
Mechanisms of Fibroblast Activation and Myocardial Fibrosis: Lessons Learned from FB-Specific Conditional Mouse Models



Abstract
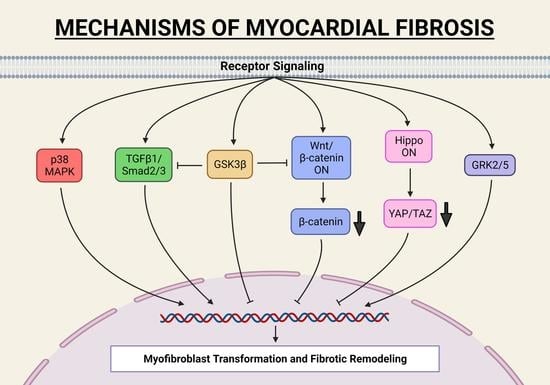
1. Introduction
2. Studies of FB-Specific In Vivo Mouse Models
2.1. TGF-β1 Signaling Pathway in Myocardial Fibrosis
2.2. Co-Operation between Canonical Wnt/β-Catenin and TGF-β1-SMAD3 Signaling in Fibrosis
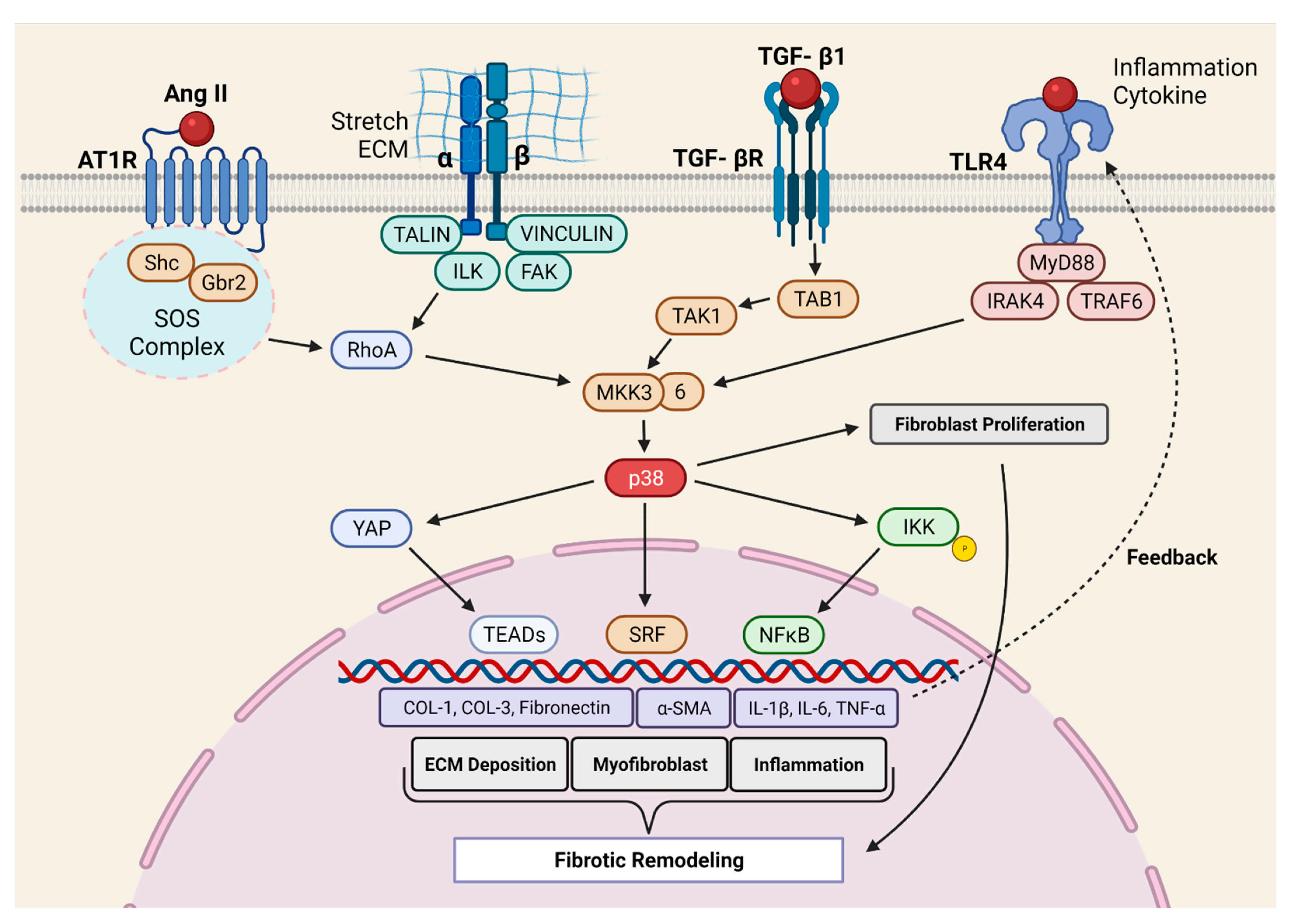
2.3. Molecular Mechanism of p38 MAPK Mediated Pro-Fibrotic Signaling
2.4. GPCR-Mediated Myocardial Pro-Fibrotic Signaling
2.5. Hippo Signaling Pathway in Myocardial Fibrosis
3. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kramann, R.; Schneider, R.K.; DiRocco, D.P.; Machado, F.; Fleig, S.; Bondzie, P.A.; Henderson, J.M.; Ebert, B.L.; Humphreys, B.D. Perivascular Gli1+ progenitors are key contributors to injury-induced organ fibrosis. Cell Stem Cell 2015, 16, 51–66. [Google Scholar] [CrossRef]
- Ieronimakis, N.; Hays, A.L.; Janebodin, K.; Mahoney, W.M., Jr.; Duffield, J.S.; Majesky, M.W.; Reyes, M. Coronary adventitial cells are linked to perivascular cardiac fibrosis via TGFbeta1 signaling in the mdx mouse model of Duchenne muscular dystrophy. J. Mol. Cell. Cardiol. 2013, 63, 122–134. [Google Scholar] [CrossRef]
- Van Wijk, B.; Gunst, Q.D.; Moorman, A.F.; van den Hoff, M.J. Cardiac regeneration from activated epicardium. PLoS ONE 2012, 7, e44692. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Honor, L.B.; He, H.; Ma, Q.; Oh, J.H.; Butterfield, C.; Lin, R.Z.; Melero-Martin, J.M.; Dolmatova, E.; Duffy, H.S.; et al. Adult mouse epicardium modulates myocardial injury by secreting paracrine factors. J. Clin. Investig. 2011, 121, 1894–1904. [Google Scholar] [CrossRef] [PubMed]
- Widyantoro, B.; Emoto, N.; Nakayama, K.; Anggrahini, D.W.; Adiarto, S.; Iwasa, N.; Yagi, K.; Miyagawa, K.; Rikitake, Y.; Suzuki, T.; et al. Endothelial cell-derived endothelin-1 promotes cardiac fibrosis in diabetic hearts through stimulation of endothelial-to-mesenchymal transition. Circulation 2010, 121, 2407–2418. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, E.M.; Tarnavski, O.; Zeisberg, M.; Dorfman, A.L.; McMullen, J.R.; Gustafsson, E.; Chandraker, A.; Yuan, X.; Pu, W.T.; Roberts, A.B.; et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat. Med. 2007, 13, 952–961. [Google Scholar] [CrossRef]
- Haudek, S.B.; Xia, Y.; Huebener, P.; Lee, J.M.; Carlson, S.; Crawford, J.R.; Pilling, D.; Gomer, R.H.; Trial, J.; Frangogiannis, N.G.; et al. Bone marrow-derived fibroblast precursors mediate ischemic cardiomyopathy in mice. Proc. Natl. Acad. Sci. USA 2006, 103, 18284–18289. [Google Scholar] [CrossRef] [PubMed]
- Mollmann, H.; Nef, H.M.; Kostin, S.; von Kalle, C.; Pilz, I.; Weber, M.; Schaper, J.; Hamm, C.W.; Elsasser, A. Bone marrow-derived cells contribute to infarct remodelling. Cardiovasc. Res. 2006, 71, 661–671. [Google Scholar] [CrossRef]
- Kanisicak, O.; Khalil, H.; Ivey, M.J.; Karch, J.; Maliken, B.D.; Correll, R.N.; Brody, M.J.; SC, J.L.; Aronow, B.J.; Tallquist, M.D.; et al. Genetic lineage tracing defines myofibroblast origin and function in the injured heart. Nat. Commun. 2016, 7, 12260. [Google Scholar] [CrossRef]
- Ruiz-Villalba, A.; Simon, A.M.; Pogontke, C.; Castillo, M.I.; Abizanda, G.; Pelacho, B.; Sanchez-Dominguez, R.; Segovia, J.C.; Prosper, F.; Perez-Pomares, J.M. Interacting resident epicardium-derived fibroblasts and recruited bone marrow cells form myocardial infarction scar. J. Am. Coll. Cardiol. 2015, 65, 2057–2066. [Google Scholar] [CrossRef]
- Ali, S.R.; Ranjbarvaziri, S.; Talkhabi, M.; Zhao, P.; Subat, A.; Hojjat, A.; Kamran, P.; Muller, A.M.; Volz, K.S.; Tang, Z.; et al. Developmental heterogeneity of cardiac fibroblasts does not predict pathological proliferation and activation. Circ. Res. 2014, 115, 625–635. [Google Scholar] [CrossRef] [PubMed]
- Moore-Morris, T.; Guimaraes-Camboa, N.; Banerjee, I.; Zambon, A.C.; Kisseleva, T.; Velayoudon, A.; Stallcup, W.B.; Gu, Y.; Dalton, N.D.; Cedenilla, M.; et al. Resident fibroblast lineages mediate pressure overload-induced cardiac fibrosis. J. Clin. Investig. 2014, 124, 2921–2934. [Google Scholar] [CrossRef]
- Tallquist, M.D. Cardiac Fibroblast Diversity. Annu. Rev. Physiol. 2020, 82, 63–78. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Khalil, H.; Kanisicak, O.; Boyer, J.G.; Vagnozzi, R.J.; Maliken, B.D.; Sargent, M.A.; Prasad, V.; Valiente-Alandi, I.; Blaxall, B.C.; et al. Specialized fibroblast differentiated states underlie scar formation in the infarcted mouse heart. J. Clin. Investig. 2018, 128, 2127–2143. [Google Scholar] [CrossRef] [PubMed]
