
Feedback Signaling between Cholangiopathies, Ductular Reaction, and Non-Alcoholic Fatty Liver Disease

 ,
,  ,
, 

Abstract
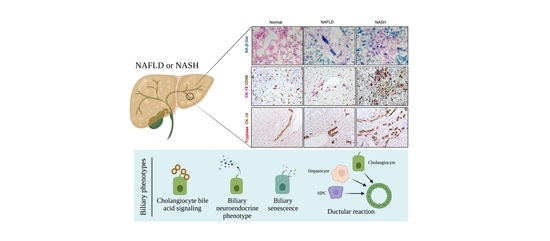
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Cholangiocytes—The Fundamentals
1.1. Structure, Function, and Heterogeneity
1.2. Cholestasis and Cholangiopathies
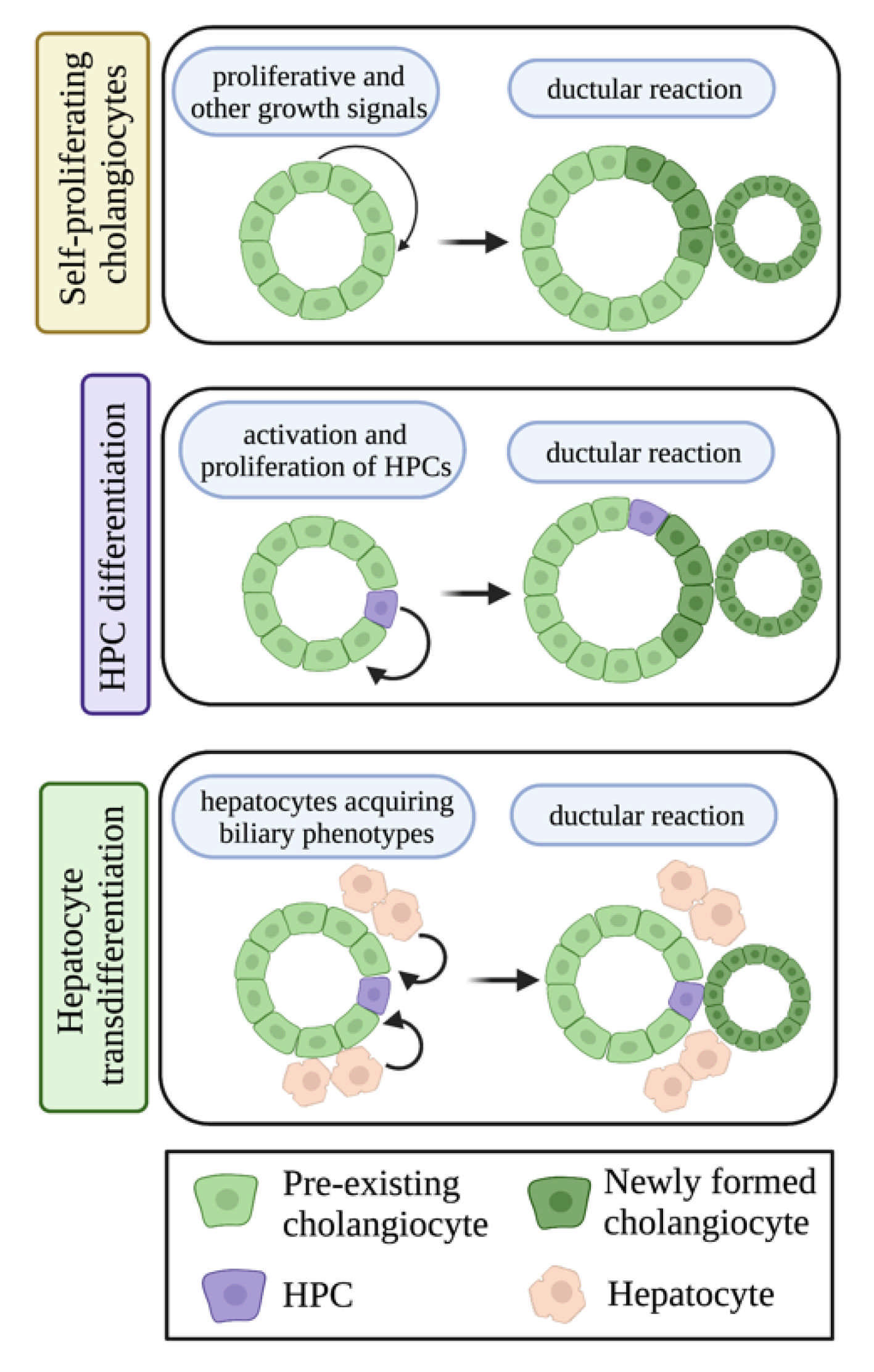
1.3. Ductular Reaction
1.4. Bile Acid Signaling
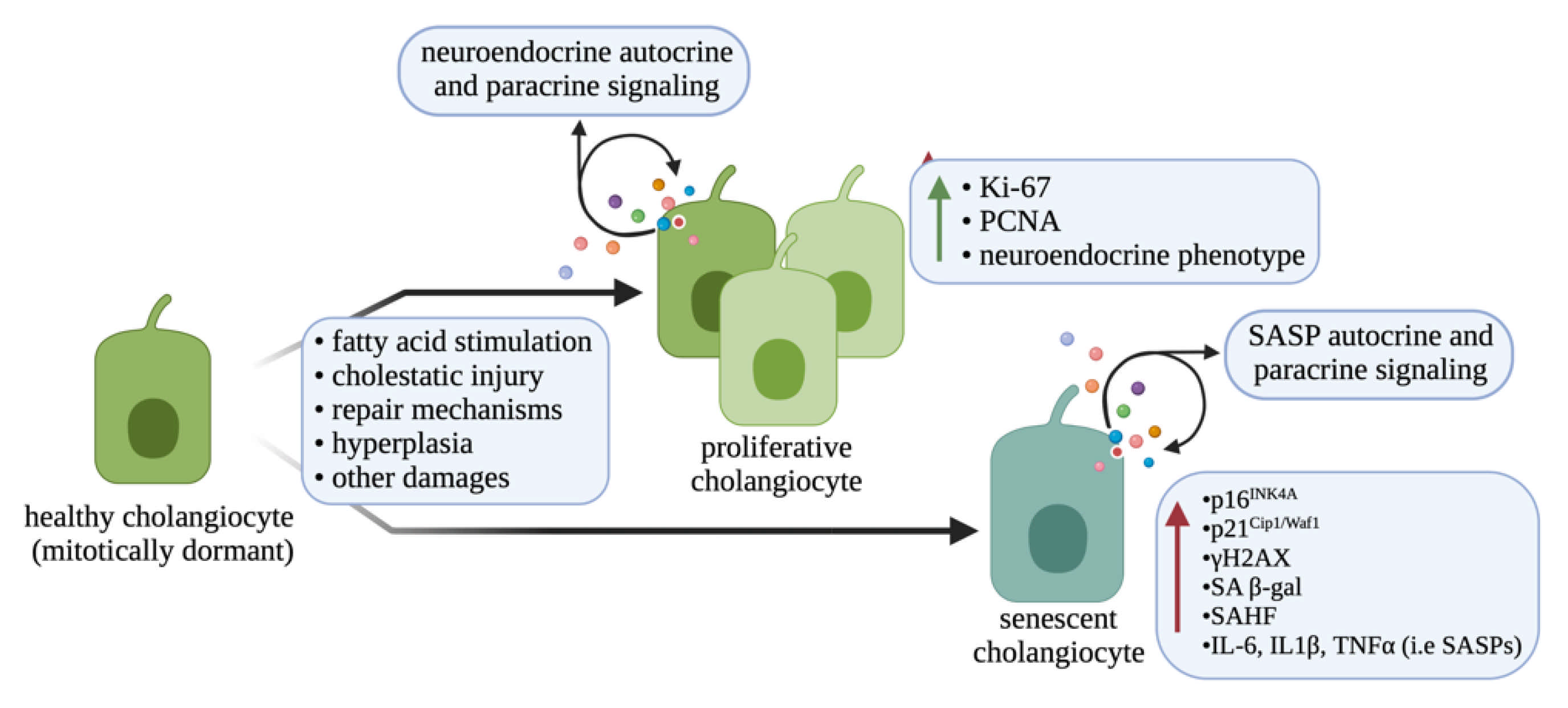
1.5. Biliary Neuroendocrine Phenotype
2. Non-Alcoholic Fatty Liver Disease
3. Role of Cholangiocytes in NAFLD
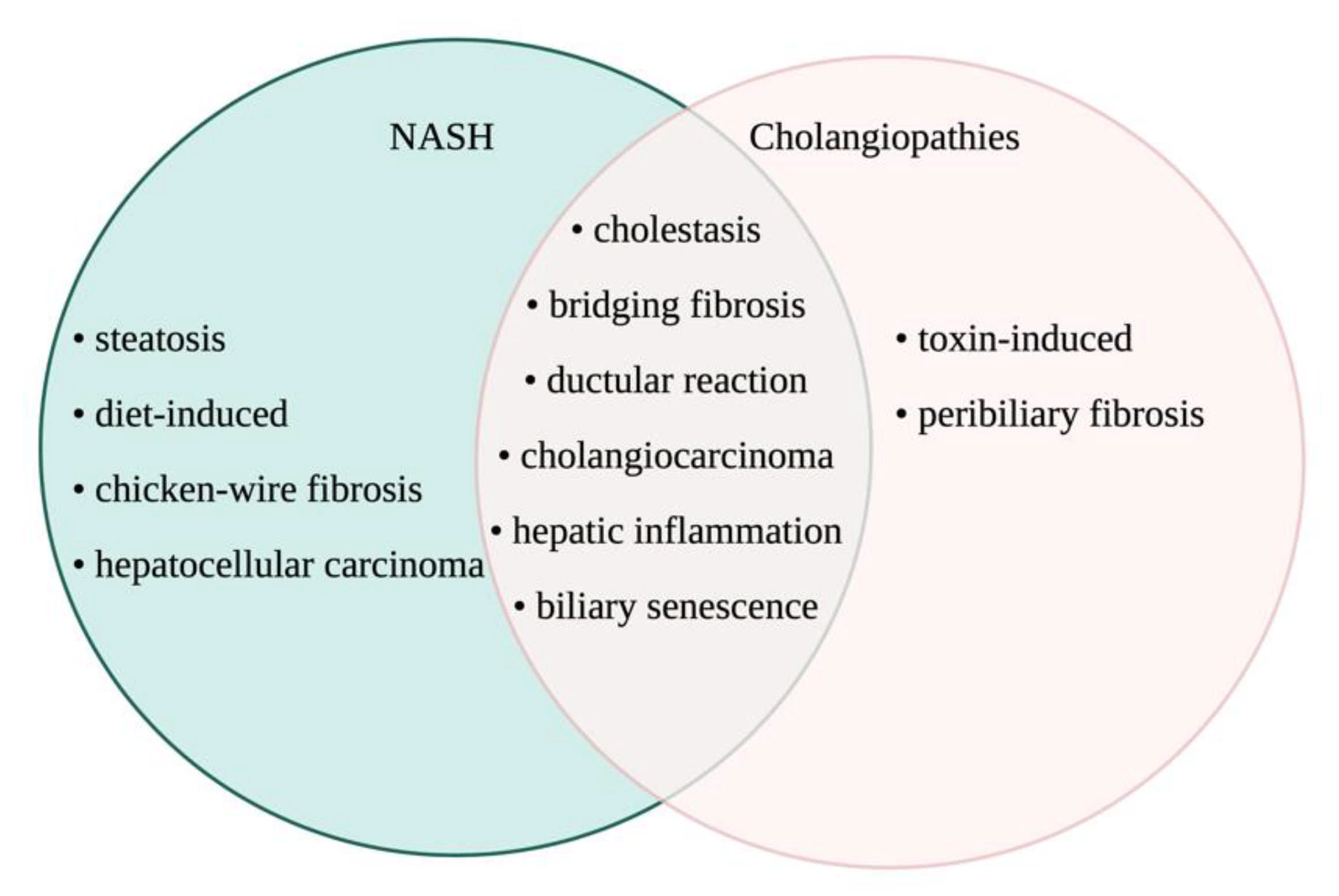
3.1. Cholestasis and Cholangiopathies Associated with NAFLD and NASH
3.2. Ductular Reaction and Biliary Damage in NAFLD/NASH
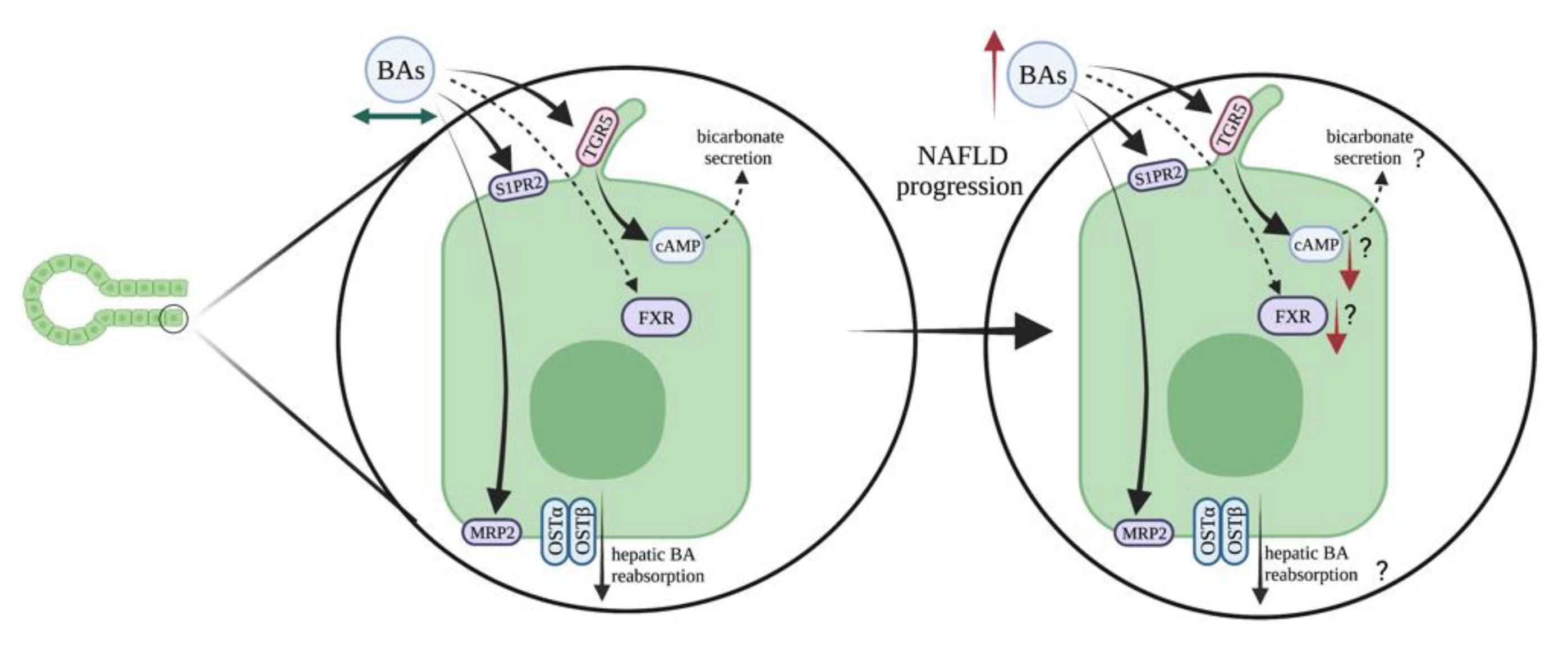
3.3. Bile Acid Signaling
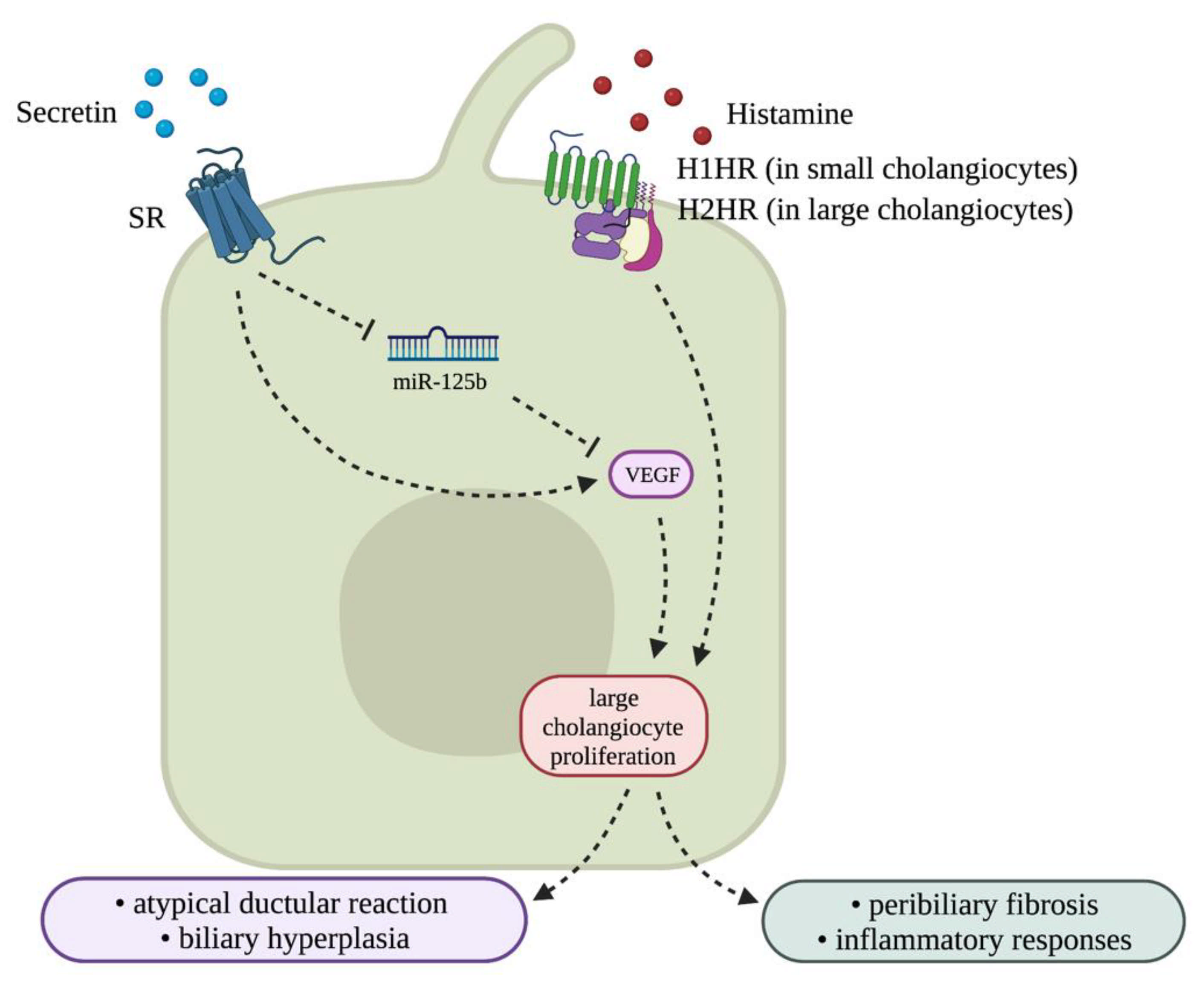
3.4. Biliary Neuroendocrine Phenotype
3.4.1. Vascular Endothelial Growth Factor (VEGF)
3.4.2. Cannabinoid Signaling
3.4.3. Histamine/Histamine Receptor (HR) Axis and Mast Cells (MCs)
3.4.4. Toll-like Receptor 4 (TLR4)
3.4.5. Osteopontin (OPN)
3.4.6. Secretin/SR Signaling and Associated CFTR Activation
4. Conclusions/Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Boyer, J.L. Bile formation and secretion. Compr. Physiol. 2013, 3, 1035–1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maroni, L.; Haibo, B.; Ray, D.; Zhou, T.; Wan, Y.; Meng, F.; Marzioni, M.; Alpini, G. Functional and structural features of cholangiocytes in health and disease. Cell. Mol. Gastroenterol. Hepatol. 2015, 1, 368–380. [Google Scholar] [CrossRef] [Green Version]
- Sato, K.; Meng, F.; Giang, T.; Glaser, S.; Alpini, G. Mechanisms of cholangiocyte responses to injury. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1262–1269. [Google Scholar] [CrossRef] [PubMed]
- Marzioni, M.; Fava, G.; Alvaro, D.; Alpini, G.; Benedetti, A. Control of cholangiocyte adaptive responses by visceral hormones and neuropeptides. Clin. Rev. Allergy Immunol. 2009, 36, 13–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanno, N.; LeSage, G.; Glaser, S.; Alvaro, D.; Alpini, G. Functional heterogeneity of the intrahepatic biliary epithelium. Hepatology 2000, 31, 555–561. [Google Scholar] [CrossRef] [PubMed]
- Mancinelli, R.; Franchitto, A.; Glaser, S.; Meng, F.; Onori, P.; Demorrow, S.; Francis, H.; Venter, J.; Carpino, G.; Baker, K.; et al. GABA induces the differentiation of small into large cholangiocytes by activation of Ca(2+) /CaMK I-dependent adenylyl cyclase 8. Hepatology 2013, 58, 251–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alpini, G.; Glaser, S.; Robertson, W.; Rodgers, R.E.; Phinizy, J.L.; Lasater, J.; LeSage, G.D. Large but not small intrahepatic bile ducts are involved in secretin-regulated ductal bile secretion. Am. J. Physiol. 1997, 272, G1064–G1074. [Google Scholar] [CrossRef]
- Saint-Criq, V.; Gray, M.A. Role of CFTR in epithelial physiology. Cell Mol. Life Sci. 2017, 74, 93–115. [Google Scholar] [CrossRef] [Green Version]
- Huda, N.; Liu, G.; Hong, H.; Yan, S.; Khambu, B.; Yin, X.M. Hepatic senescence, the good and the bad. World J. Gastroenterol. 2019, 25, 5069–5081. [Google Scholar] [CrossRef]
- Reichel, C.; Meier-Abt, P.J. Cholestatic liver diseases. Ther. Umsch. 1997, 54, 639–644. [Google Scholar]
- Padda, M.S.; Sanchez, M.; Akhtar, A.J.; Boyer, J.L. Drug-induced cholestasis. Hepatology 2011, 53, 1377–1387. [Google Scholar] [CrossRef] [Green Version]
- Glaser, S.; Lam, I.P.; Franchitto, A.; Gaudio, E.; Onori, P.; Chow, B.K.; Wise, C.; Kopriva, S.; Venter, J.; White, M.; et al. Knockout of secretin receptor reduces large cholangiocyte hyperplasia in mice with extrahepatic cholestasis induced by bile duct ligation. Hepatology 2010, 52, 204–214. [Google Scholar] [CrossRef] [Green Version]
- Zhou, T.; Wu, N.; Meng, F.; Venter, J.; Giang, T.K.; Francis, H.; Kyritsi, K.; Wu, C.; Franchitto, A.; Alvaro, D.; et al. Knockout of secretin receptor reduces biliary damage and liver fibrosis in Mdr2(-/-) mice by diminishing senescence of cholangiocytes. Lab. Investig. 2018, 98, 1449–1464. [Google Scholar] [CrossRef] [PubMed]
