
Remodeling of Ion Channel Trafficking and Cardiac Arrhythmias


{kind=link}
{kind=link}
Abstract
:1. Introduction
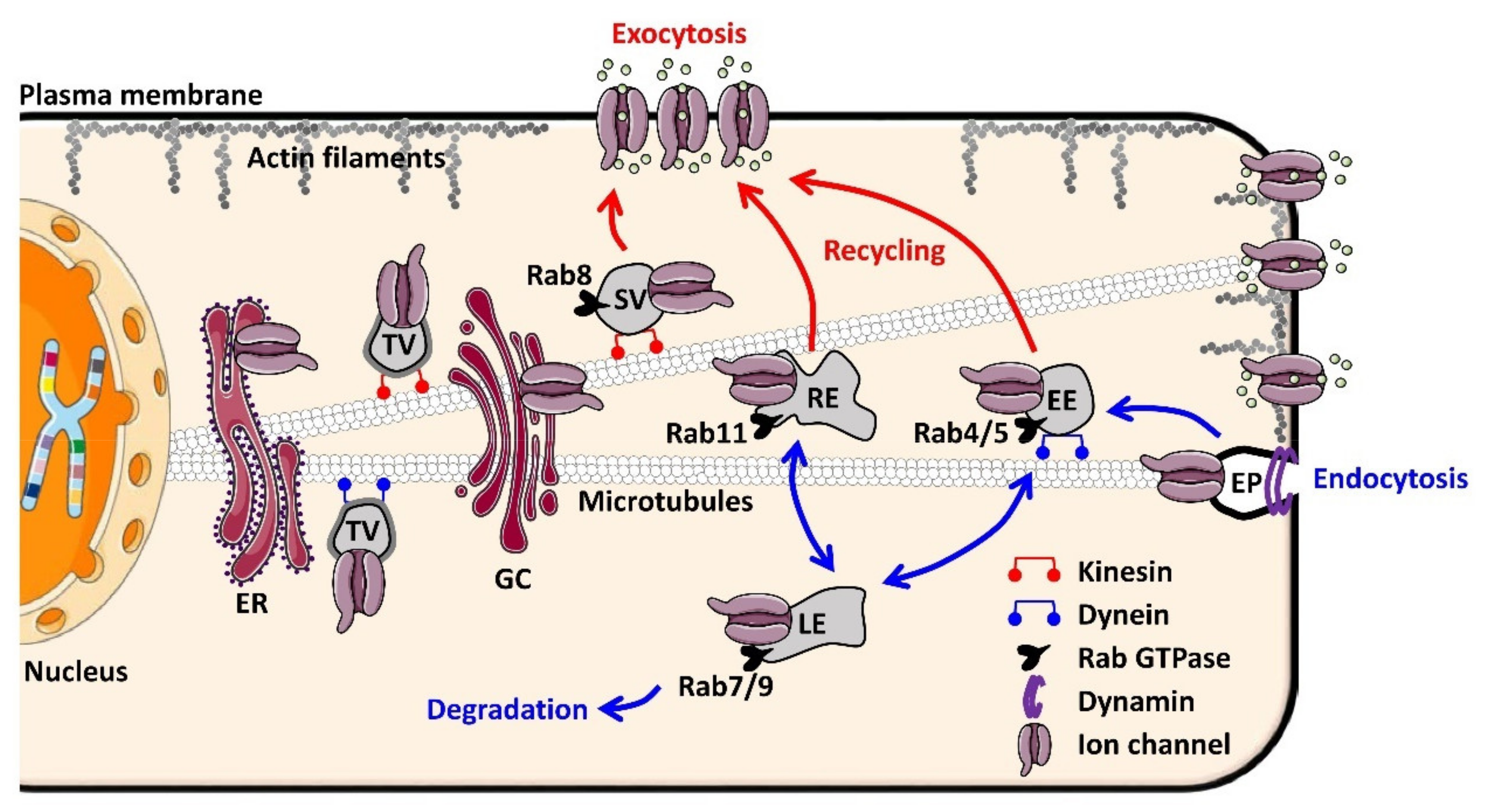
2. The Trafficking Machinery of Cardiomyocytes
2.1. Molecular Basis of Ion Channel Trafficking
2.2. Why Ion Channel Trafficking Is Crucial for Normal Cardiac Electrical Properties
3. Tribute to Inherited Cardiac Arrhythmias for Knowledge on Channel Trafficking
3.1. Long QT Syndrome and Ion Channel Partners
3.1.1. Long QT 4 (Ankyrin Mutations)
3.1.2. Long QT 9 (Caveolin-3 Mutations)
3.2. Brugada Syndrome and Ion Channel Partners
3.2.1. SCN1-3B Mutations
3.2.2. Ankyrin-G
3.2.3. Other Trafficking Partner Mutations in Brugada Syndrome
4. Can Acquired Cardiac Arrhythmias Be Caused by Default of Channel Trafficking?
4.1. When Tissue Remodeling Disorganizes Cardiac Channel Trafficking
4.2. Keep Ion Channel Trafficking Balanced
4.2.1. hERG, Channel at Demand
4.2.2. The Restless KV1.5 Channel
- a.
- KV1.5 channel trafficking needs fat
- b.
