
Unraveling the IGF System Interactome in Sarcomas Exploits Novel Therapeutic Options




{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. The IGF System: Major Components and Signal Transduction
3. The IGF System in Cancer: A Complex Network of Interactions
4. Therapeutic Approaches to Block the IGF System
4.1. Anti-IGF1R mAbs
4.2. Tyrosine Kinase Inhibitor Small Molecules
4.3. IGFs Neutralizing Antibodies and Ligands TRAP
5. The IGF System in Sarcomas: Functions and Response to Therapies
5.1. Osteosarcoma
5.2. Ewing Sarcoma
5.3. Rhabdomyosarcoma
5.4. Synovial Sarcoma
5.5. Desmoplastic Small Round Cell Tumor
6. Perspectives for Novel Therapeutic Combinations in Sarcomas
6.1. Ephrin Receptors
6.2. Hippo Pathway
6.3. BET Proteins
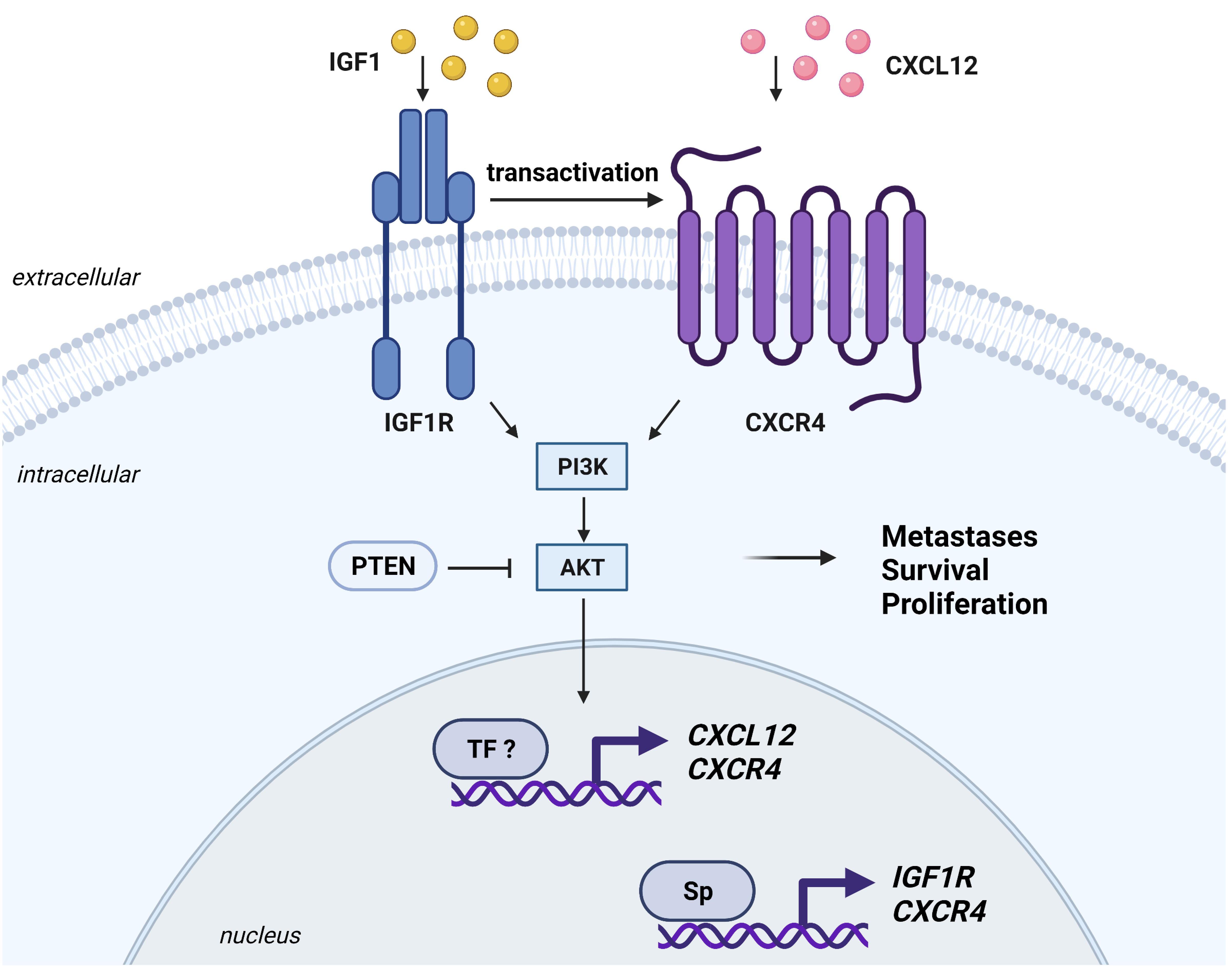
6.4. CXCR4
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Stanley, T.L.; Fourman, L.T.; Zheng, I.; McClure, C.M.; Feldpausch, M.N.; Torriani, M.; Corey, K.E.; Chung, R.T.; Lee, H.; Kleiner, D.E.; et al. Relationship of IGF-1 and IGF-Binding Proteins to Disease Severity and Glycemia in Nonalcoholic Fatty Liver Disease. J. Clin. Endocrinol. Metab. 2021, 106, e520–e533. [Google Scholar] [CrossRef] [PubMed]
- Hakuno, F.; Takahashi, S.I. IGF1 receptor signaling pathways. J. Mol. Endocrinol. 2018, 61, T69–T86. [Google Scholar] [CrossRef] [Green Version]
- Frystyk, J.; Teran, E.; Gude, M.F.; Bjerre, M.; Hjortebjerg, R. Pregnancy-associated plasma proteins and Stanniocalcin-2—Novel players controlling IGF-I physiology. Growth Horm. IGF Res. Off. J. Growth Horm. Res. Soc. Int. IGF Res. Soc. 2020, 53–54, 101330. [Google Scholar] [CrossRef] [PubMed]
- Osher, E.; Macaulay, V.M. Therapeutic Targeting of the IGF Axis. Cells 2019, 8, 895. [Google Scholar] [CrossRef] [Green Version]
- de Groot, S.; Rottgering, B.; Gelderblom, H.; Pijl, H.; Szuhai, K.; Kroep, J.R. Unraveling the Resistance of IGF-Pathway Inhibition in Ewing Sarcoma. Cancers 2020, 12, 3568. [Google Scholar] [CrossRef] [PubMed]
- Simpson, A.; Petnga, W.; Macaulay, V.M.; Weyer-Czernilofsky, U.; Bogenrieder, T. Insulin-Like Growth Factor (IGF) Pathway Targeting in Cancer: Role of the IGF Axis and Opportunities for Future Combination Studies. Target. Oncol. 2017, 12, 571–597. [Google Scholar] [CrossRef] [Green Version]
- Cao, J.; Yee, D. Disrupting Insulin and IGF Receptor Function in Cancer. Int. J. Mol. Sci. 2021, 22, 555. [Google Scholar] [CrossRef]
- Hua, H.; Kong, Q.; Yin, J.; Zhang, J.; Jiang, Y. Insulin-like growth factor receptor signaling in tumorigenesis and drug resistance: A challenge for cancer therapy. J. Hematol. Oncol. 2020, 13, 64. [Google Scholar] [CrossRef]
- Mancarella, C.; Scotlandi, K. IGF system in sarcomas: A crucial pathway with many unknowns to exploit for therapy. J. Mol. Endocrinol. 2018, 61, T45–T60. [Google Scholar] [CrossRef] [PubMed]
- Mancarella, C.; Scotlandi, K. IGF2BP3 From Physiology to Cancer: Novel Discoveries, Unsolved Issues, and Future Perspectives. Front. Cell Dev. Biol. 2019, 7, 363. [Google Scholar] [CrossRef]
- Chen, B.; Li, J.; Chi, D.; Sahnoune, I.; Calin, S.; Girnita, L.; Calin, G.A. Non-Coding RNAs in IGF-1R Signaling Regulation: The Underlying Pathophysiological Link between Diabetes and Cancer. Cells 2019, 8, 1638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mancarella, C.; Morrione, A.; Scotlandi, K. Novel Regulators of the IGF System in Cancer. Biomolecules 2021, 11, 273. [Google Scholar] [CrossRef] [PubMed]
- Goryashchenko, A.S.; Mozhaev, A.A.; Serova, O.V.; Erokhina, T.N.; Orsa, A.N.; Deyev, I.E.; Petrenko, A.G. Probing Structure and Function of Alkali Sensor IRR with Monoclonal Antibodies. Biomolecules 2020, 10, 1060. [Google Scholar] [CrossRef] [PubMed]
- Deyev, I.E.; Mitrofanova, A.V.; Zhevlenev, E.S.; Radionov, N.; Berchatova, A.A.; Popova, N.V.; Serova, O.V.; Petrenko, A.G. Structural determinants of the insulin receptor-related receptor activation by alkali. J. Biol. Chem. 2013, 288, 33884–33893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Meyts, P.; Whittaker, J. Structural biology of insulin and IGF1 receptors: Implications for drug design. Nat. Rev. Drug Discov. 2002, 1, 769–783. [Google Scholar] [CrossRef] [PubMed]
- LeRoith, D.; Werner, H.; Beitner-Johnson, D.; Roberts, C.T., Jr. Molecular and cellular aspects of the insulin-like growth factor I receptor. Endocr. Rev. 1995, 16, 143–163. [Google Scholar] [CrossRef]
- Ullrich, A.; Gray, A.; Tam, A.W.; Yang-Feng, T.; Tsubokawa, M.; Collins, C.; Henzel, W.; Le Bon, T.; Kathuria, S.; Chen, E.; et al. Insulin-like growth factor I receptor primary structure: Comparison with insulin receptor suggests structural determinants that define functional specificity. EMBO J. 1986, 5, 2503–2512. [Google Scholar] [CrossRef]
- Belfiore, A.; Malaguarnera, R.; Vella, V.; Lawrence, M.C.; Sciacca, L.; Frasca, F.; Morrione, A.; Vigneri, R. Insulin Receptor Isoforms in Physiology and Disease: An Updated View. Endocr. Rev. 2017, 38, 379–431. [Google Scholar] [CrossRef]
- Pandini, G.; Frasca, F.; Mineo, R.; Sciacca, L.; Vigneri, R.; Belfiore, A. Insulin/insulin-like growth factor I hybrid receptors have different biological characteristics depending on the insulin receptor isoform involved. J. Biol. Chem. 2002, 277, 39684–39695. [Google Scholar] [CrossRef] [Green Version]
- Belfiore, A.; Frasca, F.; Pandini, G.; Sciacca, L.; Vigneri, R. Insulin receptor isoforms and insulin receptor/insulin-like growth factor receptor hybrids in physiology and disease. Endocr. Rev. 2009, 30, 586–623. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Nagle, A.M.; Wang, Y.F.; Boone, D.N.; Lee, A.V. Controlled dimerization of insulin-like growth factor-1 and insulin receptors reveals shared and distinct activities of holo and hybrid receptors. J. Biol. Chem. 2018, 293, 3700–3709. [Google Scholar] [CrossRef] [Green Version]
- Torrente, Y.; Bella, P.; Tripodi, L.; Villa, C.; Farini, A. Role of Insulin-Like Growth Factor Receptor 2 across Muscle Homeostasis: Implications for Treating Muscular Dystrophy. Cells 2020, 9, 441. [Google Scholar] [CrossRef] [Green Version]
- Deyev, I.E.; Popova, N.V.; Serova, O.V.; Zhenilo, S.V.; Regoli, M.; Bertelli, E.; Petrenko, A.G. Alkaline pH induces IRR-mediated phosphorylation of IRS-1 and actin cytoskeleton remodeling in a pancreatic beta cell line. Biochimie 2017, 138, 62–69. [Google Scholar] [CrossRef]
- Batishchev, O.V.; Kuzmina, N.V.; Mozhaev, A.A.; Goryashchenko, A.S.; Mileshina, E.D.; Orsa, A.N.; Bocharov, E.V.; Deyev, I.E.; Petrenko, A.G. Activity-dependent conformational transitions of the insulin receptor-related receptor. J. Biol. Chem. 2021, 296, 100534. [Google Scholar] [CrossRef]
- Takahashi, S.I. IGF research 2016–2018. Growth Horm. IGF Res. Off. J. Growth Horm. Res. Soc. Int. IGF Res. Soc. 2019, 48–49, 65–69. [Google Scholar] [CrossRef]
- Bergman, D.; Halje, M.; Nordin, M.; Engstrom, W. Insulin-like growth factor 2 in development and disease: A mini-review. Gerontology 2013, 59, 240–249. [Google Scholar] [CrossRef]
- Bach, L.A. IGF-binding proteins. J. Mol. Endocrinol. 2018, 61, T11–T28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haywood, N.J.; Slater, T.A.; Matthews, C.J.; Wheatcroft, S.B. The insulin like growth factor and binding protein family: Novel therapeutic targets in obesity & diabetes. Mol. Metab. 2019, 19, 86–96. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, A.; Pathak, K.A.; Shrivastav, A.; Varma Shrivastav, S. Insulin-Like Growth Factor Binding Protein-3 Binds to Histone 3. Int. J. Mol. Sci. 2021, 22, 407. [Google Scholar] [CrossRef]
- Cai, Q.; Dozmorov, M.; Oh, Y. IGFBP-3/IGFBP-3 Receptor System as an Anti-Tumor and Anti-Metastatic Signaling in Cancer. Cells 2020, 9, 1261. [Google Scholar] [CrossRef] [PubMed]
- Fruman, D.A.; Chiu, H.; Hopkins, B.D.; Bagrodia, S.; Cantley, L.C.; Abraham, R.T. The PI3K Pathway in Human Disease. Cell 2017, 170, 605–635. [Google Scholar] [CrossRef] [Green Version]
- Liu, F.; Yang, X.; Geng, M.; Huang, M. Targeting ERK, an Achilles’ Heel of the MAPK pathway, in cancer therapy. Acta Pharm. Sin. B 2018, 8, 552–562. [Google Scholar] [CrossRef] [PubMed]
- Morrione, A. Grb10 adapter protein as regulator of insulin-like growth factor receptor signaling. J. Cell. Physiol. 2003, 197, 307–311. [Google Scholar] [CrossRef]
- Gual, P.; Baron, V.; Lequoy, V.; Van Obberghen, E. Interaction of Janus kinases JAK-1 and JAK-2 with the insulin receptor and the insulin-like growth factor-1 receptor. Endocrinology 1998, 139, 884–893. [Google Scholar] [CrossRef] [PubMed]
- Samani, A.A.; Yakar, S.; LeRoith, D.; Brodt, P. The role of the IGF system in cancer growth and metastasis: Overview and recent insights. Endocr. Rev. 2007, 28, 20–47. [Google Scholar] [CrossRef] [PubMed]
- Harrison, D.A. The Jak/STAT pathway. Cold Spring Harb. Perspect. Biol. 2012, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vishwamitra, D.; George, S.K.; Shi, P.; Kaseb, A.O.; Amin, H.M. Type I insulin-like growth factor receptor signaling in hematological malignancies. Oncotarget 2017, 8, 1814–1844. [Google Scholar] [CrossRef] [Green Version]