- Tallquist, M.D.; Molkentin, J.D. Redefining the identity of cardiac fibroblasts. Nat. Rev. Cardiol. 2017, 14, 484–491. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Iyer, R.P.; Jung, M.; Czubryt, M.P.; Lindsey, M.L. Cardiac Fibroblast Activation Post-Myocardial Infarction: Current Knowledge Gaps. Trends Pharmacol. Sci. 2017, 38, 448–458. [Google Scholar] [CrossRef]
- Bhowmick, N.A.; Chytil, A.; Plieth, D.; Gorska, A.E.; Dumont, N.; Shappell, S.; Washington, M.K.; Neilson, E.G.; Moses, H.L. TGF-beta signaling in fibroblasts modulates the oncogenic potential of adjacent epithelia. Science 2004, 303, 848–851. [Google Scholar] [CrossRef] [PubMed]
- Kong, P.; Christia, P.; Saxena, A.; Su, Y.; Frangogiannis, N.G. Lack of specificity of fibroblast-specific protein 1 in cardiac remodeling and fibrosis. Am. J. Physiol. Heart Circ. Physiol. 2013, 305, H1363–H1372. [Google Scholar] [CrossRef]
- Osterreicher, C.H.; Penz-Osterreicher, M.; Grivennikov, S.I.; Guma, M.; Koltsova, E.K.; Datz, C.; Sasik, R.; Hardiman, G.; Karin, M.; Brenner, D.A. Fibroblast-specific protein 1 identifies an inflammatory subpopulation of macrophages in the liver. Proc. Natl. Acad. Sci. USA 2011, 108, 308–313. [Google Scholar] [CrossRef]
- Acharya, A.; Baek, S.T.; Huang, G.; Eskiocak, B.; Goetsch, S.; Sung, C.Y.; Banfi, S.; Sauer, M.F.; Olsen, G.S.; Duffield, J.S.; et al. The bHLH transcription factor Tcf21 is required for lineage-specific EMT of cardiac fibroblast progenitors. Development 2012, 139, 2139–2149. [Google Scholar] [CrossRef]
- Kaur, H.; Takefuji, M.; Ngai, C.Y.; Carvalho, J.; Bayer, J.; Wietelmann, A.; Poetsch, A.; Hoelper, S.; Conway, S.J.; Mollmann, H.; et al. Targeted Ablation of Periostin-Expressing Activated Fibroblasts Prevents Adverse Cardiac Remodeling in Mice. Circ. Res. 2016, 118, 1906–1917. [Google Scholar] [CrossRef]
- Ivey, M.J.; Tallquist, M.D. Defining the Cardiac Fibroblast. Circ. J. 2016, 80, 2269–2276. [Google Scholar] [CrossRef] [PubMed]
- Swonger, J.M.; Liu, J.S.; Ivey, M.J.; Tallquist, M.D. Genetic tools for identifying and manipulating fibroblasts in the mouse. Differentiation 2016, 92, 66–83. [Google Scholar] [CrossRef]
- Mohamed, T.M.A.; Abou-Leisa, R.; Stafford, N.; Maqsood, A.; Zi, M.; Prehar, S.; Baudoin-Stanley, F.; Wang, X.; Neyses, L.; Cartwright, E.J.; et al. The plasma membrane calcium ATPase 4 signalling in cardiac fibroblasts mediates cardiomyocyte hypertrophy. Nat. Commun. 2016, 7, 11074. [Google Scholar] [CrossRef] [PubMed]
- Travers, J.G.; Kamal, F.A.; Robbins, J.; Yutzey, K.E.; Blaxall, B.C. Cardiac Fibrosis: The Fibroblast Awakens. Circ. Res. 2016, 118, 1021–1040. [Google Scholar] [CrossRef]
- Lal, H.; Ahmad, F.; Zhou, J.; Yu, J.E.; Vagnozzi, R.J.; Guo, Y.; Yu, D.; Tsai, E.J.; Woodgett, J.; Gao, E.; et al. Cardiac fibroblast glycogen synthase kinase-3beta regulates ventricular remodeling and dysfunction in ischemic heart. Circulation 2014, 130, 419–430. [Google Scholar] [CrossRef] [PubMed]
- Takeda, N.; Manabe, I.; Uchino, Y.; Eguchi, K.; Matsumoto, S.; Nishimura, S.; Shindo, T.; Sano, M.; Otsu, K.; Snider, P.; et al. Cardiac fibroblasts are essential for the adaptive response of the murine heart to pressure overload. J. Clin. Investig. 2010, 120, 254–265. [Google Scholar] [CrossRef]
- Snider, P.; Standley, K.N.; Wang, J.; Azhar, M.; Doetschman, T.; Conway, S.J. Origin of cardiac fibroblasts and the role of periostin. Circ. Res. 2009, 105, 934–947. [Google Scholar] [CrossRef] [PubMed]
- Oka, T.; Xu, J.; Kaiser, R.A.; Melendez, J.; Hambleton, M.; Sargent, M.A.; Lorts, A.; Brunskill, E.W.; Dorn, G.W., 2nd; Conway, S.J.; et al. Genetic manipulation of periostin expression reveals a role in cardiac hypertrophy and ventricular remodeling. Circ. Res. 2007, 101, 313–321. [Google Scholar] [CrossRef]
- Bugg, D.; Bretherton, R.; Kim, P.; Olszewski, E.; Nagle, A.; Schumacher, A.E.; Chu, N.; Gunaje, J.; DeForest, C.A.; Stevens, K.; et al. Infarct Collagen Topography Regulates Fibroblast Fate via p38-Yes-Associated Protein Transcriptional Enhanced Associate Domain Signals. Circ. Res. 2020, 127, 1306–1322. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.; Salomonis, N.; Ghearing, N.; Lin, S.C.; Kwong, J.Q.; Mohan, A.; Swanson, M.S.; Molkentin, J.D. MBNL1-mediated regulation of differentiation RNAs promotes myofibroblast transformation and the fibrotic response. Nat. Commun. 2015, 6, 10084. [Google Scholar] [CrossRef]
- Dufeys, C.; Daskalopoulos, E.P.; Castanares-Zapatero, D.; Conway, S.J.; Ginion, A.; Bouzin, C.; Ambroise, J.; Bearzatto, B.; Gala, J.L.; Heymans, S.; et al. AMPKalpha1 deletion in myofibroblasts exacerbates post-myocardial infarction fibrosis by a connexin 43 mechanism. Basic Res. Cardiol. 2021, 116, 10. [Google Scholar] [CrossRef]