- Do not stress the KV1.5 channel
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Hatem, S.N.; Coulombe, A.; Balse, E. Specificities of Atrial Electrophysiology: Clues to a Better Understanding of Cardiac Function and the Mechanisms of Arrhythmias. J. Mol. Cell. Cardiol. 2010, 48, 90–95. [Google Scholar] [CrossRef]
- Douglas, P.M.; Cyr, D.M. Interplay Between Protein Homeostasis Networks in Protein Aggregation and Proteotoxicity. Biopolymers 2010, 93, 229–236. [Google Scholar] [CrossRef] [Green Version]
- Shikano, S.; Coblitz, B.; Wu, M.; Li, M. 14-3-3 Proteins: Regulation of Endoplasmic Reticulum Localization and Surface Expression of Membrane Proteins. Trends Cell Biol. 2006, 16, 370–375. [Google Scholar] [CrossRef]
- Yuan, H.; Michelsen, K.; Schwappach, B. 14-3-3 Dimers Probe the Assembly Status of Multimeric Membrane Proteins. Curr. Biol. CB 2003, 13, 638–646. [Google Scholar] [CrossRef]
- Glembotski, C.C. Endoplasmic Reticulum Stress in the Heart. Circ. Res. 2007, 101, 975–984. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Shen, J.; Arenzana, N.; Tirasophon, W.; Kaufman, R.J.; Prywes, R. Activation of ATF6 and an ATF6 DNA Binding Site by the Endoplasmic Reticulum Stress Response. J. Biol. Chem. 2000, 275, 27013–27020. [Google Scholar] [CrossRef]
- Carvalho, P.; Goder, V.; Rapoport, T.A. Distinct Ubiquitin-Ligase Complexes Define Convergent Pathways for the Degradation of ER Proteins. Cell 2006, 126, 361–373. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Watanabe, I.; Gomez, B.; Thornhill, W.B. Trafficking of Kv1.4 Potassium Channels: Interdependence of a Pore Region Determinant and a Cytoplasmic C-Terminal VXXSL Determinant in Regulating Cell-Surface Trafficking. Biochem. J. 2003, 375, 761–768. [Google Scholar] [CrossRef]
- Fath, S.; Mancias, J.D.; Bi, X.; Goldberg, J. Structure and Organization of Coat Proteins in the COPII Cage. Cell 2007, 129, 1325–1336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowman, P.R.T.; Smith, G.L.; Gould, G.W. Cardiac SNARE Expression in Health and Disease. Front. Endocrinol. 2019, 10, 881. [Google Scholar] [CrossRef]
- Zerial, M.; McBride, H. Rab Proteins as Membrane Organizers. Nat. Rev. Mol. Cell Biol. 2001, 2, 107–117. [Google Scholar] [CrossRef]
- Sönnichsen, B.; De Renzis, S.; Nielsen, E.; Rietdorf, J.; Zerial, M. Distinct Membrane Domains on Endosomes in the Recycling Pathway Visualized by Multicolor Imaging of Rab4, Rab5, and Rab11. J. Cell Biol. 2000, 149, 901–914. [Google Scholar] [CrossRef]
- Satoh, A.K.; O’Tousa, J.E.; Ozaki, K.; Ready, D.F. Rab11 Mediates Post-Golgi Trafficking of Rhodopsin to the Photosensitive Apical Membrane of Drosophila Photoreceptors. Dev. Camb. Engl. 2005, 132, 1487–1497. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Feng, Y.; Chen, D.; Wandinger-Ness, A. Rab11 Is Required for Trans-Golgi Network-to-Plasma Membrane Transport and a Preferential Target for GDP Dissociation Inhibitor. Mol. Biol. Cell 1998, 9, 3241–3257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Press, B.; Feng, Y.; Hoflack, B.; Wandinger-Ness, A. Mutant Rab7 Causes the Accumulation of Cathepsin D and Cation-Independent Mannose 6-Phosphate Receptor in an Early Endocytic Compartment. J. Cell Biol. 1998, 140, 1075–1089. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; Press, B.; Wandinger-Ness, A. Rab 7: An Important Regulator of Late Endocytic Membrane Traffic. J. Cell Biol. 1995, 131, 1435–1452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, J.; Chen, W.; Welford, A.; Wandinger-Ness, A. The Proteasome Alpha-Subunit XAPC7 Interacts Specifically with Rab7 and Late Endosomes. J. Biol. Chem. 2004, 279, 21334–21342. [Google Scholar] [CrossRef] [Green Version]
- Karcher, R.L.; Deacon, S.W.; Gelfand, V.I. Motor-Cargo Interactions: The Key to Transport Specificity. Trends Cell Biol. 2002, 12, 21–27. [Google Scholar] [CrossRef]
- Abi-Char, J.; El-Haou, S.; Balse, E.; Neyroud, N.; Vranckx, R.; Coulombe, A.; Hatem, S.N. The Anchoring Protein SAP97 Retains Kv1.5 Channels in the Plasma Membrane of Cardiac Myocytes. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H1851–H1861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Godreau, D.; Vranckx, R.; Maguy, A.; Rücker-Martin, C.; Goyenvalle, C.; Abdelshafy, S.; Tessier, S.; Couétil, J.P.; Hatem, S.N. Expression, Regulation and Role of the MAGUK Protein SAP-97 in Human Atrial Myocardium. Cardiovasc. Res. 2002, 56, 433–442. [Google Scholar] [CrossRef] [Green Version]
- El-Haou, S.; Balse, E.; Neyroud, N.; Dilanian, G.; Gavillet, B.; Abriel, H.; Coulombe, A.; Jeromin, A.; Hatem, S.N. Kv4 Potassium Channels Form a Tripartite Complex with the Anchoring Protein SAP97 and CaMKII in Cardiac Myocytes. Circ. Res. 2009, 104, 758–769. [Google Scholar] [CrossRef] [Green Version]
