
The Saga of Endocrine FGFs

 ,
, 
Abstract
:1. Introduction
1.1. Endocrine FGFs—A Historical Perspective
1.1.1. Discovery of FGF19
1.1.2. Identification of FGF21
1.1.3. History of FGF23
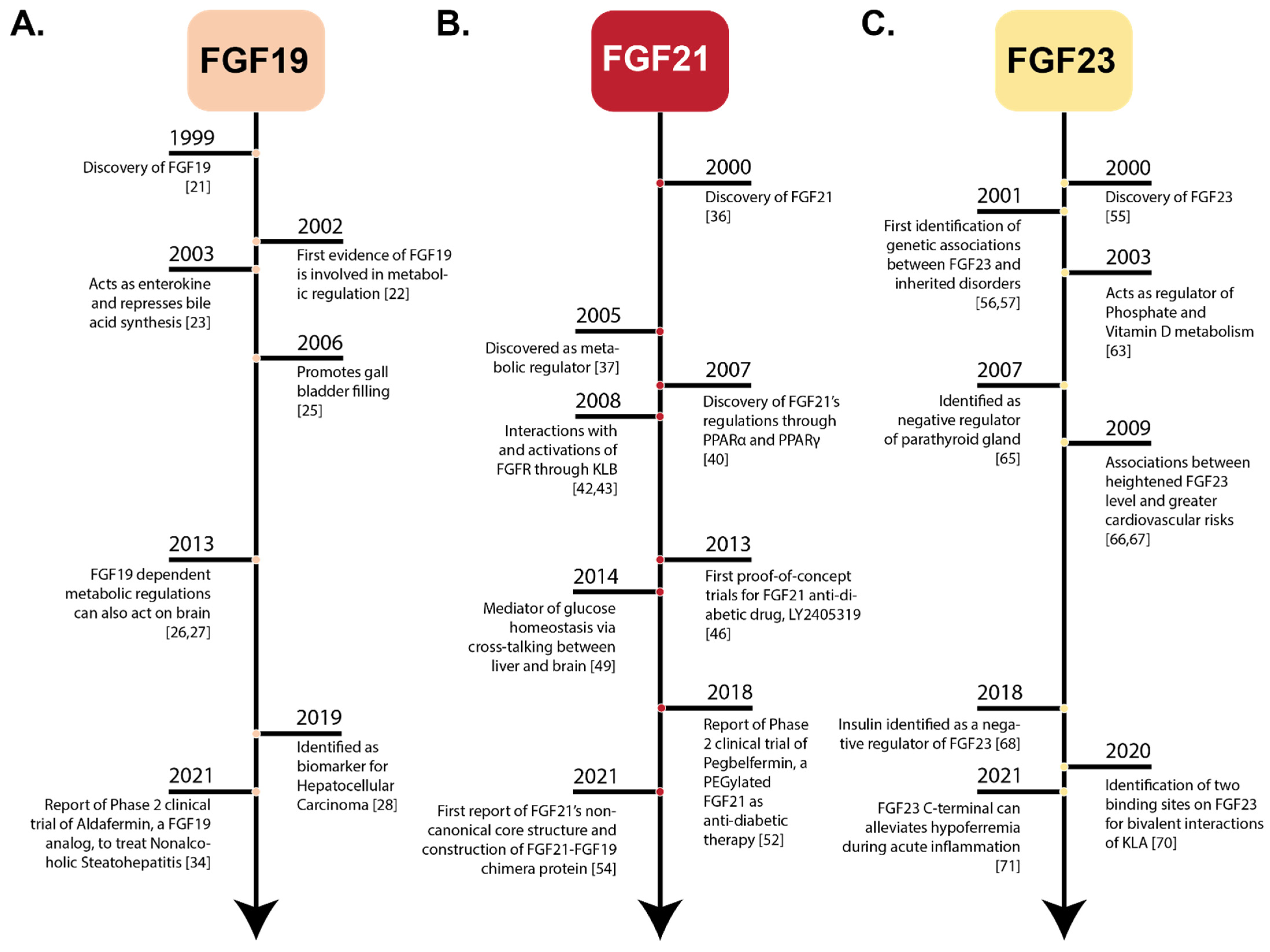
2. Physiology and Clinical Significance of Endocrine FGFs
2.1. Physiology of FGF19
2.2. Clinical Significance of FGF19
2.3. Physiology of FGF21
2.4. Clinical Significance of FGF21
2.5. Physiology of FGF23
2.6. Clinical Significance of FGF23
3. Comparison of Amino Acid Sequences of FGF2 with Endocrine FGFs
4. Structural Features of Paracrine and Endocrine FGFs
4.1. Structural Features of Paracrine Secretions
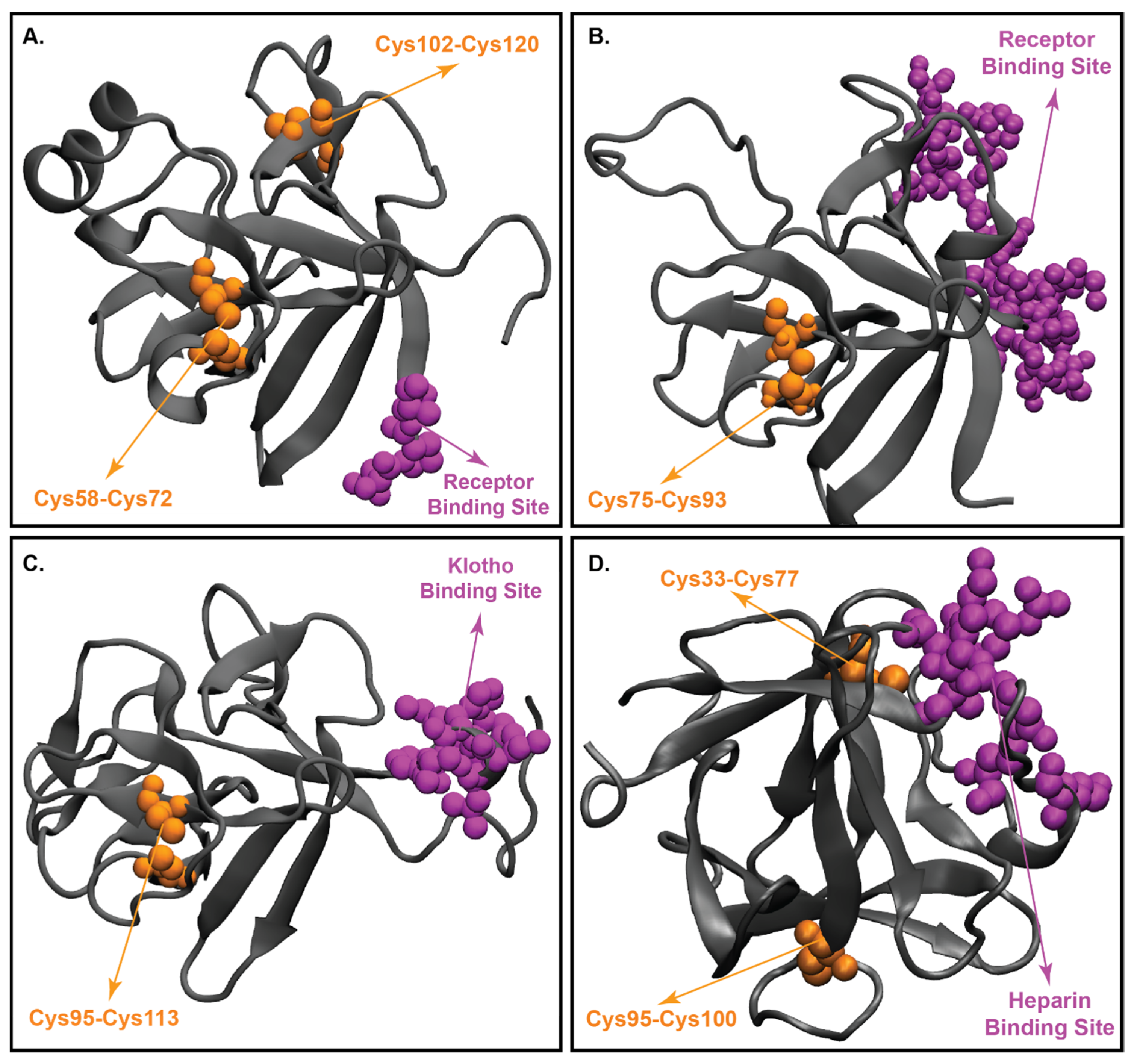
4.2. Structural Characteristics of FGF19
4.3. Three-Dimensional Structure of FGF21
4.4. Structure of FGF23
5. Klotho Proteins
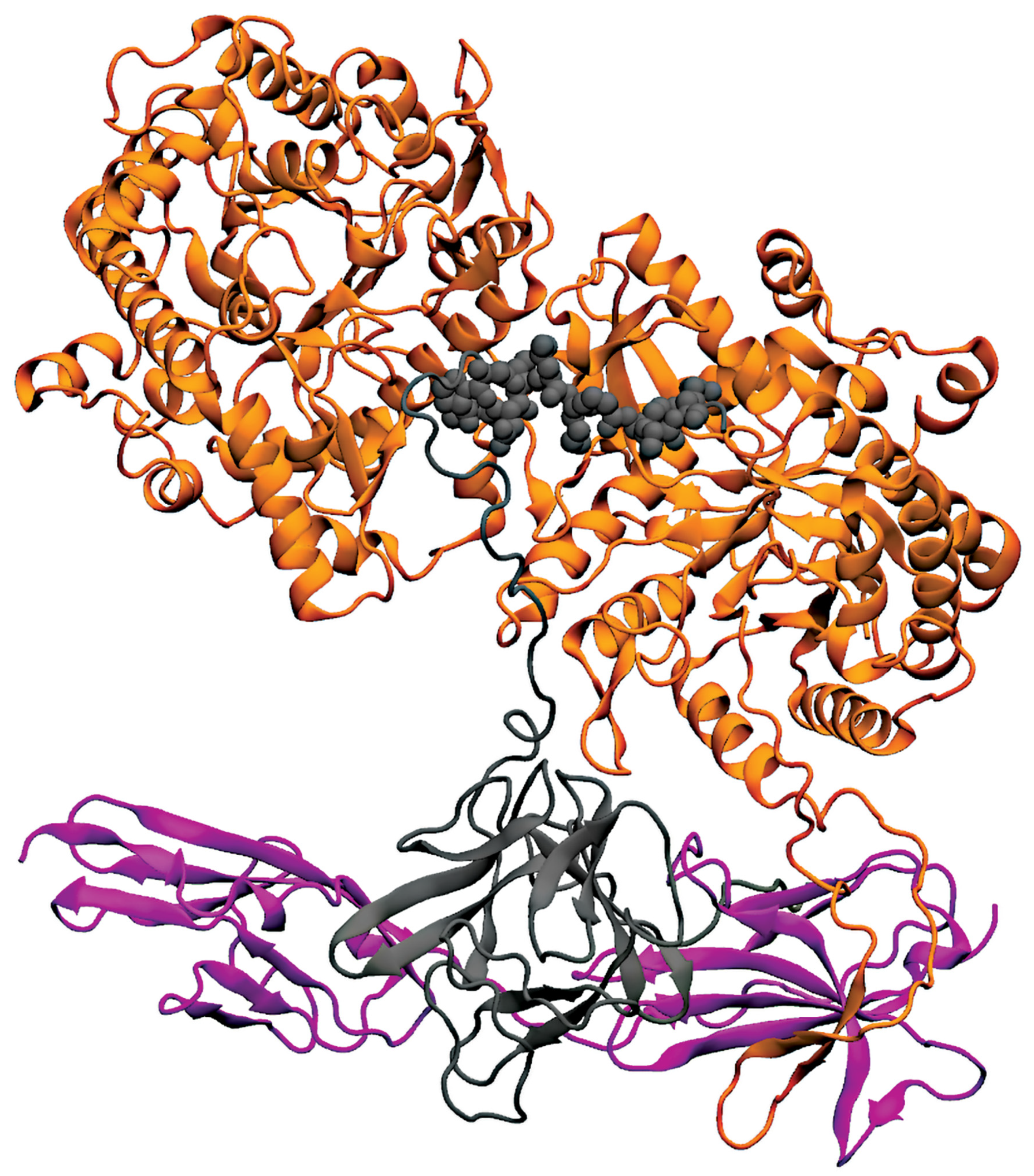
6. Interaction of Endocrine FGFs with FGFR1c and Klotho Proteins
6.1. FGF19/21-Receptor Interaction
6.2. FGF23 Forming Complex with FGFR1c and KLA
7. Applications and Clinical Trials Involving Endocrine FGFs

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trial Identifier | Status | Disease/Condition | Intervention | Phase | Outcomes |
|---|---|---|---|---|---|
| FGF19 | |||||
| NCT01943045 | Completed | Diabetes mellitus | NGM282 | Phase 2 | An ideal strategy for the replacement of bariatric surgery with equally effective and less invasive treatment for NASH [232]. |
| NCT01585025 | Completed | Chronic diarrhea | Obeticholic acid | Phase 2 | Obeticholic acid was beneficial in the treatment of bile acid diarrhea [228]. |
| NCT02443116 | Completed | NASH | NGM282 or (Aldafermin) | Phase 2 | The administration of 3 mg or 6 mg of NGM282 was well tolerated in patients and it reduced liver fat content as well as liver inflammation and fibrosis [229]. |
| NCT01265498 | Completed | NAFLD, NASH | Obeticholic acid | Phase 2 | Obeticholic acid improves the histological features of NASH [227]. |
| NCT02713243 | Completed | Primary bile acid diarrhea | LJN452 | Phase 2 | Tropifexor at a daily dose of 60 μg for 14 days had an acceptable safety and tolerability profile in patients [226]. |
| NCT02834780 | Active, not recruiting | Advanced HCC | H3B-657 | Phase 1 | Ongoing |
| NCT02508467 | Active, not recruiting | HCC | Fisogatinib (BLU-554) | Phase 1 | Ongoing |
| FGF21 | |||||
| NCT02097277 | Completed | T2DM | BMS-986036 | Phase 2 | Pegbelfermin is generally safe, well-tolerated, in patients with obesity and type 2 diabetes mellitus [52]. |
| NCT01673178 | Completed | T2DM | PF-05231023 | Phase 1 | PF-05231023 is essential in lowering TG in obese hypertriglyceridemic adult subjects with or without T2DM [238]. |
| NCT02413372 | Completed | NASH | BMS-986036 | Phase 2 | A 16 week well-tolerable dose of Pegbelfermin can reduce hepatic fat fraction in patient with NASH [51]. |
| NCT04541186 | Recruiting | Severe hypertriglyceridemia | BIO89-100D | Phase 2 | Ongoing |
| NCT03486899 | Active, not recruiting | Liver fibrosis, NAFLD, NASH | BMS-986036 | Phase 2 | Ongoing |
| NCT03486912 | Active, not recruiting | Hepatic cirrhosis, Liver fibrosis, NAFLD, NASH | BMS-986036 | Phase 2 | Ongoing |
| FGF23 | |||||
| NCT00830674 | Completed | XLH | KRN23 | Phase 1 | A favorable safety profile with Burosumab increased serum phosphorus levels, improved rickets and prevented early declines in growth in children with X-linked hypophosphataemia [239]. |
| NCT02163577 | Completed | XLH | Burosumab | Phase 2 | Burosumab improved renal tubular phosphate reabsorption, serum phosphorus levels in children with X-linked hypophosphatemia [243]. |
| NCT02750618 | Completed | XLH | Burosumab | Phase 2 | Treatment with Burosumab had a favorable safety profile, increased serum phosphorus, and improved rickets and prevented early declines in growth in children aged 1–4 years with X-linked hypophosphataemia [244]. |
| NCT02915705 | Completed | XLH | Burosumab, oral phosphate supplement, active vitamin D | Phase 3 | Significantly greater clinical improvements in rickets severity, growth among children with X-linked hypophosphataemia treated with Burosumab versus conventional therapy [194]. |
| NCT03920072 | Recruiting | XLH | Burosumab | Phase 3 | Ongoing |
| NCT04695860 | Recruiting | XLH | Burosumab | Phase 3 | Ongoing |
| NCT04320316 | Active, not recruiting | Epidermal nevus syndrome | Crysvita | Phase 4 | Ongoing |
8. Future Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Kharitonenkov, A.; Adams, A.C. Inventing new medicines: The FGF21 story. Mol. Metab. 2013, 3, 221–229. [Google Scholar] [CrossRef]
- Agrawal, S.; Maity, S.; Al-Raawi, Z.; Al-Ameer, M.; Kumar, T.K.S. Targeting Drugs Against Fibroblast Growth Factor(s)-Induced Cell Signaling. Curr. Drug Targets 2021, 22, 214–240. [Google Scholar] [CrossRef]
- Ornitz, D.M.; Itoh, N. Fibroblast growth factors. Genome Biol. 2001, 2, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Itoh, N.; Ornitz, D.M. Fibroblast growth factors: From molecular evolution to roles in development, metabolism and disease. J. Biochem. 2011, 149, 121–130. [Google Scholar] [CrossRef] [Green Version]
