
Biology before the SOS Response—DNA Damage Mechanisms at Chromosome Fragile Sites


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Bacterial to Human Biology, per Miroslav Radman: A Personal Introduction
- (1)
- Not only could Miro try lots of wild ideas, but others did too. Miro’s lab was like permission to play in a raucous, exciting game—with Miro!
- (2)
- If he had dozens of bad ideas and one good one per day, overall, that was a win. Eradicating bad ideas is easier, and, arguably, can be taught. Some ideas that prove useful were just gifts, and it was ok to give them to oneself and others, who would tolerate the clunkers.
- (3)
- The media kitchen would not make my media, nor let me into their domain to make them myself, because American postdocs are inconsequential in the French system of tenured technicians. So, six months produced only a modest hole in my CV.
2. DNA-Damage and -Repair Intermediates in Bacteria Illuminate Cancer
2.1. Trapping Four-Way DNA Junctions
2.2. Proteins That Promote or Reduce Stalled-Fork Structures
2.3. Bacterial Proteins Suggest Reversed-Fork RNA Signature in Cancer Transcriptomes
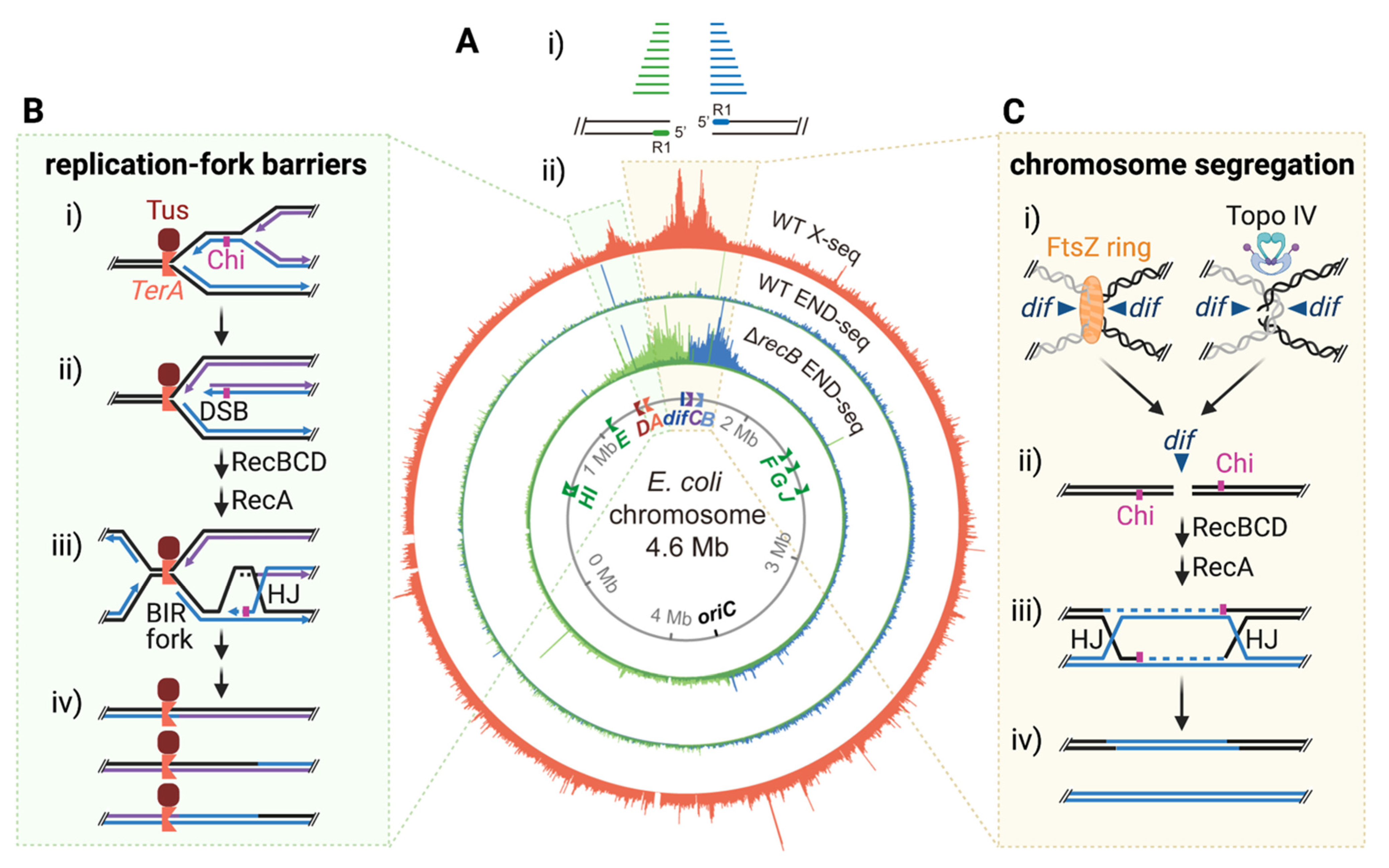
2.4. Seeing Holliday-Junction Genomic Landscapes with X-seq
3. Spontaneous “Fragile Sites” in the E. coli Genome
3.1. HJs at E. coli Genomic Fragile Sites
3.2. DSB Ends near the HJs
3.3. Fragility at Replication Barriers
3.4. Fragility at the Site of Chromosome Decatenation
3.5. Previous Observations of Terminus Pathology
3.6. Possible Genome Instability
4. Human Fragility Mechanisms and the Bacterial Models
4.1. “Stop-and-Wait” Model at Replication Barriers
4.2. Dangerous Decatenation Model
5. Concluding Thoughts
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Glickman, B.W.; Radman, M. Escherichia coli mutator mutants deficient in methylation-instructed DNA mismatch repair. Proc. Natl. Acad. Sci. USA 1980, 77, 1063–1067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glickman, B.; van den Elsen, P.; Radman, M. Induced mutagenesis in dam- mutants of Escherichia coli: A role for 6-methyladenine residues in mutation avoidance. Mol Gen Genet 1978, 163, 307–312. [Google Scholar] [CrossRef]
- Pukkila, P.J.; Peterson, J.; Herman, G.; Modrich, P.; Meselson, M. Effects of high levels of DNA adenine methylation on methyl-directed mismatch repair in Escherichia coli. Genetics 1983, 104, 571–582. [Google Scholar] [CrossRef] [PubMed]
- Lu, A.L.; Clark, S.; Modrich, P. Methyl-directed repair of DNA base-pair mismatches in vitro. Proc. Natl. Acad. Sci. USA 1983, 80, 4639–4643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rayssiguier, C.; Thaler, D.S.; Radman, M. The barrier to recombination between Escherichia coli and Salmonella typhimurium is disrupted in mismatch repair mutants. Nature 1989, 342, 396–401. [Google Scholar] [CrossRef]