- Pandit, S.; Samant, H. Primary biliary cholangitis. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- European Association for the Study of the Liver. EASL Clinical Practice Guidelines: The diagnosis and management of patients with primary biliary cholangitis. J. Hepatol. 2017, 67, 145–172. [Google Scholar] [CrossRef]
- Kjærgaard, K.; Frisch, K.; Sørensen, M.; Munk, O.L.; Hofmann, A.F.; Horsager, J.; Schacht, A.C.; Erickson, M.; Shapiro, D.; Keiding, S. Obeticholic acid improves hepatic bile acid excretion in patients with primary biliary cholangitis. J. Hepatol. 2021, 74, 58–65. [Google Scholar] [CrossRef]
- Lazaridis, K.N.; LaRusso, N.F. Primary sclerosing cholangitis. N. Engl. J. Med. 2016, 375, 1161–1170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dyson, J.K.; Beuers, U.; Jones, D.E.J.; Lohse, A.W.; Hudson, M. Primary sclerosing cholangitis. Lancet 2018, 391, 2547–2559. [Google Scholar] [CrossRef]
- Karlsen, T.H.; Folseraas, T.; Thorburn, D.; Vesterhus, M. Primary sclerosing cholangitis—A comprehensive review. J. Hepatol. 2017, 67, 1298–1323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blechacz, B. Cholangiocarcinoma: Current knowledge and new developments. Gut Liver 2017, 11, 13–26. [Google Scholar] [CrossRef]
- Gatto, M.; Bragazzi, M.C.; Semeraro, R.; Napoli, C.; Gentile, R.; Torrice, A.; Gaudio, E.; Alvaro, D. Cholangiocarcinoma: Update and future perspectives. Dig. Liver Dis. 2010, 42, 253–260. [Google Scholar] [CrossRef]
- Sato, K.; Marzioni, M.; Meng, F.; Francis, H.; Glaser, S.; Alpini, G. Ductular Reaction in liver diseases: Pathological mechanisms and translational significances. Hepatology 2019, 69, 420–430. [Google Scholar] [CrossRef] [Green Version]
- Bria, A.; Marda, J.; Zhou, J.; Sun, X.; Cao, Q.; Petersen, B.E.; Pi, L. Hepatic progenitor cell activation in liver repair. Liver Res. 2017, 1, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Nagahama, Y.; Sone, M.; Chen, X.; Okada, Y.; Yamamoto, M.; Xin, B.; Matsuo, Y.; Komatsu, M.; Suzuki, A.; Enomoto, K.; et al. Contributions of hepatocytes and bile ductular cells in ductular reactions and remodeling of the biliary system after chronic liver injury. Am. J. Pathol. 2014, 184, 3001–3012. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, Y.; Sone, M.; Nagahama, Y.; Kumagai, E.; Doi, Y.; Omori, Y.; Yoshioka, T.; Tokairin, T.; Yoshida, M.; Yamamoto, Y.; et al. Tumor necrosis factor-α promotes bile ductular transdifferentiation of mature rat hepatocytes in vitro. J. Cell. Biochem. 2013, 114, 831–843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Wu, N.; Kennedy, L.; Francis, H.; Ceci, L.; Zhou, T.; Samala, N.; Kyritsi, K.; Wu, C.; Sybenga, A.; et al. Inhibition of secretin/secretin receptor axis ameliorates non-alcoholic fatty liver disease phenotypes. Hepatology 2021. [Google Scholar] [CrossRef] [PubMed]
- Richardson, M.M.; Jonsson, J.R.; Powell, E.E.; Brunt, E.M.; Neuschwander-Tetri, B.A.; Bhathal, P.S.; Dixon, J.B.; Weltman, M.D.; Tilg, H.; Moschen, A.R.; et al. Progressive fibrosis in nonalcoholic steatohepatitis: Association with altered regeneration and a ductular reaction. Gastroenterology 2007, 133, 80–90. [Google Scholar] [CrossRef]
- Zhao, L.; Westerhoff, M.; Pai, R.K.; Choi, W.T.; Gao, Z.H.; Hart, J. Centrilobular ductular reaction correlates with fibrosis stage and fibrosis progression in non-alcoholic steatohepatitis. Mod. Pathol. 2018, 31, 150–159. [Google Scholar] [CrossRef]
- Staels, B.; Fonseca, V.A. Bile acids and metabolic regulation: Mechanisms and clinical responses to bile acid sequestration. Diabetes Care 2009, 32, S237–S245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagahashi, M.; Yuza, K.; Hirose, Y.; Nakajima, M.; Ramanathan, R.; Hait, N.C.; Hylemon, P.B.; Zhou, H.; Takabe, K.; Wakai, T. The roles of bile acids and sphingosine-1-phosphate signaling in the hepatobiliary diseases. J. Lipid Res. 2016, 57, 1636–1643. [Google Scholar] [CrossRef] [Green Version]