- Mitsiades, C.S.; Mitsiades, N.S.; McMullan, C.J.; Poulaki, V.; Shringarpure, R.; Akiyama, M.; Hideshima, T.; Chauhan, D.; Joseph, M.; Libermann, T.A.; et al. Inhibition of the insulin-like growth factor receptor-1 tyrosine kinase activity as a therapeutic strategy for multiple myeloma, other hematologic malignancies, and solid tumors. Cancer Cell 2004, 5, 221–230. [Google Scholar] [CrossRef] [Green Version]
- Knuppel, A.; Fensom, G.K.; Watts, E.L.; Gunter, M.J.; Murphy, N.; Papier, K.; Perez-Cornago, A.; Schmidt, J.A.; Smith Byrne, K.; Travis, R.C.; et al. Circulating Insulin-like Growth Factor-I Concentrations and Risk of 30 Cancers: Prospective Analyses in UK Biobank. Cancer Res. 2020, 80, 4014–4021. [Google Scholar] [CrossRef]
- Werner, H.; Lapkina-Gendler, L.; Achlaug, L.; Nagaraj, K.; Somri, L.; Yaron-Saminsky, D.; Pasmanik-Chor, M.; Sarfstein, R.; Laron, Z.; Yakar, S. Genome-Wide Profiling of Laron Syndrome Patients Identifies Novel Cancer Protection Pathways. Cells 2019, 8, 596. [Google Scholar] [CrossRef] [Green Version]
- Swerdlow, A.J.; Cooke, R.; Beckers, D.; Borgstrom, B.; Butler, G.; Carel, J.C.; Cianfarani, S.; Clayton, P.; Coste, J.; Deodati, A.; et al. Cancer Risks in Patients Treated With Growth Hormone in Childhood: The SAGhE European Cohort Study. J. Clin. Endocrinol. Metab. 2017, 102, 1661–1672. [Google Scholar] [CrossRef]
- Sciacca, L.; Vella, V.; Frittitta, L.; Tumminia, A.; Manzella, L.; Squatrito, S.; Belfiore, A.; Vigneri, R. Long-acting insulin analogs and cancer. Nutr. Metab. Cardiovasc. Dis. NMCD 2018, 28, 436–443. [Google Scholar] [CrossRef]
- Holly, J.M.P.; Biernacka, K.; Perks, C.M. The Neglected Insulin: IGF-II, a Metabolic Regulator with Implications for Diabetes, Obesity, and Cancer. Cells 2019, 8, 1207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livingstone, C. IGF2 and cancer. Endocr.-Relat. Cancer 2013, 20, R321–R339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LeRoith, D.; Holly, J.M.P.; Forbes, B.E. Insulin-like growth factors: Ligands, binding proteins, and receptors. Mol. Metab. 2021. [Google Scholar] [CrossRef] [PubMed]
- Scalia, P.; Giordano, A.; Williams, S.J. The IGF-II-Insulin Receptor Isoform-A Autocrine Signal in Cancer: Actionable Perspectives. Cancers 2020, 12, 366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scalia, P.; Pandini, G.; Carnevale, V.; Giordano, A.; Williams, S.J. Correction: Identification of a novel EphB4 phosphodegron regulated by the autocrine IGFII/IRA axis in malignant mesothelioma. Oncogene 2021, 40, 1551. [Google Scholar] [CrossRef] [PubMed]
- Dynkevich, Y.; Rother, K.I.; Whitford, I.; Qureshi, S.; Galiveeti, S.; Szulc, A.L.; Danoff, A.; Breen, T.L.; Kaviani, N.; Shanik, M.H.; et al. Tumors, IGF-2, and hypoglycemia: Insights from the clinic, the laboratory, and the historical archive. Endocr. Rev. 2013, 34, 798–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- de-Freitas-Junior, J.C.M.; Andrade-da-Costa, J.; Silva, M.C.; Pinho, S.S. Glycans as Regulatory Elements of the Insulin/IGF System: Impact in Cancer Progression. Int. J. Mol. Sci. 2017, 18, 1921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasprzak, A.; Kwasniewski, W.; Adamek, A.; Gozdzicka-Jozefiak, A. Insulin-like growth factor (IGF) axis in cancerogenesis. Mutat. Res. Rev. Mutat. Res. 2017, 772, 78–104. [Google Scholar] [CrossRef]
- Sell, C.; Rubini, M.; Rubin, R.; Liu, J.P.; Efstratiadis, A.; Baserga, R. Simian virus 40 large tumor antigen is unable to transform mouse embryonic fibroblasts lacking type 1 insulin-like growth factor receptor. Proc. Natl. Acad. Sci. USA 1993, 90, 11217–11221. [Google Scholar] [CrossRef] [Green Version]
- Frittitta, L.; Vigneri, R.; Stampfer, M.R.; Goldfine, I.D. Insulin receptor overexpression in 184B5 human mammary epithelial cells induces a ligand-dependent transformed phenotype. J. Cell. Biochem. 1995, 57, 666–669. [Google Scholar] [CrossRef]
- Moorehead, R.A.; Sanchez, O.H.; Baldwin, R.M.; Khokha, R. Transgenic overexpression of IGF-II induces spontaneous lung tumors: A model for human lung adenocarcinoma. Oncogene 2003, 22, 853–857. [Google Scholar] [CrossRef] [Green Version]
- Bates, P.; Fisher, R.; Ward, A.; Richardson, L.; Hill, D.J.; Graham, C.F. Mammary cancer in transgenic mice expressing insulin-like growth factor II (IGF-II). Br. J. Cancer 1995, 72, 1189–1193. [Google Scholar] [CrossRef] [Green Version]
- Christofori, G.; Naik, P.; Hanahan, D. A second signal supplied by insulin-like growth factor II in oncogene-induced tumorigenesis. Nature 1994, 369, 414–418. [Google Scholar] [CrossRef]
- Rogler, C.E.; Yang, D.; Rossetti, L.; Donohoe, J.; Alt, E.; Chang, C.J.; Rosenfeld, R.; Neely, K.; Hintz, R. Altered body composition and increased frequency of diverse malignancies in insulin-like growth factor-II transgenic mice. J. Biol. Chem. 1994, 269, 13779–13784. [Google Scholar] [CrossRef]
- Tarn, C.; Rink, L.; Merkel, E.; Flieder, D.; Pathak, H.; Koumbi, D.; Testa, J.R.; Eisenberg, B.; von Mehren, M.; Godwin, A.K. Insulin-like growth factor 1 receptor is a potential therapeutic target for gastrointestinal stromal tumors. Proc. Natl. Acad. Sci. USA 2008, 105, 8387–8392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Trent, J.M.; Meltzer, P.S. Rapid isolation and characterization of amplified DNA by chromosome microdissection: Identification of IGF1R amplification in malignant melanoma. Oncogene 1993, 8, 2827–2831. [Google Scholar] [PubMed]
- Behjati, S.; Tarpey, P.S.; Haase, K.; Ye, H.; Young, M.D.; Alexandrov, L.B.; Farndon, S.J.; Collord, G.; Wedge, D.C.; Martincorena, I.; et al. Recurrent mutation of IGF signalling genes and distinct patterns of genomic rearrangement in osteosarcoma. Nat. Commun. 2017, 8, 15936. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wang, W.; Li, Y.; Sun, F.; Lin, J.; Li, L. CKS1BP7, a Pseudogene of CKS1B, is Co-Amplified with IGF1R in Breast Cancers. Pathol. Oncol. Res. POR 2018, 24, 223–229. [Google Scholar] [CrossRef]
- Armengol, G.; Knuutila, S.; Lluis, F.; Capella, G.; Miro, R.; Caballin, M.R. DNA copy number changes and evaluation of MYC, IGF1R, and FES amplification in xenografts of pancreatic adenocarcinoma. Cancer Genet. Cytogenet. 2000, 116, 133–141. [Google Scholar] [CrossRef]
- Piarulli, G.; Puls, F.; Wangberg, B.; Fagman, H.; Hansson, M.; Nilsson, J.; Arbajian, E.; Mertens, F. Gene fusion involving the insulin-like growth factor 1 receptor in an ALK-negative inflammatory myofibroblastic tumour. Histopathology 2019, 74, 1098–1102. [Google Scholar] [CrossRef] [PubMed]
- Wise, T.L.; Pravtcheva, D.D. Delayed onset of Igf2-induced mammary tumors in Igf2r transgenic mice. Cancer Res. 2006, 66, 1327–1336. [Google Scholar] [CrossRef] [Green Version]
- Gao, T.; Liu, X.; He, B.; Pan, Y.; Wang, S. IGF2 loss of imprinting enhances colorectal cancer stem cells pluripotency by promoting tumor autophagy. Aging 2020, 12, 21236–21252. [Google Scholar] [CrossRef]
- Vella, V.; Milluzzo, A.; Scalisi, N.M.; Vigneri, P.; Sciacca, L. Insulin Receptor Isoforms in Cancer. Int. J. Mol. Sci. 2018, 19, 3615. [Google Scholar] [CrossRef] [Green Version]
- Aiello, A.; Pandini, G.; Sarfstein, R.; Werner, H.; Manfioletti, G.; Vigneri, R.; Belfiore, A. HMGA1 protein is a positive regulator of the insulin-like growth factor-I receptor gene. Eur. J. Cancer 2010, 46, 1919–1926. [Google Scholar] [CrossRef] [PubMed]
- Foti, D.; Iuliano, R.; Chiefari, E.; Brunetti, A. A nucleoprotein complex containing Sp1, C/EBP beta, and HMGI-Y controls human insulin receptor gene transcription. Mol. Cell. Biol. 2003, 23, 2720–2732. [Google Scholar] [CrossRef] [Green Version]
- Mancarella, C.; Casanova-Salas, I.; Calatrava, A.; Ventura, S.; Garofalo, C.; Rubio-Briones, J.; Magistroni, V.; Manara, M.C.; Lopez-Guerrero, J.A.; Scotlandi, K. ERG deregulation induces IGF-1R expression in prostate cancer cells and affects sensitivity to anti-IGF-1R agents. Oncotarget 2015, 6, 16611–16622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meisel Sharon, S.; Pozniak, Y.; Geiger, T.; Werner, H. TMPRSS2-ERG fusion protein regulates insulin-like growth factor-1 receptor (IGF1R) gene expression in prostate cancer: Involvement of transcription factor Sp1. Oncotarget 2016, 7, 51375–51392. [Google Scholar] [CrossRef] [PubMed]
- Werner, H.; Idelman, G.; Rubinstein, M.; Pattee, P.; Nagalla, S.R.; Roberts, C.T., Jr. A novel EWS-WT1 gene fusion product in desmoplastic small round cell tumor is a potent transactivator of the insulin-like growth factor-I receptor (IGF-IR) gene. Cancer Lett. 2007, 247, 84–90. [Google Scholar] [CrossRef]
- Ayalon, D.; Glaser, T.; Werner, H. Transcriptional regulation of IGF-I receptor gene expression by the PAX3-FKHR oncoprotein. Growth Horm. IGF Res. Off. J. Growth Horm. Res. Soc. Int. IGF Res. Soc. 2001, 11, 289–297. [Google Scholar] [CrossRef]
- Toretsky, J.A.; Kalebic, T.; Blakesley, V.; LeRoith, D.; Helman, L.J. The insulin-like growth factor-I receptor is required for EWS/FLI-1 transformation of fibroblasts. J. Biol. Chem. 1997, 272, 30822–30827. [Google Scholar] [CrossRef] [Green Version]
