
Insulin Sensitivity Is Retained in Mice with Endothelial Loss of Carcinoembryonic Antigen Cell Adhesion Molecule 1

 , ,
, ,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Generation of Null Mice
2.2. Biochemical Parameters
2.3. Glucose and Insulin Tolerance Tests
2.4. Hyperinsulinemic-Euglycemic Clamp Analysis
2.5. Isolation of Primary Cells
2.6. Insulin Treatment and Media Analysis
2.7. Western Blot Analysis
2.8. Semi-Quantitative Real-Time PCR (qRT-PCR) Analysis of mRNA
2.9. Gomori’s Trichrome Staining
2.10. Immunofluorescence of Visceral White Adipose Tissue
2.11. Statistical Analysis
3. Results
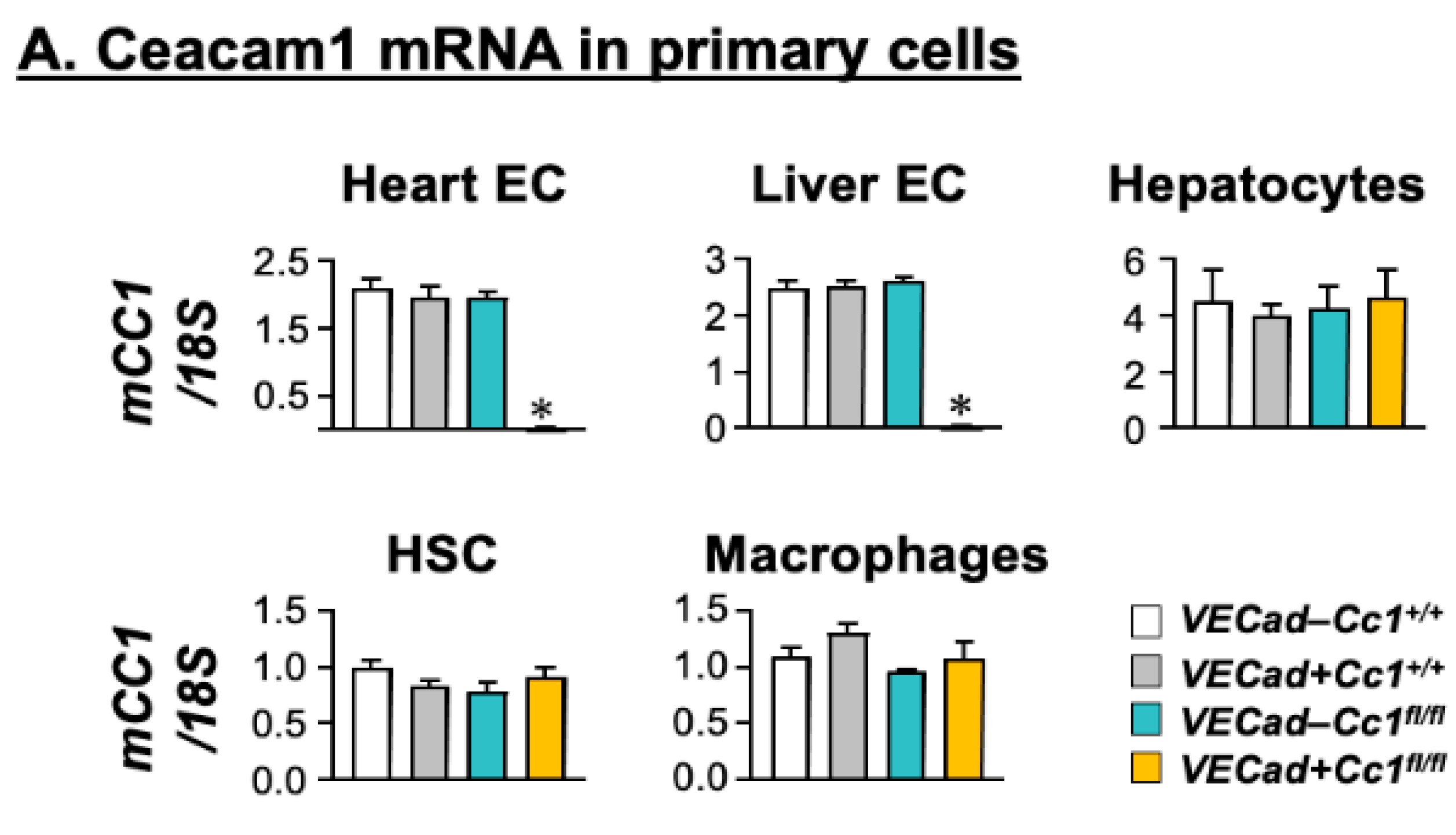
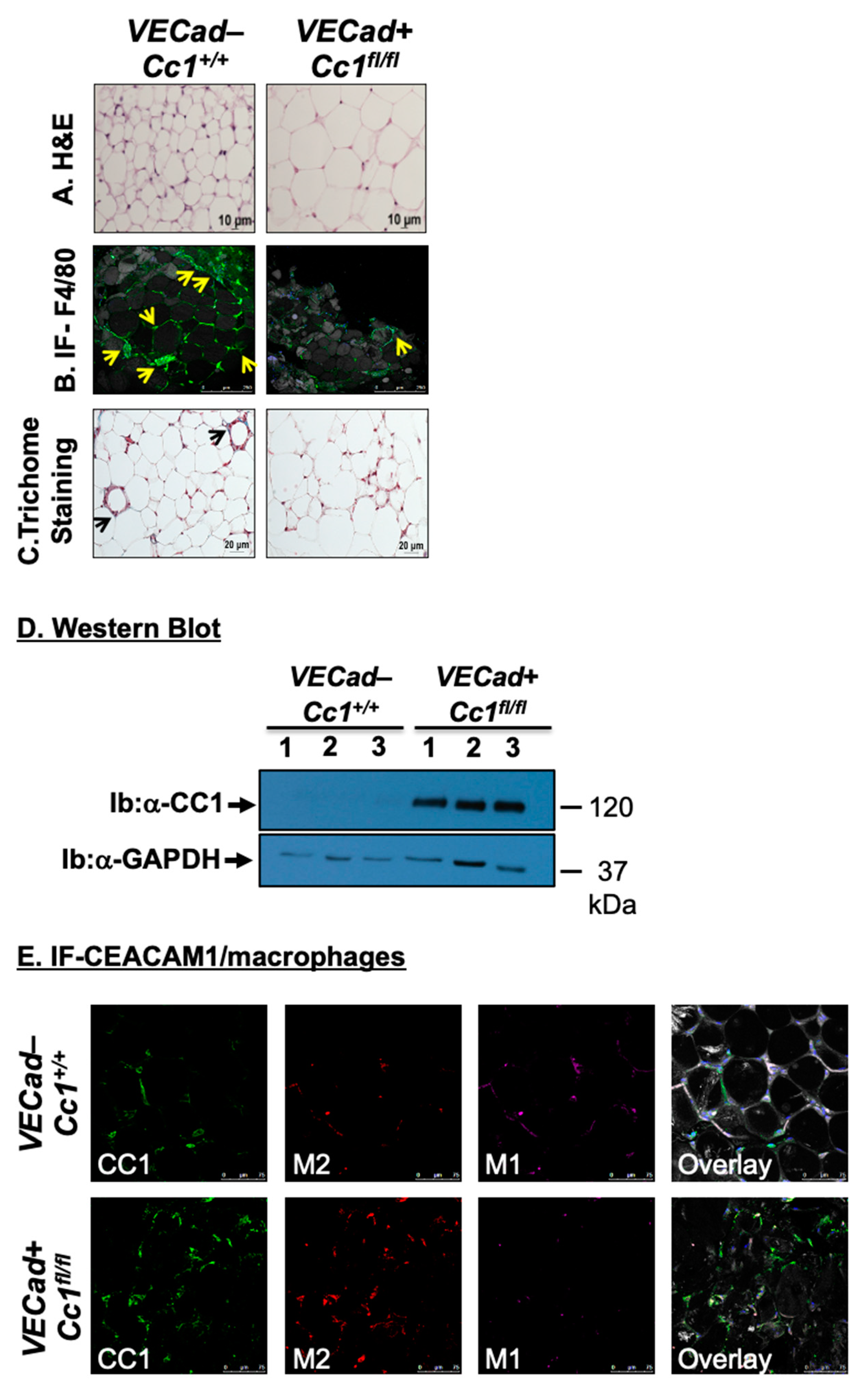
3.1. Specific Deletion of Ceacam1 in Endothelial Cells of VECadCre+Cc1fl/fl Mice
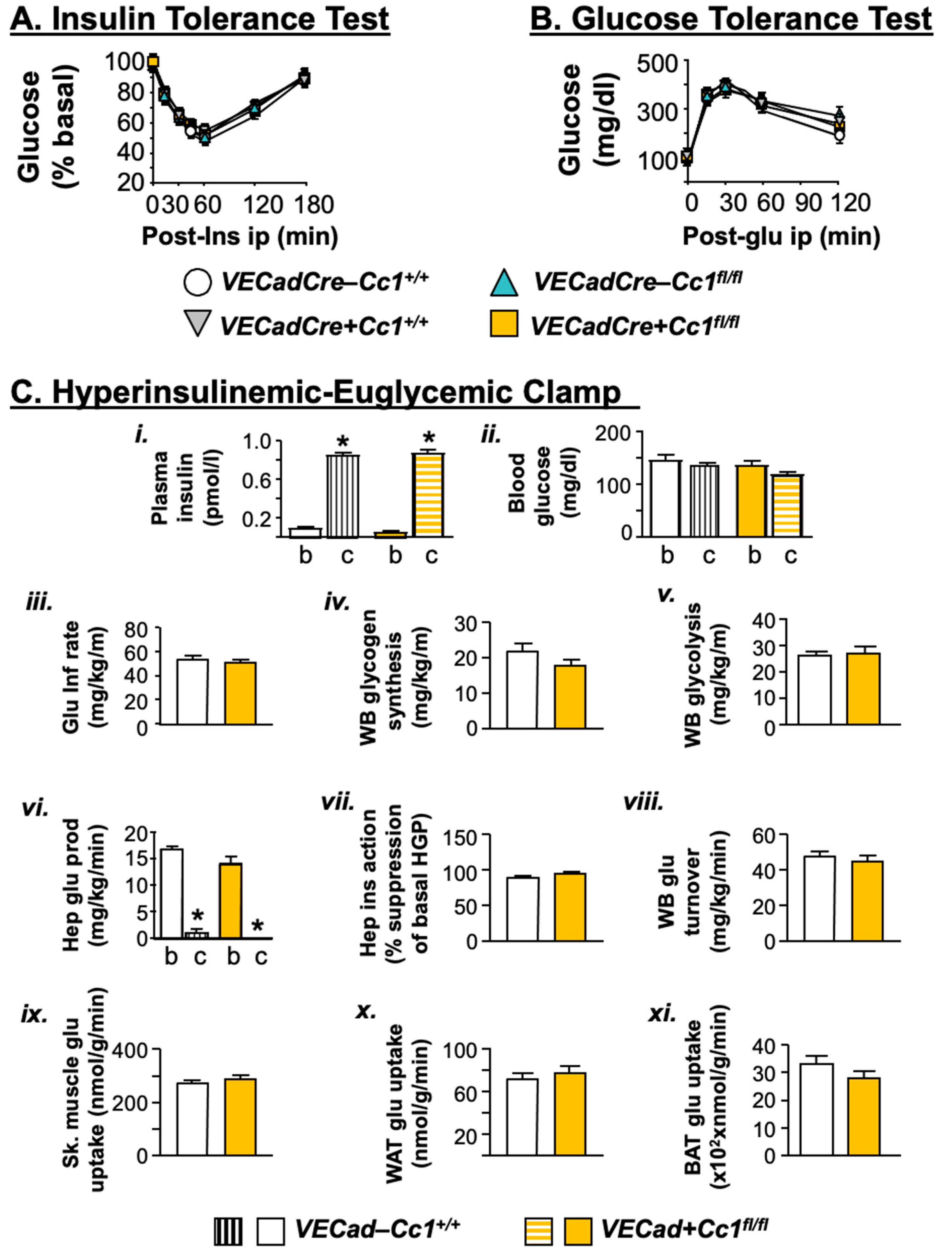
3.2. VECadCre+Cc1fl/fl Mice Display Insulin Sensitivity
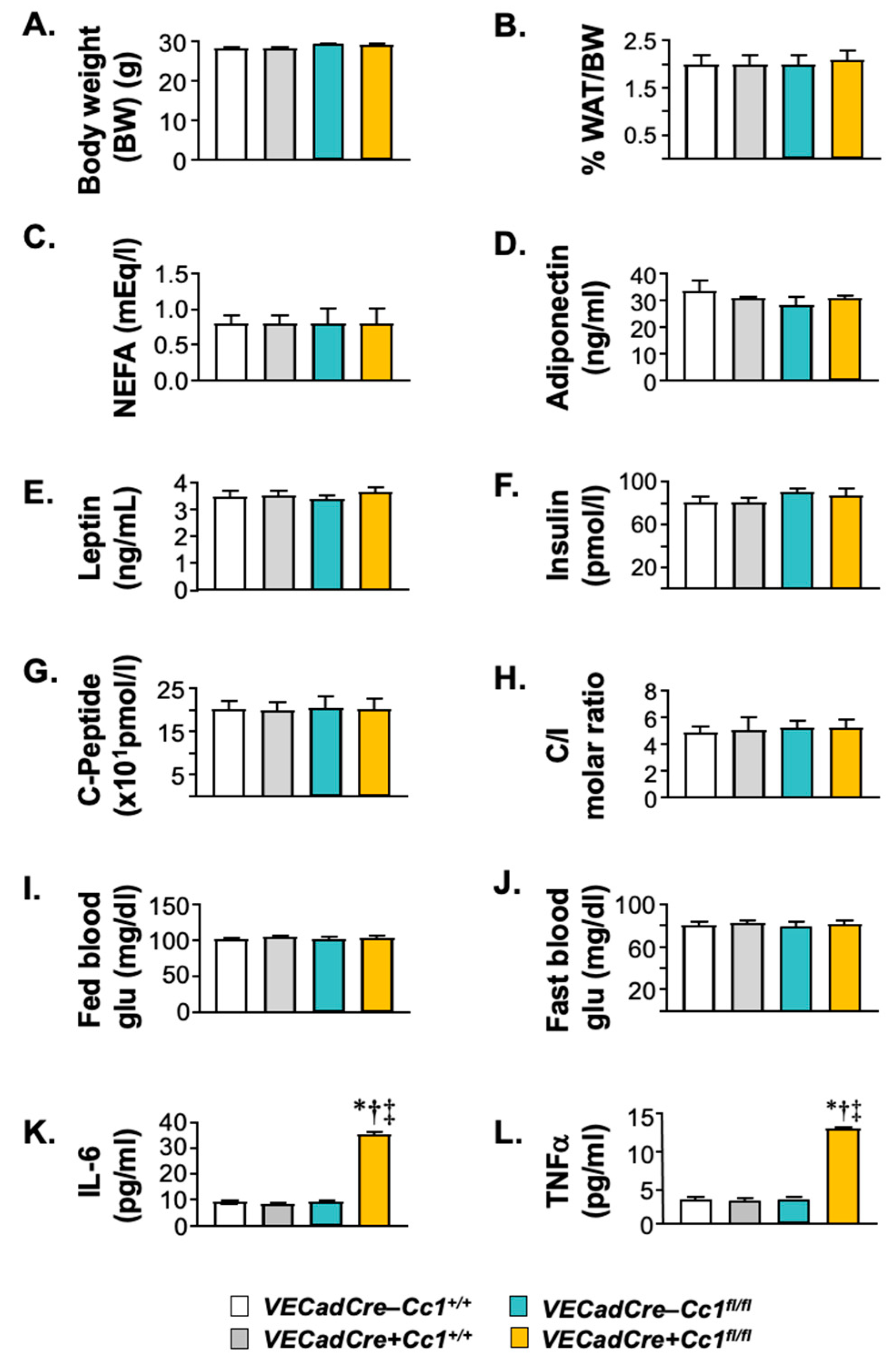
3.3. Increased NF-κB-Mediated Systemic Inflammation in VECadCre+Cc1fl/fl Mice
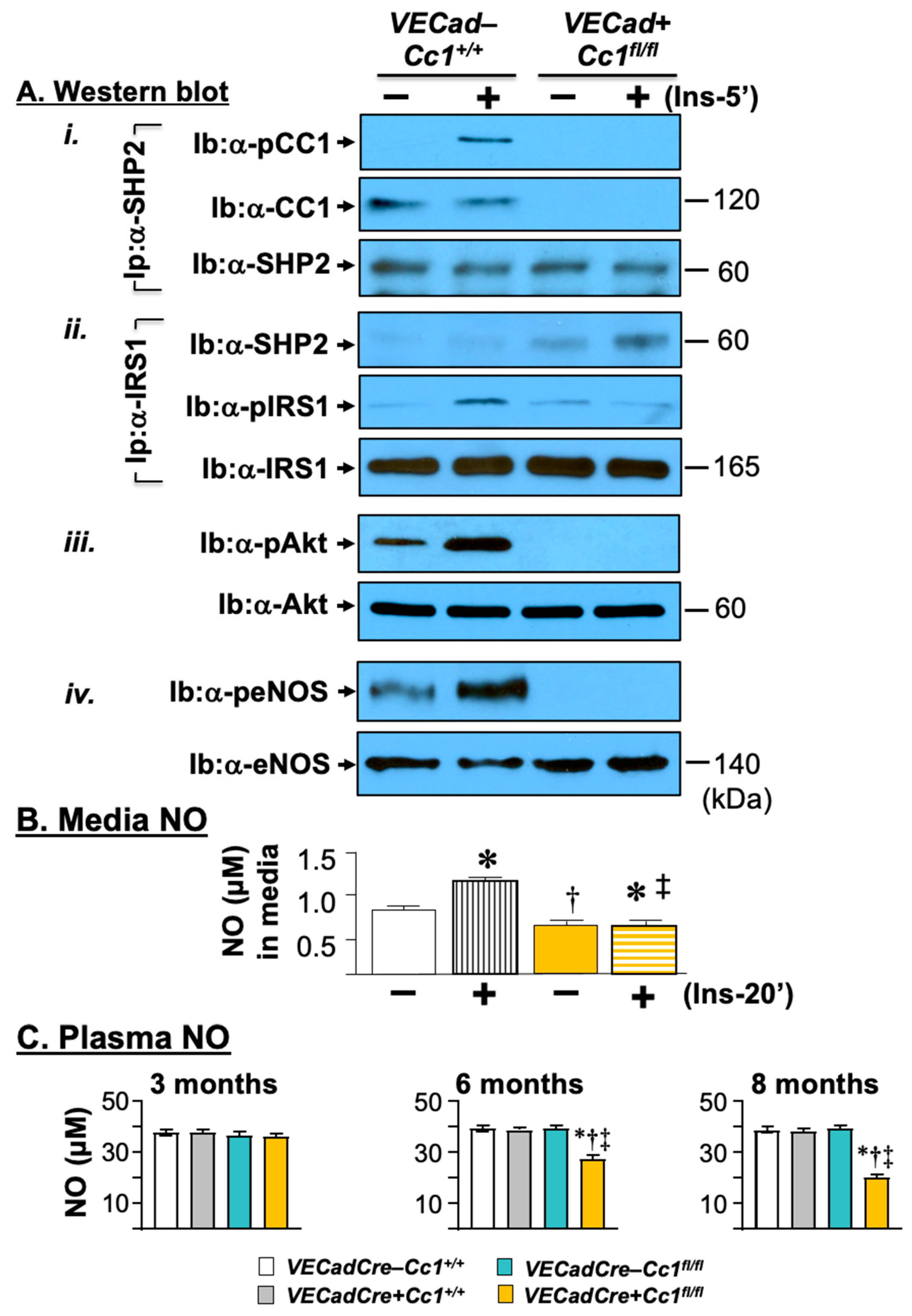
3.4. Compromised Endothelial Barrier and Lower Nitric Oxide Production in VECadCre+Cc1fl/fl Mice
3.5. Increased Fat Accumulation and Insulin Sensitivity in Adipocytes of VECadCre+Cc1fl/fl Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Najjar, S.M.; Philippe, N.; Suzuki, Y.; Ignacio, G.A.; Formisano, P.; Accili, D.; Taylor, S.I. Insulin-stimulated phosphorylation of recombinant pp120/HA4, an endogenous substrate of the insulin receptor tyrosine kinase. Biochemistry 1995, 34, 9341–9349. [Google Scholar] [CrossRef]
- Nouvion, A.L.; Oubaha, M.; Leblanc, S.; Davis, E.C.; Jastrow, H.; Kammerer, R.; Breton, V.; Turbide, C.; Ergun, S.; Gratton, J.-P.; et al. CEACAM1: A key regulator of vascular permeability. J. Cell Sci. 2010, 123, 4221–4230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Najjar, S.M.; Perdomo, G. Hepatic Insulin Clearance: Mechanism and Physiology. Physiology (Bethesda) 2019, 34, 198–215. [Google Scholar] [CrossRef]