- Grzych, G.; Chavez-Talavera, O.; Descat, A.; Thuillier, D.; Verrijken, A.; Kouach, M.; Legry, V.; Verkindt, H.; Raverdy, V.; Legendre, B.; et al. NASH-related increases in plasma bile acid levels depend on insulin resistance. JHEP Rep. 2021, 3, 100222. [Google Scholar] [CrossRef] [PubMed]
- Boyer, J.L.; Trauner, M.; Mennone, A.; Soroka, C.J.; Cai, S.Y.; Moustafa, T.; Zollner, G.; Lee, J.Y.; Ballatori, N. Upregulation of a basolateral FXR-dependent bile acid efflux transporter OSTalpha-OSTbeta in cholestasis in humans and rodents. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, G1124–G1130. [Google Scholar] [CrossRef]
- Ballatori, N.; Christian, W.V.; Lee, J.Y.; Dawson, P.A.; Soroka, C.J.; Boyer, J.L.; Madejczyk, M.S.; Li, N. OSTalpha-OSTbeta: A major basolateral bile acid and steroid transporter in human intestinal, renal, and biliary epithelia. Hepatology 2005, 42, 1270–1279. [Google Scholar] [CrossRef] [PubMed]
- Meier, P.J.; Stieger, B. Bile salt transporters. Annu. Rev. Physiol. 2002, 64, 635–661. [Google Scholar] [CrossRef] [PubMed]
- Gonzales, E.; Grosse, B.; Schuller, B.; Davit-Spraul, A.; Conti, F.; Guettier, C.; Cassio, D.; Jacquemin, E. Targeted pharmacotherapy in progressive familial intrahepatic cholestasis type 2: Evidence for improvement of cholestasis with 4-phenylbutyrate. Hepatology 2015, 62, 558–566. [Google Scholar] [CrossRef] [PubMed]
- Kenna, J.G.; Taskar, K.S.; Battista, C.; Bourdet, D.L.; Brouwer, K.L.R.; Brouwer, K.R.; Dai, D.; Funk, C.; Hafey, M.J.; Lai, Y.; et al. Can bile salt export pump inhibition testing in drug discovery and development reduce liver injury risk? An international transporter consortium perspective. Clin. Pharmacol. Ther. 2018, 104, 916–932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makishima, M.; Okamoto, A.Y.; Repa, J.J.; Tu, H.; Learned, R.M.; Luk, A.; Hull, M.V.; Lustig, K.D.; Mangelsdorf, D.J.; Shan, B. Identification of a nuclear receptor for bile acids. Science 1999, 284, 1362–1365. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Yang, L.; Wang, Z.; Huang, W. Bile acid nuclear receptor FXR and digestive system diseases. Acta Pharm. Sin. B 2015, 5, 135–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chignard, N.; Mergey, M.; Barbu, V.; Finzi, L.; Tiret, E.; Paul, A.; Housset, C. VPAC1 expression is regulated by FXR agonists in the human gallbladder epithelium. Hepatology 2005, 42, 549–557. [Google Scholar] [CrossRef]
- Cariello, M.; Piccinin, E.; Moschetta, A. Transcriptional Regulation of metabolic pathways via lipid-sensing nuclear receptors PPARs, FXR, and LXR in NASH. Cell. Mol. Gastroenterol. Hepatol. 2021, 11, 1519–1539. [Google Scholar] [CrossRef]
- Halilbasic, E.; Baghdasaryan, A.; Trauner, M. Nuclear receptors as drug targets in cholestatic liver diseases. Clin. Liver Dis. 2013, 17, 161–189. [Google Scholar] [CrossRef] [Green Version]
- Jones, H.; Alpini, G.; Francis, H. Bile acid signaling and biliary functions. Acta Pharm. Sin. B 2015, 5, 123–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Aoki, H.; Yang, J.; Peng, K.; Liu, R.; Li, X.; Qiang, X.; Sun, L.; Gurley, E.C.; Lai, G.; et al. The role of sphingosine 1-phosphate receptor 2 in bile-acid-induced cholangiocyte proliferation and cholestasis-induced liver injury in mice. Hepatology 2017, 65, 2005–2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Studer, E.; Zhou, X.; Zhao, R.; Wang, Y.; Takabe, K.; Nagahashi, M.; Pandak, W.M.; Dent, P.; Spiegel, S.; Shi, R.; et al. Conjugated bile acids activate the sphingosine-1-phosphate receptor 2 in primary rodent hepatocytes. Hepatology 2012, 55, 267–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, K.; Francis, H.; Zhou, T.; Meng, F.; Kennedy, L.; Ekser, B.; Baiocchi, L.; Onori, P.; Mancinelli, R.; Gaudio, E.; et al. Neuroendocrine Changes in Cholangiocarcinoma Growth. Cells 2020, 9, 436. [Google Scholar] [CrossRef] [Green Version]
