
Communications between Mitochondria and Endoplasmic Reticulum in the Regulation of Metabolic Homeostasis


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Structure of MAM
3. MAM and Lipids Biogenesis
4. MAM and Interorganellar Calcium Flux
5. MAM and ROS Signaling
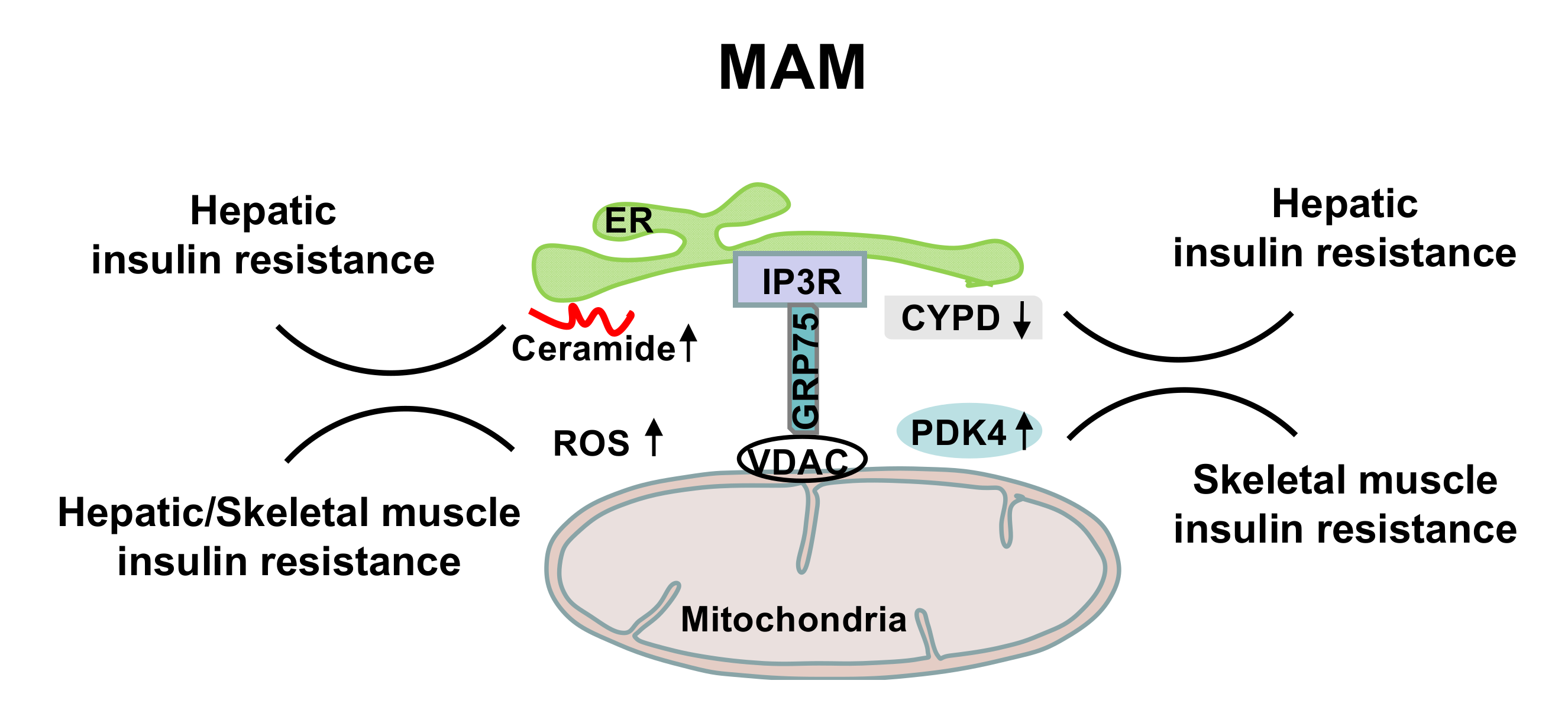
6. MAM in the Regulation of Insulin Sensitivity and Energy Homeostasis
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Spinelli, J.B.; Haigis, M.C. The multifaceted contributions of mitochondria to cellular metabolism. Nat. Cell Biol. 2018, 20, 745–754. [Google Scholar] [CrossRef] [PubMed]
- Bhatti, J.S.; Bhatti, G.K.; Reddy, P.H. Mitochondrial dysfunction and oxidative stress in metabolic disorders—A step towards mitochondria based therapeutic strategies. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 1066–1077. [Google Scholar] [CrossRef] [PubMed]
- Bock, F.J.; Tait, S.W. Mitochondria as multifaceted regulators of cell death. Nat. Rev. Mol. Cell Biol. 2020, 21, 85–100. [Google Scholar] [CrossRef]
- Javadov, S.; Kozlov, A.V.; Camara, A.K. Mitochondria in health and diseases. Cells 2020, 9, 1177. [Google Scholar] [CrossRef] [PubMed]
- Kummer, E.; Ban, N. Mechanisms and regulation of protein synthesis in mitochondria. Nat. Rev. Mol. Cell Biol. 2021, 22, 1–19. [Google Scholar] [CrossRef]
- Haroon, S.; Vermulst, M. Linking mitochondrial dynamics to mitochondrial protein quality control. Curr. Opin. Genet. Dev. 2016, 38, 68–74. [Google Scholar] [CrossRef]
- Ni, H.-M.; Williams, J.A.; Ding, W.-X. Mitochondrial dynamics and mitochondrial quality control. Redox Biol. 2015, 4, 6–13. [Google Scholar] [CrossRef] [Green Version]
- Ristow, M.; Zarse, K. How increased oxidative stress promotes longevity and metabolic health: The concept of mitochondrial hormesis (mitohormesis). Exp. Gerontol. 2010, 45, 410–418. [Google Scholar] [CrossRef]
- Ristow, M.; Schmeisser, K. Mitohormesis: Promoting health and lifespan by increased levels of reactive oxygen species (ROS). Dose Response 2014, 12, 13–035. [Google Scholar] [CrossRef]
- Arruda, A.P.; Hotamisligil, G.S. Calcium homeostasis and organelle function in the pathogenesis of obesity and diabetes. Cell Metab. 2015, 22, 381–397. [Google Scholar] [CrossRef] [Green Version]
- Fu, S.; Watkins, S.M.; Hotamisligil, G.S. The role of endoplasmic reticulum in hepatic lipid homeostasis and stress signaling. Cell Metab. 2012, 15, 623–634. [Google Scholar] [CrossRef] [Green Version]
- Hotamisligil, G.S. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell 2010, 140, 900–917. [Google Scholar] [CrossRef] [Green Version]
- Lemmer, I.L.; Willemsen, N.; Hilal, N.; Bartelt, A. A guide to understanding endoplasmic reticulum stress in metabolic disorders. Mol. Metab. 2021, 47, 101169. [Google Scholar] [CrossRef] [PubMed]
- Walter, P.; Ron, D. The unfolded protein response: From stress pathway to homeostatic regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Rapoport, T.A. Mechanistic insights into ER-associated protein degradation. Curr. Opin. Cell Biol. 2018, 53, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Maamoun, H.; Abdelsalam, S.S.; Zeidan, A.; Korashy, H.M.; Agouni, A. Endoplasmic reticulum stress: A critical molecular driver of endothelial dysfunction and cardiovascular disturbances associated with diabetes. Int. J. Mol. Sci. 2019, 20, 1658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramos-Lopez, O.; Riezu-Boj, J.I.; Milagro, F.I.; Moreno-Aliaga, M.J.; Martinez, J.A.; MENA, p. Endoplasmic reticulum stress epigenetics is related to adiposity, dyslipidemia, and insulin resistance. Adipocyte 2018, 7, 137–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Özcan, U.; Cao, Q.; Yilmaz, E.; Lee, A.-H.; Iwakoshi, N.N.; Özdelen, E.; Tuncman, G.; Görgün, C.; Glimcher, L.H.; Hotamisligil, G.S. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science 2004, 306, 457–461. [Google Scholar] [CrossRef] [Green Version]
- Engin, F. ER stress and development of type 1 diabetes. J. Investig. Med. Off. Publ. Am. Fed. Clin. Res. 2016, 64, 2–6. [Google Scholar] [CrossRef] [PubMed]
- Csordás, G.; Weaver, D.; Hajnóczky, G. Endoplasmic reticulum–mitochondrial contactology: Structure and signaling functions. Trends Cell Biol. 2018, 28, 523–540. [Google Scholar] [CrossRef] [PubMed]
- Jackisch, L.; Murphy, A.M.; Kumar, S.; Randeva, H.; Tripathi, G.; McTernan, P.G. Tunicamycin-induced Endoplasmic Reticulum stress mediates mitochondrial dysfunction in human adipocytes. J. Clin. Endocrinol. Metab. 2020, 105, 2905–2918. [Google Scholar] [CrossRef]
- Lim, J.H.; Lee, H.J.; Jung, M.H.; Song, J. Coupling mitochondrial dysfunction to endoplasmic reticulum stress response: A molecular mechanism leading to hepatic insulin resistance. Cell. Signal. 2009, 21, 169–177. [Google Scholar] [CrossRef]