- Potthoff, M.J.; Kliewer, S.A.; Mangelsdorf, D.J. Endocrine fibroblast growth factors 15/19 and 21: From feast to famine. Genes Dev. 2012, 26, 312–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Degirolamo, C.; Sabbà, C.; Moschetta, A. Therapeutic potential of the endocrine fibroblast growth factors FGF19, FGF21 and FGF23. Nat. Rev. Drug Discov. 2015, 15, 51–69. [Google Scholar] [CrossRef]
- Dolegowska, K.; Marchelek-Mysliwiec, M.; Nowosiad-Magda, M.; Slawinski, M.; Dolegowska, B. FGF19 subfamily members: FGF19 and FGF21. J. Physiol. Biochem. 2019, 75, 229–240. [Google Scholar] [CrossRef] [Green Version]
- Kurosu, H.; Kuro-o, M. The Klotho gene family as a regulator of endocrine fibroblast growth factors. Mol. Cell. Endocrinol. 2009, 299, 72–78. [Google Scholar] [CrossRef]
- Itoh, N.; Ohta, H.; Konishi, M. Endocrine FGFs: Evolution, Physiology, Pathophysiology, and Pharmacotherapy. Front. Endocrinol. 2015, 6, 154. [Google Scholar] [CrossRef] [Green Version]
- Beenken, A.; Mohammadi, M. The FGF family: Biology, pathophysiology and therapy. Nat. Rev. Drug Discov. 2009, 8, 235–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babaknejad, N.; Nayeri, H.; Hemmati, R.; Bahrami, S.; Esmaillzadeh, A. An Overview of FGF19 and FGF21: The Therapeutic Role in the Treatment of the Metabolic Disorders and Obesity. Horm. Metab. Res. 2018, 50, 441–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, T.J.; Ladher, R.; McWhirter, J.; Murre, C.; Schoenwolf, G.C.; Mansour, S.L. Mouse FGF15 is the ortholog of human and chick FGF19, but is not uniquely required for otic induction. Dev. Biol. 2004, 269, 264–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harmer, N.J.; Pellegrini, L.; Chirgadze, D.; Fernandez-Recio, J.; Blundell, T.L. The crystal structure of fibroblast growth factor (FGF) 19 reveals novel features of the FGF family and offers a structural basis for its unusual receptor affinity. Biochemistry 2004, 43, 629–640. [Google Scholar] [CrossRef] [Green Version]
- Schaap, F.G. Role of fibroblast growth factor 19 in the control of glucose homeostasis. Curr. Opin. Clin. Nutr. Metab. Care 2012, 15, 386–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Owen, B.M.; Mangelsdorf, D.J.; Kliewer, S.A. Tissue-specific actions of the metabolic hormones FGF15/19 and FGF21. Trends Endocrinol. Metab. TEM 2015, 26, 22–29. [Google Scholar] [CrossRef] [Green Version]
- Hui, Q.; Jin, Z.; Li, X.; Liu, C.; Wang, X. FGF Family: From Drug Development to Clinical Application. Int. J. Mol. Sci. 2018, 19, 1875. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Li, Y. Therapeutic utilities of fibroblast growth factor 19. Expert Opin. Ther. Targets 2011, 15, 1307–1316. [Google Scholar] [CrossRef]
- Rysz, J.; Gluba-Brzózka, A.; Mikhailidis, D.P.; Banach, M. Fibroblast growth factor 19-targeted therapies for the treatment of metabolic disease. Expert Opin. Investig. Drugs 2015, 24, 603–610. [Google Scholar] [CrossRef] [PubMed]
- Kurosu, H.; Kuro-o, M. Endocrine fibroblast growth factors as regulators of metabolic homeostasis. BioFactors 2009, 35, 52–60. [Google Scholar] [CrossRef]
- Zhang, J.; Li, Y. Fibroblast growth factor 21, the endocrine FGF pathway and novel treatments for metabolic syndrome. Drug Discov. Today 2014, 19, 579–589. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, T.; Utsunomiya, Y.; Hoshikawa, M.; Ohuchi, H.; Itoh, N. Structure and expression of a novel human FGF, FGF-19, expressed in the fetal brain. Biochim. Biophys. Acta Gene Struct. Expr. 1999, 1444, 148–151. [Google Scholar] [CrossRef]
- Tomlinson, E.; Fu, L.; John, L.; Hultgren, B.; Huang, X.; Renz, M.; Stephan, J.P.; Tsai, S.P.; Powell-Braxton, L.; French, D.; et al. Transgenic Mice Expressing Human Fibroblast Growth Factor-19 Display Increased Metabolic Rate and Decreased Adiposity. Endocrinology 2002, 143, 1741–1747. [Google Scholar] [CrossRef]
- Holt, J.A.; Luo, G.; Billin, A.N.; Bisi, J.; McNeill, Y.Y.; Kozarsky, K.F.; Donahee, M.; Wang, D.Y.; Mansfield, T.A.; Kliewer, S.A.; et al. Definition of a novel growth factor-dependent signal cascade for the suppression of bile acid biosynthesis. Genes Dev. 2003, 17, 1581–1591. [Google Scholar] [CrossRef] [Green Version]
- Choi, M.; Moschetta, A.; Bookout, A.L.; Peng, L.; Umetani, M.; Holmstrom, S.R.; Suino-Powell, K.; Xu, H.E.; Richardson, J.A.; Gerard, R.D.; et al. Identification of a hormonal basis for gallbladder filling. Nat. Med. 2006, 12, 1253–1255. [Google Scholar] [CrossRef] [PubMed]
- Ryan, K.K.; Kohli, R.; Gutierrez-Aguilar, R.; Gaitonde, S.G.; Woods, S.C.; Seeley, R.J. Fibroblast growth factor-19 action in the brain reduces food intake and body weight and improves glucose tolerance in male rats. Endocrinology 2013, 154, 9–15. [Google Scholar] [CrossRef]
- Morton, G.J.; Matsen, M.E.; Bracy, D.P.; Meek, T.H.; Nguyen, H.T.; Stefanovski, D.; Bergman, R.N.; Wasserman, D.H.; Schwartz, M.W. FGF19 action in the brain induces insulin-independent glucose lowering. J. Clin. Investig. 2013, 123, 4799–4808. [Google Scholar] [CrossRef] [Green Version]
- Maeda, T.; Kanzaki, H.; Chiba, T.; Ao, J.; Kanayama, K.; Maruta, S.; Kusakabe, Y.; Saito, T.; Kobayashi, K.; Kiyono, S.; et al. Serum fibroblast growth factor 19 serves as a potential novel biomarker for hepatocellular carcinoma. BMC Cancer 2019, 19, 1088. [Google Scholar] [CrossRef] [PubMed]
- Myojin, Y.; Kodama, T.; Maesaka, K.; Motooka, D.; Sato, Y.; Tanaka, S.; Abe, Y.; Ohkawa, K.; Mita, E.; Hayashi, Y.; et al. ST6GAL1 Is a Novel Serum Biomarker for Lenvatinib-Susceptible FGF19-Driven Hepatocellular Carcinoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2021, 27, 1150–1161. [Google Scholar] [CrossRef] [PubMed]