- Werner, H.; Re, G.G.; Drummond, I.A.; Sukhatme, V.P.; Rauscher, F.J., 3rd; Sens, D.A.; Garvin, A.J.; LeRoith, D.; Roberts, C.T., Jr. Increased expression of the insulin-like growth factor I receptor gene, IGF1R, in Wilms tumor is correlated with modulation of IGF1R promoter activity by the WT1 Wilms tumor gene product. Proc. Natl. Acad. Sci. USA 1993, 90, 5828–5832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abramovitch, S.; Glaser, T.; Ouchi, T.; Werner, H. BRCA1-Sp1 interactions in transcriptional regulation of the IGF-IR gene. FEBS Lett. 2003, 541, 149–154. [Google Scholar] [CrossRef] [Green Version]
- Yuen, J.S.; Cockman, M.E.; Sullivan, M.; Protheroe, A.; Turner, G.D.; Roberts, I.S.; Pugh, C.W.; Werner, H.; Macaulay, V.M. The VHL tumor suppressor inhibits expression of the IGF1R and its loss induces IGF1R upregulation in human clear cell renal carcinoma. Oncogene 2007, 26, 6499–6508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarfstein, R.; Werner, H. Tumor suppressor p53 regulates insulin receptor (INSR) gene expression via direct binding to the INSR promoter. Oncotarget 2020, 11, 2424–2437. [Google Scholar] [CrossRef] [PubMed]
- Werner, H.; Karnieli, E.; Rauscher, F.J.; LeRoith, D. Wild-type and mutant p53 differentially regulate transcription of the insulin-like growth factor I receptor gene. Proc. Natl. Acad. Sci. USA 1996, 93, 8318–8323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haley, V.L.; Barnes, D.J.; Sandovici, I.; Constancia, M.; Graham, C.F.; Pezzella, F.; Buhnemann, C.; Carter, E.J.; Hassan, A.B. Igf2 pathway dependency of the Trp53 developmental and tumour phenotypes. EMBO Mol. Med. 2012, 4, 705–718. [Google Scholar] [CrossRef]
- Zhang, L.; Kashanchi, F.; Zhan, Q.; Zhan, S.; Brady, J.N.; Fornace, A.J.; Seth, P.; Helman, L.J. Regulation of insulin-like growth factor II P3 promotor by p53: A potential mechanism for tumorigenesis. Cancer Res. 1996, 56, 1367–1373. [Google Scholar]
- Zhang, L.; Zhan, Q.; Zhan, S.; Kashanchi, F.; Fornace, A.J., Jr.; Seth, P.; Helman, L.J. p53 regulates human insulin-like growth factor II gene expression through active P4 promoter in rhabdomyosarcoma cells. DNA Cell Biol. 1998, 17, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Clermont, F.; Nittner, D.; Marine, J.C. IGF2: The Achilles’ heel of p53-deficiency? EMBO Mol. Med. 2012, 4, 688–690. [Google Scholar] [CrossRef] [PubMed]
- Scalia, P.; Giordano, A.; Martini, C.; Williams, S.J. Isoform- and Paralog-Switching in IR-Signaling: When Diabetes Opens the Gates to Cancer. Biomolecules 2020, 10, 1617. [Google Scholar] [CrossRef] [PubMed]
- Sen, S.; Langiewicz, M.; Jumaa, H.; Webster, N.J. Deletion of serine/arginine-rich splicing factor 3 in hepatocytes predisposes to hepatocellular carcinoma in mice. Hepatology 2015, 61, 171–183. [Google Scholar] [CrossRef]
- Huang, G.; Song, C.; Wang, N.; Qin, T.; Sui, S.; Obr, A.; Zeng, L.; Wood, T.L.; Leroith, D.; Li, M.; et al. RNA-binding protein CUGBP1 controls the differential INSR splicing in molecular subtypes of breast cancer cells and affects cell aggressiveness. Carcinogenesis 2020, 41, 1294–1305. [Google Scholar] [CrossRef] [PubMed]
- Garofalo, C.; Mancarella, C.; Grilli, A.; Manara, M.C.; Astolfi, A.; Marino, M.T.; Conte, A.; Sigismund, S.; Care, A.; Belfiore, A.; et al. Identification of common and distinctive mechanisms of resistance to different anti-IGF-IR agents in Ewing’s sarcoma. Mol. Endocrinol. 2012, 26, 1603–1616. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Mu, Q.; Huang, H. The Roles of Insulin-Like Growth Factor 2 mRNA-Binding Protein 2 in Cancer and Cancer Stem Cells. Stem Cells Int. 2018, 2018, 4217259. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Zhang, H.; Guo, X.; Zhu, Z.; Cai, H.; Kong, X. Insulin-like growth factor 2 mRNA-binding protein 1 (IGF2BP1) in cancer. J. Hematol. Oncol. 2018, 11, 88. [Google Scholar] [CrossRef] [Green Version]
- Fawzy, I.O.; Hamza, M.T.; Hosny, K.A.; Esmat, G.; Abdelaziz, A.I. Abrogating the interplay between IGF2BP1, 2 and 3 and IGF1R by let-7i arrests hepatocellular carcinoma growth. Growth Factors 2016, 34, 42–50. [Google Scholar] [CrossRef]
- Mochizuki, S.; Shimoda, M.; Abe, H.; Miyamae, Y.; Kuramoto, J.; Aramaki-Hattori, N.; Ishii, K.; Ueno, H.; Miyakoshi, A.; Kojoh, K.; et al. Selective Inhibition of ADAM28 Suppresses Lung Carcinoma Cell Growth and Metastasis. Mol. Cancer Ther. 2018, 17, 2427–2438. [Google Scholar] [CrossRef] [Green Version]
- Torres, D.; Hou, X.; Bale, L.; Heinzen, E.P.; Maurer, M.J.; Zanfagnin, V.; Oberg, A.L.; Conover, C.; Weroha, S.J. Overcoming platinum resistance in ovarian cancer by targeting pregnancy-associated plasma protein-A. PLoS ONE 2019, 14, e0224564. [Google Scholar] [CrossRef]
- Heitzeneder, S.; Sotillo, E.; Shern, J.F.; Sindiri, S.; Xu, P.; Jones, R.; Pollak, M.; Noer, P.R.; Lorette, J.; Fazli, L.; et al. Pregnancy-Associated Plasma Protein-A (PAPP-A) in Ewing Sarcoma: Role in Tumor Growth and Immune Evasion. J. Natl. Cancer Inst. 2019, 111, 970–982. [Google Scholar] [CrossRef] [PubMed]
- Morcavallo, A.; Buraschi, S.; Xu, S.Q.; Belfiore, A.; Schaefer, L.; Iozzo, R.V.; Morrione, A. Decorin differentially modulates the activity of insulin receptor isoform A ligands. Matrix Biol. J. Int. Soc. Matrix Biol. 2014, 35, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Iozzo, R.V.; Buraschi, S.; Genua, M.; Xu, S.Q.; Solomides, C.C.; Peiper, S.C.; Gomella, L.G.; Owens, R.C.; Morrione, A. Decorin antagonizes IGF receptor I (IGF-IR) function by interfering with IGF-IR activity and attenuating downstream signaling. J. Biol. Chem. 2011, 286, 34712–34721. [Google Scholar] [CrossRef] [Green Version]
- Morrione, A.; Neill, T.; Iozzo, R.V. Dichotomy of decorin activity on the insulin-like growth factor-I system. FEBS J. 2013, 280, 2138–2149. [Google Scholar] [CrossRef] [Green Version]
- Fernandez, S.; Genis, L.; Torres-Aleman, I. A phosphatase-independent gain-of-function mutation in PTEN triggers aberrant cell growth in astrocytes through an autocrine IGF-1 loop. Oncogene 2014, 33, 4114–4122. [Google Scholar] [CrossRef] [Green Version]
- Phadngam, S.; Castiglioni, A.; Ferraresi, A.; Morani, F.; Follo, C.; Isidoro, C. PTEN dephosphorylates AKT to prevent the expression of GLUT1 on plasmamembrane and to limit glucose consumption in cancer cells. Oncotarget 2016, 7, 84999–85020. [Google Scholar] [CrossRef] [Green Version]
- Kuijjer, M.L.; Paulson, J.N.; Salzman, P.; Ding, W.; Quackenbush, J. Cancer subtype identification using somatic mutation data. Br. J. Cancer 2018, 118, 1492–1501. [Google Scholar] [CrossRef] [Green Version]
- Chung, W.; Kim, M.; de la Monte, S.; Longato, L.; Carlson, R.; Slagle, B.L.; Dong, X.; Wands, J.R. Activation of signal transduction pathways during hepatic oncogenesis. Cancer Lett. 2016, 370, 1–9. [Google Scholar] [CrossRef]
- Fernandes, J.C.; Rodrigues Alves, A.P.N.; Machado-Neto, J.A.; Scopim-Ribeiro, R.; Fenerich, B.A.; da Silva, F.B.; Simoes, B.P.; Rego, E.M.; Traina, F. IRS1/beta-Catenin Axis Is Activated and Induces MYC Expression in Acute Lymphoblastic Leukemia Cells. J. Cell. Biochem. 2017, 118, 1774–1781. [Google Scholar] [CrossRef] [PubMed]
- Chang, Q.; Li, Y.; White, M.F.; Fletcher, J.A.; Xiao, S. Constitutive activation of insulin receptor substrate 1 is a frequent event in human tumors: Therapeutic implications. Cancer Res. 2002, 62, 6035–6038. [Google Scholar] [PubMed]
- Sachdev, D.; Li, S.L.; Hartell, J.S.; Fujita-Yamaguchi, Y.; Miller, J.S.; Yee, D. A chimeric humanized single-chain antibody against the type I insulin-like growth factor (IGF) receptor renders breast cancer cells refractory to the mitogenic effects of IGF-I. Cancer Res. 2003, 63, 627–635. [Google Scholar]
- Sachdev, D.; Singh, R.; Fujita-Yamaguchi, Y.; Yee, D. Down-regulation of insulin receptor by antibodies against the type I insulin-like growth factor receptor: Implications for anti-insulin-like growth factor therapy in breast cancer. Cancer Res. 2006, 66, 2391–2402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arteaga, C.L.; Kitten, L.J.; Coronado, E.B.; Jacobs, S.; Kull, F.C., Jr.; Allred, D.C.; Osborne, C.K. Blockade of the type I somatomedin receptor inhibits growth of human breast cancer cells in athymic mice. J. Clin. Investig. 1989, 84, 1418–1423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, T.; Shen, H.; Dong, W.; Qu, X.; Liu, Q.; Du, J. Antitumor effects and molecular mechanisms of figitumumab, a humanized monoclonal antibody to IGF-1 receptor, in esophageal carcinoma. Sci. Rep. 2014, 4, 6855. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.J.; Angelo, L.S.; Rodon, J.; Sun, M.; Kuenkele, K.P.; Parsons, H.A.; Trent, J.C.; Kurzrock, R. R1507, an anti-insulin-like growth factor-1 receptor (IGF-1R) antibody, and EWS/FLI-1 siRNA in Ewing’s sarcoma: Convergence at the IGF/IGFR/Akt axis. PLoS ONE 2011, 6, e26060. [Google Scholar] [CrossRef] [Green Version]
- Shin, D.H.; Min, H.Y.; El-Naggar, A.K.; Lippman, S.M.; Glisson, B.; Lee, H.Y. Akt/mTOR counteract the antitumor activities of cixutumumab, an anti-insulin-like growth factor I receptor monoclonal antibody. Mol. Cancer Ther. 2011, 10, 2437–2448. [Google Scholar] [CrossRef] [Green Version]
- Leiphrakpam, P.D.; Agarwal, E.; Mathiesen, M.; Haferbier, K.L.; Brattain, M.G.; Chowdhury, S. In vivo analysis of insulin-like growth factor type 1 receptor humanized monoclonal antibody MK-0646 and small molecule kinase inhibitor OSI-906 in colorectal cancer. Oncol. Rep. 2014, 31, 87–94. [Google Scholar] [CrossRef] [Green Version]