- Gillet, L.; Rougier, J.-S.; Shy, D.; Sonntag, S.; Mougenot, N.; Essers, M.; Shmerling, D.; Balse, E.; Hatem, S.N.; Abriel, H. Cardiac-Specific Ablation of Synapse-Associated Protein SAP97 in Mice Decreases Potassium Currents but Not Sodium Current. Heart Rhythm 2015, 12, 181–192. [Google Scholar] [CrossRef]
- Leonoudakis, D.; Mailliard, W.; Wingerd, K.; Clegg, D.; Vandenberg, C. Inward Rectifier Potassium Channel Kir2.2 Is Associated with Synapse-Associated Protein SAP97. J. Cell Sci. 2001, 114, 987–998. [Google Scholar] [CrossRef] [PubMed]
- Milstein, M.L.; Musa, H.; Balbuena, D.P.; Anumonwo, J.M.B.; Auerbach, D.S.; Furspan, P.B.; Hou, L.; Hu, B.; Schumacher, S.M.; Vaidyanathan, R.; et al. Dynamic Reciprocity of Sodium and Potassium Channel Expression in a Macromolecular Complex Controls Cardiac Excitability and Arrhythmia. Proc. Natl. Acad. Sci. USA 2012, 109, E2134–E2143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matamoros, M.; Pérez-Hernández, M.; Guerrero-Serna, G.; Amorós, I.; Barana, A.; Núñez, M.; Ponce-Balbuena, D.; Sacristán, S.; Gómez, R.; Tamargo, J.; et al. Nav1.5 N-Terminal Domain Binding to A1-Syntrophin Increases Membrane Density of Human Kir2.1, Kir2.2 and Nav1.5 Channels. Cardiovasc. Res. 2016, 110, 279–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petitprez, S.; Zmoos, A.-F.; Ogrodnik, J.; Balse, E.; Raad, N.; El-Haou, S.; Albesa, M.; Bittihn, P.; Luther, S.; Lehnart, S.E.; et al. SAP97 and Dystrophin Macromolecular Complexes Determine Two Pools of Cardiac Sodium Channels Nav1.5 in Cardiomyocytes. Circ. Res. 2011, 108, 294–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eichel, C.A.; Beuriot, A.; Chevalier, M.Y.E.; Rougier, J.-S.; Louault, F.; Dilanian, G.; Amour, J.; Coulombe, A.; Abriel, H.; Hatem, S.N.; et al. Lateral Membrane-Specific MAGUK CASK Down-Regulates NaV1.5 Channel in Cardiac Myocytes. Circ. Res. 2016, 119, 544–556. [Google Scholar] [CrossRef] [Green Version]
- Beuriot, A.; Eichel, C.A.; Dilanian, G.; Louault, F.; Melgari, D.; Doisne, N.; Coulombe, A.; Hatem, S.N.; Balse, E. Distinct Calcium/Calmodulin-Dependent Serine Protein Kinase Domains Control Cardiac Sodium Channel Membrane Expression and Focal Adhesion Anchoring. Heart Rhythm 2020, 17, 786–794. [Google Scholar] [CrossRef]
- Leonoudakis, D.; Conti, L.R.; Radeke, C.M.; McGuire, L.M.M.; Vandenberg, C.A. A Multiprotein Trafficking Complex Composed of SAP97, CASK, Veli, and Mint1 Is Associated with Inward Rectifier Kir2 Potassium Channels. J. Biol. Chem. 2004, 279, 19051–19063. [Google Scholar] [CrossRef] [Green Version]
- Verkerk, A.O.; van Ginneken, A.C.G.; van Veen, T.A.B.; Tan, H.L. Effects of Heart Failure on Brain-Type Na+ Channels in Rabbit Ventricular Myocytes. EP Europace 2007, 9, 571–577. [Google Scholar] [CrossRef]
- Lin, X.; Liu, N.; Lu, J.; Zhang, J.; Anumonwo, J.M.; Isom, L.L.; Fishman, G.I.; Delmar, M. Subcellular Heterogeneity of Sodium Current Properties in Adult Cardiac Ventricular Myocytes. Heart Rhythm 2011, 8, 1923–1930. [Google Scholar] [CrossRef] [Green Version]
- Lab, M.J.; Bhargava, A.; Wright, P.T.; Gorelik, J. The Scanning Ion Conductance Microscope for Cellular Physiology. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H1–H11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vermij, S.H.; Rougier, J.-S.; Agulló-Pascual, E.; Rothenberg, E.; Delmar, M.; Abriel, H. Single-Molecule Localization of the Cardiac Voltage-Gated Sodium Channel Reveals Different Modes of Reorganization at Cardiomyocyte Membrane Domains. Circ. Arrhythm. Electrophysiol. 2020, 13, e008241. [Google Scholar] [CrossRef] [PubMed]
- Balse, E.; Eichel, C. The Cardiac Sodium Channel and Its Protein Partners. Handb. Exp. Pharmacol. 2018, 246, 73–99. [Google Scholar] [CrossRef] [PubMed]
- Wilson, Z.T.; Jiang, M.; Geng, J.; Kaur, S.; Workman, S.W.; Hao, J.; Bernas, T.; Tseng, G.-N. Delayed KCNQ1/KCNE1 Assembly on the Cell Surface Helps IKs Fulfil Its Function as a Repolarization Reserve in the Heart. J. Physiol. 2021, 599, 3337–3361. [Google Scholar] [CrossRef] [PubMed]
- Furutani, M.; Trudeau, M.C.; Hagiwara, N.; Seki, A.; Gong, Q.; Zhou, Z.; Imamura, S.; Nagashima, H.; Kasanuki, H.; Takao, A.; et al. Novel Mechanism Associated with an Inherited Cardiac Arrhythmia: Defective Protein Trafficking by the Mutant HERG (G601S) Potassium Channel. Circulation 1999, 99, 2290–2294. [Google Scholar] [CrossRef] [PubMed]
- Gouas, L.; Bellocq, C.; Berthet, M.; Potet, F.; Demolombe, S.; Forhan, A.; Lescasse, R.; Simon, F.; Balkau, B.; Denjoy, I.; et al. New KCNQ1 Mutations Leading to Haploinsufficiency in a General Population; Defective Trafficking of a KvLQT1 Mutant. Cardiovasc. Res. 2004, 63, 60–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valdivia, C.R.; Tester, D.J.; Rok, B.A.; Porter, C.-B.J.; Munger, T.M.; Jahangir, A.; Makielski, J.C.; Ackerman, M.J. A Trafficking Defective, Brugada Syndrome-Causing SCN5A Mutation Rescued by Drugs. Cardiovasc. Res. 2004, 62, 53–62. [Google Scholar] [CrossRef]