- Worth, L., Jr.; Clark, S.; Radman, M.; Modrich, P. Mismatch repair proteins MutS and MutL inhibit RecA-catalyzed strand transfer between diverged DNAs. Proc. Natl. Acad. Sci. USA 1994, 91, 3238–3241. [Google Scholar] [CrossRef] [Green Version]
- Radman, M. SOS Repair Hypothesis: Phenomenology of an Inducible DNA Repair Which Is Accomplanied by Mutagenesis. In Molecular Mechanisms for Repair of DNA; Hanawalt, P., Ed.; Plenum Press: New York, NY, USA, 1975. [Google Scholar]
- Matic, I.; Rayssiguier, C.; Radman, M. Interspecies gene exchange in bacteria: The role of SOS and mismatch repair systems in evolution of species. Cell 1995, 80, 507–515. [Google Scholar] [CrossRef] [Green Version]
- Petit, M.A.; Dimpfl, J.; Radman, M.; Echols, H. Control of large chromosomal duplications in Escherichia coli by the mismatch repair system. Genetics 1991, 129, 327–332. [Google Scholar] [CrossRef]
- Dimpfl, J.; Echols, H. Duplication mutation as an SOS response in Escherichia coli: Enhanced duplication formation by a constitutively activated RecA. Genetics 1989, 123, 255–260. [Google Scholar] [CrossRef]
- Caillet-Fauquet, P.; Maenhaut-Michel, G.; Radman, M. SOS mutator effect in E. coli mutants deficient in mismatch correction. EMBO J. 1984, 3, 707–712. [Google Scholar] [CrossRef] [PubMed]
- de Wind, N.; Dekker, M.; Berns, A.; Radman, M.; te Riele, H. Inactivation of the mouse Msh2 gene results in mismatch repair deficiency, methylation tolerance, hyperrecombination, and predisposition to cancer. Cell 1995, 82, 321–330. [Google Scholar] [CrossRef] [Green Version]
- Varlet, I.; Pallard, C.; Radman, M.; Moreau, J.; de Wind, N. Cloning and expression of the Xenopus and mouse Msh2 mismatch repair genes. Nucleic Acids Res. 1994, 22, 5723–5728. [Google Scholar] [CrossRef] [Green Version]
- Kricker, M.C.; Drake, J.W.; Radman, M. Duplication-targeted DNA methylation and mutagenesis in the evolution of eukaryotic chromosomes. Proc. Natl. Acad. Sci. USA 1992, 89, 1075–1079. [Google Scholar] [CrossRef] [Green Version]
- Fishel, R.; Lescoe, M.K.; Rao, M.R.S.; Copeland, N.G.; Jenkins, N.A.; Garber, J.; Kane, M.; Kolodner, R. The human mutator gene homolog Msh2 and its association with hereditary non-polyposis colon cancer. Cell 1993, 75, 1027–1038. [Google Scholar] [CrossRef]
- Mei, Q.; Fitzgerald, D.M.; Liu, J.; Xia, J.; Pribis, J.P.; Zhai, Y.; Nehring, R.B.; Paiano, J.; Li, H.; Nussenzweig, A.; et al. Two mechanisms of chromosome fragility at replication-termination sites in bacteria. Sci. Adv. 2021, 7. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Chiu, L.Y.; Nehring, R.B.; Bravo Nunez, M.A.; Mei, Q.; Perez, M.; Zhai, Y.; Fitzgerald, D.M.; Pribis, J.P.; Wang, Y.; et al. Bacteria-to-Human Protein Networks Reveal Origins of Endogenous DNA Damage. Cell 2019, 176, 127–143 e124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, J.; Chen, L.T.; Mei, Q.; Ma, C.H.; Halliday, J.A.; Lin, H.Y.; Magnan, D.; Pribis, J.P.; Fitzgerald, D.M.; Hamilton, H.M.; et al. Holliday junction trap shows how cells use recombination and a junction-guardian role of RecQ helicase. Sci. Adv. 2016, 2, e1601605. [Google Scholar] [CrossRef] [Green Version]