- Beltran, P.J.; Calzone, F.J.; Mitchell, P.; Chung, Y.A.; Cajulis, E.; Moody, G.; Belmontes, B.; Li, C.M.; Vonderfecht, S.; Velculescu, V.E.; et al. Ganitumab (AMG 479) inhibits IGF-II-dependent ovarian cancer growth and potentiates platinum-based chemotherapy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 2947–2958. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Lipari, P.; Wang, X.; Hailey, J.; Liang, L.; Ramos, R.; Liu, M.; Pachter, J.A.; Bishop, W.R.; Wang, Y. A fully human insulin-like growth factor-I receptor antibody SCH 717454 (Robatumumab) has antitumor activity as a single agent and in combination with cytotoxics in pediatric tumor xenografts. Mol. Cancer Ther. 2010, 9, 410–418. [Google Scholar] [CrossRef] [Green Version]
- Schmitz, S.; Kaminsky-Forrett, M.C.; Henry, S.; Zanetta, S.; Geoffrois, L.; Bompas, E.; Moxhon, A.; Mignion, L.; Guigay, J.; Knoops, L.; et al. Phase II study of figitumumab in patients with recurrent and/or metastatic squamous cell carcinoma of the head and neck: Clinical activity and molecular response (GORTEC 2008-02). Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2012, 23, 2153–2161. [Google Scholar] [CrossRef] [PubMed]
- Pandini, G.; Wurch, T.; Akla, B.; Corvaia, N.; Belfiore, A.; Goetsch, L. Functional responses and in vivo anti-tumour activity of h7C10: A humanised monoclonal antibody with neutralising activity against the insulin-like growth factor-1 (IGF-1) receptor and insulin/IGF-1 hybrid receptors. Eur. J. Cancer 2007, 43, 1318–1327. [Google Scholar] [CrossRef] [PubMed]
- Buck, E.; Gokhale, P.C.; Koujak, S.; Brown, E.; Eyzaguirre, A.; Tao, N.; Rosenfeld-Franklin, M.; Lerner, L.; Chiu, M.I.; Wild, R.; et al. Compensatory insulin receptor (IR) activation on inhibition of insulin-like growth factor-1 receptor (IGF-1R): Rationale for cotargeting IGF-1R and IR in cancer. Mol. Cancer Ther. 2010, 9, 2652–2664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desbois-Mouthon, C.; Baron, A.; Blivet-Van Eggelpoel, M.J.; Fartoux, L.; Venot, C.; Bladt, F.; Housset, C.; Rosmorduc, O. Insulin-like growth factor-1 receptor inhibition induces a resistance mechanism via the epidermal growth factor receptor/HER3/AKT signaling pathway: Rational basis for cotargeting insulin-like growth factor-1 receptor and epidermal growth factor receptor in hepatocellular carcinoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2009, 15, 5445–5456. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.; Tang, W.; Han, X.; Geng, R.; Wang, C.; Zhang, Z. Hepatocyte growth factor-induced mesenchymal-epithelial transition factor activation leads to insulin-like growth factor 1 receptor inhibitor unresponsiveness in gastric cancer cells. Oncol. Lett. 2018, 16, 5983–5991. [Google Scholar] [CrossRef]
- Solomon, V.R.; Alizadeh, E.; Bernhard, W.; Makhlouf, A.; Hartimath, S.V.; Hill, W.; El-Sayed, A.; Barreto, K.; Geyer, C.R.; Fonge, H. Development and preclinical evaluation of cixutumumab drug conjugates in a model of insulin growth factor receptor I (IGF-1R) positive cancer. Sci. Rep. 2020, 10, 18549. [Google Scholar] [CrossRef]
- Akla, B.; Broussas, M.; Loukili, N.; Robert, A.; Beau-Larvor, C.; Malissard, M.; Boute, N.; Champion, T.; Haeuw, J.F.; Beck, A.; et al. Efficacy of the Antibody-Drug Conjugate W0101 in Preclinical Models of IGF-1 Receptor Overexpressing Solid Tumors. Mol. Cancer Ther. 2020, 19, 168–177. [Google Scholar] [CrossRef] [Green Version]
- Favelyukis, S.; Till, J.H.; Hubbard, S.R.; Miller, W.T. Structure and autoregulation of the insulin-like growth factor 1 receptor kinase. Nat. Struct. Biol. 2001, 8, 1058–1063. [Google Scholar] [CrossRef] [PubMed]
- Mulvihill, M.J.; Cooke, A.; Rosenfeld-Franklin, M.; Buck, E.; Foreman, K.; Landfair, D.; O’Connor, M.; Pirritt, C.; Sun, Y.; Yao, Y.; et al. Discovery of OSI-906: A selective and orally efficacious dual inhibitor of the IGF-1 receptor and insulin receptor. Future Med. Chem. 2009, 1, 1153–1171. [Google Scholar] [CrossRef]
- Awasthi, N.; Zhang, C.; Ruan, W.; Schwarz, M.A.; Schwarz, R.E. BMS-754807, a small-molecule inhibitor of insulin-like growth factor-1 receptor/insulin receptor, enhances gemcitabine response in pancreatic cancer. Mol. Cancer Ther. 2012, 11, 2644–2653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carrasco-Garcia, E.; Martinez-Lacaci, I.; Mayor-Lopez, L.; Tristante, E.; Carballo-Santana, M.; Garcia-Morales, P.; Ventero Martin, M.P.; Fuentes-Baile, M.; Rodriguez-Lescure, A.; Saceda, M. PDGFR and IGF-1R Inhibitors Induce a G2/M Arrest and Subsequent Cell Death in Human Glioblastoma Cell Lines. Cells 2018, 7, 131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- George, B.; George, S.K.; Shi, W.; Haque, A.; Shi, P.; Eskandari, G.; Axelson, M.; Larsson, O.; Kaseb, A.O.; Amin, H.M. Dual inhibition of IGF-IR and ALK as an effective strategy to eradicate NPM-ALK(+) T-cell lymphoma. J. Hematol. Oncol. 2019, 12, 80. [Google Scholar] [CrossRef] [Green Version]
- Aiken, R.; Axelson, M.; Harmenberg, J.; Klockare, M.; Larsson, O.; Wassberg, C. Phase I clinical trial of AXL1717 for treatment of relapsed malignant astrocytomas: Analysis of dose and response. Oncotarget 2017, 8, 81501–81510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergqvist, M.; Holgersson, G.; Bondarenko, I.; Grechanaya, E.; Maximovich, A.; Andor, G.; Klockare, M.; Thureson, M.; Jerling, M.; Harmenberg, J. Phase II randomized study of the IGF-1R pathway modulator AXL1717 compared to docetaxel in patients with previously treated, locally advanced or metastatic non-small cell lung cancer. Acta Oncol. 2017, 56, 441–447. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Chesebrough, J.W.; Cartlidge, S.A.; Ricketts, S.A.; Incognito, L.; Veldman-Jones, M.; Blakey, D.C.; Tabrizi, M.; Jallal, B.; Trail, P.A.; et al. Dual IGF-I/II-neutralizing antibody MEDI-573 potently inhibits IGF signaling and tumor growth. Cancer Res. 2011, 71, 1029–1040. [Google Scholar] [CrossRef] [Green Version]
- Zhong, H.; Fazenbaker, C.; Chen, C.; Breen, S.; Huang, J.; Yao, X.; Ren, P.; Yao, Y.; Herbst, R.; Hollingsworth, R.E. Overproduction of IGF-2 drives a subset of colorectal cancer cells, which specifically respond to an anti-IGF therapeutic antibody and combination therapies. Oncogene 2017, 36, 797–806. [Google Scholar] [CrossRef]
- Haluska, P.; Menefee, M.; Plimack, E.R.; Rosenberg, J.; Northfelt, D.; LaVallee, T.; Shi, L.; Yu, X.Q.; Burke, P.; Huang, J.; et al. Phase I dose-escalation study of MEDI-573, a bispecific, antiligand monoclonal antibody against IGFI and IGFII, in patients with advanced solid tumors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 4747–4757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parra-Guillen, Z.P.; Schmid, U.; Janda, A.; Freiwald, M.; Troconiz, I.F. Model-Informed Dose Selection for Xentuzumab, a Dual Insulin-Like Growth Factor-I/II-Neutralizing Antibody. Clin. Pharmacol. Ther. 2020, 107, 597–606. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Quetglas, I.; Pinyol, R.; Dauch, D.; Torrecilla, S.; Tovar, V.; Moeini, A.; Alsinet, C.; Portela, A.; Rodriguez-Carunchio, L.; Sole, M.; et al. IGF2 Is Up-regulated by Epigenetic Mechanisms in Hepatocellular Carcinomas and Is an Actionable Oncogene Product in Experimental Models. Gastroenterology 2016, 151, 1192–1205. [Google Scholar] [CrossRef]
- Weyer-Czernilofsky, U.; Hofmann, M.H.; Friedbichler, K.; Baumgartinger, R.; Adam, P.J.; Solca, F.; Kraut, N.; Nguyen, H.M.; Corey, E.; Liu, G.; et al. Antitumor Activity of the IGF-1/IGF-2-Neutralizing Antibody Xentuzumab (BI 836845) in Combination with Enzalutamide in Prostate Cancer Models. Mol. Cancer Ther. 2020, 19, 1059–1069. [Google Scholar] [CrossRef] [Green Version]
- de Bono, J.; Lin, C.C.; Chen, L.T.; Corral, J.; Michalarea, V.; Rihawi, K.; Ong, M.; Lee, J.H.; Hsu, C.H.; Yang, J.C.; et al. Two first-in-human studies of xentuzumab, a humanised insulin-like growth factor (IGF)-neutralising antibody, in patients with advanced solid tumours. Br. J. Cancer 2020, 122, 1324–1332. [Google Scholar] [CrossRef] [Green Version]
- Schmid, P.; Sablin, M.P.; Bergh, J.; Im, S.A.; Lu, Y.S.; Martinez, N.; Neven, P.; Lee, K.S.; Morales, S.; Perez-Fidalgo, J.A.; et al. A phase Ib/II study of xentuzumab, an IGF-neutralising antibody, combined with exemestane and everolimus in hormone receptor-positive, HER2-negative locally advanced/metastatic breast cancer. Breast Cancer Res. BCR 2021, 23, 8. [Google Scholar] [CrossRef]
- Chen, Y.M.; Qi, S.; Perrino, S.; Hashimoto, M.; Brodt, P. Targeting the IGF-Axis for Cancer Therapy: Development and Validation of an IGF-Trap as a Potential Drug. Cells 2020, 9, 1098. [Google Scholar] [CrossRef] [PubMed]
- Vaniotis, G.; Moffett, S.; Sulea, T.; Wang, N.; Elahi, S.M.; Lessard, E.; Baardsnes, J.; Perrino, S.; Durocher, Y.; Frystyk, J.; et al. Enhanced anti-metastatic bioactivity of an IGF-TRAP re-engineered to improve physicochemical properties. Sci. Rep. 2018, 8, 17361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, W.J.; Doyle, L.A. Updates from the 2020 World Health Organization Classification of Soft Tissue and Bone Tumours. Histopathology 2021, 78, 644–657. [Google Scholar] [CrossRef]