- Steffensen, A.B.; Refsgaard, L.; Andersen, M.N.; Vallet, C.; Mujezinovic, A.; Haunsø, S.; Svendsen, J.H.; Olesen, S.-P.; Olesen, M.S.; Schmitt, N. IKs Gain- and Loss-of-Function in Early-Onset Lone Atrial Fibrillation. J. Cardiovasc. Electrophysiol. 2015, 26, 715–723. [Google Scholar] [CrossRef]
- Huang, H.; Chamness, L.M.; Vanoye, C.G.; Kuenze, G.; Meiler, J.; George, A.L.; Schlebach, J.P.; Sanders, C.R. Disease-Linked Supertrafficking of a Potassium Channel. J. Biol. Chem. 2021, 296, 100423. [Google Scholar] [CrossRef]
- Olesen, M.S.; Refsgaard, L.; Holst, A.G.; Larsen, A.P.; Grubb, S.; Haunsø, S.; Svendsen, J.H.; Olesen, S.-P.; Schmitt, N.; Calloe, K. A Novel KCND3 Gain-of-Function Mutation Associated with Early-Onset of Persistent Lone Atrial Fibrillation. Cardiovasc. Res. 2013, 98, 488–495. [Google Scholar] [CrossRef]
- Kruse, M.; Schulze-Bahr, E.; Corfield, V.; Beckmann, A.; Stallmeyer, B.; Kurtbay, G.; Ohmert, I.; Schulze-Bahr, E.; Brink, P.; Pongs, O. Impaired Endocytosis of the Ion Channel TRPM4 Is Associated with Human Progressive Familial Heart Block Type I. J. Clin. Invest. 2009, 119, 2737–2744. [Google Scholar] [CrossRef]
- El-Sherif, N.; Turitto, G.; Boutjdir, M. Congenital Long QT Syndrome and Torsade de Pointes. Ann. Noninvasive Electrocardiol. 2017, 22, e12481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahida, S.; Hogarth, A.J.; Cowan, C.; Tayebjee, M.H.; Graham, L.N.; Pepper, C.B. Genetics of Congenital and Drug-Induced Long QT Syndromes: Current Evidence and Future Research Perspectives. J. Interv. Card. Electrophysiol. 2013, 37, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Cunha, S.R.; Mohler, P.J. Cardiac Ankyrins: Essential Components for Development and Maintenance of Excitable Membrane Domains in Heart. Cardiovasc. Res. 2006, 71, 22–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohler, P.J.; Schott, J.-J.; Gramolini, A.O.; Dilly, K.W.; Guatimosim, S.; du Bell, W.H.; Song, L.-S.; Haurogné, K.; Kyndt, F.; Ali, M.E.; et al. Ankyrin-B Mutation Causes Type 4 Long-QT Cardiac Arrhythmia and Sudden Cardiac Death. Nature 2003, 421, 634–639. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.A.; Sturm, A.C.; Curran, J.; Kline, C.F.; Little, S.C.; Bonilla, I.M.; Long, V.P.; Makara, M.; Polina, I.; Hughes, L.D.; et al. Dysfunction in the ΒII Spectrin-Dependent Cytoskeleton Underlies Human Arrhythmia. Circulation 2015, 131, 695–708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohler, P.J.; Splawski, I.; Napolitano, C.; Bottelli, G.; Sharpe, L.; Timothy, K.; Priori, S.G.; Keating, M.T.; Bennett, V. A Cardiac Arrhythmia Syndrome Caused by Loss of Ankyrin-B Function. Proc. Natl. Acad. Sci. USA 2004, 101, 9137–9142. [Google Scholar] [CrossRef] [Green Version]
- Swayne, L.A.; Murphy, N.P.; Asuri, S.; Chen, L.; Xu, X.; McIntosh, S.; Wang, C.; Lancione, P.J.; Roberts, J.D.; Kerr, C.; et al. Novel Variant in the ANK2 Membrane-Binding Domain Is Associated with Ankyrin-B Syndrome and Structural Heart Disease in a First Nations Population with a High Rate of Long QT Syndrome. Circ. Cardiovasc. Genet. 2017, 10, e001537. [Google Scholar] [CrossRef] [Green Version]
- Ichikawa, M.; Aiba, T.; Ohno, S.; Shigemizu, D.; Ozawa, J.; Sonoda, K.; Fukuyama, M.; Itoh, H.; Miyamoto, Y.; Tsunoda, T.; et al. Phenotypic Variability of ANK2 Mutations in Patients with Inherited Primary Arrhythmia Syndromes. Circ. J. Off. J. Jpn. Circ. Soc. 2016, 80, 2435–2442. [Google Scholar] [CrossRef] [Green Version]
- Cunha, S.R.; Hund, T.J.; Hashemi, S.; Voigt, N.; Li, N.; Wright, P.; Koval, O.; Li, J.; Gudmundsson, H.; Gumina, R.J.; et al. Defects in Ankyrin-Based Membrane Protein Targeting Pathways Underlie Atrial Fibrillation. Circulation 2011, 124, 1212–1222. [Google Scholar] [CrossRef] [Green Version]
- Palade, G.E. An Electron Microscope Study of the Mitochondrial Structure. J. Histochem. Cytochem. Off. J. Histochem. Soc. 1953, 1, 188–211. [Google Scholar] [CrossRef]
- Parton, R.G.; Tillu, V.A.; Collins, B.M. Caveolae. Curr. Biol. CB 2018, 28, R402–R405. [Google Scholar] [CrossRef] [Green Version]
- Bastiani, M.; Parton, R.G. Caveolae at a Glance. J. Cell Sci. 2010, 123, 3831–3836. [Google Scholar] [CrossRef] [Green Version]
- Del Pozo, M.A.; Lolo, F.-N.; Echarri, A. Caveolae: Mechanosensing and Mechanotransduction Devices Linking Membrane Trafficking to Mechanoadaptation. Curr. Opin. Cell Biol. 2021, 68, 113–123. [Google Scholar] [CrossRef]