- Shee, C.; Cox, B.D.; Gu, F.; Luengas, E.M.; Joshi, M.C.; Chiu, L.Y.; Magnan, D.; Halliday, J.A.; Frisch, R.L.; Gibson, J.L.; et al. Engineered proteins detect spontaneous DNA breakage in human and bacterial cells. Elife 2013, 2, e01222. [Google Scholar] [CrossRef]
- Harris, R.S.; Longerich, S.; Rosenberg, S.M. Recombination in Adaptive Mutation. Science 1994, 264, 258–260. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.M.; Longerich, S.; Gee, P.; Harris, R.S. Adaptive Mutation by Deletions in Small Mononucleotide Repeats. Science 1994, 265, 405–407. [Google Scholar] [CrossRef]
- Fitzgerald, D.M.; Hastings, P.J.; Rosenberg, S.M. Stress-Induced Mutagenesis: Implications in Cancer and Drug Resistance. Annu. Rev. Cancer Biol. 2017, 1, 119–140. [Google Scholar] [CrossRef] [Green Version]
- Pribis, J.P.; Garcia-Villada, L.; Zhai, Y.; Lewin-Epstein, O.; Wang, A.Z.; Liu, J.; Xia, J.; Mei, Q.; Fitzgerald, D.M.; Bos, J.; et al. Gamblers: An Antibiotic-Induced Evolvable Cell Subpopulation Differentiated by Reactive-Oxygen-Induced General Stress Response. Mol. Cell 2019. [Google Scholar] [CrossRef] [PubMed]
- Radman, M. Cellular parabiosis and the latency of age-related diseases. Open Biol. 2019, 9, 180250. [Google Scholar] [CrossRef] [Green Version]
- Krisko, A.; Radman, M. Protein damage, ageing and age-related diseases. Open Biol. 2019, 9, 180249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cirz, R.T.; Chin, J.K.; Andes, D.R.; de Crecy-Lagard, V.; Craig, W.A.; Romesberg, F.E. Inhibition of mutation and combating the evolution of antibiotic resistance. PLoS Biol. 2005, 3, e176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenberg, S.M.; Queitsch, C. Combating Evolution to Fight Disease. Science 2014, 343, 1088–1089. [Google Scholar] [CrossRef] [Green Version]
- Rosenberg, S.M. Evolving Responsively: Adaptive Mutation. Nat. Rev. Genet. 2001, 2, 504–515. [Google Scholar] [CrossRef]
- Skene, P.J.; Henikoff, S. An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. Elife 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Kaya-Okur, H.S.; Wu, S.J.; Codomo, C.A.; Pledger, E.S.; Bryson, T.D.; Henikoff, J.G.; Ahmad, K.; Henikoff, S. CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat. Commun. 2019, 10, 1930. [Google Scholar] [CrossRef] [Green Version]
- Michel, B.; Ehrlich, S.D.; Uzest, M. DNA double-strand breaks caused by replication arrest. EMBO J. 1997, 16, 430–438. [Google Scholar] [CrossRef] [Green Version]
- Seigneur, M.; Bidnenko, V.; Ehrlich, S.D.; Michel, B. RuvAB acts at arrested replication forks. Cell 1998, 95, 419–430. [Google Scholar] [CrossRef] [Green Version]