- Raja, A.; Park, I.; Haq, F.; Ahn, S.M. FGF19- FGFR4 Signaling in Hepatocellular Carcinoma. Cells 2019, 8, 536. [Google Scholar] [CrossRef] [Green Version]
- Kim, R.D.; Sarker, D.; Meyer, T.; Yau, T.; Macarulla, T.; Park, J.W.; Choo, S.P.; Hollebecque, A.; Sung, M.W.; Lim, H.Y.; et al. First-in-Human Phase I Study of Fisogatinib (BLU-554) Validates Aberrant FGF19 Signaling as a Driver Event in Hepatocellular Carcinoma. Cancer Discov. 2019, 9, 1696–1707. [Google Scholar] [CrossRef] [Green Version]
- Gao, L.; Wang, X.; Tang, Y.; Huang, S.; Hu, C.A.; Teng, Y. FGF19/FGFR4 signaling contributes to the resistance of hepatocellular carcinoma to sorafenib. J. Exp. Clin. Cancer Res. CR 2017, 36, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joshi, J.J.; Coffey, H.; Corcoran, E.; Tsai, J.; Huang, C.L.; Ichikawa, K.; Prajapati, S.; Hao, M.H.; Bailey, S.; Wu, J.; et al. H3B-6527 Is a Potent and Selective Inhibitor of FGFR4 in FGF19-Driven Hepatocellular Carcinoma. Cancer Res. 2017, 77, 6999–7013. [Google Scholar] [CrossRef] [Green Version]
- Harrison, S.A.; Neff, G.; Guy, C.D.; Bashir, M.R.; Paredes, A.H.; Frias, J.P.; Younes, Z.; Trotter, J.F.; Gunn, N.T.; Moussa, S.E.; et al. Efficacy and Safety of Aldafermin, an Engineered FGF19 Analog, in a Randomized, Double-Blind, Placebo-Controlled Trial of Patients With Nonalcoholic Steatohepatitis. Gastroenterology 2021, 160, 219–231. [Google Scholar] [CrossRef]
- Sanyal, A.J.; Ling, L.; Beuers, U.; DePaoli, A.M.; Lieu, H.D.; Harrison, S.A.; Hirschfield, G.M. Potent suppression of hydrophobic bile acids by aldafermin, an FGF19 analogue, across metabolic and cholestatic liver diseases. JHEP Rep. Innov. Hepatol. 2021, 3, 100255. [Google Scholar] [CrossRef]
- Nishimura, T.; Nakatake, Y.; Konishi, M.; Itoh, N. Identification of a novel FGF, FGF-21, preferentially expressed in the liver. Biochim. Biophys. Acta Gene Struct. Expr. 2000, 1492, 203–206. [Google Scholar] [CrossRef]
- Muise, E.S.; Azzolina, B.; Kuo, D.W.; El-Sherbeini, M.; Tan, Y.; Yuan, X.; Mu, J.; Thompson, J.R.; Berger, J.P.; Wong, K.K. Adipose fibroblast growth factor 21 is up-regulated by peroxisome proliferator-activated receptor gamma and altered metabolic states. Mol. Pharmacol. 2008, 74, 403–412. [Google Scholar] [CrossRef] [PubMed]
- Gälman, C.; Lundåsen, T.; Kharitonenkov, A.; Bina, H.A.; Eriksson, M.; Hafström, I.; Dahlin, M.; Amark, P.; Angelin, B.; Rudling, M. The circulating metabolic regulator FGF21 is induced by prolonged fasting and PPARalpha activation in man. Cell Metab. 2008, 8, 169–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lundåsen, T.; Hunt, M.C.; Nilsson, L.M.; Sanyal, S.; Angelin, B.; Alexson, S.E.; Rudling, M. PPARalpha is a key regulator of hepatic FGF21. Biochem. Biophys. Res. Commun. 2007, 360, 437–440. [Google Scholar] [CrossRef] [Green Version]
- Badman, M.K.; Pissios, P.; Kennedy, A.R.; Koukos, G.; Flier, J.S.; Maratos-Flier, E. Hepatic Fibroblast Growth Factor 21 Is Regulated by PPARα and Is a Key Mediator of Hepatic Lipid Metabolism in Ketotic States. Cell Metab. 2007, 5, 426–437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kharitonenkov, A.; Shiyanova, T.L.; Koester, A.; Ford, A.M.; Micanovic, R.; Galbreath, E.J.; Sandusky, G.E.; Hammond, L.J.; Moyers, J.S.; Owens, R.A.; et al. FGF-21 as a novel metabolic regulator. J. Clin. Investig. 2005, 115, 1627–1635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kharitonenkov, A.; Dunbar, J.D.; Bina, H.A.; Bright, S.; Moyers, J.S.; Zhang, C.; Ding, L.; Micanovic, R.; Mehrbod, S.F.; Knierman, M.D.; et al. FGF-21/FGF-21 receptor interaction and activation is determined by betaKlotho. J. Cell. Physiol. 2008, 215, 1–7. [Google Scholar] [CrossRef]
- Suzuki, M.; Uehara, Y.; Motomura-Matsuzaka, K.; Oki, J.; Koyama, Y.; Kimura, M.; Asada, M.; Komi-Kuramochi, A.; Oka, S.; Imamura, T. betaKlotho is required for fibroblast growth factor (FGF) 21 signaling through FGF receptor (FGFR) 1c and FGFR3c. Mol. Endocrinol. 2008, 22, 1006–1014. [Google Scholar] [CrossRef] [Green Version]
- Yie, J.; Hecht, R.; Patel, J.; Stevens, J.; Wang, W.; Hawkins, N.; Steavenson, S.; Smith, S.; Winters, D.; Fisher, S.; et al. FGF21 N- and C-termini play different roles in receptor interaction and activation. FEBS Lett. 2009, 583, 19–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Micanovic, R.; Raches, D.W.; Dunbar, J.D.; Driver, D.A.; Bina, H.A.; Dickinson, C.D.; Kharitonenkov, A. Different roles of N- and C-termini in the functional activity of FGF21. J. Cell. Physiol. 2009, 219, 227–234. [Google Scholar] [CrossRef]
- Kharitonenkov, A.; Beals, J.M.; Micanovic, R.; Strifler, B.A.; Rathnachalam, R.; Wroblewski, V.J.; Li, S.; Koester, A.; Ford, A.M.; Coskun, T.; et al. Rational design of a fibroblast growth factor 21-based clinical candidate, LY2405319. PLoS ONE 2013, 8, e58575. [Google Scholar] [CrossRef] [Green Version]