- Yarbrough, T.L.; Lu, T.; Lee, H.-C.; Shibata, E.F. Localization of Cardiac Sodium Channels in Caveolin-Rich Membrane Domains: Regulation of Sodium Current Amplitude. Circ. Res. 2002, 90, 443–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vatta, M.; Ackerman, M.J.; Ye, B.; Makielski, J.C.; Ughanze, E.E.; Taylor, E.W.; Tester, D.J.; Balijepalli, R.C.; Foell, J.D.; Li, Z.; et al. Mutant Caveolin-3 Induces Persistent Late Sodium Current and Is Associated with Long-QT Syndrome. Circulation 2006, 114, 2104–2112. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.; Valdivia, C.R.; Vaidyanathan, R.; Balijepalli, R.C.; Ackerman, M.J.; Makielski, J.C. Caveolin-3 Suppresses Late Sodium Current by Inhibiting NNOS-Dependent S-Nitrosylation of SCN5A. J. Mol. Cell. Cardiol. 2013, 61, 102–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cronk, L.B.; Ye, B.; Kaku, T.; Tester, D.J.; Vatta, M.; Makielski, J.C.; Ackerman, M.J. Novel Mechanism for Sudden Infant Death Syndrome: Persistent Late Sodium Current Secondary to Mutations in Caveolin-3. Heart Rhythm 2007, 4, 161–166. [Google Scholar] [CrossRef] [Green Version]
- Tyan, L.; Foell, J.D.; Vincent, K.P.; Woon, M.T.; Mesquitta, W.T.; Lang, D.; Best, J.M.; Ackerman, M.J.; McCulloch, A.D.; Glukhov, A.V.; et al. Long QT Syndrome Caveolin-3 Mutations Differentially Modulate Kv 4 and Cav 1.2 Channels to Contribute to Action Potential Prolongation. J. Physiol. 2019, 597, 1531–1551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaidyanathan, R.; Vega, A.L.; Song, C.; Zhou, Q.; Tan, B.-H.; Tan, B.; Berger, S.; Makielski, J.C.; Eckhardt, L.L. The Interaction of Caveolin 3 Protein with the Potassium Inward Rectifier Channel Kir2.1: Physiology and Pathology Related to Long Qt Syndrome 9 (LQT9). J. Biol. Chem. 2013, 288, 17472–17480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isom, L.L. Sodium Channel β Subunits: Anything but Auxiliary. The Neuroscientist 2001, 7, 42–54. [Google Scholar] [CrossRef]
- Watanabe, H.; Koopmann, T.T.; Le Scouarnec, S.; Yang, T.; Ingram, C.R.; Schott, J.-J.; Demolombe, S.; Probst, V.; Anselme, F.; Escande, D.; et al. Sodium Channel Β1 Subunit Mutations Associated with Brugada Syndrome and Cardiac Conduction Disease in Humans. J. Clin. Invest. 2008, 118, 2260–2268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edokobi, N.; Isom, L.L. Voltage-Gated Sodium Channel Β1/Β1B Subunits Regulate Cardiac Physiology and Pathophysiology. Front. Physiol. 2018, 9, 351. [Google Scholar] [CrossRef] [Green Version]
- Holst, A.G.; Saber, S.; Houshmand, M.; Zaklyazminskaya, E.V.; Wang, Y.; Jensen, H.K.; Refsgaard, L.; Haunsø, S.; Svendsen, J.H.; Olesen, M.S.; et al. Sodium Current and Potassium Transient Outward Current Genes in Brugada Syndrome: Screening and Bioinformatics. Can. J. Cardiol. 2012, 28, 196–200. [Google Scholar] [CrossRef] [PubMed]
- Olesen, M.S.; Holst, A.G.; Svendsen, J.H.; Haunsø, S.; Tfelt-Hansen, J. SCN1Bb R214Q Found in 3 Patients: 1 with Brugada Syndrome and 2 with Lone Atrial Fibrillation. Heart Rhythm 2012, 9, 770–773. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.; Barajas-Martínez, H.; Medeiros-Domingo, A.; Crotti, L.; Veltmann, C.; Schimpf, R.; Urrutia, J.; Alday, A.; Casis, O.; Pfeiffer, R.; et al. A Novel Rare Variant in SCN1Bb Linked to Brugada Syndrome and SIDS by Combined Modulation of Nav1.5 and Kv4.3 Channel Currents. Heart Rhythm 2012, 9, 760–769. [Google Scholar] [CrossRef] [Green Version]
- Yuan, L.; Koivumäki, J.T.; Liang, B.; Lorentzen, L.G.; Tang, C.; Andersen, M.N.; Svendsen, J.H.; Tfelt-Hansen, J.; Maleckar, M.; Schmitt, N.; et al. Investigations of the Navβ1b Sodium Channel Subunit in Human Ventricle; Functional Characterization of the H162P Brugada Syndrome Mutant. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H1204–H1212. [Google Scholar] [CrossRef] [Green Version]
- Ricci, M.T.; Menegon, S.; Vatrano, S.; Mandrile, G.; Cerrato, N.; Carvalho, P.; De Marchi, M.; Gaita, F.; Giustetto, C.; Giachino, D.F. SCN1B Gene Variants in Brugada Syndrome: A Study of 145 SCN5A-Negative Patients. Sci. Rep. 2014, 4, 6470. [Google Scholar] [CrossRef]
- Wang, L.; Han, Z.; Dai, J.; Cao, K. Brugada Syndrome Caused by Sodium Channel Dysfunction Associated with a SCN1B Variant A197V. Arch. Med. Res. 2020, 51, 245–253. [Google Scholar] [CrossRef]
- Hu, D.; Barajas-Martinez, H.; Burashnikov, E.; Springer, M.; Wu, Y.; Varro, A.; Pfeiffer, R.; Koopmann, T.T.; Cordeiro, J.M.; Guerchicoff, A.; et al. A Mutation in the Β3 Subunit of the Cardiac Sodium Channel Associated with Brugada ECG Phenotype. Circ. Cardiovasc. Genet. 2009, 2, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, T.; Takahashi, N.; Ohno, S.; Sakurada, H.; Nakamura, K.; On, Y.K.; Park, J.E.; Makiyama, T.; Horie, M.; Arimura, T.; et al. Novel SCN3B Mutation Associated With Brugada Syndrome Affects Intracellular Trafficking and Function of Nav1.5. Circ. J. 2013, 77, 959–967. [Google Scholar] [CrossRef] [Green Version]