- Alvaro, D.; Mancino, M.G.; Glaser, S.; Gaudio, E.; Marzioni, M.; Francis, H.; Alpini, G. Proliferating cholangiocytes: A neuroendocrine compartment in the diseased liver. Gastroenterology 2007, 132, 415–431. [Google Scholar] [CrossRef]
- Glaser, S.; Meng, F.; Han, Y.; Onori, P.; Chow, B.K.; Francis, H.; Venter, J.; McDaniel, K.; Marzioni, M.; Invernizzi, P.; et al. Secretin stimulates biliary cell proliferation by regulating expression of microRNA 125b and microRNA let7a in mice. Gastroenterology 2014, 146, 1795–1808.e1712. [Google Scholar] [CrossRef] [Green Version]
- Francis, H.L.; Demorrow, S.; Franchitto, A.; Venter, J.K.; Mancinelli, R.A.; White, M.A.; Meng, F.; Ueno, Y.; Carpino, G.; Renzi, A.; et al. Histamine stimulates the proliferation of small and large cholangiocytes by activation of both IP3/Ca2+ and cAMP-dependent signaling mechanisms. Lab. Investig. 2012, 92, 282–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francis, H.; Glaser, S.; Demorrow, S.; Gaudio, E.; Ueno, Y.; Venter, J.; Dostal, D.; Onori, P.; Franchitto, A.; Marzioni, M.; et al. Small mouse cholangiocytes proliferate in response to H1 histamine receptor stimulation by activation of the IP3/CaMK I/CREB pathway. Am. J. Physiol. Cell. Physiol. 2008, 295, C499–C513. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, L.; Meadows, V.; Kyritsi, K.; Pham, L.; Kundu, D.; Kulkarni, R.; Cerritos, K.; Demieville, J.; Hargrove, L.; Glaser, S.; et al. Amelioration of large bile duct damage by histamine-2 receptor vivo-morpholino treatment. Am. J. Pathol. 2020, 190, 1018–1029. [Google Scholar] [CrossRef]
- Kennedy, L.; Meadows, V.; Demieville, J.; Hargrove, L.; Virani, S.; Glaser, S.; Zhou, T.; Rinehart, E.; Jaeger, V.; Kyritsi, K.; et al. Biliary damage and liver fibrosis are ameliorated in a novel mouse model lacking l-histidine decarboxylase/histamine signaling. Lab. Investig. 2020, 100, 837–848. [Google Scholar] [CrossRef]
- Kennedy, L.; Hargrove, L.; Demieville, J.; Karstens, W.; Jones, H.; DeMorrow, S.; Meng, F.; Invernizzi, P.; Bernuzzi, F.; Alpini, G.; et al. Blocking H1/H2 histamine receptors inhibits damage/fibrosis in Mdr2(-/-) mice and human cholangiocarcinoma tumorigenesis. Hepatology 2018, 68, 1042–1056. [Google Scholar] [CrossRef] [Green Version]
- Kanwar, P.; Kowdley, K.V. The metabolic syndrome and its influence on nonalcoholic steatohepatitis. Clin. Liver Dis. 2016, 20, 225–243. [Google Scholar] [CrossRef] [PubMed]
- Benedict, M.; Zhang, X. Non-alcoholic fatty liver disease: An expanded review. World J. Hepatol. 2017, 9, 715–732. [Google Scholar] [CrossRef] [PubMed]
- Cotter, T.G.; Rinella, M. Nonalcoholic fatty liver disease 2020: The state of the disease. Gastroenterology 2020, 158, 1851–1864. [Google Scholar] [CrossRef] [PubMed]
- Nobili, V.; Alisi, A.; Valenti, L.; Miele, L.; Feldstein, A.E.; Alkhouri, N. NAFLD in children: New genes, new diagnostic modalities and new drugs. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 517–530. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, S.K.; Ingham, S.A.; Mohr, A.M.; Wehrkamp, C.J.; Ray, A.; Roy, S.; Cazanave, S.C.; Phillippi, M.A.; Mott, J.L. Saturated free fatty acids induce cholangiocyte lipoapoptosis. Hepatology 2014, 60, 1942–1956. [Google Scholar] [CrossRef] [Green Version]
- Sorrentino, P.; Tarantino, G.; Perrella, A.; Micheli, P.; Perrella, O.; Conca, P. A clinical-morphological study on cholestatic presentation of nonalcoholic fatty liver disease. Dig. Dis. Sci. 2005, 50, 1130–1135. [Google Scholar] [CrossRef] [PubMed]
- Skoien, R.; Richardson, M.M.; Jonsson, J.R.; Powell, E.E.; Brunt, E.M.; Neuschwander-Tetri, B.A.; Bhathal, P.S.; Dixon, J.B.; O’Brien, P.E.; Tilg, H.; et al. Heterogeneity of fibrosis patterns in non-alcoholic fatty liver disease supports the presence of multiple fibrogenic pathways. Liver Int. 2013, 33, 624–632. [Google Scholar] [CrossRef]
- Neuschwander-Tetri, B.A.; Clark, J.M.; Bass, N.M.; Van Natta, M.L.; Unalp-Arida, A.; Tonascia, J.; Zein, C.O.; Brunt, E.M.; Kleiner, D.E.; McCullough, A.J.; et al. Clinical, laboratory and histological associations in adults with nonalcoholic fatty liver disease. Hepatology 2010, 52, 913–924. [Google Scholar] [CrossRef]