- Gaich, G.; Chien, J.Y.; Fu, H.; Glass, L.C.; Deeg, M.A.; Holland, W.L.; Kharitonenkov, A.; Bumol, T.; Schilske, H.K.; Moller, D.E. The effects of LY2405319, an FGF21 analog, in obese human subjects with type 2 diabetes. Cell Metab. 2013, 18, 333–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, A.C.; Halstead, C.A.; Hansen, B.C.; Irizarry, A.R.; Martin, J.A.; Myers, S.R.; Reynolds, V.L.; Smith, H.W.; Wroblewski, V.J.; Kharitonenkov, A. LY2405319, an Engineered FGF21 Variant, Improves the Metabolic Status of Diabetic Monkeys. PLoS ONE 2013, 8, e65763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, Q.; Zhong, L.; Zhang, J.; Wang, Y.; Bornstein, S.R.; Triggle, C.R.; Ding, H.; Lam, K.S.; Xu, A. FGF21 maintains glucose homeostasis by mediating the cross talk between liver and brain during prolonged fasting. Diabetes 2014, 63, 4064–4075. [Google Scholar] [CrossRef] [Green Version]
- Inagaki, T.; Dutchak, P.; Zhao, G.; Ding, X.; Gautron, L.; Parameswara, V.; Li, Y.; Goetz, R.; Mohammadi, M.; Esser, V.; et al. Endocrine Regulation of the Fasting Response by PPARα-Mediated Induction of Fibroblast Growth Factor 21. Cell Metab. 2007, 5, 415–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verzijl, C.R.C.; Van De Peppel, I.P.; Struik, D.; Jonker, J.W. Pegbelfermin (BMS-986036): An investigational PEGylated fibroblast growth factor 21 analogue for the treatment of nonalcoholic steatohepatitis. Expert Opin. Investig. Drugs 2020, 29, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, A.; Charles, E.D.; Neuschwander-Tetri, B.A.; Loomba, R.; Harrison, S.A.; Abdelmalek, M.F.; Lawitz, E.J.; Halegoua-DeMarzio, D.; Kundu, S.; Noviello, S.; et al. Pegbelfermin (BMS-986036), a PEGylated fibroblast growth factor 21 analogue, in patients with non-alcoholic steatohepatitis: A randomised, double-blind, placebo-controlled, phase 2a trial. Lancet 2018, 392, 2705–2717. [Google Scholar] [CrossRef]
- Charles, E.D.; Neuschwander-Tetri, B.A.; Pablo Frias, J.; Kundu, S.; Luo, Y.; Tirucherai, G.S.; Christian, R. Pegbelfermin (BMS-986036), PEGylated FGF21, in Patients with Obesity and Type 2 Diabetes: Results from a Randomized Phase 2 Study. Obesity 2019, 27, 41–49. [Google Scholar] [CrossRef]
- Zhu, L.; Zhao, H.; Liu, J.; Cai, H.; Wu, B.; Liu, Z.; Zhou, S.; Liu, Q.; Li, X.; Bao, B.; et al. Dynamic folding modulation generates FGF21 variant against diabetes. EMBO Rep. 2021, 22, e51352. [Google Scholar] [CrossRef]
- Yamashita, T.; Yoshioka, M.; Itoh, N. Identification of a novel fibroblast growth factor, FGF-23, preferentially expressed in the ventrolateral thalamic nucleus of the brain. Biochem. Biophys. Res. Commun. 2000, 277, 494–498. [Google Scholar] [CrossRef] [PubMed]
- Shimada, T.; Mizutani, S.; Muto, T.; Yoneya, T.; Hino, R.; Takeda, S.; Takeuchi, Y.; Fujita, T.; Fukumoto, S.; Yamashita, T. Cloning and characterization of FGF23 as a causative factor of tumor-induced osteomalacia. Proc. Natl. Acad. Sci. USA 2001, 98, 6500–6505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, K.E.; Carn, G.; Lorenz-Depiereux, B.; Benet-Pages, A.; Strom, T.M.; Econs, M.J. Autosomal-dominant hypophosphatemic rickets (ADHR) mutations stabilize FGF-23. Kidney Int. 2001, 60, 2079–2086. [Google Scholar] [CrossRef] [Green Version]
- Michigami, T.; Ozono, K. Roles of Phosphate in Skeleton. Front. Endocrinol. 2019, 10, 180. [Google Scholar] [CrossRef]
- Takeda, E.; Taketani, Y.; Morita, K.; Tatsumi, S.; Katai, K.; Nii, T.; Yamamoto, H.; Miyamoto, K. Molecular mechanisms of mammalian inorganic phosphate homeostasis. Adv. Enzym. Regul. 2000, 40, 285–302. [Google Scholar] [CrossRef]
- Liu, S.; Tang, W.; Zhou, J.; Stubbs, J.R.; Luo, Q.; Pi, M.; Quarles, L.D. Fibroblast growth factor 23 is a counter-regulatory phosphaturic hormone for vitamin D. J. Am. Soc. Nephrol. JASN 2006, 17, 1305–1315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Figurek, A.; Rroji, M.; Spasovski, G. The Complexity of FGF23 Effects on Cardiomyocytes in Normal and Uremic Milieu. Cells 2021, 10, 1266. [Google Scholar] [CrossRef] [PubMed]
- Erben, R.G.; Andrukhova, O. FGF23-Klotho signaling axis in the kidney. Bone 2017, 100, 62–68. [Google Scholar] [CrossRef] [Green Version]
- Shimada, T.; Hasegawa, H.; Yamazaki, Y.; Muto, T.; Hino, R.; Takeuchi, Y.; Fujita, T.; Nakahara, K.; Fukumoto, S.; Yamashita, T. FGF-23 Is a Potent Regulator of Vitamin D Metabolism and Phosphate Homeostasis. J. Bone Miner. Res. 2003, 19, 429–435. [Google Scholar] [CrossRef] [Green Version]
- Yamashita, T.; Konishi, M.; Miyake, A.; Inui, K.; Itoh, N. Fibroblast growth factor (FGF)-23 inhibits renal phosphate reabsorption by activation of the mitogen-activated protein kinase pathway. J. Biol. Chem. 2002, 277, 173–176. [Google Scholar] [CrossRef] [Green Version]
- Krajisnik, T.; Björklund, P.; Marsell, R.; Ljunggren, O.; Akerström, G.; Jonsson, K.B.; Westin, G.; Larsson, T.E. Fibroblast growth factor-23 regulates parathyroid hormone and 1alpha-hydroxylase expression in cultured bovine parathyroid cells. J. Endocrinol. 2007, 195, 125–131. [Google Scholar] [CrossRef] [Green Version]
- Mirza, M.A.; Larsson, A.; Melhus, H.; Lind, L.; Larsson, T.E. Serum intact FGF23 associate with left ventricular mass, hypertrophy and geometry in an elderly population. Atherosclerosis 2009, 207, 546–551. [Google Scholar] [CrossRef]
- Mirza, M.A.; Hansen, T.; Johansson, L.; Ahlström, H.; Larsson, A.; Lind, L.; Larsson, T.E. Relationship between circulating FGF23 and total body atherosclerosis in the community. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transpl. Assoc. Eur. Ren. Assoc. 2009, 24, 3125–3131. [Google Scholar] [CrossRef]