- Khalil, H.; Kanisicak, O.; Prasad, V.; Correll, R.N.; Fu, X.; Schips, T.; Vagnozzi, R.J.; Liu, R.; Huynh, T.; Lee, S.J.; et al. Fibroblast-specific TGF-beta-Smad2/3 signaling underlies cardiac fibrosis. J. Clin. Investig. 2017, 127, 3770–3783. [Google Scholar] [CrossRef]
- Khalil, H.; Kanisicak, O.; Vagnozzi, R.J.; Johansen, A.K.; Maliken, B.D.; Prasad, V.; Boyer, J.G.; Brody, M.J.; Schips, T.; Kilian, K.K.; et al. Cell-specific ablation of Hsp47 defines the collagen-producing cells in the injured heart. JCI Insight 2019, 4, e128722. [Google Scholar] [CrossRef]
- Kong, P.; Shinde, A.V.; Su, Y.; Russo, I.; Chen, B.; Saxena, A.; Conway, S.J.; Graff, J.M.; Frangogiannis, N.G. Opposing Actions of Fibroblast and Cardiomyocyte Smad3 Signaling in the Infarcted Myocardium. Circulation 2018, 137, 707–724. [Google Scholar] [CrossRef]
- Meng, Q.; Bhandary, B.; Bhuiyan, M.S.; James, J.; Osinska, H.; Valiente-Alandi, I.; Shay-Winkler, K.; Gulick, J.; Molkentin, J.D.; Blaxall, B.C.; et al. Myofibroblast-Specific TGFbeta Receptor II Signaling in the Fibrotic Response to Cardiac Myosin Binding Protein C-Induced Cardiomyopathy. Circ. Res. 2018, 123, 1285–1297. [Google Scholar] [CrossRef] [PubMed]
- Molkentin, J.D.; Bugg, D.; Ghearing, N.; Dorn, L.E.; Kim, P.; Sargent, M.A.; Gunaje, J.; Otsu, K.; Davis, J. Fibroblast-Specific Genetic Manipulation of p38 Mitogen-Activated Protein Kinase In Vivo Reveals Its Central Regulatory Role in Fibrosis. Circulation 2017, 136, 549–561. [Google Scholar] [CrossRef] [PubMed]
- Russo, I.; Cavalera, M.; Huang, S.; Su, Y.; Hanna, A.; Chen, B.; Shinde, A.V.; Conway, S.J.; Graff, J.; Frangogiannis, N.G. Protective Effects of Activated Myofibroblasts in the Pressure-Overloaded Myocardium Are Mediated Through Smad-Dependent Activation of a Matrix-Preserving Program. Circ. Res. 2019, 124, 1214–1227. [Google Scholar] [CrossRef] [PubMed]
- Scharf, G.M.; Kilian, K.; Cordero, J.; Wang, Y.; Grund, A.; Hofmann, M.; Froese, N.; Wang, X.; Kispert, A.; Kist, R.; et al. Inactivation of Sox9 in fibroblasts reduces cardiac fibrosis and inflammation. JCI Insight 2019, 5, e126721. [Google Scholar] [CrossRef]
- Shimizu, T.; Narang, N.; Chen, P.; Yu, B.; Knapp, M.; Janardanan, J.; Blair, J.; Liao, J.K. Fibroblast deletion of ROCK2 attenuates cardiac hypertrophy, fibrosis, and diastolic dysfunction. JCI Insight 2017, 2, e93187. [Google Scholar] [CrossRef]
- Snider, J.C.; Riley, L.A.; Mallory, N.T.; Bersi, M.R.; Umbarkar, P.; Gautam, R.; Zhang, Q.; Mahadevan-Jansen, A.; Hatzopoulos, A.K.; Maroteaux, L.; et al. Targeting 5-HT2B Receptor Signaling Prevents Border Zone Expansion and Improves Microstructural Remodeling After Myocardial Infarction. Circulation 2021, 143, 1317–1330. [Google Scholar] [CrossRef] [PubMed]
- Travers, J.G.; Kamal, F.A.; Valiente-Alandi, I.; Nieman, M.L.; Sargent, M.A.; Lorenz, J.N.; Molkentin, J.D.; Blaxall, B.C. Pharmacological and Activated Fibroblast Targeting of Gbetagamma-GRK2 After Myocardial Ischemia Attenuates Heart Failure Progression. J. Am. Coll. Cardiol. 2017, 70, 958–971. [Google Scholar] [CrossRef] [PubMed]
- Xiang, F.L.; Fang, M.; Yutzey, K.E. Loss of beta-catenin in resident cardiac fibroblasts attenuates fibrosis induced by pressure overload in mice. Nat. Commun. 2017, 8, 712. [Google Scholar] [CrossRef]
- Umbarkar, P.; Tousif, S.; Singh, A.P.; Anderson, J.C.; Zhang, Q.; Lal, H. Cardiac fibroblast GSK-3α mediates adverse myocardial fibrosis via IL-11 and ERK pathway. bioRxiv 2021. [Google Scholar] [CrossRef]
- Pinto, A.R.; Ilinykh, A.; Ivey, M.J.; Kuwabara, J.T.; D′Antoni, M.L.; Debuque, R.; Chandran, A.; Wang, L.; Arora, K.; Rosenthal, N.A.; et al. Revisiting Cardiac Cellular Composition. Circ. Res. 2016, 118, 400–409. [Google Scholar] [CrossRef] [PubMed]
- Francisco, J.; Zhang, Y.; Jeong, J.I.; Mizushima, W.; Ikeda, S.; Ivessa, A.; Oka, S.; Zhai, P.; Tallquist, M.D.; Del Re, D.P. Blockade of Fibroblast YAP Attenuates Cardiac Fibrosis and Dysfunction Through MRTF-A Inhibition. JACC Basic Transl. Sci. 2020, 5, 931–945. [Google Scholar] [CrossRef]
- Xiao, Y.; Hill, M.C.; Li, L.; Deshmukh, V.; Martin, T.J.; Wang, J.; Martin, J.F. Hippo pathway deletion in adult resting cardiac fibroblasts initiates a cell state transition with spontaneous and self-sustaining fibrosis. Genes. Dev. 2019, 33, 1491–1505. [Google Scholar] [CrossRef]
- Abraham, D.M.; Lee, T.E.; Watson, L.J.; Mao, L.; Chandok, G.; Wang, H.G.; Frangakis, S.; Pitt, G.S.; Shah, S.H.; Wolf, M.J.; et al. The two-pore domain potassium channel TREK-1 mediates cardiac fibrosis and diastolic dysfunction. J. Clin. Investig. 2018, 128, 4843–4855. [Google Scholar] [CrossRef]