- Riuró, H.; Beltran-Alvarez, P.; Tarradas, A.; Selga, E.; Campuzano, O.; Vergés, M.; Pagans, S.; Iglesias, A.; Brugada, J.; Brugada, P.; et al. A Missense Mutation in the Sodium Channel Β2 Subunit Reveals SCN2B as a New Candidate Gene for Brugada Syndrome. Hum. Mutat. 2013, 34, 961–966. [Google Scholar] [CrossRef]
- Riuró, H.; Campuzano, O.; Arbelo, E.; Iglesias, A.; Batlle, M.; Pérez-Villa, F.; Brugada, J.; Pérez, G.J.; Scornik, F.S.; Brugada, R. A Missense Mutation in the Sodium Channel Β1b Subunit Reveals SCN1B as a Susceptibility Gene Underlying Long QT Syndrome. Heart Rhythm 2014, 11, 1202–1209. [Google Scholar] [CrossRef]
- Lowe, J.S.; Palygin, O.; Bhasin, N.; Hund, T.J.; Boyden, P.A.; Shibata, E.; Anderson, M.E.; Mohler, P.J. Voltage-Gated Nav Channel Targeting in the Heart Requires an Ankyrin-G–Dependent Cellular Pathway. J. Cell Biol. 2008, 180, 173–186. [Google Scholar] [CrossRef] [PubMed]
- Sato, P.Y.; Coombs, W.; Lin, X.; Nekrasova, O.; Green, K.; Isom, L.L.; Taffet, S.; Delmar, M. Interactions between Ankyrin-G, Plakophilin-2 and Connexin43 at the Cardiac Intercalated Disc. Circ. Res. 2011, 109, 193–201. [Google Scholar] [CrossRef] [Green Version]
- Casini, S.; Tan, H.L.; Demirayak, I.; Remme, C.A.; Amin, A.S.; Scicluna, B.P.; Chatyan, H.; Ruijter, J.M.; Bezzina, C.R.; van Ginneken, A.C.G.; et al. Tubulin Polymerization Modifies Cardiac Sodium Channel Expression and Gating. Cardiovasc. Res. 2010, 85, 691–700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chkourko, H.S.; Guerrero-Serna, G.; Lin, X.; Darwish, N.; Pohlmann, J.R.; Cook, K.E.; Martens, J.R.; Rothenberg, E.; Musa, H.; Delmar, M. Remodeling of Mechanical Junctions and of Microtubule-Associated Proteins Accompany Cardiac Connexin43 Lateralization. Heart Rhythm 2012, 9, 1133–1140.e6. [Google Scholar] [CrossRef] [Green Version]
- Makara, M.A.; Curran, J.; Little, S.C.; Musa, H.; Polina, I.; Smith, S.A.; Wright, P.J.; Unudurthi, S.D.; Snyder, J.; Bennett, V.; et al. Ankyrin-G Coordinates Intercalated Disc Signaling Platform to Regulate Cardiac Excitability In Vivo. Circ. Res. 2014, 115, 929–938. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.-Q.; Pérez-Hernández, M.; Sanchez-Alonso, J.; Shevchuk, A.; Gorelik, J.; Rothenberg, E.; Delmar, M.; Coetzee, W.A. Ankyrin-G Mediates Targeting of Both Na+ and KATP Channels to the Rat Cardiac Intercalated Disc. eLife 2020, 9, e52373. [Google Scholar] [CrossRef]
- Cerrone, M.; Lin, X.; Zhang, M.; Agullo-Pascual, E.; Pfenniger, A.; Chkourko Gusky, H.; Novelli, V.; Kim, C.; Tirasawadichai, T.; Judge, D.P.; et al. Missense Mutations in Plakophilin-2 Cause Sodium Current Deficit and Associate with a Brugada Syndrome Phenotype. Circulation 2014, 129, 1092–1103. [Google Scholar] [CrossRef] [Green Version]
- Musa, H.; Marcou, C.A.; Herron, T.J.; Makara, M.A.; Tester, D.J.; O’Connell, R.P.; Rosinski, B.; Guerrero-Serna, G.; Milstein, M.L.; Monteiro da Rocha, A.; et al. Abnormal Myocardial Expression of SAP97 Is Associated with Arrhythmogenic Risk. Am. J. Physiol. Heart Circ. Physiol. 2020, 318, H1357–H1370. [Google Scholar] [CrossRef]
- Shy, D.; Gillet, L.; Ogrodnik, J.; Albesa, M.; Verkerk, A.O.; Wolswinkel, R.; Rougier, J.-S.; Barc, J.; Essers, M.C.; Syam, N.; et al. PDZ Domain-Binding Motif Regulates Cardiomyocyte Compartment-Specific NaV1.5 Channel Expression and Function. Circulation 2014, 130, 147–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ai, X.; Pogwizd, S.M. Connexin 43 Downregulation and Dephosphorylation in Nonischemic Heart Failure Is Associated with Enhanced Colocalized Protein Phosphatase Type 2A. Circ. Res. 2005, 96, 54–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rucker-Martin, C.; Milliez, P.; Tan, S.; Decrouy, X.; Recouvreur, M.; Vranckx, R.; Delcayre, C.; Renaud, J.-F.; Dunia, I.; Segretain, D.; et al. Chronic Hemodynamic Overload of the Atria Is an Important Factor for Gap Junction Remodeling in Human and Rat Hearts. Cardiovasc. Res. 2006, 72, 69–79. [Google Scholar] [CrossRef]
- Sasano, C.; Honjo, H.; Takagishi, Y.; Uzzaman, M.; Emdad, L.; Shimizu, A.; Murata, Y.; Kamiya, K.; Kodama, I. Internalization and Dephosphorylation of Connexin43 in Hypertrophied Right Ventricles of Rats with Pulmonary Hypertension. Circ. J. 2007, 71, 382–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hesketh, G.G.; Shah, M.H.; Halperin, V.L.; Cooke, C.A.; Akar, F.G.; Yen, T.E.; Kass, D.A.; Machamer, C.E.; Van Eyk, J.E.; Tomaselli, G.F. Ultrastructure and Regulation of Lateralized Connexin43 in the Failing Heart. Circ. Res. 2010, 106, 1153–1163. [Google Scholar] [CrossRef] [Green Version]
- Rücker-Martin, C.; Pecker, F.; Godreau, D.; Hatem, S.N. Dedifferentiation of Atrial Myocytes during Atrial Fibrillation: Role of Fibroblast Proliferation in Vitro. Cardiovasc. Res. 2002, 55, 38–52. [Google Scholar] [CrossRef]