- Bär, L.; Feger, M.; Fajol, A.; Klotz, L.-O.; Zeng, S.; Lang, F.; Hocher, B.; Föller, M. Insulin suppresses the production of fibroblast growth factor 23 (FGF23). Proc. Natl. Acad. Sci. USA 2018, 115, 5804–5809. [Google Scholar] [CrossRef] [Green Version]
- Agrawal, A.; Ni, P.; Agoro, R.; White, K.E.; DiMarchi, R.D. Identification of a second Klotho interaction site in the C terminus of FGF23. Cell Rep. 2021, 34, 108665. [Google Scholar] [CrossRef]
- Suzuki, Y.; Kuzina, E.; An, S.J.; Tome, F.; Mohanty, J.; Li, W.; Lee, S.; Liu, Y.; Lax, I.; Schlessinger, J. FGF23 contains two distinct high-affinity binding sites enabling bivalent interactions with α-Klotho. Proc. Natl. Acad. Sci. USA 2020, 117, 31800–31807. [Google Scholar] [CrossRef]
- Agoro, R.; Park, M.Y.; Le Henaff, C.; Jankauskas, S.; Gaias, A.; Chen, G.; Mohammadi, M.; Sitara, D. C-FGF23 peptide alleviates hypoferremia during acute inflammation. Haematologica 2021, 106, 391–403. [Google Scholar] [CrossRef] [Green Version]
- Kurose, H.; Bito, T.; Adachi, T.; Shimizu, M.; Noji, S.; Ohuchi, H. Expression of Fibroblast growth factor 19 (Fgf19) during chicken embryogenesis and eye development, compared with Fgf15 expression in the mouse. Gene Expr. Patterns GEP 2004, 4, 687–693. [Google Scholar] [CrossRef] [PubMed]
- Somm, E.; Jornayvaz, F.R. Fibroblast Growth Factor 15/19: From Basic Functions to Therapeutic Perspectives. Endocr. Rev. 2018, 39, 960–989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krejci, P.; Kunova, M.; Kubikova, I.; Trantirek, L.; Kozubik, A.; Dvorak, P. Expression of FGF19 in human embryonic stem cells. Stem Cells 2013, 31, 2582–2584. [Google Scholar] [CrossRef]
- Xie, M.H.; Holcomb, I.; Deuel, B.; Dowd, P.; Huang, A.; Vagts, A.; Foster, J.; Liang, J.; Brush, J.; Gu, Q.; et al. FGF-19, a novel fibroblast growth factor with unique specificity for FGFR4. Cytokine 1999, 11, 729–735. [Google Scholar] [CrossRef]
- Naugler, W.E.; Tarlow, B.D.; Fedorov, L.M.; Taylor, M.; Pelz, C.; Li, B.; Darnell, J.; Grompe, M. Fibroblast Growth Factor Signaling Controls Liver Size in Mice With Humanized Livers. Gastroenterology 2015, 149, 728–740. [Google Scholar] [CrossRef] [Green Version]
- Jansen, P.L. Fibroblast growth factor 19, a double-edged sword. Hepat. Oncol. 2017, 4, 1–4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofmann, A.F. The continuing importance of bile acids in liver and intestinal disease. Arch. Intern. Med. 1999, 159, 2647–2658. [Google Scholar] [CrossRef] [PubMed]
- van Erpecum, K.J.; Schaap, F.G. Intestinal failure to produce FGF19: A culprit in intestinal failure-associated liver disease? J. Hepatol. 2015, 62, 1231–1233. [Google Scholar] [CrossRef] [Green Version]
- Kir, S.; Kliewer, S.A.; Mangelsdorf, D.J. Roles of FGF19 in liver metabolism. Cold Spring Harb. Symp. Quant. Biol. 2011, 76, 139–144. [Google Scholar] [CrossRef] [Green Version]
- Kir, S.; Beddow, S.A.; Samuel, V.T.; Miller, P.; Previs, S.F.; Suino-Powell, K.; Xu, H.E.; Shulman, G.I.; Kliewer, S.A.; Mangelsdorf, D.J. FGF19 as a postprandial, insulin-independent activator of hepatic protein and glycogen synthesis. Science 2011, 331, 1621–1624. [Google Scholar] [CrossRef] [Green Version]
- Sinal, C.J.; Tohkin, M.; Miyata, M.; Ward, J.M.; Lambert, G.; Gonzalez, F.J. Targeted Disruption of the Nuclear Receptor FXR/BAR Impairs Bile Acid and Lipid Homeostasis. Cell 2000, 102, 731–744. [Google Scholar] [CrossRef] [Green Version]
- Al-Khaifi, A.; Rudling, M.; Angelin, B. An FXR Agonist Reduces Bile Acid Synthesis Independently of Increases in FGF19 in Healthy Volunteers. Gastroenterology 2018, 155, 1012–1016. [Google Scholar] [CrossRef]
- Pozo, M.J.; Camello, P.J.; Mawe, G.M. Chemical mediators of gallbladder dysmotility. Curr. Med. Chem. 2004, 11, 1801–1812. [Google Scholar] [CrossRef]
- Shaffer, E.A. Review article: Control of gall-bladder motor function. Aliment. Pharmacol. Ther. 2000, 14 (Suppl. 2), 2–8. [Google Scholar] [CrossRef]
- Huang, X.; Yang, C.; Luo, Y.; Jin, C.; Wang, F.; McKeehan, W.L. FGFR4 prevents hyperlipidemia and insulin resistance but underlies high-fat diet induced fatty liver. Diabetes 2007, 56, 2501–2510. [Google Scholar] [CrossRef] [Green Version]
- Jensen-Cody, S.O.; Flippo, K.H.; Claflin, K.E.; Yavuz, Y.; Sapouckey, S.A.; Walters, G.C.; Usachev, Y.M.; Atasoy, D.; Gillum, M.P.; Potthoff, M.J. FGF21 Signals to Glutamatergic Neurons in the Ventromedial Hypothalamus to Suppress Carbohydrate Intake. Cell Metab. 2020, 32, 273–286. [Google Scholar] [CrossRef] [PubMed]
- Bodine, S.C.; Stitt, T.N.; Gonzalez, M.; Kline, W.O.; Stover, G.L.; Bauerlein, R.; Zlotchenko, E.; Scrimgeour, A.; Lawrence, J.C.; Glass, D.J.; et al. Akt/mTOR pathway is a crucial regulator of skeletal muscle hypertrophy and can prevent muscle atrophy in vivo. Nat. Cell Biol. 2001, 3, 1014–1019. [Google Scholar] [CrossRef] [PubMed]
- Ohanna, M.; Sobering, A.K.; Lapointe, T.; Lorenzo, L.; Praud, C.; Petroulakis, E.; Sonenberg, N.; Kelly, P.A.; Sotiropoulos, A.; Pende, M. Atrophy of S6K1(-/-) skeletal muscle cells reveals distinct mTOR effectors for cell cycle and size control. Nat. Cell Biol. 2005, 7, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Benoit, B.; Meugnier, E.; Castelli, M.; Chanon, S.; Vieille-Marchiset, A.; Durand, C.; Bendridi, N.; Pesenti, S.; Monternier, P.A.; Durieux, A.C.; et al. Fibroblast growth factor 19 regulates skeletal muscle mass and ameliorates muscle wasting in mice. Nat. Med. 2017, 23, 990–996. [Google Scholar] [CrossRef] [PubMed]