- Biczo, G.; Vegh, E.T.; Shalbueva, N.; Mareninova, O.A.; Elperin, J.; Lotshaw, E.; Gretler, S.; Lugea, A.; Malla, S.R.; Dawson, D. Mitochondrial dysfunction, through impaired autophagy, leads to endoplasmic reticulum stress, deregulated lipid metabolism, and pancreatitis in animal models. Gastroenterology 2018, 154, 689–703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rieusset, J. Contribution of mitochondria and endoplasmic reticulum dysfunction in insulin resistance: Distinct or interrelated roles? Diabetes Metab. 2015, 41, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Herrera-Cruz, M.S.; Simmen, T. Over six decades of discovery and characterization of the architecture at mitochondria-associated membranes (MAMs). Organelle Contact Sites 2017, 13–31. [Google Scholar] [CrossRef]
- Scorrano, L.; De Matteis, M.A.; Emr, S.; Giordano, F.; Hajnóczky, G.; Kornmann, B.; Lackner, L.L.; Levine, T.P.; Pellegrini, L.; Reinisch, K. Coming together to define membrane contact sites. Nat. Commun. 2019, 10, 1–11. [Google Scholar] [CrossRef]
- Area-Gomez, E. Assessing the function of mitochondria-associated ER membranes. Methods Enzymol. 2014, 547, 181–197. [Google Scholar] [CrossRef]
- Friedman, J.R.; Lackner, L.L.; West, M.; DiBenedetto, J.R.; Nunnari, J.; Voeltz, G.K. ER tubules mark sites of mitochondrial division. Science 2011, 334, 358–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murley, A.; Lackner, L.L.; Osman, C.; West, M.; Voeltz, G.K.; Walter, P.; Nunnari, J. ER-associated mitochondrial division links the distribution of mitochondria and mitochondrial DNA in yeast. Elife 2013, 2, e00422. [Google Scholar] [CrossRef] [PubMed]
- Lewis, S.C.; Uchiyama, L.F.; Nunnari, J. ER-mitochondria contacts couple mtDNA synthesis with mitochondrial division in human cells. Science 2016, 353, aaf5549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Copeland, D.; Dalton, A. An association between mitochondria and the endoplasmic reticulum in cells of the pseudobranch gland of a teleost. J. Cell Biol. 1959, 5, 393–396. [Google Scholar] [CrossRef]
- Bernhard, W.; Rouiller, C. Close topographical relationship between mitochondria and ergastoplasm of liver cells in a definite phase of cellular activity. J. Cell Biol. 1956, 2, 73–78. [Google Scholar] [CrossRef]
- Shiao, Y.-J.; Lupo, G.; Vance, J.E. Evidence that phosphatidylserine is imported into mitochondria via a mitochondria-associated membrane and that the majority of mitochondrial phosphatidylethanolamine is derived from decarboxylation of phosphatidylserine. J. Biol. Chem. 1995, 270, 11190–11198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vance, J.E. Phospholipid synthesis in a membrane fraction associated with mitochondria. J. Biol. Chem. 1990, 265, 7248–7256. [Google Scholar] [CrossRef]
- Zinser, E.; Sperka-Gottlieb, C.; Fasch, E.-V.; Kohlwein, S.D.; Paltauf, F.; Daum, G. Phospholipid synthesis and lipid composition of subcellular membranes in the unicellular eukaryote Saccharomyces cerevisiae. J. Bacteriol. 1991, 173, 2026–2034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rusinol, A.E.; Cui, Z.; Chen, M.H.; Vance, J.E. A unique mitochondria-associated membrane fraction from rat liver has a high capacity for lipid synthesis and contains pre-Golgi secretory proteins including nascent lipoproteins. J. Biol. Chem. 1994, 269, 27494–27502. [Google Scholar] [CrossRef]
- Soltys, S.M.; Kashani, J.H.; Dandoy, A.C.; Vaidya, A.F.; Reid, J.C. Comorbidity for disruptive behavior disorders in psychiatrically hospitalized children. Child Psychiatry Hum. Dev. 1992, 23, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Csordás, G.r.; Renken, C.; Várnai, P.; Walter, L.; Weaver, D.; Buttle, K.F.; Balla, T.; Mannella, C.A.; Hajnóczky, G.r. Structural and functional features and significance of the physical linkage between ER and mitochondria. J. Cell Biol. 2006, 174, 915–921. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.T.; Garcin, P.O.; Fu, M.; Masoudi, M.; St-Pierre, P.; Panté, N.; Nabi, I.R. Distinct mechanisms controlling rough and smooth endoplasmic reticulum contacts with mitochondria. J. Cell Sci. 2015, 128, 2759–2765. [Google Scholar] [CrossRef] [Green Version]
- Giacomello, M.; Pellegrini, L. The coming of age of the mitochondria–ER contact: A matter of thickness. Cell Death Differ. 2016, 23, 1417–1427. [Google Scholar] [CrossRef]
- Csordás, G.; Várnai, P.; Golenár, T.; Roy, S.; Purkins, G.; Schneider, T.G.; Balla, T.; Hajnóczky, G. Imaging interorganelle contacts and local calcium dynamics at the ER-mitochondrial interface. Mol. Cell 2010, 39, 121–132. [Google Scholar] [CrossRef]
- Lee, S.; Min, K.-T. The interface between ER and mitochondria: Molecular compositions and functions. Mol. Cells 2018, 41, 1000. [Google Scholar] [CrossRef]
- Poston, C.N.; Krishnan, S.C.; Bazemore-Walker, C.R. In-depth proteomic analysis of mammalian mitochondria-associated membranes (MAM). J. Proteom. 2013, 79, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wen, Y.; Dong, J.; Cao, C.; Yuan, S. Systematic In-Depth Proteomic Analysis of Mitochondria-Associated Endoplasmic Reticulum Membranes in Mouse and Human Testes. Proteomics 2018, 18, e1700478. [Google Scholar] [CrossRef] [PubMed]