- DeAngelis, A.M.; Heinrich, G.; Dai, T.; Bowman, T.A.; Patel, P.R.; Lee, S.J.; Hong, E.-G.; Jung, D.Y.; Assmann, A.; Kulkarni, R.N.; et al. Carcinoembryonic antigen-related cell adhesion molecule 1: A link between insulin and lipid metabolism. Diabetes 2008, 57, 2296–2303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghadieh, H.E.; Russo, L.; Muturi, H.T.; Ghanem, S.S.; Manaserh, I.H.; Noh, H.L.; Suk, S.; Kim, J.K.; Hill, J.W.; Najjar, S.M. Hyperinsulinemia drives hepatic insulin resistance in male mice with liver-specific Ceacam1 deletion independently of lipolysis. Metabolism 2019, 93, 33–43. [Google Scholar] [CrossRef]
- Poy, M.N.; Yang, Y.; Rezaei, K.; Fernstrom, M.A.; Lee, A.D.; Kido, Y.; Erickson, S.K.; Najjar, S.M. CEACAM1 regulates insulin clearance in liver. Nat Genet. 2002, 30, 270–276. [Google Scholar] [CrossRef]
- Russo, L.; Muturi, H.T.; Ghadieh, H.E.; Wisniewski, A.M.; Morgan, E.E.; Quadri, S.S.; Landesberg, G.P.; Siragy, H.M.; Vazquez, G.; Scalia, R.; et al. Liver-specific rescuing of CEACAM1 reverses endothelial and cardiovascular abnormalities in male mice with null deletion of Ceacam1 gene. Mol. Metab. 2018, 9, 98–113. [Google Scholar] [CrossRef]
- Russo, L.; Muturi, H.T.; Ghadieh, H.E.; Ghanem, S.S.; Bowman, T.A.; Noh, H.L.; Dagdeviren, S.; Dogbey, G.Y.; Kim, J.K.; Heinrich, G.; et al. Liver-specific reconstitution of CEACAM1 reverses the metabolic abnormalities caused by its global deletion in male mice. Diabetologia 2017, 60, 2463–2474. [Google Scholar] [CrossRef] [PubMed]
- Sylow, L.; Tokarz, V.L.; Richter, E.A.; Klip, A. The many actions of insulin in skeletal muscle, the paramount tissue determining glycemia. Cell Metab. 2021, 33, 758–780. [Google Scholar] [CrossRef]
- Yazdani, S.; Jaldin-Fincati, J.R.; Pereira, R.V.S.; Klip, A. Endothelial cell barriers: Transport of molecules between blood and tissues. Traffic 2019, 20, 390–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azizi, P.M.; Zyla, R.E.; Guan, S.; Wang, C.; Liu, J.; Bolz, S.S.; Heit, B.; Klip, A.; Lee, W.L. Clathrin-dependent entry and vesicle-mediated exocytosis define insulin transcytosis across microvascular endothelial cells. Mol. Biol. Cell 2015, 26, 740–750. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, A.X.; Barrett, E.J. Caveolin-1 is required for vascular endothelial insulin uptake. Am. J. Physiol. Endocrinol. Metab. 2011, 300, E134–E144. [Google Scholar] [CrossRef]
