson, U.P.; Jonsson, N.; Grubb, A.; Abrahamson, M.; Karlsson, S. Decreased metastatic spread in mice homozygous for a null allele of the cystatin C protease inhibitor gene. J. Clin. Pathol. Mol. Pathol. 1999, 52, 332–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weischenfeldt, J.; Porse, B. Bone marrow-derived macrophages (BMM): Isolation and applications. Cold Spring Harb. Protoc. 2008, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopitar-Jerala, N.; Schweiger, A.; Myers, R.M.; Turk, V.; Turk, B. Sensitization of stefin B-deficient thymocytes towards staurosporin-induced apoptosis is independent of cysteine cathepsins. FEBS Lett. 2005, 579, 2149–2155. [Google Scholar] [CrossRef] [Green Version]
- Jäättelä, M.; Nylandsted, J. Quantification of lysosomal membrane permeabilization by cytosolic cathepsin and β-N-acetyl-glucosaminidase activity measurements. Cold Spring Harb. Protoc. 2015, 2015, 1017–1023. [Google Scholar] [CrossRef] [Green Version]
- Schauvliege, R.; Vanrobaeys, J.; Schotte, P.; Beyaert, R. Caspase-11 gene expression in response to lipopolysaccharide and interferon-γ requires nuclear factor-κB and signal transducer and activator of transcription (STAT) 1. J. Biol. Chem. 2002, 277, 41624–41630. [Google Scholar] [CrossRef] [Green Version]
- Hur, J.; Kim, S.Y.; Kim, H.; Cha, S.; Lee, M.-S.; Suk, K. Induction of caspase-11 by inflammatory stimuli in rat astrocytes: Lipopolysaccharide induction through p38 mitogen-activated protein kinase pathway. FEBS Lett. 2001, 507, 157–162. [Google Scholar] [CrossRef] [Green Version]
- Puren, A.J.; Fantuzzi, G.; Dinarello, C.A. Gene expression, synthesis, and secretion of interleukin 18 and interleukin 1β are differentially regulated in human blood mononuclear cells and mouse spleen cells. Proc. Natl. Acad. Sci. USA 1999, 96, 2256–2261. [Google Scholar] [CrossRef] [Green Version]
- Spes, A.; Sobotic, B.; Turk, V.; Turk, B. Cysteine cathepsins are not critical for TRAIL-and CD95-induced apoptosis in several human cancer cell lines. In Proceedings of the Biological Chemistry; De Gruyter: Berlin, Germany, 2012; Volume 393, pp. 1417–1431. [Google Scholar]
- Campden, R.I.; Zhang, Y. The role of lysosomal cysteine cathepsins in NLRP3 inflammasome activation. Arch. Biochem. Biophys. 2019, 670, 32–42. [Google Scholar] [CrossRef]
- Edgington-Mitchell, L.E.; Wartmann, T.; Fleming, A.K.; Gocheva, V.; Van Der Linden, W.A.; Withana, N.P.; Verdoes, M.; Aurelio, L.; Edgington-Mitchell, D.; Lieu, T.; et al. Legumain is activated in macrophages during pancreatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 311, G548–G560. [Google Scholar] [CrossRef] [Green Version]
- Ross, C.; Chan, A.H.; Von Pein, J.; Boucher, D.; Schroder, K. Dimerization and auto-processing induce caspase-11 protease activation within the non-canonical inflammasome. Life Sci. Alliance 2018, 1. [Google Scholar] [CrossRef]
- Lee, B.L.; Stowe, I.B.; Gupta, A.; Kornfeld, O.S.; Roose-Girma, M.; Anderson, K.; Warming, S.; Zhang, J.; Lee, W.P.; Kayagaki, N. Caspase-11 auto-proteolysis is crucial for noncanonical inflammasome activation. J. Exp. Med. 2018, 215, 2279–2288. [Google Scholar] [CrossRef]
- Wassélius, J.; Johansson, K.; Håkansson, K.; Abrahamson, M.; Ehinger, B. Cystatin C uptake in the eye. Graefe’s Arch. Clin. Exp. Ophthalmol. 2005, 243, 583–592. [Google Scholar] [CrossRef] [Green Version]
- Gren, S.T.; Janciauskiene, S.; Sandeep, S.; Jonigk, D.; Kvist, P.H.; Gerwien, J.G.; Håkansson, K.; Grip, O. The protease inhibitor cystatin C down-regulates the release of IL-β and TNF-α in lipopolysaccharide activated monocytes. J. Leukoc. Biol. 2016, 100, 811–822. [Google Scholar] [CrossRef] [Green Version]
- Ekström, U.; Wallin, H.; Lorenzo, J.; Holmqvist, B.; Abrahamson, M.; Avilés, F.X. Internalization of cystatin C in human cell lines. FEBS J. 2008, 275, 4571–4582. [Google Scholar] [CrossRef] [PubMed]
- Wallin, H.; Abrahamson, M.; Ekström, U. Cystatin C properties crucial for uptake and inhibition of intracellular target enzymes. J. Biol. Chem. 2013, 288, 17019–17029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warfel, A.H.; Zucker-Franklin, D.; Frangione, B.; Ghiso, J. Constitutive secretion of cystatin C (γ-TRACE) by monocytes and macrophages and its downregulation after stimulation. J. Exp. Med. 1987, 166, 1912–1917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orlowski, G.M.; Colbert, J.D.; Sharma, S.; Bogyo, M.; Robertson, S.A.; Rock, K.L. Multiple Cathepsins Promote Pro–IL-1β Synthesis and NLRP3-Mediated IL-1β Activation. J. Immunol. 2015, 195, 1685–1697. [Google Scholar] [CrossRef]