- Valiente-Alandi, I.; Potter, S.J.; Salvador, A.M.; Schafer, A.E.; Schips, T.; Carrillo-Salinas, F.; Gibson, A.M.; Nieman, M.L.; Perkins, C.; Sargent, M.A.; et al. Inhibiting Fibronectin Attenuates Fibrosis and Improves Cardiac Function in a Model of Heart Failure. Circulation 2018, 138, 1236–1252. [Google Scholar] [CrossRef]
- Clement, S.; Stouffs, M.; Bettiol, E.; Kampf, S.; Krause, K.H.; Chaponnier, C.; Jaconi, M. Expression and function of alpha-smooth muscle actin during embryonic-stem-cell-derived cardiomyocyte differentiation. J. Cell Sci. 2007, 120, 229–238. [Google Scholar] [CrossRef]
- Bursac, N. Cardiac fibroblasts in pressure overload hypertrophy: The enemy within? J. Clin. Investig. 2014, 124, 2850–2853. [Google Scholar] [CrossRef]
- Hudon-David, F.; Bouzeghrane, F.; Couture, P.; Thibault, G. Thy-1 expression by cardiac fibroblasts: Lack of association with myofibroblast contractile markers. J. Mol. Cell. Cardiol. 2007, 42, 991–1000. [Google Scholar] [CrossRef]
- Vitetta, E.S.; Boyse, E.A.; Uhr, J.W. Isolation and characterization of a molecular complex containing Thy-1 antigen from the surface of murine thymocytes and T cells. Eur. J. Immunol. 1973, 3, 446–453. [Google Scholar] [CrossRef] [PubMed]
- Ubil, E.; Duan, J.; Pillai, I.C.; Rosa-Garrido, M.; Wu, Y.; Bargiacchi, F.; Lu, Y.; Stanbouly, S.; Huang, J.; Rojas, M.; et al. Mesenchymal-endothelial transition contributes to cardiac neovascularization. Nature 2014, 514, 585–590. [Google Scholar] [CrossRef] [PubMed]
- Duan, J.; Gherghe, C.; Liu, D.; Hamlett, E.; Srikantha, L.; Rodgers, L.; Regan, J.N.; Rojas, M.; Willis, M.; Leask, A.; et al. Wnt1/betacatenin injury response activates the epicardium and cardiac fibroblasts to promote cardiac repair. EMBO J. 2012, 31, 429–442. [Google Scholar] [CrossRef] [PubMed]
- Baum, J.; Duffy, H.S. Fibroblasts and myofibroblasts: What are we talking about? J. Cardiovasc. Pharmacol. 2011, 57, 376–379. [Google Scholar] [CrossRef] [PubMed]
- Zheng, B.; Zhang, Z.; Black, C.M.; de Crombrugghe, B.; Denton, C.P. Ligand-dependent genetic recombination in fibroblasts: A potentially powerful technique for investigating gene function in fibrosis. Am. J. Pathol. 2002, 160, 1609–1617. [Google Scholar] [CrossRef]
- Eguchi, A.; Coleman, R.; Gresham, K.; Gao, E.; Ibetti, J.; Chuprun, J.K.; Koch, W.J. GRK5 is a regulator of fibroblast activation and cardiac fibrosis. Proc. Natl. Acad. Sci. USA 2021, 118, e2012854118. [Google Scholar] [CrossRef]
- Bageghni, S.A.; Hemmings, K.E.; Yuldasheva, N.Y.; Maqbool, A.; Gamboa-Esteves, F.O.; Humphreys, N.E.; Jackson, M.S.; Denton, C.P.; Francis, S.; Porter, K.E.; et al. Fibroblast-specific deletion of interleukin-1 receptor-1 reduces adverse cardiac remodeling following myocardial infarction. JCI Insight 2019, 5, e125074. [Google Scholar] [CrossRef]
- Bageghni, S.A.; Hemmings, K.E.; Zava, N.; Denton, C.P.; Porter, K.E.; Ainscough, J.F.X.; Drinkhill, M.J.; Turner, N.A. Cardiac fibroblast-specific p38alpha MAP kinase promotes cardiac hypertrophy via a putative paracrine interleukin-6 signaling mechanism. FASEB J. 2018, 32, 4941–4954. [Google Scholar] [CrossRef]
- Li, Y.; Li, Z.; Zhang, C.; Li, P.; Wu, Y.; Wang, C.; Bond Lau, W.; Ma, X.L.; Du, J. Cardiac Fibroblast-Specific Activating Transcription Factor 3 Protects Against Heart Failure by Suppressing MAP2K3-p38 Signaling. Circulation 2017, 135, 2041–2057. [Google Scholar] [CrossRef] [PubMed]
- Woodall, M.C.; Woodall, B.P.; Gao, E.; Yuan, A.; Koch, W.J. Cardiac Fibroblast GRK2 Deletion Enhances Contractility and Remodeling Following Ischemia/Reperfusion Injury. Circ. Res. 2016, 119, 1116–1127. [Google Scholar] [CrossRef]
- DeLeon-Pennell, K.Y. May the fibrosis be with you: Is discoidin domain receptor 2 the receptor we have been looking for? J. Mol. Cell Cardiol. 2016, 91, 201–203. [Google Scholar] [CrossRef] [PubMed]
- Cowling, R.T.; Yeo, S.J.; Kim, I.J.; Park, J.I.; Gu, Y.; Dalton, N.D.; Peterson, K.L.; Greenberg, B.H. Discoidin domain receptor 2 germline gene deletion leads to altered heart structure and function in the mouse. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H773–H781. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Morales, M.O.; Price, R.L.; Goldsmith, E.C. Expression of Discoidin Domain Receptor 2 (DDR2) in the developing heart. Microsc. Microanal. 2005, 11, 260–267. [Google Scholar] [CrossRef]
- Strutz, F.; Okada, H.; Lo, C.W.; Danoff, T.; Carone, R.L.; Tomaszewski, J.E.; Neilson, E.G. Identification and characterization of a fibroblast marker: FSP1. J. Cell Biol. 1995, 130, 393–405. [Google Scholar] [CrossRef]