- Xiao, S.; Shimura, D.; Baum, R.; Hernandez, D.M.; Agvanian, S.; Nagaoka, Y.; Katsumata, M.; Lampe, P.D.; Kleber, A.G.; Hong, T.; et al. Auxiliary Trafficking Subunit GJA1-20k Protects Connexin-43 from Degradation and Limits Ventricular Arrhythmias. J. Clin. Invest. 2020, 130, 4858–4870. [Google Scholar] [CrossRef]
- Shaw, R.M.; Fay, A.J.; Puthenveedu, M.A.; von Zastrow, M.; Jan, Y.-N.; Jan, L.Y. Microtubule Plus-End-Tracking Proteins Target Gap Junctions Directly from the Cell Interior to Adherens Junctions. Cell 2007, 128, 547–560. [Google Scholar] [CrossRef] [Green Version]
- Hong, T.-T.; Smyth, J.W.; Chu, K.Y.; Vogan, J.M.; Fong, T.S.; Jensen, B.C.; Fang, K.; Halushka, M.K.; Russell, S.D.; Colecraft, H.; et al. BIN1 Is Reduced and Cav1.2 Trafficking Is Impaired in Human Failing Cardiomyocytes. Heart Rhythm Off. J. Heart Rhythm Soc. 2012, 9, 812–820. [Google Scholar] [CrossRef] [Green Version]
- Hong, T.; Yang, H.; Zhang, S.-S.; Cho, H.C.; Kalashnikova, M.; Sun, B.; Zhang, H.; Bhargava, A.; Grabe, M.; Olgin, J.; et al. Cardiac BIN1 Folds T-Tubule Membrane, Controlling Ion Flux and Limiting Arrhythmia. Nat. Med. 2014, 20, 624–632. [Google Scholar] [CrossRef] [Green Version]
- Mustroph, J.; Sag, C.M.; Bähr, F.; Schmidtmann, A.-L.; Gupta, S.N.; Dietz, A.; Islam, M.M.T.; Lücht, C.; Beuthner, B.E.; Pabel, S.; et al. Loss of CASK Accelerates Heart Failure Development. Circ. Res. 2021, 128, 1139–1155. [Google Scholar] [CrossRef]
- Balse, E.; Steele, D.F.; Abriel, H.; Coulombe, A.; Fedida, D.; Hatem, S.N. Dynamic of Ion Channel Expression at the Plasma Membrane of Cardiomyocytes. Physiol. Rev. 2012, 92, 1317–1358. [Google Scholar] [CrossRef]
- Sanguinetti, M.C.; Jiang, C.; Curran, M.E.; Keating, M.T. A Mechanistic Link between an Inherited and an Acquired Cardiac Arrhythmia: HERG Encodes the IKr Potassium Channel. Cell 1995, 81, 299–307. [Google Scholar] [CrossRef] [Green Version]
- Trudeau, M.C.; Warmke, J.W.; Ganetzky, B.; Robertson, G.A. HERG, a Human Inward Rectifier in the Voltage-Gated Potassium Channel Family. Science 1995, 269, 92–95. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.L.; Delisle, B.P.; Anson, B.D.; Kilby, J.A.; Will, M.L.; Tester, D.J.; Gong, Q.; Zhou, Z.; Ackerman, M.J.; January, C.T. Most LQT2 Mutations Reduce Kv11.1 (HERG) Current by a Class 2 (Trafficking-Deficient) Mechanism. Circulation 2006, 113, 365–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanguinetti, M.C.; Tristani-Firouzi, M. HERG Potassium Channels and Cardiac Arrhythmia. Nature 2006, 440, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Dennis, A.T.; Nassal, D.; Deschenes, I.; Thomas, D.; Ficker, E. Antidepressant-Induced Ubiquitination and Degradation of the Cardiac Potassium Channel HERG. J. Biol. Chem. 2011, 286, 34413–34425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Compton, S.J.; Lux, R.L.; Ramsey, M.R.; Strelich, K.R.; Sanguinetti, M.C.; Green, L.S.; Keating, M.T.; Mason, J.W. Genetically Defined Therapy of Inherited Long-QT Syndrome. Correction of Abnormal Repolarization by Potassium. Circulation 1996, 94, 1018–1022. [Google Scholar] [CrossRef]
- Guo, J.; Massaeli, H.; Xu, J.; Jia, Z.; Wigle, J.T.; Mesaeli, N.; Zhang, S. Extracellular K+ Concentration Controls Cell Surface Density of IKr in Rabbit Hearts and of the HERG Channel in Human Cell Lines. J. Clin. Invest. 2009, 119, 2745–2757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.-Q.; Yan, M.; Liu, L.-R.; Zhang, X.; Wang, X.; Geng, H.-Z.; Zhao, X.; Li, B.-X. High Glucose Represses HERG K+ Channel Expression through Trafficking Inhibition. Cell. Physiol. Biochem. 2015, 37, 284–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melgari, D.; Barbier, C.; Dilanian, G.; Rücker-Martin, C.; Doisne, N.; Coulombe, A.; Hatem, S.N.; Balse, E. Microtubule Polymerization State and Clathrin-Dependent Internalization Regulate Dynamics of Cardiac Potassium Channel: Microtubule and Clathrin Control of KV1.5 Channel. J. Mol. Cell. Cardiol. 2020, 144, 127–139. [Google Scholar] [CrossRef] [PubMed]
- Fedida, D.; Wible, B.; Wang, Z.; Fermini, B.; Faust, F.; Nattel, S.; Brown, A.M. Identity of a Novel Delayed Rectifier Current from Human Heart with a Cloned K+ Channel Current. Circ. Res. 1993, 73, 210–216. [Google Scholar] [CrossRef] [Green Version]
- Van Wagoner, D.R.; Pond, A.L.; McCarthy, P.M.; Trimmer, J.S.; Nerbonne, J.M. Outward K+ Current Densities and Kv1.5 Expression Are Reduced in Chronic Human Atrial Fibrillation. Circ. Res. 1997, 80, 772–781. [Google Scholar] [CrossRef]
- Brundel, B.J.; Van Gelder, I.C.; Henning, R.H.; Tuinenburg, A.E.; Wietses, M.; Grandjean, J.G.; Wilde, A.A.; Van Gilst, W.H.; Crijns, H.J. Alterations in Potassium Channel Gene Expression in Atria of Patients with Persistent and Paroxysmal Atrial Fibrillation: Differential Regulation of Protein and MRNA Levels for K+ Channels. J. Am. Coll. Cardiol. 2001, 37, 926–932. [Google Scholar] [CrossRef] [Green Version]