- Kim, H.J.; Lee, H.S.; Chong, Y.H.; Kang, J.L. p38 mitogen-activated protein kinase up-regulates LPS-induced NF-κB activation in the development of lung injury and RAW 264.7 macrophages. Toxicology 2006, 225, 36–47. [Google Scholar] [CrossRef]
- Frendéus, K.H.; Wallin, H.; Janciauskiene, S.; Abrahamson, M. Macrophage responses to interferon-γ are dependent on cystatin C levels. Int. J. Biochem. Cell Biol. 2009, 41, 2262–2269. [Google Scholar] [CrossRef]
- Klotz, C.; Ziegler, T.; Figueiredo, A.S.; Rausch, S.; Hepworth, M.R.; Obsivac, N.; Sers, C.; Lang, R.; Hammerstein, P.; Lucius, R.; et al. A Helminth Immunomodulator Exploits Host Signaling Events to Regulate Cytokine Production in Macrophages. PLoS Pathog. 2011, 7, e1001248. [Google Scholar] [CrossRef] [Green Version]
- Kar, S.; Ukil, A.; Das, P.K. Cystatin cures visceral leishmaniasis by NF-κB-mediated proinflammatory response through co-ordination of TLR/MyD88 signaling with p105-Tpl2-ERK pathway. Eur. J. Immunol. 2011, 41, 116–127. [Google Scholar] [CrossRef]
- Kar, S.; Ukil, A.; Das, P.K. Signaling events leading to the curative effect of cystatin on experimental visceral leishmaniasis: Involvement of ERK1/2, NF-κB and JAK/STAT pathways. Eur. J. Immunol. 2009, 39, 741–751. [Google Scholar] [CrossRef]
- Meng, A.; Zhang, X.; Shi, Y. Role of p38 MAPK and STAT3 in lipopolysaccharide-stimulated mouse alveolar macrophages. Exp. Ther. Med. 2014, 8, 1772–1776. [Google Scholar] [CrossRef] [Green Version]
- Chanteux, H.; Guisset, A.C.; Pilette, C.; Sibille, Y. LPS induces IL-10 production by human alveolar macrophages via MAPKinases- and Sp1-dependent mechanisms. Respir. Res. 2007, 8, 71. [Google Scholar] [CrossRef] [Green Version]
- Gurung, P.; Li, B.; Subbarao Malireddi, R.K.; Lamkanfi, M.; Geiger, T.L.; Kanneganti, T.D. Chronic TLR Stimulation Controls NLRP3 Inflammasome Activation through IL-10 Mediated Regulation of NLRP3 Expression and Caspase-8 Activation. Sci. Rep. 2015, 5, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Hartmann, S.; Schonemeyer, A.; Sonnenburg, B.; Vray, B.; Lucius, R. Cystatins of filarial nematodes up-regulate the nitric oxide production of interferon-gamma-activated murine macrophages. Parasite Immunol. 2002, 24, 253–262. [Google Scholar] [CrossRef]
- Verdot, L.; Lalmanach, G.; Vercruysse, V.; Hartmann, S.; Lucius, R.; Hoebeke, J.; Gauthier, F.; Vray, B. Cystatins up-regulate nitric oxide release from interferon-γ-activated mouse peritoneal macrophages. J. Biol. Chem. 1996, 271, 28077–28081. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Sultana, N.; Siraj, N.; Ward, L.J.; Pawlik, M.; Levy, E.; Jovinge, S.; Bengtsson, E.; Yuan, X.M. Autophagy dysfunction and regulatory cystatin C in macrophage death of atherosclerosis. J. Cell. Mol. Med. 2016, 20, 1664–1672. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Hayakawa, T.; Wakasugi, K.; Yamanaka, K. Cystatin C protects neuronal cells against mutant copper-zinc superoxide dismutase-mediated toxicity. Cell Death Dis. 2014, 5, e1497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Watanabe, S.; Komine, O.; Endo, F.; Wakasugi, K.; Yamanaka, K. Intracerebroventricular administration of Cystatin C ameliorates disease in SOD1-linked amyotrophic lateral sclerosis mice. J. Neurochem. 2018, 145, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Biasizzo, M.; Trstenjak-Prebanda, M.; Dolinar, K.; Pirkmajer, S.; Završnik, J.; Turk, B.; Kopitar-Jerala, N. Cystatin C Deficiency Increases LPS-Induced Sepsis and NLRP3 Inflammasome Activation in Mice. Cells 2021, 10, 2071. https://doi.org/10.3390/cells10082071
Biasizzo M, Trstenjak-Prebanda M, Dolinar K, Pirkmajer S, Završnik J, Turk B, Kopitar-Jerala N. Cystatin C Deficiency Increases LPS-Induced Sepsis and NLRP3 Inflammasome Activation in Mice. Cells. 2021; 10(8):2071. https://doi.org/10.3390/cells10082071
Chicago/Turabian StyleBiasizzo, Monika, Mojca Trstenjak-Prebanda, Klemen Dolinar, Sergej Pirkmajer, Janja Završnik, Boris Turk, and Nataša Kopitar-Jerala. 2021. "Cystatin C Deficiency Increases LPS-Induced Sepsis and NLRP3 Inflammasome Activation in Mice" Cells 10, no. 8: 2071. https://doi.org/10.3390/cells10082071