- Kuzminov, A. Recombinational repair of DNA damage in Escherichia coli and bacteriophage lambda. Microbiol. Mol. Biol. Rev. 1999, 63, 751–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robu, M.E.; Inman, R.B.; Cox, M.M. RecA protein promotes the regression of stalled replication forks in vitro. Proc. Natl. Acad. Sci. USA 2001, 98, 8211–8218. [Google Scholar] [CrossRef] [Green Version]
- Larsen, N.B.; Hickson, I.D. RecQ Helicases: Conserved Guardians of Genomic Integrity. Adv. Exp. Med. Biol. 2013, 767, 161–184. [Google Scholar] [CrossRef]
- Klein, H.L. The consequences of Rad51 overexpression for normal and tumor cells. DNA Repair 2008, 7, 686–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Churchill, J.J.; Anderson, D.G.; Kowalczykowski, S.C. The RecBC enzyme loads RecA protein onto ssDNA asymmetrically and independently of c, resulting in constitutive recombination activation. Genes Dev. 1999, 13, 901–911. [Google Scholar] [CrossRef] [Green Version]
- Canela, A.; Sridharan, S.; Sciascia, N.; Tubbs, A.; Meltzer, P.; Sleckman, B.P.; Nussenzweig, A. DNA Breaks and End Resection Measured Genome-wide by End Sequencing. Mol. Cell 2016, 63, 898–911. [Google Scholar] [CrossRef] [Green Version]
- Motamedi, M.R.; Szigety, S.K.; Rosenberg, S.M. Double-strand-break repair recombination in Escherichia coli: Physical evidence for a DNA replication mechanism in vivo. Genes Dev. 1999, 13, 2889–2903. [Google Scholar] [CrossRef] [Green Version]
- Louarn, J.-M.; Louarn, J.; Francois, V.; Patte, J. Analysis and possible role of hyperrecombination in the terminus region of the Escherichia coli chromosome. J. Bacteriol. 1991, 173, 5096–5104. [Google Scholar] [CrossRef] [Green Version]
- Tubbs, A.; Sridharan, S.; van Wietmarschen, N.; Maman, Y.; Callen, E.; Stanlie, A.; Wu, W.; Wu, X.; Day, A.; Wong, N.; et al. Dual Roles of Poly(dA:dT) Tracts in Replication Initiation and Fork Collapse. Cell 2018, 174, 1127–1142 e1119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Espeli, O.; Lee, C.; Marians, K.J. A physical and functional interaction between Escherichia coli FtsK and topoisomerase IV. J. Biol. Chem. 2003, 278, 44639–44644. [Google Scholar] [CrossRef] [Green Version]
- Aussel, L.; Barre, F.X.; Aroyo, M.; Stasiak, A.; Stasiak, A.Z.; Sherratt, D. FtsK Is a DNA motor protein that activates chromosome dimer resolution by switching the catalytic state of the XerC and XerD recombinases. Cell 2002, 108, 195–205. [Google Scholar] [CrossRef] [Green Version]
- El Sayyed, H.; Le Chat, L.; Lebailly, E.; Vickridge, E.; Pages, C.; Cornet, F.; Cosentino Lagomarsino, M.; Espeli, O. Mapping Topoisomerase IV Binding and Activity Sites on the E. coli Genome. PLoS Genet. 2016, 12, e1006025. [Google Scholar] [CrossRef]
- Hendricks, E.C.; Szerlong, H.; Hill, T.; Kuempel, P. Cell division, guillotining of dimer chromosomes and SOS induction in resolution mutants (dif, xerC and xerD) of Escherichia coli. Mol. Microbiol. 2000, 36, 973–981. [Google Scholar] [CrossRef]