- Baudier, J. ATAD3 proteins: Brokers of a mitochondria-endoplasmic reticulum connection in mammalian cells. Biol. Rev. Camb. Philos. Soc. 2018, 93, 827–844. [Google Scholar] [CrossRef]
- De Vos, K.J.; Morotz, G.M.; Stoica, R.; Tudor, E.L.; Lau, K.-F.; Ackerley, S.; Warley, A.; Shaw, C.E.; Miller, C.C. VAPB interacts with the mitochondrial protein PTPIP51 to regulate calcium homeostasis. Hum. Mol. Genet. 2012, 21, 1299–1311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez-Suaga, P.; Paillusson, S.; Stoica, R.; Noble, W.; Hanger, D.P.; Miller, C.C. The ER-mitochondria tethering complex VAPB-PTPIP51 regulates autophagy. Curr. Biol. 2017, 27, 371–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoica, R.; De Vos, K.J.; Paillusson, S.; Mueller, S.; Sancho, R.M.; Lau, K.-F.; Vizcay-Barrena, G.; Lin, W.-L.; Xu, Y.-F.; Lewis, J. ER–mitochondria associations are regulated by the VAPB–PTPIP51 interaction and are disrupted by ALS/FTD-associated TDP-43. Nat. Commun. 2014, 5, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Szabadkai, G.; Bianchi, K.; Várnai, P.; De Stefani, D.; Wieckowski, M.R.; Cavagna, D.; Nagy, A.I.; Balla, T.; Rizzuto, R. Chaperone-mediated coupling of endoplasmic reticulum and mitochondrial Ca2+ channels. J. Cell Biol. 2006, 175, 901–911. [Google Scholar] [CrossRef] [Green Version]
- Rosencrans, W.M.; Rajendran, M.; Bezrukov, S.M.; Rostovtseva, T.K. VDAC regulation of mitochondrial calcium flux: From channel biophysics to disease. Cell Calcium 2021, 94, 102356. [Google Scholar] [CrossRef]
- Cárdenas, C.; Miller, R.A.; Smith, I.; Bui, T.; Molgó, J.; Müller, M.; Vais, H.; Cheung, K.-H.; Yang, J.; Parker, I. Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell 2010, 142, 270–283. [Google Scholar] [CrossRef] [Green Version]
- Adachi, Y.; Kato, T.; Yamada, T.; Murata, D.; Arai, K.; Stahelin, R.V.; Chan, D.C.; Iijima, M.; Sesaki, H. Drp1 Tubulates the ER in a GTPase-Independent Manner. Mol. Cell 2020, 80, 621–632.e626. [Google Scholar] [CrossRef] [PubMed]
- De Brito, O.M.; Scorrano, L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 2008, 456, 605–610. [Google Scholar] [CrossRef] [PubMed]
- Naon, D.; Zaninello, M.; Giacomello, M.; Varanita, T.; Grespi, F.; Lakshminaranayan, S.; Serafini, A.; Semenzato, M.; Herkenne, S.; Hernández-Alvarez, M.I. Critical reappraisal confirms that Mitofusin 2 is an endoplasmic reticulum–mitochondria tether. Proc. Natl. Acad. Sci. USA 2016, 113, 11249–11254. [Google Scholar] [CrossRef] [Green Version]
- Sugiura, A.; Nagashima, S.; Tokuyama, T.; Amo, T.; Matsuki, Y.; Ishido, S.; Kudo, Y.; McBride, H.M.; Fukuda, T.; Matsushita, N. MITOL regulates endoplasmic reticulum-mitochondria contacts via Mitofusin2. Mol. Cell 2013, 51, 20–34. [Google Scholar] [CrossRef] [Green Version]
- McLelland, G.-L.; Goiran, T.; Yi, W.; Dorval, G.; Chen, C.X.; Lauinger, N.D.; Krahn, A.I.; Valimehr, S.; Rakovic, A.; Rouiller, I. Mfn2 ubiquitination by PINK1/parkin gates the p97-dependent release of ER from mitochondria to drive mitophagy. Elife 2018, 7, e32866. [Google Scholar] [CrossRef]
- Basso, V.; Marchesan, E.; Peggion, C.; Chakraborty, J.; von Stockum, S.; Giacomello, M.; Ottolini, D.; Debattisti, V.; Caicci, F.; Tasca, E.; et al. Regulation of ER-mitochondria contacts by Parkin via Mfn2. Pharmacol. Res. 2018, 138, 43–56. [Google Scholar] [CrossRef]
- Filadi, R.; Greotti, E.; Turacchio, G.; Luini, A.; Pozzan, T.; Pizzo, P. Presenilin 2 Modulates Endoplasmic Reticulum-Mitochondria Coupling by Tuning the Antagonistic Effect of Mitofusin 2. Cell Rep. 2016, 15, 2226–2238. [Google Scholar] [CrossRef] [Green Version]
- Loson, O.C.; Song, Z.; Chen, H.; Chan, D.C. Fis1, Mff, MiD49, and MiD51 mediate Drp1 recruitment in mitochondrial fission. Mol. Biol. Cell 2013, 24, 659–667. [Google Scholar] [CrossRef]
- Iwasawa, R.; Mahul-Mellier, A.L.; Datler, C.; Pazarentzos, E.; Grimm, S. Fis1 and Bap31 bridge the mitochondria–ER interface to establish a platform for apoptosis induction. EMBO J. 2011, 30, 556–568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Namba, T. BAP31 regulates mitochondrial function via interaction with Tom40 within ER-mitochondria contact sites. Sci. Adv. 2019, 5, eaaw1386. [Google Scholar] [CrossRef] [Green Version]
- Vance, J.E. MAM (mitochondria-associated membranes) in mammalian cells: Lipids and beyond. Biochim. Biophys. Acta 2014, 1841, 595–609. [Google Scholar] [CrossRef]
- Prinz, W.A. Bridging the gap: Membrane contact sites in signaling, metabolism, and organelle dynamics. J. Cell Biol. 2014, 205, 759–769. [Google Scholar] [CrossRef] [Green Version]