- Ghavampour, S.; Kleefeldt, F.; Bommel, H.; Volland, J.; Paus, A.; Horst, A.; Pfeiffer, V.; Hubner, S.; Wagner, N.; Rueckschloss, U.; et al. Endothelial barrier function is differentially regulated by CEACAM1-mediated signaling. FASEB J. 2018, 32, 5612–5625. [Google Scholar] [CrossRef] [PubMed]
- Horst, A.K.; Ito, W.D.; Dabelstein, J.; Schumacher, U.; Sander, H.; Turbide, C.; Brümmer, J.; Meinertz, T.; Beauchemin, N.; Wagener, C. Carcinoembryonic antigen-related cell adhesion molecule 1 modulates vascular remodeling in vitro and in vivo. J. Clin. Investig. 2006, 116, 1596–1605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rueckschloss, U.; Kuerten, S.; Ergun, S. The role of CEA-related cell adhesion molecule-1 (CEACAM1) in vascular homeostasis. Histochem. Cell Biol. 2016, 146, 657–671. [Google Scholar] [CrossRef] [PubMed]
- Najjar, S.M.; Ledford, K.J.; Abdallah, S.L.; Paus, A.; Russo, L.; Kaw, M.K.; Ramakrishnan, S.K.; Muturi, H.T.; Raphael, C.K.; Lester, S.G.; et al. Ceacam1 deletion causes vascular alterations in large vessels. Am. J. Physiol. Endocrinol. Metab. 2013, 305, E519–E529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerstel, D.; Wegwitz, F.; Jannasch, K.; Ludewig, P.; Scheike, K.; Alves, F.; Beauchemin, N.; Deppert, W.; Wagener, C.; Horst, A.K. CEACAM1 creates a pro-angiogenic tumor microenvironment that supports tumor vessel maturation. Oncogene 2011, 30, 4275–4288. [Google Scholar] [CrossRef] [PubMed]
- Al-Share, Q.Y.; DeAngelis, A.M.; Lester, S.G.; Bowman, T.A.; Ramakrishnan, S.K.; Abdallah, S.L.; Russo, L.; Patel, P.R.; Kaw, M.K.; Raphael, C.K.; et al. Forced Hepatic Overexpression of CEACAM1 Curtails Diet-Induced Insulin Resistance. Diabetes 2015, 64, 2780–2790. [Google Scholar] [CrossRef] [Green Version]
- Ghadieh, H.E.; Abu Helal, R.; Muturi, H.T.; Issa, D.D.; Russo, L.; Abdallah, S.L.; Najjar, J.A.; Benencia, F.; Vazquez, G.; Li, W.; et al. Loss of Hepatic Carcinoembryonic Antigen-Related Cell Adhesion Molecule 1 Links Nonalcoholic Steatohepatitis to Atherosclerosis. Hepatol. Commun. 2020, 4, 1591–1609. [Google Scholar] [CrossRef]
- Mederacke, I.; Dapito, D.H.; Affo, S.; Uchinami, H.; Schwabe, R.F. High-yield and high-purity isolation of hepatic stellate cells from normal and fibrotic mouse livers. Nat. Protoc. 2015, 10, 305–315. [Google Scholar] [CrossRef] [Green Version]