- Pennington, J.M.; Rosenberg, S.M. Spontaneous DNA breakage in single living Escherichia coli cells. Nat. Genet. 2007, 39, 797–802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudolph, C.J.; Upton, A.L.; Stockum, A.; Nieduszynski, C.A.; Lloyd, R.G. Avoiding chromosome pathology when replication forks collide. Nature 2013, 500, 608–611. [Google Scholar] [CrossRef] [Green Version]
- Wendel, B.M.; Cole, J.M.; Courcelle, C.T.; Courcelle, J. SbcC-SbcD and ExoI process convergent forks to complete chromosome replication. Proc. Natl. Acad. Sci. USA 2018, 115, 349–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wendel, B.M.; Courcelle, C.T.; Courcelle, J. Completion of DNA replication in Escherichia coli. Proc. Natl. Acad. Sci. USA 2014, 111, 16454–16459. [Google Scholar] [CrossRef] [Green Version]
- Sinha, A.K.; Possoz, C.; Durand, A.; Desfontaines, J.M.; Barre, F.X.; Leach, D.R.F.; Michel, B. Broken replication forks trigger heritable DNA breaks in the terminus of a circular chromosome. PLoS Genet. 2018, 14, e1007256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinha, A.K.; Durand, A.; Desfontaines, J.M.; Iurchenko, I.; Auger, H.; Leach, D.R.F.; Barre, F.X.; Michel, B. Division-induced DNA double strand breaks in the chromosome terminus region of Escherichia coli lacking RecBCD DNA repair enzyme. PLoS Genet. 2017, 13, e1006895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durkin, S.G.; Glover, T.W. Chromosome fragile sites. Annu. Rev. Genet. 2007, 41, 169–192. [Google Scholar] [CrossRef] [PubMed]
- Glover, T.W.; Wilson, T.E.; Arlt, M.F. Fragile sites in cancer: More than meets the eye. Nat. Rev. Cancer 2017, 17, 489–501. [Google Scholar] [CrossRef]
- Rothstein, R.; Michel, B.; Gangloff, S. Replication fork pausing and recombination or "gimme a break". Genes Dev. 2000, 14, 1–10. [Google Scholar] [PubMed]
- Zhang, H.; Freudenreich, C.H. An AT-rich sequence in human common fragile site FRA16D causes fork stalling and chromosome breakage in S. cerevisiae. Mol. Cell 2007, 27, 367–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Wietmarschen, N.; Sridharan, S.; Nathan, W.J.; Tubbs, A.; Chan, E.M.; Callen, E.; Wu, W.; Belinky, F.; Tripathi, V.; Wong, N.; et al. Repeat expansions confer WRN dependence in microsatellite-unstable cancers. Nature 2020, 586, 292–298. [Google Scholar] [CrossRef]
- Bertolin, A.P.; Hoffmann, J.S.; Gottifredi, V. Under-Replicated DNA: The Byproduct of Large Genomes? Cancers 2020, 12, 2764. [Google Scholar] [CrossRef]
- Reusswig, K.U.; Pfander, B. Control of Eukaryotic DNA Replication Initiation-Mechanisms to Ensure Smooth Transitions. Genes 2019, 10, 99. [Google Scholar] [CrossRef] [Green Version]
- Nitiss, J.L. DNA topoisomerase II and its growing repertoire of biological functions. Nat. Rev. Cancer 2009, 9, 327–337. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Nielsen, C.F.; Yao, Q.; Hickson, I.D. The origins and processing of ultra fine anaphase DNA bridges. Curr. Opin. Genet. 2014, 26, 1–5. [Google Scholar] [CrossRef]