- Cui, Z.; Vance, J.E.; Chen, M.H.; Voelker, D.R.; Vance, D.E. Cloning and expression of a novel phosphatidylethanolamine N-methyltransferase. A specific biochemical and cytological marker for a unique membrane fraction in rat liver. J. Biol. Chem. 1993, 268, 16655–16663. [Google Scholar] [CrossRef]
- Petrungaro, C.; Kornmann, B. Lipid exchange at ER-mitochondria contact sites: A puzzle falling into place with quite a few pieces missing. Curr. Opin. Cell Biol. 2019, 57, 71–76. [Google Scholar] [CrossRef]
- Kornmann, B.; Currie, E.; Collins, S.R.; Schuldiner, M.; Nunnari, J.; Weissman, J.S.; Walter, P. An ER-mitochondria tethering complex revealed by a synthetic biology screen. Science 2009, 325, 477–481. [Google Scholar] [CrossRef] [Green Version]
- Elbaz-Alon, Y.; Guo, Y.; Segev, N.; Harel, M.; Quinnell, D.E.; Geiger, T.; Avinoam, O.; Li, D.; Nunnari, J. PDZD8 interacts with Protrudin and Rab7 at ER-late endosome membrane contact sites associated with mitochondria. Nat. Commun. 2020, 11, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Kulinski, A.; Rustaeus, S.; Vance, J.E. Microsomal triacylglycerol transfer protein is required for lumenal accretion of triacylglycerol not associated with ApoB, as well as for ApoB lipidation. J. Biol. Chem. 2002, 277, 31516–31525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- AhYoung, A.P.; Jiang, J.; Zhang, J.; Khoi Dang, X.; Loo, J.A.; Zhou, Z.H.; Egea, P.F. Conserved SMP domains of the ERMES complex bind phospholipids and mediate tether assembly. Proc. Natl. Acad. Sci. USA 2015, 112, E3179–E3188. [Google Scholar] [CrossRef] [Green Version]
- Hirabayashi, Y.; Kwon, S.K.; Paek, H.; Pernice, W.M.; Paul, M.A.; Lee, J.; Erfani, P.; Raczkowski, A.; Petrey, D.S.; Pon, L.A.; et al. ER-mitochondria tethering by PDZD8 regulates Ca(2+) dynamics in mammalian neurons. Science 2017, 358, 623–630. [Google Scholar] [CrossRef] [Green Version]
- Anastasia, I.; Ilacqua, N.; Raimondi, A.; Lemieux, P.; Ghandehari-Alavijeh, R.; Faure, G.; Mekhedov, S.L.; Williams, K.J.; Caicci, F.; Valle, G.; et al. Mitochondria-rough-ER contacts in the liver regulate systemic lipid homeostasis. Cell Rep. 2021, 34, 108873. [Google Scholar] [CrossRef]
- Bartke, N.; Hannun, Y.A. Bioactive sphingolipids: Metabolism and function. J. Lipid Res. 2009, 50, S91–S96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hannun, Y.A.; Obeid, L.M. Sphingolipids and their metabolism in physiology and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 175. [Google Scholar] [CrossRef] [PubMed]
- Trayssac, M.; Hannun, Y.A.; Obeid, L.M. Role of sphingolipids in senescence: Implication in aging and age-related diseases. J. Clin. Investig. 2018, 128, 2702–2712. [Google Scholar] [CrossRef] [PubMed]
- Breslow, D.K. Sphingolipid homeostasis in the endoplasmic reticulum and beyond. Cold Spring Harb. Perspect. Biol. 2013, 5, a013326. [Google Scholar] [CrossRef] [Green Version]
- Bionda, C.; Portoukalian, J.; Schmitt, D.; Rodriguez-Lafrasse, C.; Ardail, D. Subcellular compartmentalization of ceramide metabolism: MAM (mitochondria-associated membrane) and/or mitochondria? Biochem. J. 2004, 382, 527–533. [Google Scholar] [CrossRef] [Green Version]
- Stiban, J.; Caputo, L.; Colombini, M. Ceramide synthesis in the endoplasmic reticulum can permeabilize mitochondria to proapoptotic proteins. J. Lipid Res. 2008, 49, 625–634. [Google Scholar] [CrossRef] [Green Version]
- Roszczyc-Owsiejczuk, K.; Zabielski, P. Sphingolipids as a Culprit of Mitochondrial Dysfunction in Insulin Resistance and Type 2 Diabetes. Front. Endocrinol. 2021, 12, 143. [Google Scholar] [CrossRef] [PubMed]
- Perreault, L.; Newsom, S.A.; Strauss, A.; Kerege, A.; Kahn, D.E.; Harrison, K.A.; Snell-Bergeon, J.K.; Nemkov, T.; D’Alessandro, A.; Jackman, M.R. Intracellular localization of diacylglycerols and sphingolipids influences insulin sensitivity and mitochondrial function in human skeletal muscle. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [Green Version]
- Gudz, T.I.; Tserng, K.-Y.; Hoppel, C.L. Direct inhibition of mitochondrial respiratory chain complex III by cell-permeable ceramide. J. Biol. Chem. 1997, 272, 24154–24158. [Google Scholar] [CrossRef] [Green Version]
- Jain, A.; Beutel, O.; Ebell, K.; Korneev, S.; Holthuis, J.C. Diverting CERT-mediated ceramide transport to mitochondria triggers Bax-dependent apoptosis. J. Cell Sci. 2017, 130, 360–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tait, S.W.; Green, D.R. Mitochondrial regulation of cell death. Cold Spring Harb. Perspect. Biol. 2013, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dadsena, S.; Bockelmann, S.; Mina, J.G.; Hassan, D.G.; Korneev, S.; Razzera, G.; Jahn, H.; Niekamp, P.; Müller, D.; Schneider, M. Ceramides bind VDAC2 to trigger mitochondrial apoptosis. Nat. Commun. 2019, 10, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Chavez, J.A.; Knotts, T.A.; Wang, L.-P.; Li, G.; Dobrowsky, R.T.; Florant, G.L.; Summers, S.A. A role for ceramide, but not diacylglycerol, in the antagonism of insulin signal transduction by saturated fatty acids. J. Biol. Chem. 2003, 278, 10297–10303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hajduch, E.; Turban, S.; Le Liepvre, X.; Le Lay, S.; Lipina, C.; Dimopoulos, N.; Dugail, I.; Hundal, H.S. Targeting of PKCζ and PKB to caveolin-enriched microdomains represents a crucial step underpinning the disruption in PKB-directed signalling by ceramide. Biochem. J. 2008, 410, 369–379. [Google Scholar] [CrossRef] [Green Version]