- Muratori, P.; Muratori, L.; Gershwin, M.E.; Czaja, A.J.; Pappas, G.; MacCariello, S.; Granito, A.; Cassani, F.; Loria, P.; Lenzi, M.; et al. ‘True’ antimitochondrial antibody-negative primary biliary cirrhosis, low sensitivity of the routine assays, or both? Clin. Exp. Immunol. 2004, 135, 154–158. [Google Scholar] [CrossRef]
- Hindi, M.; Levy, C.; Couto, C.A.; Bejarano, P.; Mendes, F. Primary biliary cirrhosis is more severe in overweight patients. J. Clin. Gastroenterol. 2013, 47, e28–e32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wongjarupong, N.; Assavapongpaiboon, B.; Susantitaphong, P.; Cheungpasitporn, W.; Treeprasertsuk, S.; Rerknimitr, R.; Chaiteerakij, R. Non-alcoholic fatty liver disease as a risk factor for cholangiocarcinoma: A systematic review and meta-analysis. BMC Gastroenterol. 2017, 17, 149. [Google Scholar] [CrossRef] [Green Version]
- Yugawa, K.; Itoh, S.; Iseda, N.; Kurihara, T.; Kitamura, Y.; Toshima, T.; Harada, N.; Kohashi, K.; Baba, S.; Ishigami, K.; et al. Obesity is a risk factor for intrahepatic cholangiocarcinoma progression associated with alterations of metabolic activity and immune status. Sci. Rep. 2021, 11, 5845. [Google Scholar] [CrossRef] [PubMed]
- De Lorenzo, S.; Tovoli, F.; Mazzotta, A.; Vasuri, F.; Edeline, J.; Malvi, D.; Boudjema, K.; Renzulli, M.; Jeddou, H.; D’Errico, A.; et al. Non-alcoholic steatohepatitis as a risk factor for intrahepatic cholangiocarcinoma and its prognostic role. Cancers 2020, 12, 3182. [Google Scholar] [CrossRef] [PubMed]
- Bosch, D.E.; Yeh, M.M. Primary sclerosing cholangitis is protective against nonalcoholic fatty liver disease in inflammatory bowel disease. Hum. Pathol. 2017, 69, 55–62. [Google Scholar] [CrossRef]
- Jain, D.; Nayak, N.C. Bile duct changes in different etiologic types of end-stage chronic liver disease: A study on native explant livers. J. Clin. Pathol. 2012, 65, 348–351. [Google Scholar] [CrossRef]
- Chiba, M.; Sasaki, M.; Kitamura, S.; Ikeda, H.; Sato, Y.; Nakanuma, Y. Participation of bile ductular cells in the pathological progression of non-alcoholic fatty liver disease. J. Clin. Pathol. 2011, 64, 564–570. [Google Scholar] [CrossRef]
- Nguyen, D.; Smith, L.M.; Lagasse, L.L.; Derauf, C.; Grant, P.; Shah, R.; Arria, A.; Huestis, M.A.; Haning, W.; Strauss, A.; et al. Intrauterine growth of infants exposed to prenatal methamphetamine: Results from the infant development, environment, and lifestyle study. J. Pediatr. 2010, 157, 337–339. [Google Scholar] [CrossRef] [Green Version]
- Tabibian, J.H.; O’Hara, S.P.; Splinter, P.L.; Trussoni, C.E.; LaRusso, N.F. Cholangiocyte senescence by way of N-ras activation is a characteristic of primary sclerosing cholangitis. Hepatology 2014, 59, 2263–2275. [Google Scholar] [CrossRef] [Green Version]
- Nobili, V.; Carpino, G.; Alisi, A.; Franchitto, A.; Alpini, G.; De Vito, R.; Onori, P.; Alvaro, D.; Gaudio, E. Hepatic progenitor cells activation, fibrosis, and adipokines production in pediatric nonalcoholic fatty liver disease. Hepatology 2012, 56, 2142–2153. [Google Scholar] [CrossRef]
- Schuppan, D.; Surabattula, R.; Wang, X.Y. Determinants of fibrosis progression and regression in NASH. J. Hepatol. 2018, 68, 238–250. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.W.; Wang, S.P.; Alvarez, F.; Casavant, S.; Gauthier, N.; Abed, L.; Soni, K.G.; Yang, G.; Mitchell, G.A. Deficiency of liver adipose triglyceride lipase in mice causes progressive hepatic steatosis. Hepatology 2011, 54, 122–132. [Google Scholar] [CrossRef] [PubMed]
- Helal, M.; Yan, C.; Gong, Z. Stimulation of hepatocarcinogenesis by activated cholangiocytes via Il17a/f1 pathway in kras transgenic zebrafish model. Sci. Rep. 2021, 11, 1372. [Google Scholar] [CrossRef]