- Zhang, X.; Yuan, S.; Li, H.; Zhan, J.; Wang, F.; Fan, J.; Nie, X.; Wang, Y.; Wen, Z.; Chen, Y.; et al. The double face of miR-320: Cardiomyocytes-derived miR-320 deteriorated while fibroblasts-derived miR-320 protected against heart failure induced by transverse aortic constriction. Signal Transduct. Target. Ther. 2021, 6, 69. [Google Scholar] [CrossRef] [PubMed]
- Bagchi, R.A.; Lin, J.; Wang, R.; Czubryt, M.P. Regulation of fibronectin gene expression in cardiac fibroblasts by scleraxis. Cell Tissue Res. 2016, 366, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Souders, C.A.; Bowers, S.L.; Baudino, T.A. Cardiac fibroblast: The renaissance cell. Circ. Res. 2009, 105, 1164–1176. [Google Scholar] [CrossRef] [PubMed]
- Philips, N.; Bashey, R.I.; Jimenez, S.A. Collagen and fibronectin expression in cardiac fibroblasts from hypertensive rats. Cardiovasc. Res. 1994, 28, 1342–1347. [Google Scholar] [CrossRef]
- Miwa, H.; Era, T. Generation and characterization of PDGFRalpha-GFPCreERT2 knock-In mouse line. Genesis 2015, 53, 329–336. [Google Scholar] [CrossRef]
- Chong, J.J.; Reinecke, H.; Iwata, M.; Torok-Storb, B.; Stempien-Otero, A.; Murry, C.E. Progenitor cells identified by PDGFR-alpha expression in the developing and diseased human heart. Stem Cells Dev. 2013, 22, 1932–1943. [Google Scholar] [CrossRef]
- Smith, C.L.; Baek, S.T.; Sung, C.Y.; Tallquist, M.D. Epicardial-derived cell epithelial-to-mesenchymal transition and fate specification require PDGF receptor signaling. Circ. Res. 2011, 108, e15–e26. [Google Scholar] [CrossRef] [PubMed]
- Furtado, M.B.; Costa, M.W.; Pranoto, E.A.; Salimova, E.; Pinto, A.R.; Lam, N.T.; Park, A.; Snider, P.; Chandran, A.; Harvey, R.P.; et al. Cardiogenic genes expressed in cardiac fibroblasts contribute to heart development and repair. Circ. Res. 2014, 114, 1422–1434. [Google Scholar] [CrossRef] [PubMed]
- McQualter, J.L.; Brouard, N.; Williams, B.; Baird, B.N.; Sims-Lucas, S.; Yuen, K.; Nilsson, S.K.; Simmons, P.J.; Bertoncello, I. Endogenous fibroblastic progenitor cells in the adult mouse lung are highly enriched in the sca-1 positive cell fraction. Stem Cells 2009, 27, 623–633. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Hu, Q.; Nakamura, Y.; Lee, J.; Zhang, G.; From, A.H.; Zhang, J. The role of the sca-1+/CD31- cardiac progenitor cell population in postinfarction left ventricular remodeling. Stem Cells 2006, 24, 1779–1788. [Google Scholar] [CrossRef]
- Cheng, F.; Shen, Y.; Mohanasundaram, P.; Lindstrom, M.; Ivaska, J.; Ny, T.; Eriksson, J.E. Vimentin coordinates fibroblast proliferation and keratinocyte differentiation in wound healing via TGF-beta-Slug signaling. Proc. Natl. Acad. Sci. USA 2016, 113, E4320–E4327. [Google Scholar] [CrossRef]
- Matthijs Blankesteijn, W. Has the search for a marker of activated fibroblasts finally come to an end? J. Mol. Cell. Cardiol. 2015, 88, 120–123. [Google Scholar] [CrossRef]
- Camelliti, P.; Borg, T.K.; Kohl, P. Structural and functional characterisation of cardiac fibroblasts. Cardiovasc. Res. 2005, 65, 40–51. [Google Scholar] [CrossRef]
- Lane, E.B.; Hogan, B.L.; Kurkinen, M.; Garrels, J.I. Co-expression of vimentin and cytokeratins in parietal endoderm cells of early mouse embryo. Nature 1983, 303, 701–704. [Google Scholar] [CrossRef]
- Frangogiannis, N. Transforming growth factor-beta in tissue fibrosis. J. Exp. Med. 2020, 217, e20190103. [Google Scholar] [CrossRef]
- Meng, X.M.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-beta: The master regulator of fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338. [Google Scholar] [CrossRef]
- Biernacka, A.; Dobaczewski, M.; Frangogiannis, N.G. TGF-beta signaling in fibrosis. Growth Factors 2011, 29, 196–202. [Google Scholar] [CrossRef]
- Patel, N.J.; Nassal, D.M.; Greer-Short, A.D.; Unudurthi, S.D.; Scandling, B.W.; Gratz, D.; Xu, X.; Kalyanasundaram, A.; Fedorov, V.V.; Accornero, F.; et al. betaIV-Spectrin/STAT3 complex regulates fibroblast phenotype, fibrosis, and cardiac function. JCI Insight 2019, 4, e131046. [Google Scholar] [CrossRef]
- Dobaczewski, M.; Chen, W.; Frangogiannis, N.G. Transforming growth factor (TGF)-beta signaling in cardiac remodeling. J. Mol. Cell Cardiol. 2011, 51, 600–606. [Google Scholar] [CrossRef] [PubMed]
- Schmierer, B.; Hill, C.S. TGFbeta-SMAD signal transduction: Molecular specificity and functional flexibility. Nat. Rev. Mol. Cell Biol. 2007, 8, 970–982. [Google Scholar] [CrossRef] [PubMed]
- Derynck, R.; Zhang, Y.E. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature 2003, 425, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, M.; Fatyol, K.; Jin, C.; Wang, X.; Liu, Z.; Zhang, Y.E. TRAF6 mediates Smad-independent activation of JNK and p38 by TGF-beta. Mol. Cell 2008, 31, 918–924. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.K.; Pardoux, C.; Hall, M.C.; Lee, P.S.; Warburton, D.; Qing, J.; Smith, S.M.; Derynck, R. TGF-beta activates Erk MAP kinase signalling through direct phosphorylation of ShcA. EMBO J. 2007, 26, 3957–3967. [Google Scholar] [CrossRef] [PubMed]