- Mahfouz, R.; Khoury, R.; Blachnio-Zabielska, A.; Turban, S.; Loiseau, N.; Lipina, C.; Stretton, C.; Bourron, O.; Ferré, P.; Foufelle, F. Characterising the inhibitory actions of ceramide upon insulin signaling in different skeletal muscle cell models: A mechanistic insight. PLoS ONE 2014, 9, e101865. [Google Scholar] [CrossRef]
- Powell, D.J.; Hajduch, E.; Kular, G.; Hundal, H.S. Ceramide disables 3-phosphoinositide binding to the pleckstrin homology domain of protein kinase B (PKB)/Akt by a PKCζ-dependent mechanism. Mol. Cell. Biol. 2003, 23, 7794–7808. [Google Scholar] [CrossRef] [Green Version]
- Schubert, K.M.; Scheid, M.P.; Duronio, V. Ceramide inhibits protein kinase B/Akt by promoting dephosphorylation of serine 473. J. Biol. Chem. 2000, 275, 13330–13335. [Google Scholar] [CrossRef] [Green Version]
- Chaurasia, B.; Ying, L.; Talbot, C.L.; Maschek, J.A.; Cox, J.; Schuchman, E.H.; Hirabayashi, Y.; Holland, W.L.; Summers, S.A. Ceramides are necessary and sufficient for diet-induced impairment of thermogenic adipocytes. Mol. Metab. 2021, 45, 101145. [Google Scholar] [CrossRef]
- Jiang, M.; Li, C.; Liu, Q.; Wang, A.; Lei, M. Inhibiting Ceramide Synthesis Attenuates Hepatic Steatosis and Fibrosis in Rats With Non-alcoholic Fatty Liver Disease. Front. Endocrinol. 2019, 10, 665. [Google Scholar] [CrossRef]
- Yang, G.; Badeanlou, L.; Bielawski, J.; Roberts, A.J.; Hannun, Y.A.; Samad, F. Central role of ceramide biosynthesis in body weight regulation, energy metabolism, and the metabolic syndrome. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E211–E224. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.-S.; Huh, S.; Lee, S.; Wu, Z.; Kim, A.-K.; Kang, H.-Y.; Lu, B. Altered ER–mitochondria contact impacts mitochondria calcium homeostasis and contributes to neurodegeneration in vivo in disease models. Proc. Natl. Acad. Sci. USA 2018, 115, E8844–E8853. [Google Scholar] [CrossRef] [Green Version]
- De Stefani, D.; Rizzuto, R.; Pozzan, T. Enjoy the Trip: Calcium in Mitochondria Back and Forth. Annu. Rev. Biochem. 2016, 85, 161–192. [Google Scholar] [CrossRef]
- Fleischer, S. Personal recollections on the discovery of the ryanodine receptors of muscle. Biochem. Biophys. Res. Commun. 2008, 369, 195–207. [Google Scholar] [CrossRef]
- Ludtke, S.J.; Tran, T.P.; Ngo, Q.T.; Moiseenkova-Bell, V.Y.; Chiu, W.; Serysheva, I.I. Flexible architecture of IP3R1 by Cryo-EM. Structure 2011, 19, 1192–1199. [Google Scholar] [CrossRef] [Green Version]
- Honrath, B.; Metz, I.; Bendridi, N.; Rieusset, J.; Culmsee, C.; Dolga, A.M. Glucose-regulated protein 75 determines ER-mitochondrial coupling and sensitivity to oxidative stress in neuronal cells. Cell Death Discov. 2017, 3, 17076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tubbs, E.; Theurey, P.; Vial, G.; Bendridi, N.; Bravard, A.; Chauvin, M.-A.; Ji-Cao, J.; Zoulim, F.; Bartosch, B.; Ovize, M. Mitochondria-associated endoplasmic reticulum membrane (MAM) integrity is required for insulin signaling and is implicated in hepatic insulin resistance. Diabetes 2014, 63, 3279–3294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carreras-Sureda, A.; Jaña, F.; Urra, H.; Durand, S.; Mortenson, D.E.; Sagredo, A.; Bustos, G.; Hazari, Y.; Ramos-Fernández, E.; Sassano, M.L. Non-canonical function of IRE1α determines mitochondria-associated endoplasmic reticulum composition to control calcium transfer and bioenergetics. Nat. Cell Biol. 2019, 21, 755–767. [Google Scholar] [CrossRef]
- Liu, Y.; Ma, X.; Fujioka, H.; Liu, J.; Chen, S.; Zhu, X. DJ-1 regulates the integrity and function of ER-mitochondria association through interaction with IP3R3-Grp75-VDAC1. Proc. Natl. Acad. Sci. USA 2019, 116, 25322–25328. [Google Scholar] [CrossRef]
- Bonneau, B.; Ando, H.; Kawaai, K.; Hirose, M.; Takahashi-Iwanaga, H.; Mikoshiba, K. IRBIT controls apoptosis by interacting with the Bcl-2 homolog, Bcl2l10, and by promoting ER-mitochondria contact. Elife 2016, 5. [Google Scholar] [CrossRef]
- Wu, S.; Lu, Q.; Wang, Q.; Ding, Y.; Ma, Z.; Mao, X.; Huang, K.; Xie, Z.; Zou, M.H. Binding of FUN14 Domain Containing 1 With Inositol 1,4,5-Trisphosphate Receptor in Mitochondria-Associated Endoplasmic Reticulum Membranes Maintains Mitochondrial Dynamics and Function in Hearts in Vivo. Circulation 2017, 136, 2248–2266. [Google Scholar] [CrossRef] [PubMed]