- Soni, P.; Lakkis, M.; Poy, M.N.; Fernstrom, M.A.; Najjar, S.M. The differential effects of pp120 (Ceacam 1) on the mitogenic action of insulin and insulin-like growth factor 1 are regulated by the nonconserved tyrosine 1316 in the insulin receptor. Mol. Cell Biol. 2000, 20, 3896–3905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramakrishnan, S.K.; Russo, L.; Ghanem, S.S.; Patel, P.R.; Oyarce, A.M.; Heinrich, G.; Najjar, S.M. Fenofibrate decreases insulin clearance and insulin secretion to maintain insulin sensitivity. J. Biol. Chem. 2016, 291, 23915–23924. [Google Scholar] [CrossRef] [Green Version]
- Pereira, S.; Park, E.; Mori, Y.; Haber, C.A.; Han, P.; Uchida, T.; Stavar, L.; Oprescu, A.I.; Koulajian, K.; Ivovic, A.; et al. FFA-induced hepatic insulin resistance in vivo is mediated by PKCδ, NADPH oxidase, and oxidative stress. Am. J. Physiol. Endocrinol. Metab. 2014, 307, E34–E46. [Google Scholar] [CrossRef] [Green Version]
- Lester, S.G.; Russo, L.; Ghanem, S.S.; Khuder, S.S.; DeAngelis, A.M.; Esakov, E.L.; Bowman, T.A.; Heinrich, G.; Al-Share, Q.Y.; Mclnerner, M.F.; et al. Hepatic CEACAM1 over-expression protects against diet-induced fibrosis and inflammation in white adipose tissue. Front. Endocrinol. 2015, 6, 116–122. [Google Scholar] [CrossRef] [Green Version]
- Poy, M.N.; Ruch, R.J.; Fernstrom, M.A.; Okabayashi, Y.; Najjar, S.M. Shc and CEACAM1 interact to regulate the mitogenic action of insulin. J. Biol. Chem. 2002, 277, 1076–1084. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Alcaide, P.; Liu, L.; Sun, J.; He, A.; Luscinskas, F.W.; Shi, G.P. Regulation of endothelial cell adhesion molecule expression by mast cells, macrophages, and neutrophils. PLoS ONE 2011, 6, e14525. [Google Scholar]
- Walker, T.; Wendel, H.P.; Tetzloff, L.; Raabe, C.; Heidenreich, O.; Simon, P.; Scheule, A.M.; Ziemer, G. Inhibition of adhesion molecule expression on human venous endothelial cells by non-viral siRNA transfection. J. Cell Mol. Med. 2007, 11, 139–147. [Google Scholar] [CrossRef]
- Pan, J.; McEver, R.P. Regulation of the human P-selectin promoter by Bcl-3 and specific homodimeric members of the NF-kappa B/Rel family. J. Biol. Chem. 1995, 270, 23077–23083. [Google Scholar] [CrossRef] [Green Version]