- Lilienthal, I.; Herold, N. Targeting Molecular Mechanisms Underlying Treatment Efficacy and Resistance in Osteosarcoma: A Review of Current and Future Strategies. Int. J. Mol. Sci. 2020, 21, 6885. [Google Scholar] [CrossRef]
- Pollak, M.; Sem, A.W.; Richard, M.; Tetenes, E.; Bell, R. Inhibition of metastatic behavior of murine osteosarcoma by hypophysectomy. J. Natl. Cancer Inst. 1992, 84, 966–971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burrow, S.; Andrulis, I.L.; Pollak, M.; Bell, R.S. Expression of insulin-like growth factor receptor, IGF-1, and IGF-2 in primary and metastatic osteosarcoma. J. Surg. Oncol. 1998, 69, 21–27. [Google Scholar] [CrossRef]
- Yang, R.; Piperdi, S.; Zhang, Y.; Zhu, Z.; Neophytou, N.; Hoang, B.H.; Mason, G.; Geller, D.; Dorfman, H.; Meyers, P.A.; et al. Transcriptional Profiling Identifies the Signaling Axes of IGF and Transforming Growth Factor-b as Involved in the Pathogenesis of Osteosarcoma. Clin. Orthop. Relat. Res. 2016, 474, 178–189. [Google Scholar] [CrossRef] [Green Version]
- Cao, D.; Lei, Y.; Ye, Z.; Zhao, L.; Wang, H.; Zhang, J.; He, F.; Huang, L.; Shi, D.; Liu, Q.; et al. Blockade of IGF/IGF-1R signaling axis with soluble IGF-1R mutants suppresses the cell proliferation and tumor growth of human osteosarcoma. Am. J. Cancer Res. 2020, 10, 3248–3266. [Google Scholar]
- Denduluri, S.K.; Idowu, O.; Wang, Z.; Liao, Z.; Yan, Z.; Mohammed, M.K.; Ye, J.; Wei, Q.; Wang, J.; Zhao, L.; et al. Insulin-like growth factor (IGF) signaling in tumorigenesis and the development of cancer drug resistance. Genes Dis. 2015, 2, 13–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jentzsch, T.; Robl, B.; Husmann, M.; Bode-Lesniewska, B.; Fuchs, B. Worse prognosis of osteosarcoma patients expressing IGF-1 on a tissue microarray. Anticancer Res. 2014, 34, 3881–3889. [Google Scholar]
- Wang, Y.H.; Han, X.D.; Qiu, Y.; Xiong, J.; Yu, Y.; Wang, B.; Zhu, Z.Z.; Qian, B.P.; Chen, Y.X.; Wang, S.F.; et al. Increased expression of insulin-like growth factor-1 receptor is correlated with tumor metastasis and prognosis in patients with osteosarcoma. J. Surg. Oncol. 2012, 105, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Benini, S.; Baldini, N.; Manara, M.C.; Chano, T.; Serra, M.; Rizzi, S.; Lollini, P.L.; Picci, P.; Scotlandi, K. Redundancy of autocrine loops in human osteosarcoma cells. Int. J. Cancer 1999, 80, 581–588. [Google Scholar] [CrossRef]
- Duan, Z.; Choy, E.; Harmon, D.; Yang, C.; Ryu, K.; Schwab, J.; Mankin, H.; Hornicek, F.J. Insulin-like growth factor-I receptor tyrosine kinase inhibitor cyclolignan picropodophyllin inhibits proliferation and induces apoptosis in multidrug resistant osteosarcoma cell lines. Mol. Cancer Ther. 2009, 8, 2122–2130. [Google Scholar] [CrossRef] [Green Version]
- Kolb, E.A.; Kamara, D.; Zhang, W.; Lin, J.; Hingorani, P.; Baker, L.; Houghton, P.; Gorlick, R. R1507, a fully human monoclonal antibody targeting IGF-1R, is effective alone and in combination with rapamycin in inhibiting growth of osteosarcoma xenografts. Pediatr. Blood Cancer 2010, 55, 67–75. [Google Scholar] [CrossRef] [Green Version]
- Anderson, P.M.; Bielack, S.S.; Gorlick, R.G.; Skubitz, K.; Daw, N.C.; Herzog, C.E.; Monge, O.R.; Lassaletta, A.; Boldrini, E.; Papai, Z.; et al. A phase II study of clinical activity of SCH 717454 (robatumumab) in patients with relapsed osteosarcoma and Ewing sarcoma. Pediatr. Blood Cancer 2016, 63, 1761–1770. [Google Scholar] [CrossRef]
- Landuzzi, L.; Manara, M.C.; Lollini, P.L.; Scotlandi, K. Patient Derived Xenografts for Genome-Driven Therapy of Osteosarcoma. Cells 2021, 10, 416. [Google Scholar] [CrossRef] [PubMed]
- Zvi, Y.; Ugur, E.; Batko, B.; Gill, J.; Roth, M.; Gorlick, R.; Hall, D.; Tingling, J.; Barkauskas, D.A.; Zhang, J.; et al. Prognostic and Therapeutic Utility of Variably Expressed Cell Surface Receptors in Osteosarcoma. Sarcoma 2021, 2021, 8324348. [Google Scholar] [CrossRef]
- Wang, S.; Wei, H.; Huang, Z.; Wang, X.; Shen, R.; Wu, Z.; Lin, J. Epidermal growth factor receptor promotes tumor progression and contributes to gemcitabine resistance in osteosarcoma. Acta Biochim. Biophys. Sin. 2021, 53, 317–324. [Google Scholar] [CrossRef]
- Schwartz, G.K.; Tap, W.D.; Qin, L.X.; Livingston, M.B.; Undevia, S.D.; Chmielowski, B.; Agulnik, M.; Schuetze, S.M.; Reed, D.R.; Okuno, S.H.; et al. Cixutumumab and temsirolimus for patients with bone and soft-tissue sarcoma: A multicentre, open-label, phase 2 trial. Lancet Oncol. 2013, 14, 371–382. [Google Scholar] [CrossRef] [Green Version]
- Mansky, P.J.; Liewehr, D.J.; Steinberg, S.M.; Chrousos, G.P.; Avila, N.A.; Long, L.; Bernstein, D.; Mackall, C.L.; Hawkins, D.S.; Helman, L.J. Treatment of metastatic osteosarcoma with the somatostatin analog OncoLar: Significant reduction of insulin-like growth factor-1 serum levels. J. Pediatr. Hematol. Oncol. 2002, 24, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Ameline, B.; Kovac, M.; Nathrath, M.; Barenboim, M.; Witt, O.; Krieg, A.H.; Baumhoer, D. Overactivation of the IGF signalling pathway in osteosarcoma: A potential therapeutic target? J. Pathol. Clin. Res. 2021, 7, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Habel, N.; Stefanovska, B.; Carene, D.; Patino-Garcia, A.; Lecanda, F.; Fromigue, O. CYR61 triggers osteosarcoma metastatic spreading via an IGF1Rbeta-dependent EMT-like process. BMC Cancer 2019, 19, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Wang, Q.; Wang, G.D.; Wang, H.S.; Huang, Y.; Liu, X.M.; Cai, X.H. miR-16 inhibits cell proliferation by targeting IGF1R and the Raf1-MEK1/2-ERK1/2 pathway in osteosarcoma. FEBS Lett. 2013, 587, 1366–1372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brohl, A.S.; Solomon, D.A.; Chang, W.; Wang, J.; Song, Y.; Sindiri, S.; Patidar, R.; Hurd, L.; Chen, L.; Shern, J.F.; et al. The genomic landscape of the Ewing Sarcoma family of tumors reveals recurrent STAG2 mutation. PLoS Genet. 2014, 10, e1004475. [Google Scholar] [CrossRef] [Green Version]
- Prieur, A.; Tirode, F.; Cohen, P.; Delattre, O. EWS/FLI-1 silencing and gene profiling of Ewing cells reveal downstream oncogenic pathways and a crucial role for repression of insulin-like growth factor binding protein 3. Mol. Cell. Biol. 2004, 24, 7275–7283. [Google Scholar] [CrossRef] [Green Version]
- Tirode, F.; Laud-Duval, K.; Prieur, A.; Delorme, B.; Charbord, P.; Delattre, O. Mesenchymal stem cell features of Ewing tumors. Cancer Cell 2007, 11, 421–429. [Google Scholar] [CrossRef] [Green Version]
- Scotlandi, K.; Benini, S.; Sarti, M.; Serra, M.; Lollini, P.L.; Maurici, D.; Picci, P.; Manara, M.C.; Baldini, N. Insulin-like growth factor I receptor-mediated circuit in Ewing’s sarcoma/peripheral neuroectodermal tumor: A possible therapeutic target. Cancer Res. 1996, 56, 4570–4574. [Google Scholar] [PubMed]
- Scotlandi, K.; Manara, M.C.; Nicoletti, G.; Lollini, P.L.; Lukas, S.; Benini, S.; Croci, S.; Perdichizzi, S.; Zambelli, D.; Serra, M.; et al. Antitumor activity of the insulin-like growth factor-I receptor kinase inhibitor NVP-AEW541 in musculoskeletal tumors. Cancer Res. 2005, 65, 3868–3876. [Google Scholar] [CrossRef] [Green Version]
- Scotlandi, K.; Benini, S.; Nanni, P.; Lollini, P.L.; Nicoletti, G.; Landuzzi, L.; Serra, M.; Manara, M.C.; Picci, P.; Baldini, N. Blockage of insulin-like growth factor-I receptor inhibits the growth of Ewing’s sarcoma in athymic mice. Cancer Res. 1998, 58, 4127–4131. [Google Scholar]
- Mancarella, C.; Pasello, M.; Manara, M.C.; Toracchio, L.; Sciandra, E.F.; Picci, P.; Scotlandi, K. Insulin-Like Growth Factor 2 mRNA-Binding Protein 3 Influences Sensitivity to Anti-IGF System Agents Through the Translational Regulation of IGF1R. Front. Endocrinol. 2018, 9, 178. [Google Scholar] [CrossRef] [PubMed]
- Tap, W.D.; Demetri, G.; Barnette, P.; Desai, J.; Kavan, P.; Tozer, R.; Benedetto, P.W.; Friberg, G.; Deng, H.; McCaffery, I.; et al. Phase II study of ganitumab, a fully human anti-type-1 insulin-like growth factor receptor antibody, in patients with metastatic Ewing family tumors or desmoplastic small round cell tumors. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2012, 30, 1849–1856. [Google Scholar] [CrossRef]
- Pappo, A.S.; Patel, S.R.; Crowley, J.; Reinke, D.K.; Kuenkele, K.P.; Chawla, S.P.; Toner, G.C.; Maki, R.G.; Meyers, P.A.; Chugh, R.; et al. R1507, a monoclonal antibody to the insulin-like growth factor 1 receptor, in patients with recurrent or refractory Ewing sarcoma family of tumors: Results of a phase II Sarcoma Alliance for Research through Collaboration study. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2011, 29, 4541–4547. [Google Scholar] [CrossRef]
- Juergens, H.; Daw, N.C.; Geoerger, B.; Ferrari, S.; Villarroel, M.; Aerts, I.; Whelan, J.; Dirksen, U.; Hixon, M.L.; Yin, D.; et al. Preliminary efficacy of the anti-insulin-like growth factor type 1 receptor antibody figitumumab in patients with refractory Ewing sarcoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2011, 29, 4534–4540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olmos, D.; Postel-Vinay, S.; Molife, L.R.; Okuno, S.H.; Schuetze, S.M.; Paccagnella, M.L.; Batzel, G.N.; Yin, D.; Pritchard-Jones, K.; Judson, I.; et al. Safety, pharmacokinetics, and preliminary activity of the anti-IGF-1R antibody figitumumab (CP-751,871) in patients with sarcoma and Ewing’s sarcoma: A phase 1 expansion cohort study. Lancet Oncol. 2010, 11, 129–135. [Google Scholar] [CrossRef] [Green Version]