- Paillard, M.; Tubbs, E.; Thiebaut, P.-A.; Gomez, L.; Fauconnier, J.; Crola Da Silva, C.; Teixeira, G.; Mewton, N.; Belaidi, E.; Durand, A. Depressing mitochondria-reticulum interactions protects cardiomyocytes from lethal hypoxia-reoxygenation injury. Circulation 2013, 128, 1555–1565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rieusset, J.; Fauconnier, J.; Paillard, M.; Belaidi, E.; Tubbs, E.; Chauvin, M.-A.; Durand, A.; Bravard, A.; Teixeira, G.; Bartosch, B. Disruption of calcium transfer from ER to mitochondria links alterations of mitochondria-associated ER membrane integrity to hepatic insulin resistance. Diabetologia 2016, 59, 614–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thoudam, T.; Ha, C.-M.; Leem, J.; Chanda, D.; Park, J.-S.; Kim, H.-J.; Jeon, J.-H.; Choi, Y.-K.; Liangpunsakul, S.; Huh, Y.H. PDK4 augments ER–mitochondria contact to dampen skeletal muscle insulin signaling during obesity. Diabetes 2019, 68, 571–586. [Google Scholar] [CrossRef]
- Boncompagni, S.; Rossi, A.E.; Micaroni, M.; Beznoussenko, G.V.; Polishchuk, R.S.; Dirksen, R.T.; Protasi, F. Mitochondria are linked to calcium stores in striated muscle by developmentally regulated tethering structures. Mol. Biol. Cell 2009, 20, 1058–1067. [Google Scholar] [CrossRef]
- Chen, Y.; Csordas, G.; Jowdy, C.; Schneider, T.G.; Csordas, N.; Wang, W.; Liu, Y.; Kohlhaas, M.; Meiser, M.; Bergem, S.; et al. Mitofusin 2-containing mitochondrial-reticular microdomains direct rapid cardiomyocyte bioenergetic responses via interorganelle Ca(2+) crosstalk. Circ. Res. 2012, 111, 863–875. [Google Scholar] [CrossRef] [Green Version]
- Pacher, P.; Csordas, P.; Schneider, T.; Hajnoczky, G. Quantification of calcium signal transmission from sarco-endoplasmic reticulum to the mitochondria. J. Physiol. 2000, 529, 553–564. [Google Scholar] [CrossRef]
- Szalai, G.; Csordás, G.; Hantash, B.M.; Thomas, A.P.; Hajnóczky, G. Calcium signal transmission between ryanodine receptors and mitochondria. J. Biol. Chem. 2000, 275, 15305–15313. [Google Scholar] [CrossRef] [Green Version]
- Rapizzi, E.; Pinton, P.; Szabadkai, G.; Wieckowski, M.R.; Vandecasteele, G.; Baird, G.; Tuft, R.A.; Fogarty, K.E.; Rizzuto, R. Recombinant expression of the voltage-dependent anion channel enhances the transfer of Ca2+ microdomains to mitochondria. J. Cell Biol. 2002, 159, 613–624. [Google Scholar] [CrossRef] [Green Version]
- Dulhunty, A.F.; Wei-LaPierre, L.; Casarotto, M.G.; Beard, N.A. Core skeletal muscle ryanodine receptor calcium release complex. Clin. Exp. Pharmacol. Physiol. 2017, 44, 3–12. [Google Scholar] [CrossRef] [Green Version]
- Marty, I. Triadin regulation of the ryanodine receptor complex. J. Physiol. 2015, 593, 3261–3266. [Google Scholar] [CrossRef] [Green Version]
- Ríos, E.; Györke, S. Calsequestrin, triadin and more: The molecules that modulate calcium release in cardiac and skeletal muscle. J. Physiol. 2009, 587, 3069. [Google Scholar] [CrossRef]
- Prole, D.L.; Taylor, C.W. Inositol 1, 4, 5-trisphosphate receptors and their protein partners as signalling hubs. J. Physiol. 2016, 594, 2849–2866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fourest-Lieuvin, A.; Rendu, J.; Osseni, A.; Pernet-Gallay, K.; Rossi, D.; Oddoux, S.; Brocard, J.; Sorrentino, V.; Marty, I.; Fauré, J. Role of triadin in the organization of reticulum membrane at the muscle triad. J. Cell Sci. 2012, 125, 3443–3453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filadi, R.; Leal, N.S.; Schreiner, B.; Rossi, A.; Dentoni, G.; Pinho, C.M.; Wiehager, B.; Cieri, D.; Calì, T.; Pizzo, P. TOM70 sustains cell bioenergetics by promoting IP3R3-mediated ER to mitochondria Ca2+ transfer. Curr. Biol. 2018, 28, 369–382.e366. [Google Scholar] [CrossRef] [Green Version]
- Csordas, G.; Hajnoczky, G. Sorting of calcium signals at the junctions of endoplasmic reticulum and mitochondria. Cell Calcium 2001, 29, 249–262. [Google Scholar] [CrossRef] [PubMed]
- Roderick, H.L.; Lechleiter, J.D.; Camacho, P. Cytosolic phosphorylation of calnexin controls intracellular Ca2+ oscillations via an interaction with SERCA2b. J. Cell Biol. 2000, 149, 1235–1248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raturi, A.; Gutiérrez, T.; Ortiz-Sandoval, C.; Ruangkittisakul, A.; Herrera-Cruz, M.S.; Rockley, J.P.; Gesson, K.; Ourdev, D.; Lou, P.-H.; Lucchinetti, E. TMX1 determines cancer cell metabolism as a thiol-based modulator of ER–mitochondria Ca2+ flux. J. Cell Biol. 2016, 214, 433–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krols, M.; Bultynck, G.; Janssens, S. ER–Mitochondria contact sites: A new regulator of cellular calcium flux comes into play. J. Cell Biol. 2016, 214, 367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoboue, E.D.; Rimessi, A.; Anelli, T.; Pinton, P.; Sitia, R. Regulation of calcium fluxes by GPX8, a type-II transmembrane peroxidase enriched at the mitochondria-associated endoplasmic reticulum membrane. Antioxid. Redox Signal. 2017, 27, 583–595. [Google Scholar] [CrossRef]