- Min, C.; Eddy, S.F.; Sherr, D.H.; Sonenshein, G.E. NF-kappaB and epithelial to mesenchymal transition of cancer. J. Cell Biochem. 2008, 104, 733–744. [Google Scholar] [CrossRef]
- Alexander, J.S.; Elrod, J.W. Extracellular matrix, junctional integrity and matrix metalloproteinase interactions in endothelial permeability regulation. J. Anat. 2002, 200, 561–574. [Google Scholar] [CrossRef] [PubMed]
- Schulz, B.; Pruessmeyer, J.; Maretzky, T.; Ludwig, A.; Blobel, C.P.; Saftig, P.; Reiss, K. ADAM10 regulates endothelial permeability and T-Cell transmigration by proteolysis of vascular endothelial cadherin. Circ. Res. 2008, 102, 1192–1201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Easwaran, V.; Lee, S.H.; Inge, L.; Guo, L.; Goldbeck, C.; Garrett, E.; Wiesmann, M.; Garcia, P.D.; Fuller, J.H.; Chan, V.; et al. beta-Catenin regulates vascular endothelial growth factor expression in colon cancer. Cancer Res. 2003, 63, 3145–3153. [Google Scholar]
- Muenzner, P.; Naumann, M.; Meyer, T.F.; Gray-Owen, S.D. Pathogenic Neisseria trigger expression of their carcinoembryonic antigen-related cellular adhesion molecule 1 (CEACAM1; previously CD66a) receptor on primary endothelial cells by activating the immediate early response transcription factor, nuclear factor-kappaB. J. Biol. Chem. 2001, 276, 24331–24340. [Google Scholar] [PubMed] [Green Version]
- Najjar, S.M. Regulation of insulin action by CEACAM1. Trends Endocrinol. Metab. 2002, 13, 240–245. [Google Scholar] [CrossRef]
- Najjar, S.M.; Accili, D.; Philippe, N.; Jernberg, J.; Margolis, R.; Taylor, S.I. pp120/ecto-ATPase, an endogenous substrate of the insulin receptor tyrosine kinase, is expressed as two variably spliced isoforms. J. Biol. Chem. 1993, 268, 1201–1206. [Google Scholar] [CrossRef]
- Dery, K.J.; Kujawski, M.; Grunert, D.; Wu, X.; Ngyuen, T.; Cheung, C.; Yim, J.H.; Shively, J.E. IRF-1 regulates alternative mRNA splicing of carcinoembryonic antigen-related cell adhesion molecule 1 (CEACAM1) in breast epithelial cells generating an immunoreceptor tyrosine-based inhibition motif (ITIM) containing isoform. Mol. Cancer 2014, 13, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diab, M.; Vitenshtein, A.; Drori, Y.; Yamin, R.; Danziger, O.; Zamostiano, R.; Mandelboim, M.; Bacharach, E.; Mandelboim, O. Suppression of human metapneumovirus (HMPV) infection by the innate sensing gene CEACAM1. Oncotarget 2016, 7, 66468–66479. [Google Scholar] [CrossRef] [Green Version]