- Bhowmick, N.A.; Ghiassi, M.; Bakin, A.; Aakre, M.; Lundquist, C.A.; Engel, M.E.; Arteaga, C.L.; Moses, H.L. Transforming growth factor-beta1 mediates epithelial to mesenchymal transdifferentiation through a RhoA-dependent mechanism. Mol. Biol. Cell 2001, 12, 27–36. [Google Scholar] [CrossRef]
- Bakin, A.V.; Tomlinson, A.K.; Bhowmick, N.A.; Moses, H.L.; Arteaga, C.L. Phosphatidylinositol 3-kinase function is required for transforming growth factor beta-mediated epithelial to mesenchymal transition and cell migration. J. Biol. Chem. 2000, 275, 36803–36810. [Google Scholar] [CrossRef]
- Yamaguchi, K.; Nagai, S.; Ninomiya-Tsuji, J.; Nishita, M.; Tamai, K.; Irie, K.; Ueno, N.; Nishida, E.; Shibuya, H.; Matsumoto, K. XIAP, a cellular member of the inhibitor of apoptosis protein family, links the receptors to TAB1-TAK1 in the BMP signaling pathway. EMBO J. 1999, 18, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Woodgett, J.R. Molecular cloning and expression of glycogen synthase kinase-3/factor A. EMBO J. 1990, 9, 2431–2438. [Google Scholar] [CrossRef] [PubMed]
- Doble, B.W.; Woodgett, J.R. GSK-3: Tricks of the trade for a multi-tasking kinase. J. Cell Sci. 2003, 116, 1175–1186. [Google Scholar] [CrossRef] [PubMed]
- Frame, S.; Cohen, P. GSK3 takes centre stage more than 20 years after its discovery. Biochem. J. 2001, 359, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Lal, H.; Ahmad, F.; Woodgett, J.; Force, T. The GSK-3 family as therapeutic target for myocardial diseases. Circ. Res. 2015, 116, 138–149. [Google Scholar] [CrossRef] [PubMed]
- Woulfe, K.C.; Gao, E.; Lal, H.; Harris, D.; Fan, Q.; Vagnozzi, R.; DeCaul, M.; Shang, X.; Patel, S.; Woodgett, J.R.; et al. Glycogen synthase kinase-3beta regulates post-myocardial infarction remodeling and stress-induced cardiomyocyte proliferation in vivo. Circ. Res. 2010, 106, 1635–1645. [Google Scholar] [CrossRef]
- Matsuda, T.; Zhai, P.; Maejima, Y.; Hong, C.; Gao, S.; Tian, B.; Goto, K.; Takagi, H.; Tamamori-Adachi, M.; Kitajima, S.; et al. Distinct roles of GSK-3alpha and GSK-3beta phosphorylation in the heart under pressure overload. Proc. Natl. Acad. Sci. USA 2008, 105, 20900–20905. [Google Scholar] [CrossRef]
- Kerkela, R.; Kockeritz, L.; Macaulay, K.; Zhou, J.; Doble, B.W.; Beahm, C.; Greytak, S.; Woulfe, K.; Trivedi, C.M.; Woodgett, J.R.; et al. Deletion of GSK-3beta in mice leads to hypertrophic cardiomyopathy secondary to cardiomyoblast hyperproliferation. J. Clin. Investig. 2008, 118, 3609–3618. [Google Scholar] [CrossRef]
- Michael, A.; Haq, S.; Chen, X.; Hsich, E.; Cui, L.; Walters, B.; Shao, Z.; Bhattacharya, K.; Kilter, H.; Huggins, G.; et al. Glycogen synthase kinase-3beta regulates growth, calcium homeostasis, and diastolic function in the heart. J. Biol. Chem. 2004, 279, 21383–21393. [Google Scholar] [CrossRef]
- Hardt, S.E.; Sadoshima, J. Glycogen synthase kinase-3beta: A novel regulator of cardiac hypertrophy and development. Circ. Res. 2002, 90, 1055–1063. [Google Scholar] [CrossRef] [PubMed]
- Haq, S.; Choukroun, G.; Kang, Z.B.; Ranu, H.; Matsui, T.; Rosenzweig, A.; Molkentin, J.D.; Alessandrini, A.; Woodgett, J.; Hajjar, R.; et al. Glycogen synthase kinase-3beta is a negative regulator of cardiomyocyte hypertrophy. J. Cell Biol. 2000, 151, 117–130. [Google Scholar] [CrossRef]
- Guo, Y.; Gupte, M.; Umbarkar, P.; Singh, A.P.; Sui, J.Y.; Force, T.; Lal, H. Entanglement of GSK-3beta, beta-catenin and TGF-beta1 signaling network to regulate myocardial fibrosis. J. Mol. Cell. Cardiol. 2017, 110, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Kim, M.; Jho, E.H. Wnt/beta-catenin signalling: From plasma membrane to nucleus. Biochem. J. 2013, 450, 9–21. [Google Scholar] [CrossRef]
- Hernandez, G.; Lal, H.; Fidalgo, M.; Guerrero, A.; Zalvide, J.; Force, T.; Pombo, C.M. A novel cardioprotective p38-MAPK/mTOR pathway. Exp. Cell Res. 2011, 317, 2938–2949. [Google Scholar] [CrossRef]
- Cuadrado, A.; Nebreda, A.R. Mechanisms and functions of p38 MAPK signalling. Biochem. J. 2010, 429, 403–417. [Google Scholar] [CrossRef]
- Lal, H.; Verma, S.K.; Golden, H.B.; Foster, D.M.; Smith, M.; Dostal, D.E. Stretch-induced regulation of angiotensinogen gene expression in cardiac myocytes and fibroblasts: Opposing roles of JNK1/2 and p38alpha MAP kinases. J. Mol. Cell. Cardiol. 2008, 45, 770–778. [Google Scholar] [CrossRef][Green Version]
- Turner, N.A.; Blythe, N.M. Cardiac Fibroblast p38 MAPK: A Critical Regulator of Myocardial Remodeling. J. Cardiovasc. Dev. Dis. 2019, 6, 27. [Google Scholar] [CrossRef]