- Tubbs, E.; Rieusset, J. Metabolic signaling functions of ER–mitochondria contact sites: Role in metabolic diseases. J. Mol. Endocrinol. 2017, 58, R87–R106. [Google Scholar] [CrossRef] [Green Version]
- Zhao, R.Z.; Jiang, S.; Zhang, L.; Yu, Z.B. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mammucari, C. In the right place at the right time: ROS and Ca2+ are allies in the battle for survival. Cell Calcium 2021, 95, 102354. [Google Scholar] [CrossRef]
- Booth, D.M.; Enyedi, B.; Geiszt, M.; Várnai, P.; Hajnóczky, G. Redox nanodomains are induced by and control calcium signaling at the ER-mitochondrial interface. Mol. Cell 2016, 63, 240–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nunes, P.; Demaurex, N. Redox regulation of store-operated Ca2+ entry. Antioxid. Redox Signal. 2014, 21, 915–932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreadou, I.; Schulz, R.; Papapetropoulos, A.; Turan, B.; Ytrehus, K.; Ferdinandy, P.; Daiber, A.; Di Lisa, F. The role of mitochondrial reactive oxygen species, NO and H2S in ischaemia/reperfusion injury and cardioprotection. J. Cell. Mol. Med. 2020, 24, 6510–6522. [Google Scholar] [CrossRef]
- Beretta, M.; Santos, C.X.; Molenaar, C.; Hafstad, A.D.; Miller, C.C.; Revazian, A.; Betteridge, K.; Schröder, K.; Streckfuß-Bömeke, K.; Doroshow, J.H. Nox4 regulates InsP3 receptor-dependent Ca2+ release into mitochondria to promote cell survival. EMBO J. 2020, 39, e103530. [Google Scholar] [CrossRef]
- Lu, L.-Q.; Tian, J.; Luo, X.-J.; Peng, J. Targeting the pathways of regulated necrosis: A potential strategy for alleviation of cardio-cerebrovascular injury. Cell. Mol. Life Sci. 2020, 1–16. [Google Scholar] [CrossRef]
- Wei, X.; Wei, X.; Lu, Z.; Li, L.; Hu, Y.; Sun, F.; Jiang, Y.; Ma, H.; Zheng, H.; Yang, G. Activation of TRPV1 channel antagonizes diabetic nephropathy through inhibiting endoplasmic reticulum-mitochondria contact in podocytes. Metabolism 2020, 105, 154182. [Google Scholar] [CrossRef]
- Gilady, S.Y.; Bui, M.; Lynes, E.M.; Benson, M.D.; Watts, R.; Vance, J.E.; Simmen, T. Ero1α requires oxidizing and normoxic conditions to localize to the mitochondria-associated membrane (MAM). Cell Stress Chaperones 2010, 15, 619–629. [Google Scholar] [CrossRef] [Green Version]
- Bánsághi, S.; Golenár, T.; Madesh, M.; Csordás, G.; RamachandraRao, S.; Sharma, K.; Yule, D.I.; Joseph, S.K.; Hajnóczky, G. Isoform-and species-specific control of inositol 1, 4, 5-trisphosphate (IP3) receptors by reactive oxygen species. J. Biol. Chem. 2014, 289, 8170–8181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchi, S.; Corricelli, M.; Branchini, A.; Vitto, V.A.M.; Missiroli, S.; Morciano, G.; Perrone, M.; Ferrarese, M.; Giorgi, C.; Pinotti, M. Akt-mediated phosphorylation of MICU 1 regulates mitochondrial Ca2+ levels and tumor growth. EMBO J. 2019, 38, e99435. [Google Scholar] [CrossRef]
- Anelli, T.; Bergamelli, L.; Margittai, E.; Rimessi, A.; Fagioli, C.; Malgaroli, A.; Pinton, P.; Ripamonti, M.; Rizzuto, R.; Sitia, R. Ero1α regulates Ca2+ fluxes at the endoplasmic reticulum–mitochondria interface (MAM). Antioxid. Redox Signal. 2012, 16, 1077–1087. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Mongillo, M.; Chin, K.-T.; Harding, H.; Ron, D.; Marks, A.R.; Tabas, I. Role of ERO1-α–mediated stimulation of inositol 1, 4, 5-triphosphate receptor activity in endoplasmic reticulum stress–induced apoptosis. J. Cell Biol. 2009, 186, 783–792. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Li, S.J.; Sidhu, A.; Zhu, L.; Liang, Y.; Freedman, R.B.; Wang, C.C. Reconstitution of human Ero1-Lalpha/protein-disulfide isomerase oxidative folding pathway in vitro. Position-dependent differences in role between the a and a′ domains of protein-disulfide isomerase. J. Biol. Chem. 2009, 284, 199–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.J.; Lee, S.B.; Suh, Y.; Kim, S.-J.; Lee, N.; Hong, J.-H.; Park, C.; Woo, Y.; Ishizuka, K.; Kim, J.-H. DISC1 modulates neuronal stress responses by gate-keeping ER-mitochondria Ca2+ transfer through the MAM. Cell Rep. 2017, 21, 2748–2759. [Google Scholar] [CrossRef] [Green Version]
- Hou, T.; Zhang, X.; Xu, J.; Jian, C.; Huang, Z.; Ye, T.; Hu, K.; Zheng, M.; Gao, F.; Wang, X.; et al. Synergistic triggering of superoxide flashes by mitochondrial Ca2+ uniport and basal reactive oxygen species elevation. J. Biol. Chem. 2013, 288, 4602–4612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theurey, P.; Rieusset, J. Mitochondria-associated membranes response to nutrient availability and role in metabolic diseases. Trends Endocrinol. Metab. 2017, 28, 32–45. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Torres, M.; Sha, H.; Halbrook, C.J.; Van den Bergh, F.; Reinert, R.B.; Yamada, T.; Wang, S.; Luo, Y.; Hunter, A.H. Endoplasmic reticulum–associated degradation regulates mitochondrial dynamics in brown adipocytes. Science 2020, 368, 54–60. [Google Scholar] [CrossRef]