- Kim, W.M.; Huang, Y.H.; Gandhi, A.; Blumberg, R.S. CEACAM1 structure and function in immunity and its therapeutic implications. Semin. Immunol. 2019, 42, 101296. [Google Scholar] [CrossRef]
- Vicent, D.; Ilany, J.; Kondo, T.; Naruse, K.; Fisher, S.J.; Kisanuki, Y.Y.; Bursell, S.; Yanagisawa, M.; King, G.L.; Kahn, C.R. The role of endothelial insulin signaling in the regulation of vascular tone and insulin resistance. J. Clin. Investig. 2003, 111, 1373–1380. [Google Scholar] [CrossRef] [Green Version]
- Kolka, C.M.; Bergman, R.N. The barrier within: Endothelial transport of hormones. Physiology (Bethesda) 2012, 27, 237–247. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Wang, A.X.; Aylor, K.; Barrett, E.J. Nitric oxide directly promotes vascular endothelial insulin transport. Diabetes 2013, 62, 4030–4042. [Google Scholar] [CrossRef] [Green Version]
- Marcos-Ramiro, B.; Garcia-Weber, D.; Millan, J. TNF-induced endothelial barrier disruption: Beyond actin and Rho. Thromb. Haemost. 2014, 112, 1088–1102. [Google Scholar] [CrossRef] [PubMed]
- Preston, K.J.; Rom, I.; Vrakas, C.; Landesberg, G.; Etwebi, Z.; Muraoka, S.; Autieri, M.; Eguchi, S.; Scalia, R. Postprandial activation of leukocyte-endothelium interaction by fatty acids in the visceral adipose tissue microcirculation. FASEB J. 2019, 33, 11993–12007. [Google Scholar] [CrossRef] [Green Version]
- Goldberg, I.J.; Cabodevilla, A.G.; Samovski, D.; Cifarelli, V.; Basu, D.; Abumrad, N.A. Lipolytic enzymes and free fatty acids at the endothelial interface. Atherosclerosis 2021, 329, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Saltiel, A.R.; Olefsky, J.M. Inflammatory mechanisms linking obesity and metabolic disease. J. Clin. Investig. 2017, 127, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Bajaj, M.; Suraamornkul, S.; Kashyap, S.; Cusi, K.; Mandarino, L.; DeFronzo, R.A. Sustained reduction in plasma free fatty acid concentration improves insulin action without altering plasma adipocytokine levels in subjects with strong family history of type 2 diabetes. J. Clin. Endocrinol. Metab. 