- Asmane, I.; Watkin, E.; Alberti, L.; Duc, A.; Marec-Berard, P.; Ray-Coquard, I.; Cassier, P.; Decouvelaere, A.V.; Ranchere, D.; Kurtz, J.E.; et al. Insulin-like growth factor type 1 receptor (IGF-1R) exclusive nuclear staining: A predictive biomarker for IGF-1R monoclonal antibody (Ab) therapy in sarcomas. Eur. J. Cancer 2012, 48, 3027–3035. [Google Scholar] [CrossRef] [PubMed]
- Lamhamedi-Cherradi, S.E.; Menegaz, B.A.; Ramamoorthy, V.; Vishwamitra, D.; Wang, Y.; Maywald, R.L.; Buford, A.S.; Fokt, I.; Skora, S.; Wang, J.; et al. IGF-1R and mTOR Blockade: Novel Resistance Mechanisms and Synergistic Drug Combinations for Ewing Sarcoma. J. Natl. Cancer Inst. 2016, 108, djw182. [Google Scholar] [CrossRef] [PubMed]
- Amin, H.M.; Morani, A.C.; Daw, N.C.; Lamhamedi-Cherradi, S.E.; Subbiah, V.; Menegaz, B.A.; Vishwamitra, D.; Eskandari, G.; George, B.; Benjamin, R.S.; et al. IGF-1R/mTOR Targeted Therapy for Ewing Sarcoma: A Meta-Analysis of Five IGF-1R-Related Trials Matched to Proteomic and Radiologic Predictive Biomarkers. Cancers 2020, 12, 1768. [Google Scholar] [CrossRef]
- Naing, A.; LoRusso, P.; Fu, S.; Hong, D.S.; Anderson, P.; Benjamin, R.S.; Ludwig, J.; Chen, H.X.; Doyle, L.A.; Kurzrock, R. Insulin growth factor-receptor (IGF-1R) antibody cixutumumab combined with the mTOR inhibitor temsirolimus in patients with refractory Ewing’s sarcoma family tumors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2012, 18, 2625–2631. [Google Scholar] [CrossRef] [Green Version]
- Guenther, L.M.; Dharia, N.V.; Ross, L.; Conway, A.; Robichaud, A.L.; Catlett, J.L., 2nd; Wechsler, C.S.; Frank, E.S.; Goodale, A.; Church, A.J.; et al. A Combination CDK4/6 and IGF1R Inhibitor Strategy for Ewing Sarcoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 1343–1357. [Google Scholar] [CrossRef] [Green Version]
- Amaral, A.T.; Garofalo, C.; Frapolli, R.; Manara, M.C.; Mancarella, C.; Uboldi, S.; Di Giandomenico, S.; Ordonez, J.L.; Sevillano, V.; Malaguarnera, R.; et al. Trabectedin efficacy in Ewing sarcoma is greatly increased by combination with anti-IGF signaling agents. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2015, 21, 1373–1382. [Google Scholar] [CrossRef] [Green Version]
- Ramadan, F.; Fahs, A.; Ghayad, S.E.; Saab, R. Signaling pathways in Rhabdomyosarcoma invasion and metastasis. Cancer Metastasis Rev. 2020, 39, 287–301. [Google Scholar] [CrossRef]
- Cao, L.; Yu, Y.; Bilke, S.; Walker, R.L.; Mayeenuddin, L.H.; Azorsa, D.O.; Yang, F.; Pineda, M.; Helman, L.J.; Meltzer, P.S. Genome-wide identification of PAX3-FKHR binding sites in rhabdomyosarcoma reveals candidate target genes important for development and cancer. Cancer Res. 2010, 70, 6497–6508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blandford, M.C.; Barr, F.G.; Lynch, J.C.; Randall, R.L.; Qualman, S.J.; Keller, C. Rhabdomyosarcomas utilize developmental, myogenic growth factors for disease advantage: A report from the Children’s Oncology Group. Pediatr. Blood Cancer 2006, 46, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.; Gordon, A.; McManus, A.; Shipley, J.; Pritchard-Jones, K. Disruption of imprinted genes at chromosome region 11p15.5 in paediatric rhabdomyosarcoma. Neoplasia 1999, 1, 340–348. [Google Scholar] [CrossRef] [Green Version]
- De Giovanni, C.; Nanni, P.; Landuzzi, L.; Ianzano, M.L.; Nicoletti, G.; Croci, S.; Palladini, A.; Lollini, P.L. Immune targeting of autocrine IGF2 hampers rhabdomyosarcoma growth and metastasis. BMC Cancer 2019, 19, 126. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhang, Y.; Ramanujan, K.; Ma, Y.; Kirsch, D.G.; Glass, D.J. Oncogenic NRAS, required for pathogenesis of embryonic rhabdomyosarcoma, relies upon the HMGA2-IGF2BP2 pathway. Cancer Res. 2013, 73, 3041–3050. [Google Scholar] [CrossRef] [Green Version]
- Wan, X.; Yeung, C.; Heske, C.; Mendoza, A.; Helman, L.J. IGF-1R Inhibition Activates a YES/SFK Bypass Resistance Pathway: Rational Basis for Co-Targeting IGF-1R and Yes/SFK Kinase in Rhabdomyosarcoma. Neoplasia 2015, 17, 358–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Makawita, S.; Ho, M.; Durbin, A.D.; Thorner, P.S.; Malkin, D.; Somers, G.R. Expression of insulin-like growth factor pathway proteins in rhabdomyosarcoma: IGF-2 expression is associated with translocation-negative tumors. Pediatr. Dev. Pathol. Off. J. Soc. Pediatr. Pathol. Paediatr. Pathol. Soc. 2009, 12, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Tarnowski, M.; Tkacz, M.; Zgutka, K.; Bujak, J.; Kopytko, P.; Pawlik, A. Picropodophyllin (PPP) is a potent rhabdomyosarcoma growth inhibitor both in vitro and in vivo. BMC Cancer 2017, 17, 532. [Google Scholar] [CrossRef] [Green Version]
- Pappo, A.S.; Vassal, G.; Crowley, J.J.; Bolejack, V.; Hogendoorn, P.C.; Chugh, R.; Ladanyi, M.; Grippo, J.F.; Dall, G.; Staddon, A.P.; et al. A phase 2 trial of R1507, a monoclonal antibody to the insulin-like growth factor-1 receptor (IGF-1R), in patients with recurrent or refractory rhabdomyosarcoma, osteosarcoma, synovial sarcoma, and other soft tissue sarcomas: Results of a Sarcoma Alliance for Research Through Collaboration study. Cancer 2014, 120, 2448–2456. [Google Scholar] [CrossRef] [Green Version]
- Weigel, B.; Malempati, S.; Reid, J.M.; Voss, S.D.; Cho, S.Y.; Chen, H.X.; Krailo, M.; Villaluna, D.; Adamson, P.C.; Blaney, S.M. Phase 2 trial of cixutumumab in children, adolescents, and young adults with refractory solid tumors: A report from the Children’s Oncology Group. Pediatr. Blood Cancer 2014, 61, 452–456. [Google Scholar] [CrossRef] [Green Version]
- Malempati, S.; Weigel, B.J.; Chi, Y.Y.; Tian, J.; Anderson, J.R.; Parham, D.M.; Teot, L.A.; Rodeberg, D.A.; Yock, T.I.; Shulkin, B.L.; et al. The addition of cixutumumab or temozolomide to intensive multiagent chemotherapy is feasible but does not improve outcome for patients with metastatic rhabdomyosarcoma: A report from the Children’s Oncology Group. Cancer 2019, 125, 290–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, Z.; Yu, Y.; Zhu, Y.J.; Davis, S.; Walker, R.; Meltzer, P.S.; Helman, L.J.; Cao, L. Downregulation of IGFBP2 is associated with resistance to IGF1R therapy in rhabdomyosarcoma. Oncogene 2014, 33, 5697–5705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, L.; Yu, Y.; Darko, I.; Currier, D.; Mayeenuddin, L.H.; Wan, X.; Khanna, C.; Helman, L.J. Addiction to elevated insulin-like growth factor I receptor and initial modulation of the AKT pathway define the responsiveness of rhabdomyosarcoma to the targeting antibody. Cancer Res. 2008, 68, 8039–8048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmerini, E.; Benassi, M.S.; Quattrini, I.; Pazzaglia, L.; Donati, D.; Benini, S.; Gamberi, G.; Gambarotti, M.; Picci, P.; Ferrari, S. Prognostic and predictive role of CXCR4, IGF-1R and Ezrin expression in localized synovial sarcoma: Is chemotaxis important to tumor response? Orphanet J. Rare Dis. 2015, 10, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isfort, I.; Cyra, M.; Elges, S.; Kailayangiri, S.; Altvater, B.; Rossig, C.; Steinestel, K.; Grunewald, I.; Huss, S.; Esseling, E.; et al. SS18-SSX-Dependent YAP/TAZ Signaling in Synovial Sarcoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 3718–3731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ladanyi, M. Fusions of the SYT and SSX genes in synovial sarcoma. Oncogene 2001, 20, 5755–5762. [Google Scholar] [CrossRef] [Green Version]
- Haldar, M.; Hancock, J.D.; Coffin, C.M.; Lessnick, S.L.; Capecchi, M.R. A conditional mouse model of synovial sarcoma: Insights into a myogenic origin. Cancer Cell 2007, 11, 375–388. [Google Scholar] [CrossRef] [Green Version]
- Banito, A.; Li, X.; Laporte, A.N.; Roe, J.S.; Sanchez-Vega, F.; Huang, C.H.; Dancsok, A.R.; Hatzi, K.; Chen, C.C.; Tschaharganeh, D.F.; et al. The SS18-SSX Oncoprotein Hijacks KDM2B-PRC1.1 to Drive Synovial Sarcoma. Cancer Cell 2018, 34, 346–348. [Google Scholar] [CrossRef] [Green Version]
- de Bruijn, D.R.; Allander, S.V.; van Dijk, A.H.; Willemse, M.P.; Thijssen, J.; van Groningen, J.J.; Meltzer, P.S.; van Kessel, A.G. The synovial-sarcoma-associated SS18-SSX2 fusion protein induces epigenetic gene (de)regulation. Cancer Res. 2006, 66, 9474–9482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedrichs, N.; Kuchler, J.; Endl, E.; Koch, A.; Czerwitzki, J.; Wurst, P.; Metzger, D.; Schulte, J.H.; Holst, M.I.; Heukamp, L.C.; et al. Insulin-like growth factor-1 receptor acts as a growth regulator in synovial sarcoma. J. Pathol. 2008, 216, 428–439. [Google Scholar] [CrossRef]
- Tornkvist, M.; Natalishvili, N.; Xie, Y.; Girnita, A.; D’Arcy, P.; Brodin, B.; Axelson, M.; Girnita, L. Differential roles of SS18-SSX fusion gene and insulin-like growth factor-1 receptor in synovial sarcoma cell growth. Biochem. Biophys. Res. Commun. 2008, 368, 793–800. [Google Scholar] [CrossRef] [PubMed]