- Guo, A.; Li, K.; Tian, H.C.; Fan, Z.; Chen, Q.N.; Yang, Y.F.; Yu, J.; Wu, Y.X.; Xiao, Q. FGF19 protects skeletal muscle against obesity-induced muscle atrophy, metabolic derangement and abnormal irisin levels via the AMPK/SIRT-1/PGC-α pathway. J. Cell. Mol. Med. 2021, 25, 3585–3600. [Google Scholar] [CrossRef] [PubMed]
- Guo, A.; Li, K.; Xiao, Q. Fibroblast growth factor 19 alleviates palmitic acid-induced mitochondrial dysfunction and oxidative stress via the AMPK/PGC-1α pathway in skeletal muscle. Biochem. Biophys. Res. Commun. 2020, 526, 1069–1076. [Google Scholar] [CrossRef] [PubMed]
- Powell, D.J.; Turban, S.; Gray, A.; Hajduch, E.; Hundal, H.S. Intracellular ceramide synthesis and protein kinase Czeta activation play an essential role in palmitate-induced insulin resistance in rat L6 skeletal muscle cells. Biochem. J. 2004, 382, 619–629. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.N.; Yang, Z.X.; Ren, F.Z.; Fang, B. FGF19 alleviates palmitate-induced atrophy in C2C12 cells by inhibiting mitochondrial overload and insulin resistance. Int. J. Biol. Macromol. 2020, 158, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Marchelek-Myśliwiec, M.; Dziedziejko, V.; Nowosiad-Magda, M.; Dołęgowska, K.; Dołęgowska, B.; Pawlik, A.; Safranow, K.; Wiśniewska, M.; Stępniewska, J.; Domański, M.; et al. Chronic Kidney Disease Is Associated with Increased Plasma Levels of Fibroblast Growth Factors 19 and 21. Kidney Blood Press. Res. 2019, 44, 1207–1218. [Google Scholar] [CrossRef] [PubMed]
- Brandl, K.; Hartmann, P.; Jih, L.J.; Pizzo, D.P.; Argemi, J.; Ventura-Cots, M.; Coulter, S.; Liddle, C.; Ling, L.; Rossi, S.J.; et al. Dysregulation of serum bile acids and FGF19 in alcoholic hepatitis. J. Hepatol. 2018, 69, 396–405. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Ley, S.H.; Hu, F.B. Global aetiology and epidemiology of type 2 diabetes mellitus and its complications. Nat. Rev. Endocrinol. 2018, 14, 88. [Google Scholar] [CrossRef]
- Hu, J.; Liu, Z.; Tong, Y.; Mei, Z.; Xu, A.; Zhou, P.; Chen, X.; Tang, W.; Zhou, Z.; Xiao, Y. Fibroblast Growth Factor 19 Levels Predict Subclinical Atherosclerosis in Men With Type 2 Diabetes. Front. Endocrinol. 2020, 11, 282. [Google Scholar] [CrossRef] [PubMed]
- Burtenshaw, D.; Kitching, M.; Redmond, E.M.; Megson, I.L.; Cahill, P.A. Reactive Oxygen Species (ROS), Intimal Thickening, and Subclinical Atherosclerotic Disease. Front. Cardiovasc. Med. 2019, 6, 89. [Google Scholar] [CrossRef]
- Zhou, M.; Learned, R.M.; Rossi, S.J.; Tian, H.; DePaoli, A.M.; Ling, L. Therapeutic FGF19 promotes HDL biogenesis and transhepatic cholesterol efflux to prevent atherosclerosis. J. Lipid Res. 2019, 60, 550–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubler, S.; Dlugash, J.; Yuceoglu, Y.Z.; Kumral, T.; Branwood, A.W.; Grishman, A. New type of cardiomyopathy associated with diabetic glomerulosclerosis. Am. J. Cardiol. 1972, 30, 595–602. [Google Scholar] [CrossRef]
- Rijzewijk, L.J.; van der Meer, R.W.; Lamb, H.J.; de Jong, H.W.; Lubberink, M.; Romijn, J.A.; Bax, J.J.; de Roos, A.; Twisk, J.W.; Heine, R.J.; et al. Altered myocardial substrate metabolism and decreased diastolic function in nonischemic human diabetic cardiomyopathy: Studies with cardiac positron emission tomography and magnetic resonance imaging. J. Am. Coll. Cardiol. 2009, 54, 1524–1532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Longo, M.; Paolini, E.; Meroni, M.; Dongiovanni, P. Remodeling of Mitochondrial Plasticity: The Key Switch from NAFLD/NASH to HCC. Int. J. Mol. Sci. 2021, 22, 4173. [Google Scholar] [CrossRef] [PubMed]
- Tan, Q.; Li, F.; Wang, G.; Xia, W.; Li, Z.; Niu, X.; Ji, W.; Yuan, H.; Xu, Q.; Luo, Q.; et al. Identification of FGF19 as a prognostic marker and potential driver gene of lung squamous cell carcinomas in Chinese smoking patients. Oncotarget 2016, 7, 18394–18402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; Li, Z.; Han, Q.; Cheng, Y.; Ji, W.; Yang, Y.; Lu, S.; Xia, W. Enhanced autocrine FGF19/FGFR4 signaling drives the progression of lung squamous cell carcinoma, which responds to mTOR inhibitor AZD2104. Oncogene 2020, 39, 3507–3521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, A.L.; Coulter, S.; Liddle, C.; Wong, A.; Eastham-Anderson, J.; French, D.M.; Peterson, A.S.; Sonoda, J. FGF19 regulates cell proliferation, glucose and bile acid metabolism via FGFR4-dependent and independent pathways. PLoS ONE 2011, 6, e17868. [Google Scholar] [CrossRef] [Green Version]
- Tiong, K.H.; Tan, B.S.; Choo, H.L.; Chung, F.F.; Hii, L.W.; Tan, S.H.; Khor, N.T.; Wong, S.F.; See, S.J.; Tan, Y.F.; et al. Fibroblast growth factor receptor 4 (FGFR4) and fibroblast growth factor 19 (FGF19) autocrine enhance breast cancer cells survival. Oncotarget 2016, 7, 57633. [Google Scholar] [CrossRef] [Green Version]
- Frigolet, M.E.; Gutiérrez-Aguilar, R. The colors of adipose tissue. Gac. Med. Mex. 2020, 156, 142–149. [Google Scholar] [CrossRef] [Green Version]
- Samms, R.J.; Smith, D.P.; Cheng, C.C.; Antonellis, P.P.; Perfield, J.W.; Kharitonenkov, A.; Gimeno, R.E.; Adams, A.C. Discrete Aspects of FGF21 In Vivo Pharmacology Do Not Require UCP1. Cell Rep. 2015, 11, 991–999. [Google Scholar] [CrossRef] [Green Version]