2004, 89, 4649–4655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scherer, P.E. The Multifaceted Roles of Adipose Tissue-Therapeutic Targets for Diabetes and Beyond: The 2015 Banting Lecture. Diabetes 2016, 65, 1452–1461. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| VECadCre−Cc1+/+ | VECadCre+Cc1fl/fl | |
|---|---|---|
| Cell adhesion | ||
| Vcam1 | 1.51 ± 0.11 | 6.24 ± 0.55 * |
| Icam1 | 1.01 ± 0.19 | 5.34 ± 0.15 * |
| P-Selectin | 1.34 ± 0.12 | 3.72 ± 0.57 * |
| Adherence/Tight junctions | ||
| Vecadherin | 5.12 ± 1.28 | 2.15 ± 0.43 * |
| β-Catenin | 7.23 ± 1.42 | 3.43 ± 0.87 * |
| Occludin | 6.75 ± 1.22 | 3.21 ± 0.51 * |
| ZO-1 | 3.75 ± 0.56 | 1.04 ± 0.19 * |
| Claudin1 | 4.68 ± 1.13 | 1.52 ± 0.76 * |
| Claudin5 | 4.89 ± 1.31 | 1.67 ± 0.64 * |
| Vascular permeability | ||
| Vegf-A | 8.37 ± 1.68 | 2.45 ± 0.78 * |
| Vegf-C | 1.56 ± 0.07 | 1.41 ± 0.09 |
| Vegf-D | 0.98 ± 0.12 | 0.95 ± 0.11 |
| Vegfr1 | 2.01 ± 0.04 | 0.97 ± 0.03 * |
| Vegfr2 | 1.75 ± 0.08 | 0.37 ± 0.06 * |
| VECadCre−Cc1+/+ | VECadCre+Cc1+/+ | VECadCre−Cc1fl/fl | VECadCre+Cc1fl/fl | |
|---|---|---|---|---|
| Heart | ||||
| Mmp2 | 1.99 ± 0.20 | 1.87 ± 0.15 | 1.80 ± 0.15 | 3.79 ± 0.20 * |
| Mmp9 | 1.11 ± 0.10 | 1.10 ± 0.11 | 1.15 ± 0.12 | 2.58 ± 0.15 * |
| ZO-1 | 2.86 ± 0.26 | 2.82 ± 0.25 | 3.79 ± 0.30 | 0.81 ± 0.20 * |
| ZO-2 | 2.99 ± 0.21 | 3.00 ± 0.18 | 3.05 ± 0.15 | 1.28 ± 0.15 * |
| Claudin1 | 1.47 ± 0.12 | 1.50 ± 0.16 | 1.40 ± 0.10 | 0.72 ± 0.09 * |
| Claudin3 | 2.58 ± 0.21 | 2.61 ± 0.25 | 2.65 ± 0.20 | 1.05 ± 0.15 * |
| Occludin | 2.33 ± 0.20 | 2.53 ± 0.20 | 2.40 ± 0.25 | 1.06 ± 0.20 * |
| Skeletal Muscle | ||||
| MMP2 | 1.35 ± 0.16 | 1.31 ± 0.12 | 1.32 ± 0.12 | 3.43 ± 0.17 * |
| MMP9 | 0.81 ± 0.12 | 0.75 ± 0.10 | 0.84 ± 0.12 | 2.25 ± 0.10 * |
| ZO-1 | 3.57 ± 0.30 | 3.45 ± 0.27 | 3.38 ± 0.35 | 0.72 ± 0.30 * |
| ZO-2 | 1.25 ± 0.13 | 1.23 ± 0.18 | 1.84 ± 0.15 | 0.57 ± 0.12 * |
| Claudin1 | 2.18 ± 0.20 | 2.22 ± 0.15 | 2.15 ± 0.24 | 0.87 ± 0.20 * |
| Claudin3 | 0.72 ± 0.23 | 0.84 ± 0.20 | 0.75 ± 0.18 | 0.27 ± 0.13 * |
| Occludin | 1.76 ± 0.18 | 1.79 ± 0.20 | 1.70 ± 0.15 | 0.60 ± 0.15 * |
| WAT | ||||
| Ceacam1 | 1.78 ± 0.15 | 1.79 ± 0.11 | 1.82 ± 0.13 | 3.00 ± 0.25 * |