- Pedersen, R.T.; Kruse, T.; Nilsson, J.; Oestergaard, V.H.; Lisby, M. TopBP1 is required at mitosis to reduce transmission of DNA damage to G1 daughter cells. J. Cell Biol. 2015, 210, 565–582. [Google Scholar] [CrossRef] [Green Version]
- Zompit, M.D.M.; Mooser, C.; Adam, S.; Rossi, S.E.; Jeanrenaud, A.; Leimbacher, P.-A.; Fink, D.; Durocher, D.; Stucki, M. The CIP2A-TOPBP1 complex safeguards chromosomal stability during mitosis. bioRxiv 2021. [Google Scholar] [CrossRef]
- Morimoto, S.; Tsuda, M.; Bunch, H.; Sasanuma, H.; Austin, C.; Takeda, S. Type II DNA Topoisomerases Cause Spontaneous Double-Strand Breaks in Genomic DNA. Genes 2019, 10, 868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmutz, I.; Timashev, L.; Xie, W.; Patel, D.J.; de Lange, T. TRF2 binds branched DNA to safeguard telomere integrity. Nat. Struct. Mol. Biol. 2017, 24, 734–742. [Google Scholar] [CrossRef]
- Hastings, P.J.; Ira, G.; Lupski, J.R. A Microhomology-Mediated Break-Induced Replication Model for the Origin of Human Copy Number Variation. PLoS Genet. 2009, 5, e1000327. [Google Scholar] [CrossRef] [Green Version]
- Slack, A.; Thornton, P.C.; Magner, D.B.; Rosenberg, S.M.; Hastings, P.J. On the mechanism of gene amplification induced under stress in Escherichia coli. PLoS Genet. 2006, 2, e48. [Google Scholar] [CrossRef] [PubMed]
- Terekhanova, N.V.; Seplyarskiy, V.B.; Soldatov, R.A.; Bazykin, G.A. Evolution of Local Mutation Rate and Its Determinants. Mol. Biol. Evol. 2017, 34, 1100–1109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Perez, A.; Sabarinathan, R.; Lopez-Bigas, N. Local Determinants of the Mutational Landscape of the Human Genome. Cell 2019, 177, 101–114. [Google Scholar] [CrossRef] [Green Version]
- Swanton, C.; McGranahan, N.; Starrett, G.J.; Harris, R.S. APOBEC Enzymes: Mutagenic Fuel for Cancer Evolution and Heterogeneity. Cancer Discov. 2015, 5, 704–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Nieto, P.E.; Schwartz, E.K.; King, D.A.; Paulsen, J.; Collas, P.; Herrera, R.E.; Morrison, A.J. Carcinogen susceptibility is regulated by genome architecture and predicts cancer mutagenesis. EMBO J. 2017, 36, 2829–2843. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fitzgerald, D.M.; Rosenberg, S.M. Biology before the SOS Response—DNA Damage Mechanisms at Chromosome Fragile Sites. Cells 2021, 10, 2275. https://doi.org/10.3390/cells10092275
Fitzgerald DM, Rosenberg SM. Biology before the SOS Response—DNA Damage Mechanisms at Chromosome Fragile Sites. Cells. 2021; 10(9):2275. https://doi.org/10.3390/cells10092275
Chicago/Turabian StyleFitzgerald, Devon M., and Susan M. Rosenberg. 2021. "Biology before the SOS Response—DNA Damage Mechanisms at Chromosome Fragile Sites" Cells 10, no. 9: 2275. https://doi.org/10.3390/cells10092275
APA StyleFitzgerald, D. M., & Rosenberg, S. M. (2021). Biology before the SOS Response—DNA Damage Mechanisms at Chromosome Fragile Sites. Cells, 10(9), 2275. https://doi.org/10.3390/cells10092275