- Chartoumpekis, D.V.; Habeos, I.G.; Ziros, P.G.; Psyrogiannis, A.I.; Kyriazopoulou, V.E.; Papavassiliou, A.G. Brown adipose tissue responds to cold and adrenergic stimulation by induction of FGF21. Mol. Med. 2011, 17, 736–740. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.; Brychta, R.J.; Linderman, J.; Smith, S.; Chen, K.Y.; Celi, F.S. Mild cold exposure modulates fibroblast growth factor 21 (FGF21) diurnal rhythm in humans: Relationship between FGF21 levels, lipolysis, and cold-induced thermogenesis. J. Clin. Endocrinol. Metab. 2013, 98, E98–E102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Onyango, I.G.; Khan, S.M.; Bennett, J.P., Jr. Mitochondria in the pathophysiology of Alzheimer’s and Parkinson’s diseases. Front. Biosci. 2017, 22, 854–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mäkelä, J.; Tselykh, T.V.; Maiorana, F.; Eriksson, O.; Do, H.T.; Mudò, G.; Korhonen, L.T.; Belluardo, N.; Lindholm, D. Fibroblast growth factor-21 enhances mitochondrial functions and increases the activity of PGC-1α in human dopaminergic neurons via Sirtuin-1. Springerplus 2014, 3, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chau, M.D.L.; Gao, J.; Yang, Q.; Wu, Z.; Gromada, J. Fibroblast growth factor 21 regulates energy metabolism by activating the AMPK-SIRT1-PGC-1alpha pathway. Proc. Natl. Acad. Sci. USA 2010, 107, 12553–12558. [Google Scholar] [CrossRef] [Green Version]
- Dushay, J.R.; Toschi, E.; Mitten, E.K.; Fisher, F.M.; Herman, M.A.; Maratos-Flier, E. Fructose ingestion acutely stimulates circulating FGF21 levels in humans. Mol. Metab. 2014, 4, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Søberg, S.; Andersen, E.S.; Dalgaard, N.B.; Jarlhelt, I.; Hansen, N.L.; Hoffmann, N.; Vilsbøll, T.; Chenchar, A.; Jensen, M.; Grevengoed, T.J.; et al. FGF21, a liver hormone that inhibits alcohol intake in mice, increases in human circulation after acute alcohol ingestion and sustained binge drinking at Oktoberfest. Mol. Metab. 2018, 11, 96–103. [Google Scholar] [CrossRef]
- Maida, A.; Zota, A.; Sjøberg, K.A.; Schumacher, J.; Sijmonsma, T.P.; Pfenninger, A.; Christensen, M.M.; Gantert, T.; Fuhrmeister, J.; Rothermel, U.; et al. A liver stress-endocrine nexus promotes metabolic integrity during dietary protein dilution. J. Clin. Investig. 2016, 126, 3263–3278. [Google Scholar] [CrossRef] [Green Version]
- von Holstein-Rathlou, S.; BonDurant, L.D.; Peltekian, L.; Naber, M.C.; Yin, T.C.; Claflin, K.E.; Urizar, A.I.; Madsen, A.N.; Ratner, C.; Holst, B.; et al. FGF21 Mediates Endocrine Control of Simple Sugar Intake and Sweet Taste Preference by the Liver. Cell Metab. 2016, 23, 335–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, C.M.; Laeger, T.; Albarado, D.C.; McDougal, D.H.; Berthoud, H.-R.; Münzberg, H.; Morrison, C.D. Low protein-induced increases in FGF21 drive UCP1-dependent metabolic but not thermoregulatory endpoints. Sci Rep. 2017, 7, 8209. [Google Scholar] [CrossRef]
- Hill, C.M.; Laeger, T.; Dehner, M.; Albarado, D.C.; Clarke, B.; Wanders, D.; Burke, S.J.; Collier, J.J.; Qualls-Creekmore, E.; Solon-Biet, S.M.; et al. FGF21 Signals Protein Status to the Brain and Adaptively Regulates Food Choice and Metabolism. Cell Rep. 2019, 27, 2934–2947. [Google Scholar] [CrossRef] [Green Version]
- Dong, Y.; Xie, M.; Jiang, Y.; Xiao, N.; Du, X.; Zhang, W.; Tosser-Klopp, G.; Wang, J.; Yang, S.; Liang, J.; et al. Sequencing and automated whole-genome optical mapping of the genome of a domestic goat (Capra hircus). Nat. Biotechnol. 2012, 31, 135–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Zhang, P.; Zhang, X.; Li, X.; Bai, Y.; Ao, Y.; Hexig, B.; Guo, X.; Liu, D. Fgf21 knockout mice generated using CRISPR/Cas9 reveal genetic alterations that may affect hair growth. Gene 2020, 733, 144242. [Google Scholar] [CrossRef]
- Mraz, M.; Bartlova, M.; Lacinova, Z.; Michalsky, D.; Kasalicky, M.; Haluzikova, D.; Matoulek, M.; Dostalova, I.; Humenanska, V.; Haluzik, M. Serum concentrations and tissue expression of a novel endocrine regulator fibroblast growth factor-21 in patients with type 2 diabetes and obesity. Clin. Endocrinol. 2009, 71, 369–375. [Google Scholar] [CrossRef]
- Zhen, E.Y.; Jin, Z.; Ackermann, B.L.; Thomas, M.K.; Gutierrez, J.A. Circulating FGF21 proteolytic processing mediated by fibroblast activation protein. Biochem. J. 2016, 473, 605–614. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Fang, Q.; Gao, F.; Fan, J.; Zhou, J.; Wang, X.; Zhang, H.; Pan, X.; Bao, Y.; Xiang, K.; et al. Fibroblast growth factor 21 levels are increased in nonalcoholic fatty liver disease patients and are correlated with hepatic triglyceride. J. Hepatol. 2010, 53, 934–940. [Google Scholar] [CrossRef] [PubMed]
- Tas, E.; Bai, S.; Ou, X.; Mercer, K.; Lin, H.; Mansfield, K.; Buchmann, R.; Diaz, E.C.; Oden, J.; Børsheim, E.; et al. Fibroblast Growth Factor-21 to Adiponectin Ratio: A Potential Biomarker to Monitor Liver Fat in Children With Obesity. Front. Endocrinol. 2020, 11, 654. [Google Scholar] [CrossRef] [PubMed]
- Suomalainen, A.; Elo, J.M.; Pietiläinen, K.H.; Hakonen, A.H.; Sevastianova, K.; Korpela, M.; Isohanni, P.; Marjavaara, S.K.; Tyni, T.; Kiuru-Enari, S.; et al. FGF-21 as a biomarker for muscle-manifesting mitochondrial respiratory chain deficiencies: A diagnostic study. Lancet Neurol. 2011, 10, 806–818. [Google Scholar] [CrossRef]