- Cassinelli, G.; Dal Bo, L.; Favini, E.; Cominetti, D.; Pozzi, S.; Tortoreto, M.; De Cesare, M.; Lecis, D.; Scanziani, E.; Minoli, L.; et al. Supersulfated low-molecular weight heparin synergizes with IGF1R/IR inhibitor to suppress synovial sarcoma growth and metastases. Cancer Lett. 2018, 415, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Michels, S.; Trautmann, M.; Sievers, E.; Kindler, D.; Huss, S.; Renner, M.; Friedrichs, N.; Kirfel, J.; Steiner, S.; Endl, E.; et al. SRC signaling is crucial in the growth of synovial sarcoma cells. Cancer Res. 2013, 73, 2518–2528. [Google Scholar] [CrossRef] [Green Version]
- Fleuren, E.D.G.; Vlenterie, M.; van der Graaf, W.T.A.; Hillebrandt-Roeffen, M.H.S.; Blackburn, J.; Ma, X.; Chan, H.; Magias, M.C.; van Erp, A.; van Houdt, L.; et al. Phosphoproteomic Profiling Reveals ALK and MET as Novel Actionable Targets across Synovial Sarcoma Subtypes. Cancer Res. 2017, 77, 4279–4292. [Google Scholar] [CrossRef] [Green Version]
- Bexelius, T.S.; Wasti, A.; Chisholm, J.C. Mini-Review on Targeted Treatment of Desmoplastic Small Round Cell Tumor. Front. Oncol. 2020, 10, 518. [Google Scholar] [CrossRef]
- Karnieli, E.; Werner, H.; Rauscher, F.J., 3rd; Benjamin, L.E.; LeRoith, D. The IGF-I receptor gene promoter is a molecular target for the Ewing’s sarcoma-Wilms’ tumor 1 fusion protein. J. Biol. Chem. 1996, 271, 19304–19309. [Google Scholar] [CrossRef] [Green Version]
- Andersson, M.K.; Aman, P.; Stenman, G. IGF2/IGF1R Signaling as a Therapeutic Target in MYB-Positive Adenoid Cystic Carcinomas and Other Fusion Gene-Driven Tumors. Cells 2019, 8, 913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almaghraby, A.; Brickman, W.J.; Goldstein, J.A.; Habiby, R.L. Refractory hypoglycemia in a pediatric patient with desmoplastic small round cell tumor. J. Pediatr. Endocrinol. Metab. JPEM 2018, 31, 947–950. [Google Scholar] [CrossRef] [PubMed]
- Barra, W.F.; Castro, G.; Hoff, A.O.; Siqueira, S.A.; Hoff, P.M. Symptomatic hypoglycemia related to inappropriately high igf-ii serum levels in a patient with desmoplastic small round cell tumor. Case Rep. Med. 2010, 2010, 684045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hingorani, P.; Dinu, V.; Zhang, X.; Lei, H.; Shern, J.F.; Park, J.; Steel, J.; Rauf, F.; Parham, D.; Gastier-Foster, J.; et al. Transcriptome analysis of desmoplastic small round cell tumors identifies actionable therapeutic targets: A report from the Children’s Oncology Group. Sci. Rep. 2020, 10, 12318. [Google Scholar] [CrossRef]
- Lindsey, R.C.; Rundle, C.H.; Mohan, S. Role of IGF1 and EFN-EPH signaling in skeletal metabolism. J. Mol. Endocrinol. 2018, 61, T87–T102. [Google Scholar] [CrossRef] [Green Version]
- Pasquale, E.B. Eph receptors and ephrins in cancer: Bidirectional signalling and beyond. Nat. Rev. Cancer 2010, 10, 165–180. [Google Scholar] [CrossRef] [Green Version]
- Xiao, T.; Xiao, Y.; Wang, W.; Tang, Y.Y.; Xiao, Z.; Su, M. Targeting EphA2 in cancer. J. Hematol. Oncol. 2020, 13, 114. [Google Scholar] [CrossRef]
- Kubo, H.; Yagyu, S.; Nakamura, K.; Yamashima, K.; Tomida, A.; Kikuchi, K.; Iehara, T.; Nakazawa, Y.; Hosoi, H. Development of non-viral, ligand-dependent, EPHB4-specific chimeric antigen receptor T cells for treatment of rhabdomyosarcoma. Mol. Ther. Oncolytics 2021, 20, 646–658. [Google Scholar] [CrossRef] [PubMed]
- Becerikli, M.; Merwart, B.; Lam, M.C.; Suppelna, P.; Rittig, A.; Mirmohammedsadegh, A.; Stricker, I.; Theiss, C.; Singer, B.B.; Jacobsen, F.; et al. EPHB4 tyrosine-kinase receptor expression and biological significance in soft tissue sarcoma. Int. J. Cancer 2015, 136, 1781–1791. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.; Li, M. The role of EphB4 and IGF-IR expression in breast cancer cells. Int. J. Clin. Exp. Pathol. 2015, 8, 5997–6004. [Google Scholar]
- Miao, H.; Li, D.Q.; Mukherjee, A.; Guo, H.; Petty, A.; Cutter, J.; Basilion, J.P.; Sedor, J.; Wu, J.; Danielpour, D.; et al. EphA2 mediates ligand-dependent inhibition and ligand-independent promotion of cell migration and invasion via a reciprocal regulatory loop with Akt. Cancer Cell 2009, 16, 9–20. [Google Scholar] [CrossRef] [Green Version]
- Jing, X.; Miyajima, M.; Sawada, T.; Chen, Q.; Iida, K.; Furushima, K.; Arai, D.; Chihara, K.; Sakaguchi, K. Crosstalk of humoral and cell-cell contact-mediated signals in postnatal body growth. Cell Rep. 2012, 2, 652–665. [Google Scholar] [CrossRef] [Green Version]
- Minami, M.; Koyama, T.; Wakayama, Y.; Fukuhara, S.; Mochizuki, N. EphrinA/EphA signal facilitates insulin-like growth factor-I-induced myogenic differentiation through suppression of the Ras/extracellular signal-regulated kinase 1/2 cascade in myoblast cell lines. Mol. Biol. Cell 2011, 22, 3508–3519. [Google Scholar] [CrossRef] [PubMed]
- Aslam, M.I.; Abraham, J.; Mansoor, A.; Druker, B.J.; Tyner, J.W.; Keller, C. PDGFRbeta reverses EphB4 signaling in alveolar rhabdomyosarcoma. Proc. Natl. Acad. Sci. USA 2014, 111, 6383–6388. [Google Scholar] [CrossRef] [Green Version]
- Fritsche-Guenther, R.; Noske, A.; Ungethum, U.; Kuban, R.J.; Schlag, P.M.; Tunn, P.U.; Karle, J.; Krenn, V.; Dietel, M.; Sers, C. De novo expression of EphA2 in osteosarcoma modulates activation of the mitogenic signalling pathway. Histopathology 2010, 57, 836–850. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Monclus, S.; Lopez-Alemany, R.; Almacellas-Rabaiget, O.; Herrero-Martin, D.; Huertas-Martinez, J.; Lagares-Tena, L.; Alba-Pavon, P.; Hontecillas-Prieto, L.; Mora, J.; de Alava, E.; et al. EphA2 receptor is a key player in the metastatic onset of Ewing sarcoma. Int. J. Cancer 2018, 143, 1188–1201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, K.; Middlemiss, S.; Saletta, F.; Gottschalk, S.; McCowage, G.B.; Kramer, B. Chimeric Antigen Receptor-modified T cells targeting EphA2 for the immunotherapy of paediatric bone tumours. Cancer Gene Ther. 2021, 28, 321–334. [Google Scholar] [CrossRef]
- Megiorni, F.; Gravina, G.L.; Camero, S.; Ceccarelli, S.; Del Fattore, A.; Desiderio, V.; Papaccio, F.; McDowell, H.P.; Shukla, R.; Pizzuti, A.; et al. Pharmacological targeting of the ephrin receptor kinase signalling by GLPG1790 in vitro and in vivo reverts oncophenotype, induces myogenic differentiation and radiosensitizes embryonal rhabdomyosarcoma cells. J. Hematol. Oncol. 2017, 10, 161. [Google Scholar] [CrossRef] [Green Version]
- Posthumadeboer, J.; Piersma, S.R.; Pham, T.V.; van Egmond, P.W.; Knol, J.C.; Cleton-Jansen, A.M.; van Geer, M.A.; van Beusechem, V.W.; Kaspers, G.J.; van Royen, B.J.; et al. Surface proteomic analysis of osteosarcoma identifies EPHA2 as receptor for targeted drug delivery. Br. J. Cancer 2013, 109, 2142–2154. [Google Scholar] [CrossRef] [Green Version]
- Randolph, M.E.; Cleary, M.M.; Bajwa, Z.; Svalina, M.N.; Young, M.C.; Mansoor, A.; Kaur, P.; Bult, C.J.; Goros, M.W.; Michalek, J.E.; et al. EphB4/EphrinB2 therapeutics in Rhabdomyosarcoma. PLoS ONE 2017, 12, e0183161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sainz-Jaspeado, M.; Huertas-Martinez, J.; Lagares-Tena, L.; Martin Liberal, J.; Mateo-Lozano, S.; de Alava, E.; de Torres, C.; Mora, J.; Del Muro, X.G.; Tirado, O.M. EphA2-induced angiogenesis in ewing sarcoma cells works through bFGF production and is dependent on caveolin-1. PLoS ONE 2013, 8, e71449. [Google Scholar] [CrossRef] [Green Version]
- Cho, Y.S.; Jiang, J. Hippo-Independent Regulation of Yki/Yap/Taz: A Non-canonical View. Front. Cell Dev. Biol. 2021, 9, 658481. [Google Scholar] [CrossRef] [PubMed]
- Moroishi, T.; Hansen, C.G.; Guan, K.L. The emerging roles of YAP and TAZ in cancer. Nat. Rev. Cancer 2015, 15, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Coffey, K. Targeting the Hippo Pathway in Prostate Cancer: What’s New? Cancers 2021, 13, 611. [Google Scholar] [CrossRef] [PubMed]
- Crose, L.E.; Galindo, K.A.; Kephart, J.G.; Chen, C.; Fitamant, J.; Bardeesy, N.; Bentley, R.C.; Galindo, R.L.; Chi, J.T.; Linardic, C.M. Alveolar rhabdomyosarcoma-associated PAX3-FOXO1 promotes tumorigenesis via Hippo pathway suppression. J. Clin. Investig. 2014, 124, 285–296. [Google Scholar] [CrossRef] [Green Version]
- Rigiracciolo, D.C.; Nohata, N.; Lappano, R.; Cirillo, F.; Talia, M.; Scordamaglia, D.; Gutkind, J.S.; Maggiolini, M. IGF-1/IGF-1R/FAK/YAP Transduction Signaling Prompts Growth Effects in Triple-Negative Breast Cancer (TNBC) Cells. Cells 2020, 9, 1010. [Google Scholar] [CrossRef] [Green Version]
- Strassburger, K.; Tiebe, M.; Pinna, F.; Breuhahn, K.; Teleman, A.A. Insulin/IGF signaling drives cell proliferation in part via Yorkie/YAP. Dev. Biol. 2012, 367, 187–196. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Chen, N.; Xu, H.; Zhou, X.; Wang, J.; Fang, X.; Zhang, Y.; Li, Y.; Yang, J.; Wang, X. Regulation of Hippo-YAP signaling by insulin-like growth factor-1 receptor in the tumorigenesis of diffuse large B-cell lymphoma. J. Hematol. Oncol. 2020, 13, 77. [Google Scholar] [CrossRef]
- Kovar, H.; Bierbaumer, L.; Radic-Sarikas, B. The YAP/TAZ Pathway in Osteogenesis and Bone Sarcoma Pathogenesis. Cells 2020, 9, 972. [Google Scholar] [CrossRef] [Green Version]