- Lundbaek, J.A.; Birn, P.; Girshman, J.; Hansen, A.J.; Andersen, O.S. Membrane Stiffness and Channel Function. Biochemistry 1996, 35, 3825–3830. [Google Scholar] [CrossRef]
- Pike, L.J. Lipid Rafts: Heterogeneity on the High Seas. Biochem. J. 2004, 378, 281–292. [Google Scholar] [CrossRef] [Green Version]
- Goonasekara, C.L.; Balse, E.; Hatem, S.; Steele, D.F.; Fedida, D. Cholesterol and Cardiac Arrhythmias. Expert Rev. Cardiovasc. Ther. 2010, 8, 965–979. [Google Scholar] [CrossRef]
- Balse, E.; El-Haou, S.; Dillanian, G.; Dauphin, A.; Eldstrom, J.; Fedida, D.; Coulombe, A.; Hatem, S.N. Cholesterol Modulates the Recruitment of Kv1.5 Channels from Rab11-Associated Recycling Endosome in Native Atrial Myocytes. Proc. Natl. Acad. Sci. USA 2009, 106, 14681–14686. [Google Scholar] [CrossRef] [Green Version]
- Parks, X.X.; Ronzier, E.; O-Uchi, J.; Lopes, C.M. Fluvastatin Inhibits Rab5-Mediated IKs Internalization Caused by Chronic Ca2+-Dependent PKC Activation. J. Mol. Cell. Cardiol. 2019, 129, 314–325. [Google Scholar] [CrossRef] [PubMed]
- Ronzier, E.; Parks, X.X.; Qudsi, H.; Lopes, C.M. Statin-Specific Inhibition of Rab-GTPase Regulates CPKC-Mediated IKs Internalization. Sci. Rep. 2019, 9, 17747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.J.; Parise, H.; Levy, D.; D’Agostino, R.B.; Wolf, P.A.; Vasan, R.S.; Benjamin, E.J. Obesity and the Risk of New-Onset Atrial Fibrillation. JAMA 2004, 292, 2471–2477. [Google Scholar] [CrossRef] [Green Version]
- Hatem, S.N. Atrial Fibrillation and Obesity: Not Just a Coincidence. J. Am. Coll. Cardiol. 2015, 66, 12–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LeGrice, I.J.; Takayama, Y.; Covell, J.W. Transverse Shear along Myocardial Cleavage Planes Provides a Mechanism for Normal Systolic Wall Thickening. Circ. Res. 1995, 77, 182–193. [Google Scholar] [CrossRef]
- Costa, K.D.; Takayama, Y.; McCulloch, A.D.; Covell, J.W. Laminar Fiber Architecture and Three-Dimensional Systolic Mechanics in Canine Ventricular Myocardium. Am. J. Physiol. 1999, 276, H595–H607. [Google Scholar] [CrossRef]
- Morad, M.; Javaheri, A.; Risius, T.; Belmonte, S. Multimodality of Ca2+ Signaling in Rat Atrial Myocytes. Ann. N. Y. Acad. Sci. 2005, 1047, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Woo, S.-H.; Risius, T.; Morad, M. Modulation of Local Ca2+ Release Sites by Rapid Fluid Puffing in Rat Atrial Myocytes. Cell Calcium 2007, 41, 397–403. [Google Scholar] [CrossRef] [Green Version]
- Lorenzen-Schmidt, I.; Schmid-Schönbein, G.W.; Giles, W.R.; McCulloch, A.D.; Chien, S.; Omens, J.H. Chronotropic Response of Cultured Neonatal Rat Ventricular Myocytes to Short-Term Fluid Shear. Cell Biochem. Biophys. 2006, 46, 113–122. [Google Scholar] [CrossRef] [Green Version]
- Kong, C.-R.; Bursac, N.; Tung, L. Mechanoelectrical Excitation by Fluid Jets in Monolayers of Cultured Cardiac Myocytes. J. Appl. Physiol. Bethesda Md. 1985 2005, 98, 2328–2336, discussion 2320. [Google Scholar] [CrossRef] [Green Version]
- Boycott, H.E.; Barbier, C.S.M.; Eichel, C.A.; Costa, K.D.; Martins, R.P.; Louault, F.; Dilanian, G.; Coulombe, A.; Hatem, S.N.; Balse, E. Shear Stress Triggers Insertion of Voltage-Gated Potassium Channels from Intracellular Compartments in Atrial Myocytes. Proc. Natl. Acad. Sci. USA 2013, 110, E3955–E3964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, S.; Mathew, M.K. Fluid Flow Modulates Electrical Activity in Cardiac HERG Potassium Channels. J. Biol. Chem. 2018, 293, 4289–4303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lisewski, U.; Koehncke, C.; Wilck, N.; Buschmeyer, B.; Pieske, B.; Roepke, T.K. Increased Aldosterone-Dependent Kv1.5 Recycling Predisposes to Pacing-Induced Atrial Fibrillation in Kcne3-/- Mice. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2016, 30, 2476–2489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dun, W.; Wright, P.; Danilo, P.; Mohler, P.J.; Boyden, P.A. SAP97 and Cortactin Remodeling in Arrhythmogenic Purkinje Cells. PLoS ONE 2014, 9, e106830. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blandin, C.E.; Gravez, B.J.; Hatem, S.N.; Balse, E. Remodeling of Ion Channel Trafficking and Cardiac Arrhythmias. Cells 2021, 10, 2417. https://doi.org/10.3390/cells10092417
Blandin CE, Gravez BJ, Hatem SN, Balse E. Remodeling of Ion Channel Trafficking and Cardiac Arrhythmias. Cells. 2021; 10(9):2417. https://doi.org/10.3390/cells10092417
Chicago/Turabian StyleBlandin, Camille E., Basile J. Gravez, Stéphane N. Hatem, and Elise Balse. 2021. "Remodeling of Ion Channel Trafficking and Cardiac Arrhythmias" Cells 10, no. 9: 2417. https://doi.org/10.3390/cells10092417