- Asai, Y.; Yamada, T.; Tsukita, S.; Takahashi, K.; Maekawa, M.; Honma, M.; Ikeda, M.; Murakami, K.; Munakata, Y.; Shirai, Y.; et al. Activation of the Hypoxia inducible factor 1alpha subunit pathway in steatotic liver contributes to formation of cholesterol gallstones. Gastroenterology 2017, 152, 1521–1535.e1528. [Google Scholar] [CrossRef] [Green Version]
- Huang, D.; Joo, H.; Song, N.; Cho, S.; Kim, W.; Shin, A. Association between gallstones and the risk of biliary tract cancer: A systematic review and meta-analysis. Epidemiol. Health 2021, 43, e2021011. [Google Scholar] [CrossRef]
- Mooli, R.G.R.; Rodriguez, J.; Takahashi, S.; Solanki, S.; Gonzalez, F.J.; Ramakrishnan, S.K.; Shah, Y.M. Hypoxia via ERK signaling inhibits hepatic PPARalpha to promote fatty liver. Cell. Mol. Gastroenterol. Hepatol. 2021, 12, 585–597. [Google Scholar] [CrossRef]
- Fracanzani, A.L.; Valenti, L.; Russello, M.; Miele, L.; Bertelli, C.; Bellia, A.; Masetti, C.; Cefalo, C.; Grieco, A.; Marchesini, G.; et al. Gallstone disease is associated with more severe liver damage in patients with non-alcoholic fatty liver disease. PLoS ONE 2012, 7, e41183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nimer, N.; Choucair, I.; Wang, Z.; Nemet, I.; Li, L.; Gukasyan, J.; Weeks, T.L.; Alkhouri, N.; Zein, N.; Tang, W.H.W.; et al. Bile acids profile, histopathological indices and genetic variants for non-alcoholic fatty liver disease progression. Metabolism 2021, 116, 154457. [Google Scholar] [CrossRef] [PubMed]
- Lionarons, D.A.; Heger, M.; van Golen, R.F.; Alles, L.K.; van der Mark, V.A.; Kloek, J.J.; de Waart, D.R.; Marsman, H.A.; Rusch, H.; Verheij, J.; et al. Simple steatosis sensitizes cholestatic rats to liver injury and dysregulates bile salt synthesis and transport. Sci. Rep. 2016, 6, 31829. [Google Scholar] [CrossRef]
- Pizarro, M.; Balasubramaniyan, N.; Solis, N.; Solar, A.; Duarte, I.; Miquel, J.F.; Suchy, F.J.; Trauner, M.; Accatino, L.; Ananthanarayanan, M.; et al. Bile secretory function in the obese Zucker rat: Evidence of cholestasis and altered canalicular transport function. Gut 2004, 53, 1837–1843. [Google Scholar] [CrossRef] [Green Version]
- Dzierlenga, A.L.; Clarke, J.D.; Klein, D.M.; Anumol, T.; Snyder, S.A.; Li, H.; Cherrington, N.J. Biliary elimination of pemetrexed is dependent on Mrp2 in rats: Potential mechanism of variable response in nonalcoholic steatohepatitis. J. Pharmacol. Exp. Ther. 2016, 358, 246–253. [Google Scholar] [CrossRef] [Green Version]
- Sookoian, S.; Castano, G.; Gianotti, T.F.; Gemma, C.; Pirola, C.J. Polymorphisms of MRP2 (ABCC2) are associated with susceptibility to nonalcoholic fatty liver disease. J. Nutr. Biochem. 2009, 20, 765–770. [Google Scholar] [CrossRef] [PubMed]
- Schioth, H.B.; Bostrom, A.; Murphy, S.K.; Erhart, W.; Hampe, J.; Moylan, C.; Mwinyi, J. A targeted analysis reveals relevant shifts in the methylation and transcription of genes responsible for bile acid homeostasis and drug metabolism in non-alcoholic fatty liver disease. BMC Genomics 2016, 17, 462. [Google Scholar] [CrossRef] [Green Version]
- Wheeler, S.G.; Hammond, C.L.; Jornayvaz, F.R.; Samuel, V.T.; Shulman, G.I.; Soroka, C.J.; Boyer, J.L.; Hinkle, P.M.; Ballatori, N. Ostalpha-/- mice exhibit altered expression of intestinal lipid absorption genes, resistance to age-related weight gain, and modestly improved insulin sensitivity. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 306, G425–G438. [Google Scholar] [CrossRef]
- Zagorova, M.; Prasnicka, A.; Kadova, Z.; Dolezelova, E.; Kazdova, L.; Cermanova, J.; Rozkydalova, L.; Hroch, M.; Mokry, J.; Micuda, S. Boldine attenuates cholestasis associated with nonalcoholic fatty liver disease in hereditary hypertriglyceridemic rats fed by high-sucrose diet. Physiol. Res. 2015, 64, S467–S476. [Google Scholar] [CrossRef] [PubMed]
- Okushin, K.; Tsutsumi, T.; Ikeuchi, K.; Kado, A.; Enooku, K.; Fujinaga, H.; Yamauchi, N.; Ushiku, T.; Moriya, K.; Yotsuyanagi, H.; et al. Heterozygous knockout of Bile salt export pump ameliorates liver steatosis in mice fed a high-fat diet. PLoS ONE 2020, 15, e0234750. [Google Scholar] [CrossRef] [PubMed]