| Ceacam1-4L | 1.02 ± 0.15 | 0.97 ± 0.17 | 1.03 ± 0.15 | 2.32 ± 0.10 * |
| Ceacam1-4S | 0.87 ± 0.09 | 0.77 ± 0.10 | 0.80 ± 0.10 | 0.82 ± 0.12 |
| Irf-1 | 2.02 ± 0.12 | 2.19 ± 0.19 | 2.09 ± 0.10 | 3.92 ± 0.20 * |
| Irf-3 | 1.29 ± 0.15 | 1.27 ± 0.17 | 1.24 ± 0.09 | 2.79 ± 0.07 * |
| Il-10 | 1.02 ± 0.17 | 1.06 ± 0.13 | 1.05 ± 0.14 | 2.97 ± 0.09 * |
| Foxp3 | 1.43 ± 0.15 | 1.47 ± 0.16 | 1.40 ± 0.15 | 1.44 ± 0.20 |
| Tnfα | 0.48 ± 0.07 | 0.55 ± 0.09 | 0.51 ± 0.06 | 0.49 ± 0.10 |
| Il-6 | 2.01 ± 0.18 | 1.98 ± 0.23 | 2.05 ± 0.21 | 2.10 ± 0.24 |
| Il-1β | 1.75 ± 0.34 | 1.78 ± 0.30 | 1.84 ± 0.27 | 1.70 ± 0.19 |
| Ifnγ | 1.91 ± 0.02 | 1.94 ± 0.08 | 1.98 ± 0.02 | 1.95 ± 0.08 |
| Smad7 | 1.00 ± 0.25 | 1.03 ± 0.31 | 1.10 ± 0.28 | 1.04 ± 0.29 |
| α-Sma | 0.63 ± 0.12 | 0.65 ± 0.10 | 0.59 ± 0.15 | 0.60 ± 0.13 |
| Col1α1 | 1.18 ± 0.05 | 1.17 ± 0.08 | 1.21 ± 0.11 | 1.21 ± 0.10 |
| Col6α3 | 1.71 ± 0.21 | 1.77 ± 0.23 | 1.79 ± 0.19 | 1.75 ± 0.22 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Muturi, H.T.; Khuder, S.S.; Ghadieh, H.E.; Esakov, E.L.; Noh, H.; Kang, H.; McInerney, M.F.; Kim, J.K.; Lee, A.D.; Najjar, S.M. Insulin Sensitivity Is Retained in Mice with Endothelial Loss of Carcinoembryonic Antigen Cell Adhesion Molecule 1. Cells 2021, 10, 2093. https://doi.org/10.3390/cells10082093
Muturi HT, Khuder SS, Ghadieh HE, Esakov EL, Noh H, Kang H, McInerney MF, Kim JK, Lee AD, Najjar SM. Insulin Sensitivity Is Retained in Mice with Endothelial Loss of Carcinoembryonic Antigen Cell Adhesion Molecule 1. Cells. 2021; 10(8):2093. https://doi.org/10.3390/cells10082093
Chicago/Turabian StyleMuturi, Harrison T., Saja S. Khuder, Hilda E. Ghadieh, Emily L. Esakov, Hyelim Noh, Heejoon Kang, Marcia F. McInerney, Jason K. Kim, Abraham D. Lee, and Sonia M. Najjar. 2021. "Insulin Sensitivity Is Retained in Mice with Endothelial Loss of Carcinoembryonic Antigen Cell Adhesion Molecule 1" Cells 10, no. 8: 2093. https://doi.org/10.3390/cells10082093
APA StyleMuturi, H. T., Khuder, S. S., Ghadieh, H. E., Esakov, E. L., Noh, H., Kang, H., McInerney, M. F., Kim, J. K., Lee, A. D., & Najjar, S. M. (2021). Insulin Sensitivity Is Retained in Mice with Endothelial Loss of Carcinoembryonic Antigen Cell Adhesion Molecule 1. Cells, 10(8), 2093. https://doi.org/10.3390/cells10082093