- Mohamed, A.D.; Tremblay, A.M.; Murray, G.I.; Wackerhage, H. The Hippo signal transduction pathway in soft tissue sarcomas. Biochim. Biophys. Acta 2015, 1856, 121–129. [Google Scholar] [CrossRef]
- Morice, S.; Mullard, M.; Brion, R.; Dupuy, M.; Renault, S.; Tesfaye, R.; Brounais-Le Royer, B.; Ory, B.; Redini, F.; Verrecchia, F. The YAP/TEAD Axis as a New Therapeutic Target in Osteosarcoma: Effect of Verteporfin and CA3 on Primary Tumor Growth. Cancers 2020, 12, 3847. [Google Scholar] [CrossRef]
- Deel, M.D.; Slemmons, K.K.; Hinson, A.R.; Genadry, K.C.; Burgess, B.A.; Crose, L.E.S.; Kuprasertkul, N.; Oristian, K.M.; Bentley, R.C.; Linardic, C.M. The Transcriptional Coactivator TAZ Is a Potent Mediator of Alveolar Rhabdomyosarcoma Tumorigenesis. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 2616–2630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanna, L.; Piredda, R.; Marchesi, I.; Bordoni, V.; Forcales, S.V.; Calvisi, D.F.; Bagella, L. Verteporfin exhibits anti-proliferative activity in embryonal and alveolar rhabdomyosarcoma cell lines. Chem.-Biol. Interact. 2019, 312, 108813. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, A.M.; Missiaglia, E.; Galli, G.G.; Hettmer, S.; Urcia, R.; Carrara, M.; Judson, R.N.; Thway, K.; Nadal, G.; Selfe, J.L.; et al. The Hippo transducer YAP1 transforms activated satellite cells and is a potent effector of embryonal rhabdomyosarcoma formation. Cancer Cell 2014, 26, 273–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohamed, A.; Sun, C.; De Mello, V.; Selfe, J.; Missiaglia, E.; Shipley, J.; Murray, G.I.; Zammit, P.S.; Wackerhage, H. The Hippo effector TAZ (WWTR1) transforms myoblasts and TAZ abundance is associated with reduced survival in embryonal rhabdomyosarcoma. J. Pathol. 2016, 240, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Bierbaumer, L.; Katschnig, A.M.; Radic-Sarikas, B.; Kauer, M.O.; Petro, J.A.; Hogler, S.; Gurnhofer, E.; Pedot, G.; Schafer, B.W.; Schwentner, R.; et al. YAP/TAZ inhibition reduces metastatic potential of Ewing sarcoma cells. Oncogenesis 2021, 10, 2. [Google Scholar] [CrossRef]
- Shorstova, T.; Foulkes, W.D.; Witcher, M. Achieving clinical success with BET inhibitors as anti-cancer agents. Br. J. Cancer 2021, 124, 1478–1490. [Google Scholar] [CrossRef] [PubMed]
- Gilan, O.; Rioja, I.; Knezevic, K.; Bell, M.J.; Yeung, M.M.; Harker, N.R.; Lam, E.Y.N.; Chung, C.W.; Bamborough, P.; Petretich, M.; et al. Selective targeting of BD1 and BD2 of the BET proteins in cancer and immunoinflammation. Science 2020, 368, 387–394. [Google Scholar] [CrossRef]
- Loganathan, S.N.; Tang, N.; Fleming, J.T.; Ma, Y.; Guo, Y.; Borinstein, S.C.; Chiang, C.; Wang, J. BET bromodomain inhibitors suppress EWS-FLI1-dependent transcription and the IGF1 autocrine mechanism in Ewing sarcoma. Oncotarget 2016, 7, 43504–43517. [Google Scholar] [CrossRef] [Green Version]
- Loganathan, S.N.; Tang, N.; Holler, A.E.; Wang, N.; Wang, J. Targeting the IGF1R/PI3K/AKT Pathway Sensitizes Ewing Sarcoma to BET Bromodomain Inhibitors. Mol. Cancer Ther. 2019, 18, 929–936. [Google Scholar] [CrossRef] [Green Version]
- Stratikopoulos, E.E.; Dendy, M.; Szabolcs, M.; Khaykin, A.J.; Lefebvre, C.; Zhou, M.M.; Parsons, R. Kinase and BET Inhibitors Together Clamp Inhibition of PI3K Signaling and Overcome Resistance to Therapy. Cancer Cell 2015, 27, 837–851. [Google Scholar] [CrossRef] [Green Version]
- Boedicker, C.; Hussong, M.; Grimm, C.; Dolgikh, N.; Meister, M.T.; Enssle, J.C.; Wanior, M.; Knapp, S.; Schweiger, M.R.; Fulda, S. Co-inhibition of BET proteins and PI3Kalpha triggers mitochondrial apoptosis in rhabdomyosarcoma cells. Oncogene 2020, 39, 3837–3852. [Google Scholar] [CrossRef]
- Lee, D.H.; Qi, J.; Bradner, J.E.; Said, J.W.; Doan, N.B.; Forscher, C.; Yang, H.; Koeffler, H.P. Synergistic effect of JQ1 and rapamycin for treatment of human osteosarcoma. Int. J. Cancer 2015, 136, 2055–2064. [Google Scholar] [CrossRef]
- Gollavilli, P.N.; Pawar, A.; Wilder-Romans, K.; Natesan, R.; Engelke, C.G.; Dommeti, V.L.; Krishnamurthy, P.M.; Nallasivam, A.; Apel, I.J.; Xu, T.; et al. EWS/ETS-Driven Ewing Sarcoma Requires BET Bromodomain Proteins. Cancer Res. 2018, 78, 4760–4773. [Google Scholar] [CrossRef] [Green Version]
- Mancarella, C.; Pasello, M.; Ventura, S.; Grilli, A.; Calzolari, L.; Toracchio, L.; Lollini, P.L.; Donati, D.M.; Picci, P.; Ferrari, S.; et al. Insulin-Like Growth Factor 2 mRNA-Binding Protein 3 is a Novel Post-Transcriptional Regulator of Ewing Sarcoma Malignancy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 3704–3716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palanichamy, J.K.; Tran, T.M.; Howard, J.M.; Contreras, J.R.; Fernando, T.R.; Sterne-Weiler, T.; Katzman, S.; Toloue, M.; Yan, W.; Basso, G.; et al. RNA-binding protein IGF2BP3 targeting of oncogenic transcripts promotes hematopoietic progenitor proliferation. J. Clin. Investig. 2016, 126, 1495–1511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, R.H.; Li, B.D.; Chu, Q.D. The role of chemokine receptor CXCR4 in the biologic behavior of human soft tissue sarcoma. Sarcoma 2011, 2011, 593708. [Google Scholar] [CrossRef]
- Britton, C.; Poznansky, M.C.; Reeves, P. Polyfunctionality of the CXCR4/CXCL12 axis in health and disease: Implications for therapeutic interventions in cancer and immune-mediated diseases. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2021, 35, e21260. [Google Scholar] [CrossRef]
- Kuo, Y.C.; Au, H.K.; Hsu, J.L.; Wang, H.F.; Lee, C.J.; Peng, S.W.; Lai, S.C.; Wu, Y.C.; Ho, H.N.; Huang, Y.H. IGF-1R Promotes Symmetric Self-Renewal and Migration of Alkaline Phosphatase(+) Germ Stem Cells through HIF-2alpha-OCT4/CXCR4 Loop under Hypoxia. Stem Cell Rep. 2018, 10, 524–537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akekawatchai, C.; Holland, J.D.; Kochetkova, M.; Wallace, J.C.; McColl, S.R. Transactivation of CXCR4 by the insulin-like growth factor-1 receptor (IGF-1R) in human MDA-MB-231 breast cancer epithelial cells. J. Biol. Chem. 2005, 280, 39701–39708. [Google Scholar] [CrossRef] [Green Version]
- Krook, M.A.; Nicholls, L.A.; Scannell, C.A.; Chugh, R.; Thomas, D.G.; Lawlor, E.R. Stress-induced CXCR4 promotes migration and invasion of ewing sarcoma. Mol. Cancer Res. MCR 2014, 12, 953–964. [Google Scholar] [CrossRef] [Green Version]
- Mancarella, C.; Caldoni, G.; Ribolsi, I.; Parra, A.; Manara, M.C.; Mercurio, A.M.; Morrione, A.; Scotlandi, K. Insulin-Like Growth Factor 2 mRNA-Binding Protein 3 Modulates Aggressiveness of Ewing Sarcoma by Regulating the CD164-CXCR4 Axis. Front. Oncol. 2020, 10, 994. [Google Scholar] [CrossRef] [PubMed]
- Pazzaglia, L.; Pollino, S.; Vitale, M.; Bientinesi, E.; Benini, S.; Ferrari, C.; Palmerini, E.; Gambarotti, M.; Picci, P.; Benassi, M.S. miR494.3p expression in synovial sarcoma: Role of CXCR4 as a potential target gene. Int. J. Oncol. 2019, 54, 361–369. [Google Scholar] [CrossRef] [Green Version]
- Pollino, S.; Palmerini, E.; Dozza, B.; Bientinesi, E.; Piccinni-Leopardi, M.; Lucarelli, E.; Righi, A.; Benassi, M.S.; Pazzaglia, L. CXCR4 in human osteosarcoma malignant progression. The response of osteosarcoma cell lines to the fully human CXCR4 antibody MDX1338. J. Bone Oncol. 2019, 17, 100239. [Google Scholar] [CrossRef] [PubMed]
- Xi, Y.; Qi, Z.; Ma, J.; Chen, Y. PTEN loss activates a functional AKT/CXCR4 signaling axis to potentiate tumor growth and lung metastasis in human osteosarcoma cells. Clin. Exp. Metastasis 2020, 37, 173–185. [Google Scholar] [CrossRef] [PubMed]
- Regenbogen, S.; Stagno, M.J.; Schleicher, S.; Schilbach, K.; Bosmuller, H.; Fuchs, J.; Schmid, E.; Seitz, G. Cytotoxic drugs in combination with the CXCR4 antagonist AMD3100 as a potential treatment option for pediatric rhabdomyosarcoma. Int. J. Oncol. 2020, 57, 289–300. [Google Scholar] [CrossRef] [PubMed]
- Vela, M.; Bueno, D.; Gonzalez-Navarro, P.; Brito, A.; Fernandez, L.; Escudero, A.; Valentin, J.; Mestre-Duran, C.; Arranz-Alvarez, M.; Perez de Diego, R.; et al. Anti-CXCR4 Antibody Combined With Activated and Expanded Natural Killer Cells for Sarcoma Immunotherapy. Front. Immunol. 2019, 10, 1814. [Google Scholar] [CrossRef] [Green Version]
- Chadalapaka, G.; Jutooru, I.; Sreevalsan, S.; Pathi, S.; Kim, K.; Chen, C.; Crose, L.; Linardic, C.; Safe, S. Inhibition of rhabdomyosarcoma cell and tumor growth by targeting specificity protein (Sp) transcription factors. Int. J. Cancer 2013, 132, 795–806. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mancarella, C.; Morrione, A.; Scotlandi, K. Unraveling the IGF System Interactome in Sarcomas Exploits Novel Therapeutic Options. Cells 2021, 10, 2075. https://doi.org/10.3390/cells10082075
Mancarella C, Morrione A, Scotlandi K. Unraveling the IGF System Interactome in Sarcomas Exploits Novel Therapeutic Options. Cells. 2021; 10(8):2075. https://doi.org/10.3390/cells10082075
Chicago/Turabian StyleMancarella, Caterina, Andrea Morrione, and Katia Scotlandi. 2021. "Unraveling the IGF System Interactome in Sarcomas Exploits Novel Therapeutic Options" Cells 10, no. 8: 2075. https://doi.org/10.3390/cells10082075
APA StyleMancarella, C., Morrione, A., & Scotlandi, K. (2021). Unraveling the IGF System Interactome in Sarcomas Exploits Novel Therapeutic Options. Cells, 10(8), 2075. https://doi.org/10.3390/cells10082075