- Donkers, J.M.; Kooijman, S.; Slijepcevic, D.; Kunst, R.F.; Roscam Abbing, R.L.; Haazen, L.; de Waart, D.R.; Levels, J.H.; Schoonjans, K.; Rensen, P.C.; et al. NTCP deficiency in mice protects against obesity and hepatosteatosis. JCI Insight 2019, 5. [Google Scholar] [CrossRef]
- Hu, Y.B.; Liu, X.Y.; Zhan, W. Farnesoid X receptor agonist INT-767 attenuates liver steatosis and inflammation in rat model of nonalcoholic steatohepatitis. Drug Des. Devel. Ther. 2018, 12, 2213–2221. [Google Scholar] [CrossRef] [Green Version]
- Pathak, P.; Liu, H.; Boehme, S.; Xie, C.; Krausz, K.W.; Gonzalez, F.; Chiang, J.Y.L. Farnesoid X receptor induces Takeda G-protein receptor 5 cross-talk to regulate bile acid synthesis and hepatic metabolism. J. Biol. Chem. 2017, 292, 11055–11069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chavez-Talavera, O.; Tailleux, A.; Lefebvre, P.; Staels, B. Bile acid control of metabolism and inflammation in obesity, type 2 diabetes, dyslipidemia, and nonalcoholic fatty liver disease. Gastroenterology 2017, 152, 1679–1694.e1673. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Ratziu, V.; Loomba, R.; Rinella, M.; Anstee, Q.M.; Goodman, Z.; Bedossa, P.; Geier, A.; Beckebaum, S.; Newsome, P.N.; et al. Obeticholic acid for the treatment of non-alcoholic steatohepatitis: Interim analysis from a multicentre, randomised, placebo-controlled phase 3 trial. Lancet 2019, 394, 2184–2196. [Google Scholar] [CrossRef] [Green Version]
- Strub, G.M.; Maceyka, M.; Hait, N.C.; Milstien, S.; Spiegel, S. Extracellular and intracellular actions of sphingosine-1-phosphate. Adv. Exp. Med. Biol. 2010, 688, 141–155. [Google Scholar] [CrossRef] [Green Version]
- Kwong, E.; Li, Y.; Hylemon, P.B.; Zhou, H. Bile acids and sphingosine-1-phosphate receptor 2 in hepatic lipid metabolism. Acta Pharm. Sin. B 2015, 5, 151–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagahashi, M.; Takabe, K.; Liu, R.; Peng, K.; Wang, X.; Wang, Y.; Hait, N.C.; Wang, X.; Allegood, J.C.; Yamada, A.; et al. Conjugated bile acid-activated S1P receptor 2 is a key regulator of sphingosine kinase 2 and hepatic gene expression. Hepatology 2015, 61, 1216–1226. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, T.; Tsuchiya, A.; Kumagai, M.; Takeuchi, S.; Nojiri, S.; Watanabe, T.; Ogawa, M.; Itoh, M.; Takamura, M.; Suganami, T.; et al. Blocking sphingosine 1-phosphate receptor 2 accelerates hepatocellular carcinoma progression in a mouse model of NASH. Biochem. Biophys. Res. Commun. 2020, 530, 665–672. [Google Scholar] [CrossRef] [PubMed]
- Coulon, S.; Francque, S.; Colle, I.; Verrijken, A.; Blomme, B.; Heindryckx, F.; De Munter, S.; Prawitt, J.; Caron, S.; Staels, B.; et al. Evaluation of inflammatory and angiogenic factors in patients with non-alcoholic fatty liver disease. Cytokine 2012, 59, 442–449. [Google Scholar] [CrossRef]
- Siddiqui, H.; Rawal, P.; Bihari, C.; Arora, N.; Kaur, S. Vascular endothelial growth factor promotes proliferation of epithelial cell adhesion molecule-positive cells in nonalcoholic steatohepatitis. J. Clin. Exp. Hepatol. 2020, 10, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Fiorotto, R.; Scirpo, R.; Trauner, M.; Fabris, L.; Hoque, R.; Spirli, C.; Strazzabosco, M. Loss of CFTR affects biliary epithelium innate immunity and causes TLR4-NF-kappaB-mediated inflammatory response in mice. Gastroenterology 2011, 141, 1498–1508.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendez-Sanchez, N.; Zamora-Valdes, D.; Pichardo-Bahena, R.; Barredo-Prieto, B.; Ponciano-Rodriguez, G.; Bermejo-Martinez, L.; Chavez-Tapia, N.C.; Baptista-Gonzalez, H.A.; Uribe, M. Endocannabinoid receptor CB2 in nonalcoholic fatty liver disease. Liver Int. 2007, 27, 215–219. [Google Scholar] [CrossRef]
- Floreani, A.; Lazzari, R.; Macchi, V.; Porzionato, A.; Variola, A.; Colavito, D.; Leon, A.; Guido, M.; Baldo, V.; De Caro, R.; et al. Hepatic expression of endocannabinoid receptors and their novel polymorphisms in primary biliary cirrhosis. J. Gastroenterol. 2010, 45, 68–76. [Google Scholar] [CrossRef]