- Gurevich, V.V.; Gurevich, E.V. GPCR Signaling Regulation: The Role of GRKs and Arrestins. Front. Pharmacol. 2019, 10, 125. [Google Scholar] [CrossRef]
- Premont, R.T.; Gainetdinov, R.R. Physiological roles of G protein-coupled receptor kinases and arrestins. Annu. Rev. Physiol. 2007, 69, 511–534. [Google Scholar] [CrossRef] [PubMed]
- Pitcher, J.A.; Freedman, N.J.; Lefkowitz, R.J. G protein-coupled receptor kinases. Annu. Rev. Biochem. 1998, 67, 653–692. [Google Scholar] [CrossRef]
- Pfleger, J.; Gresham, K.; Koch, W.J. G protein-coupled receptor kinases as therapeutic targets in the heart. Nat. Rev. Cardiol. 2019, 16, 612–622. [Google Scholar] [CrossRef]
- Belmonte, S.L.; Blaxall, B.C. G protein coupled receptor kinases as therapeutic targets in cardiovascular disease. Circ. Res. 2011, 109, 309–319. [Google Scholar] [CrossRef] [PubMed]
- Tanner, M.A.; Thomas, T.P.; Maitz, C.A.; Grisanti, L.A. beta2-Adrenergic Receptors Increase Cardiac Fibroblast Proliferation Through the Galphas/ERK1/2-Dependent Secretion of Interleukin-6. Int. J. Mol. Sci. 2020, 21, 8507. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Pan, D. The Hippo Signaling Pathway in Development and Disease. Dev. Cell 2019, 50, 264–282. [Google Scholar] [CrossRef]
- Meng, Z.; Moroishi, T.; Guan, K.L. Mechanisms of Hippo pathway regulation. Genes. Dev. 2016, 30, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Tumaneng, K.; Guan, K.L. The Hippo pathway in organ size control, tissue regeneration and stem cell self-renewal. Nat. Cell Biol. 2011, 13, 877–883. [Google Scholar] [CrossRef]
- Sun, Y.; Yong, K.M.; Villa-Diaz, L.G.; Zhang, X.; Chen, W.; Philson, R.; Weng, S.; Xu, H.; Krebsbach, P.H.; Fu, J. Hippo/YAP-mediated rigidity-dependent motor neuron differentiation of human pluripotent stem cells. Nat. Mater. 2014, 13, 599–604. [Google Scholar] [CrossRef]
- Aragona, M.; Panciera, T.; Manfrin, A.; Giulitti, S.; Michielin, F.; Elvassore, N.; Dupont, S.; Piccolo, S. A mechanical checkpoint controls multicellular growth through YAP/TAZ regulation by actin-processing factors. Cell 2013, 154, 1047–1059. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, S.; Heallen, T.; Martin, J.F. The Hippo pathway in the heart: Pivotal roles in development, disease, and regeneration. Nat. Rev. Cardiol. 2018, 15, 672–684. [Google Scholar] [CrossRef]
- Xiao, Y.; Hill, M.C.; Zhang, M.; Martin, T.J.; Morikawa, Y.; Wang, S.; Moise, A.R.; Wythe, J.D.; Martin, J.F. Hippo Signaling Plays an Essential Role in Cell State Transitions during Cardiac Fibroblast Development. Dev. Cell 2018, 45, 153–169. [Google Scholar] [CrossRef] [PubMed]
- Umbarkar, P.; Singh, A.P.; Tousif, S.; Zhang, Q.; Sethu, P.; Lal, H. Repurposing Nintedanib for pathological cardiac remodeling and dysfunction. Pharmacol. Res. 2021, 169, 105605. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Proteins/Markers | Biological Role | Expression in FB States | Expression in Other Cells | References |
|---|---|---|---|---|
| Periostin | ECM protein | Developmental stage, activated FBs | Epicardium | [9,18,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44] |
| TCF21 | Transcription factor | Resting FB, downregulate in activated FBs | Epicardium | [9,20,22,23,31,37,41,43,44,45,46,47,48,49] |
| α-SMA | Cytoskeletal protein | Activated FBs | Pericytes, VSMC, Epicardium | [6,9,12,25,50] |
| Collagen I, III | ECM protein | Resting and activated FBs | Pericytes, VSMC, Endothelial cells, Cardiomyocytes | [12,20,22,25,26,27,45,51,52,53,54,55,56,57,58,59,60,61,62] |
| CD90 | Cell-cell interaction | Resting and activated FBs | Pericytes, VSMC, immune cells, Endothelial cells | [12,27,45,51,52,53] |
| DDR2 | Cell-ECM interaction | Resting FBs | Epicardium | [22,25,63,64,65] |
| FSP1 | Calcium binding protein | Resting and activated FBs | Pericytes, VSMC, Immune cells, Endothelial cells | [9,18,19,22,23,25,66,67] |
| Fibronectin | ECM protein | Resting and activated FBs | Endothelial cells | [22,68,69,70] |
| PDGFRα | Tyrosine kinase receptor | Resting and activated FBs | Cardiac progenitor cells | [9,12,22,25,71,72,73] |
| Stem cells antigen-1 | Stem cell antigen | Resting and activated FBs | Cardiac progenitor cells | [22,74,75,76] |
| Vimentin | Cytoskeletal protein | Resting and activated FBs | Pericytes, VSMC, Endothelial cells | [22,25,77,78,79,80] |
| Target Gene | Promoter Used for Cre Expression | Major Findings | References |
|---|---|---|---|
| Tgfbr1/2, Smad2, Smad3 | Postn | FB-specific deletion of Tgfbr1/2 or Smad3, but not Smad2, markedly reduced fibrosis in pressure-overloaded mouse hearts as well as fibrosis mediated by heart-specific, latency-resistant TGF-β mutant transgene. | [33] |