- Chen, Y.-F.; Kao, C.-H.; Chen, Y.-T.; Wang, C.-H.; Wu, C.-Y.; Tsai, C.-Y.; Liu, F.-C.; Yang, C.-W.; Wei, Y.-H.; Hsu, M.-T. Cisd2 deficiency drives premature aging and causes mitochondria-mediated defects in mice. Genes Dev. 2009, 23, 1183–1194. [Google Scholar] [CrossRef] [Green Version]
- Shinjo, S.; Jiang, S.; Nameta, M.; Suzuki, T.; Kanai, M.; Nomura, Y.; Goda, N. Disruption of the mitochondria-associated ER membrane (MAM) plays a central role in palmitic acid–induced insulin resistance. Exp. Cell Res. 2017, 359, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, K.; Kang, Q.; Yoneshiro, T.; Camporez, J.P.; Maki, H.; Homma, M.; Shinoda, K.; Chen, Y.; Lu, X.; Maretich, P. UCP1-independent signaling involving SERCA2b-mediated calcium cycling regulates beige fat thermogenesis and systemic glucose homeostasis. Nat. Med. 2017, 23, 1454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theurey, P.; Tubbs, E.; Vial, G.; Jacquemetton, J.; Bendridi, N.; Chauvin, M.A.; Alam, M.R.; Le Romancer, M.; Vidal, H.; Rieusset, J. Mitochondria-associated endoplasmic reticulum membranes allow adaptation of mitochondrial metabolism to glucose availability in the liver. J. Mol. Cell Biol. 2016, 8, 129–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sood, A.; Jeyaraju, D.V.; Prudent, J.; Caron, A.; Lemieux, P.; McBride, H.M.; Laplante, M.; Tóth, K.; Pellegrini, L. A Mitofusin-2–dependent inactivating cleavage of Opa1 links changes in mitochondria cristae and ER contacts in the postprandial liver. Proc. Natl. Acad. Sci. USA 2014, 111, 16017–16022. [Google Scholar] [CrossRef] [Green Version]
- Hailey, D.W.; Rambold, A.S.; Satpute-Krishnan, P.; Mitra, K.; Sougrat, R.; Kim, P.K.; Lippincott-Schwartz, J. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell 2010, 141, 656–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giorgi, C.; Ito, K.; Lin, H.-K.; Santangelo, C.; Wieckowski, M.R.; Lebiedzinska, M.; Bononi, A.; Bonora, M.; Duszynski, J.; Bernardi, R. PML regulates apoptosis at endoplasmic reticulum by modulating calcium release. Science 2010, 330, 1247–1251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tubbs, E.; Chanon, S.; Robert, M.; Bendridi, N.; Bidaux, G.; Chauvin, M.-A.; Ji-Cao, J.; Durand, C.; Gauvrit-Ramette, D.; Vidal, H. Disruption of mitochondria-associated endoplasmic reticulum membrane (MAM) integrity contributes to muscle insulin resistance in mice and humans. Diabetes 2018, 67, 636–650. [Google Scholar] [CrossRef] [Green Version]
- Rieusset, J. The role of endoplasmic reticulum-mitochondria contact sites in the control of glucose homeostasis: An update. Cell Death Dis. 2018, 9, 388. [Google Scholar] [CrossRef] [Green Version]
- Samuel, V.T.; Shulman, G.I. The pathogenesis of insulin resistance: Integrating signaling pathways and substrate flux. J. Clin. Invest. 2016, 126, 12–22. [Google Scholar] [CrossRef] [Green Version]
- Dingreville, F.; Panthu, B.; Thivolet, C.; Ducreux, S.; Gouriou, Y.; Pesenti, S.; Chauvin, M.-A.; Chikh, K.; Errazuriz-Cerda, E.; Van Coppenolle, F.; et al. Differential Effect of Glucose on ER-Mitochondria Ca2+ Exchange Participates in Insulin Secretion and Glucotoxicity-Mediated Dysfunction of β-Cells. Diabetes 2019, 68, 1778–1794. [Google Scholar] [CrossRef]
- Thivolet, C.; Vial, G.; Cassel, R.; Rieusset, J.; Madec, A.-M. Reduction of endoplasmic reticulum- mitochondria interactions in beta cells from patients with type 2 diabetes. PLoS ONE 2017, 12, e0182027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.-H.; Chen, Y.-F.; Wu, C.-Y.; Wu, P.-C.; Huang, Y.-L.; Kao, C.-H.; Lin, C.-H.; Kao, L.-S.; Tsai, T.-F.; Wei, Y.-H. Cisd2 modulates the differentiation and functioning of adipocytes by regulating intracellular Ca2+ homeostasis. Hum. Mol. Genet. 2014, 23, 4770–4785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Townsend, L.K.; Weber, A.J.; Barbeau, P.-A.; Holloway, G.P.; Wright, D.C. Reactive oxygen species-dependent regulation of pyruvate dehydrogenase kinase-4 in white adipose tissue. Am. J. Physiol. Cell Physiol. 2020, 318, C137–C149. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, P.; Konja, D.; Zhang, Y.; Wang, Y. Communications between Mitochondria and Endoplasmic Reticulum in the Regulation of Metabolic Homeostasis. Cells 2021, 10, 2195. https://doi.org/10.3390/cells10092195
Zhang P, Konja D, Zhang Y, Wang Y. Communications between Mitochondria and Endoplasmic Reticulum in the Regulation of Metabolic Homeostasis. Cells. 2021; 10(9):2195. https://doi.org/10.3390/cells10092195
Chicago/Turabian StyleZhang, Pengcheng, Daniels Konja, Yiwei Zhang, and Yu Wang. 2021. "Communications between Mitochondria and Endoplasmic Reticulum in the Regulation of Metabolic Homeostasis" Cells 10, no. 9: 2195. https://doi.org/10.3390/cells10092195
APA StyleZhang, P., Konja, D., Zhang, Y., & Wang, Y. (2021). Communications between Mitochondria and Endoplasmic Reticulum in the Regulation of Metabolic Homeostasis. Cells, 10(9), 2195. https://doi.org/10.3390/cells10092195