- Julien, B.; Grenard, P.; Teixeira-Clerc, F.; Van Nhieu, J.T.; Li, L.; Karsak, M.; Zimmer, A.; Mallat, A.; Lotersztajn, S. Antifibrogenic role of the cannabinoid receptor CB2 in the liver. Gastroenterology 2005, 128, 742–755. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Liangpunsakul, S. Histamine H2-receptor antagonist use is associated with lower prevalence of nonalcoholic fatty liver disease: A population-based study from the national health and nutrition examination survey, 2001–2006. J. Clin. Gastroenterol. 2016, 50, 596–601. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, L.; Hargrove, L.; Demieville, J.; Bailey, J.M.; Dar, W.; Polireddy, K.; Chen, Q.; Nevah Rubin, M.I.; Sybenga, A.; DeMorrow, S.; et al. Knockout of l-histidine decarboxylase prevents cholangiocyte damage and hepatic fibrosis in mice subjected to high-fat diet feeding via disrupted histamine/leptin signaling. Am. J. Pathol. 2018, 188, 600–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennedy, L.; Meadows, V.; Sybenga, A.; Demieville, J.; Chen, L.; Hargrove, L.; Ekser, B.; Dar, W.; Ceci, L.; Kundu, D.; et al. Mast cells promote non-alcoholic fatty liver disease phenotypes and microvesicular steatosis in mice fed western diet. Hepatology 2021, 74, 164–182. [Google Scholar] [CrossRef]
- Vespasiani-Gentilucci, U.; Carotti, S.; Perrone, G.; Mazzarelli, C.; Galati, G.; Onetti-Muda, A.; Picardi, A.; Morini, S. Hepatic toll-like receptor 4 expression is associated with portal inflammation and fibrosis in patients with NAFLD. Liver Int. 2015, 35, 569–581. [Google Scholar] [CrossRef]
- Syn, W.K.; Choi, S.S.; Liaskou, E.; Karaca, G.F.; Agboola, K.M.; Oo, Y.H.; Mi, Z.; Pereira, T.A.; Zdanowicz, M.; Malladi, P.; et al. Osteopontin is induced by hedgehog pathway activation and promotes fibrosis progression in nonalcoholic steatohepatitis. Hepatology 2011, 53, 106–115. [Google Scholar] [CrossRef] [Green Version]
- Omenetti, A.; Diehl, A.M. Hedgehog signaling in cholangiocytes. Curr. Opin. Gastroenterol. 2011, 27, 268–275. [Google Scholar] [CrossRef] [Green Version]
- Sekar, R.; Chow, B.K. Secretin receptor-knockout mice are resistant to high-fat diet-induced obesity and exhibit impaired intestinal lipid absorption. FASEB J. 2014, 28, 3494–3505. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Lin, L.; Hu, X.; Li, C.; Zhang, H. Liver failure in a chinese cystic fibrosis child with homozygous R553X mutation. Front. Pediatr. 2019, 7, 36. [Google Scholar] [CrossRef]
- Ayoub, F.; Trillo-Alvarez, C.; Morelli, G.; Lascano, J. Risk factors for hepatic steatosis in adults with cystic fibrosis: Similarities to non-alcoholic fatty liver disease. World J. Hepatol. 2018, 10, 34–40. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, T.; Kundu, D.; Robles-Linares, J.; Meadows, V.; Sato, K.; Baiocchi, L.; Ekser, B.; Glaser, S.; Alpini, G.; Francis, H.; et al. Feedback Signaling between Cholangiopathies, Ductular Reaction, and Non-Alcoholic Fatty Liver Disease. Cells 2021, 10, 2072. https://doi.org/10.3390/cells10082072
Zhou T, Kundu D, Robles-Linares J, Meadows V, Sato K, Baiocchi L, Ekser B, Glaser S, Alpini G, Francis H, et al. Feedback Signaling between Cholangiopathies, Ductular Reaction, and Non-Alcoholic Fatty Liver Disease. Cells. 2021; 10(8):2072. https://doi.org/10.3390/cells10082072
Chicago/Turabian StyleZhou, Tianhao, Debjyoti Kundu, Jonathan Robles-Linares, Vik Meadows, Keisaku Sato, Leonardo Baiocchi, Burcin Ekser, Shannon Glaser, Gianfranco Alpini, Heather Francis, and et al. 2021. "Feedback Signaling between Cholangiopathies, Ductular Reaction, and Non-Alcoholic Fatty Liver Disease" Cells 10, no. 8: 2072. https://doi.org/10.3390/cells10082072
APA StyleZhou, T., Kundu, D., Robles-Linares, J., Meadows, V., Sato, K., Baiocchi, L., Ekser, B., Glaser, S., Alpini, G., Francis, H., & Kennedy, L. (2021). Feedback Signaling between Cholangiopathies, Ductular Reaction, and Non-Alcoholic Fatty Liver Disease. Cells, 10(8), 2072. https://doi.org/10.3390/cells10082072