| Smad3 | Postn | In pressure-overloaded hearts, the protective actions of the myofibroblasts were mediated through Smad3-dependent matrix-preserving program | [38] |
| Smad3 | Postn | FB-specific Smad3 loss impaired scar remodeling and increased the incidence of late rupture post-MI | [35] |
| Tgfbr2 | Postn | Tgfbr2 ablation in the myofibroblast prevented fibrosis and cardiac dysfunction in mouse model of cMyBP-C-induced cardiomyopathy | [36] |
| Gsk3b | Postn | FB-specific deletion of GSK-3β lead to the hyperactivation of SMAD-3, resulting in excessive fibrotic remodeling and cardiac dysfunction after myocardial infarction. | [26] |
| Gsk3a | Tcf21 and Postn | In pressure-overloaded hearts, FB-specific GSK-3α mediated pro-fibrotic effects through an ERK-IL-11 circuit that operated independently of TGF-β/SMAD3 signaling | [44] |
| Ctnb1 | Tcf21 and Postn | Loss of β-catenin in fibroblasts attenuated pressure-overload-induced cardiac fibrosis | [43] |
| p38 | Tcf21 and Postn | FB-specific deletion of p38 attenuated myofibroblasts transformation and fibrosis. Conversely, transgenic mice expressing constitutively active p38 in FB specific manner develops fibrosis in multiple organs. | [37] |
| p38 | Postn | Spatial variations in collagen organization regulated cardiac fibroblast phenotype through the mechanical activation of p38-YAP-TEAD signaling | [30] |
| Grk2 | Postn | Ablation of GRK2 in activated fibroblasts significantly reduced myofibroblast transformation and fibrosis and showed cardiovascular protection post-I/R injury | [42] |
| Lats1/2 | Tcf21 | FB-specific deletion of Lats1 and Lats2 initiated a self-perpetuating fibrotic response in the uninjured adult heart that was exacerbated by MI | [47] |
| Yap | Tcf21 | FB-specific deletion of YAP prevented MI-induced cardiac fibrosis and dysfunction through MRTF-A inhibition. | [46] |
| Htr2b | Tcf21 and Postn | Deletion of 5-HT2B receptor signaling in fibroblast prevented border zone expansion and improved microstructural remodeling after MI | [41] |
| Hsp47 | Postn | Myofibroblast-specific ablation of Hsp47 blocked fibrosis in mouse models of pressure overload, MI and, muscular dystrophy | [34] |
| Sox9 | Postn | FB-specific deletion of Sox9 ameliorated MI-induced left ventricular dysfunction, inflammation, and myocardial scarring | [39] |
| Kcnk2 | Tcf21 | FB-specific deletion of TREK1 prevented pressure-overload-induced deterioration in cardiac function | [48] |
| Rock2 | Postn | Deletion of ROCK2 in fibroblast attenuated cardiac hypertrophy, fibrosis, and diastolic dysfunction in mice subjected to chronic Ang-II infusion | [40] |
| Fn1 | Tcf21 | FB-specific fibronectin gene ablation ameliorated adverse cardiac remodeling and fibrosis post I/R | [49] |
| Prkaa1 | Postn | AMPKα1 deletion in myofibroblasts exacerbated post-MI adverse fibrotic remodeling | [32] |
| Sptbn4 | Postn | FB-specific deletion of βIV-spectrin aggravated Ang-II induced fibrosis and cardiac dysfunction. | [84] |
| Pmca4 | Postn | FB-deletion of PMCA4 reduced TAC-induced hypertrophy and cardiac dysfunction | [24] |
| Mbnl1 | Tcf21 and Postn | Deletion of MBNL1 impaired the fibrotic phase of wound healing in mouse models of MI. | [31] |
| Klf5 | Postn | FB–specific KLF5 deletion ameliorated TAC-induced cardiac hypertrophy and fibrosis | [27] |
| Postn | Tcf21 and Postn | Ablation periostin expressing FBs reduced collagen production and scar formation after MI. | [9] |
| Postn | Postn | Ablation of periostin expressing FBs reduced fibrosis and improved cardiac function in mice subjected to chronic Ang-II infusion as well as in mice after MI | [21] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Umbarkar, P.; Ejantkar, S.; Tousif, S.; Lal, H. Mechanisms of Fibroblast Activation and Myocardial Fibrosis: Lessons Learned from FB-Specific Conditional Mouse Models. Cells 2021, 10, 2412. https://doi.org/10.3390/cells10092412
Umbarkar P, Ejantkar S, Tousif S, Lal H. Mechanisms of Fibroblast Activation and Myocardial Fibrosis: Lessons Learned from FB-Specific Conditional Mouse Models. Cells. 2021; 10(9):2412. https://doi.org/10.3390/cells10092412
Chicago/Turabian StyleUmbarkar, Prachi, Suma Ejantkar, Sultan Tousif, and Hind Lal. 2021. "Mechanisms of Fibroblast Activation and Myocardial Fibrosis: Lessons Learned from FB-Specific Conditional Mouse Models" Cells 10, no. 9: 2412. https://doi.org/10.3390/cells10092412
APA StyleUmbarkar, P., Ejantkar, S., Tousif, S., & Lal, H. (2021). Mechanisms of Fibroblast Activation and Myocardial Fibrosis: Lessons Learned from FB-Specific Conditional Mouse Models. Cells, 10(9), 2412. https://doi.org/10.3390/cells10092412