- Suomalainen, A. Fibroblast growth factor 21: A novel biomarker for human muscle-manifesting mitochondrial disorders. Expert Opin. Med. Diagn. 2013, 7, 313–317. [Google Scholar] [CrossRef] [PubMed]
- Koene, S.; de Laat, P.; van Tienoven, D.H.; Vriens, D.; Brandt, A.M.; Sweep, F.C.; Rodenburg, R.J.; Donders, A.R.; Janssen, M.C.; Smeitink, J.A. Serum FGF21 levels in adult m.3243A>G carriers: Clinical implications. Neurology 2014, 83, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Su, S.L.; Wang, W.F.; Wu, S.L.; Wu, H.M.; Chang, J.C.; Huang, C.S.; Cheng, W.L.; Soong, B.W.; Lee, Y.C.; Li, J.Y.; et al. FGF21 in ataxia patients with spinocerebellar atrophy and mitochondrial disease. Clin. Chim. Acta 2012, 414, 225–227. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Ji, K.; Ma, X.; Liu, S.; Li, W.; Zhao, Y.; Yan, C. Accuracy of FGF-21 and GDF-15 for the diagnosis of mitochondrial disorders: A meta-analysis. Ann. Clin. Transl. Neurol. 2020, 7, 1204–1213. [Google Scholar] [CrossRef]
- Khan, N.A.; Nikkanen, J.; Yatsuga, S.; Jackson, C.; Wang, L.; Pradhan, S.; Kivelä, R.; Pessia, A.; Velagapudi, V.; Suomalainen, A. mTORC1 Regulates Mitochondrial Integrated Stress Response and Mitochondrial Myopathy Progression. Cell Metab. 2017, 26, 419–428. [Google Scholar] [CrossRef]
- Forsström, S.; Jackson, C.B.; Carroll, C.J.; Kuronen, M.; Pirinen, E.; Pradhan, S.; Marmyleva, A.; Auranen, M.; Kleine, I.-M.; Khan, N.A.; et al. Fibroblast Growth Factor 21 Drives Dynamics of Local and Systemic Stress Responses in Mitochondrial Myopathy with mtDNA Deletions. Cell Metab. 2019, 30, 1040–1054. [Google Scholar] [CrossRef]
- Kang, Y.E.; Kim, J.T.; Lim, M.; Oh, C.; Liu, L.; Jung, S.-N.; Won, H.-R.; Lee, K.; Chang, J.W.; Yi, H.-S. Association between circulating fibroblast growth factor 21 and aggressiveness in thyroid cancer. Cancers 2019, 11, 1154. [Google Scholar] [CrossRef] [Green Version]
- Knott, M.E.; Ranuncolo, S.M.; Nuñez, M.; Armanasco, E.; Puricelli, L.I.; De Lorenzo, M.S. Levels of Fibroblast Growth Factor 21 (FGF21) in serum as diagnostic biomarker in patients with breast cancer. Clin. Trials 2015, 75, 1577. [Google Scholar]
- Knott, M.E.; Minatta, J.N.; Roulet, L.; Gueglio, G.; Pasik, L.; Ranuncolo, S.M.; Nuñez, M.; Puricelli, L.; De Lorenzo, M.S. Circulating Fibroblast Growth Factor 21 (Fgf21) as Diagnostic and Prognostic Biomarker in Renal Cancer. J. Mol. Biomark. Diagn. 2016, 1, 015. [Google Scholar] [CrossRef]
- Cymbaluk-Płoska, A.; Gargulińska, P.; Chudecka-Głaz, A.; Kwiatkowski, S.; Pius-Sadowska, E.; Machaliński, B. The Suitability of FGF21 and FGF23 as New Biomarkers in Endometrial Cancer Patients. Diagnostics 2020, 10, 414. [Google Scholar] [CrossRef]
- Foltz, I.N.; Hu, S.; King, C.; Wu, X.; Yang, C.; Wang, W.; Weiszmann, J.; Stevens, J.; Chen, J.S.; Nuanmanee, N.; et al. Treating diabetes and obesity with an FGF21-mimetic antibody activating the βKlotho/FGFR1c receptor complex. Sci. Transl. Med. 2012, 4, 162ra153. [Google Scholar] [CrossRef]
- Quarles, L.D. Evidence for a bone-kidney axis regulating phosphate homeostasis. J. Clin. Investig. 2003, 112, 642–646. [Google Scholar] [CrossRef] [Green Version]
- Yoshiko, Y.; Wang, H.; Minamizaki, T.; Ijuin, C.; Yamamoto, R.; Suemune, S.; Kozai, K.; Tanne, K.; Aubin, J.E.; Maeda, N. Mineralized tissue cells are a principal source of FGF23. Bone 2007, 40, 1565–1573. [Google Scholar] [CrossRef]
- Ben-Dov, I.Z.; Galitzer, H.; Lavi-Moshayoff, V.; Goetz, R.; Kuro-o, M.; Mohammadi, M.; Sirkis, R.; Naveh-Many, T.; Silver, J. The parathyroid is a target organ for FGF23 in rats. J. Clin. Investig. 2007, 117, 4003–4008. [Google Scholar] [CrossRef] [Green Version]
- Masuyama, R.; Stockmans, I.; Torrekens, S.; Van Looveren, R.; Maes, C.; Carmeliet, P.; Bouillon, R.; Carmeliet, G. Vitamin D receptor in chondrocytes promotes osteoclastogenesis and regulates FGF23 production in osteoblasts. J. Clin. Investig. 2006, 116, 3150–3159. [Google Scholar] [CrossRef]
- Murali, S.K.; Roschger, P.; Zeitz, U.; Klaushofer, K.; Andrukhova, O.; Erben, R.G. FGF23 Regulates Bone Mineralization in a 1,25(OH)2D3 and Klotho-Independent Manner. J. Bone Miner. Res. 2015, 31, 129–142. [Google Scholar] [CrossRef]
- Addison, W.N.; Azari, F.; Sørensen, E.S.; Kaartinen, M.T.; McKee, M.D. Pyrophosphate Inhibits Mineralization of Osteoblast Cultures by Binding to Mineral, Up-regulating Osteopontin, and Inhibiting Alkaline Phosphatase Activity. J. Biol. Chem. 2007, 282, 15872–15883. [Google Scholar] [CrossRef] [Green Version]
- Clinkenbeard, E.L.; Hanudel, M.R.; Stayrook, K.R.; Appaiah, H.N.; Farrow, E.G.; Cass, T.A.; Summers, L.J.; Ip, C.S.; Hum, J.M.; Thomas, J.C.; et al. Erythropoietin stimulates murine and human fibroblast growth factor-23, revealing novel roles for bone and bone marrow. Haematologica 2017, 102, e427–e430. [Google Scholar] [CrossRef] [PubMed]
- Daryadel, A.; Bettoni, C.; Haider, T.; Imenez Silva, P.H.; Schnitzbauer, U.; Pastor-Arroyo, E.M.; Wenger, R.H.; Gassmann, M.; Wagner, C.A. Erythropoietin stimulates fibroblast growth factor 23 (FGF23) in mice and men. Pflügers Arch. Eur. J. Physiol. 2018, 470, 1569–1582. [Google Scholar] [CrossRef] [PubMed]
- Mattinzoli, D.; Rastaldi, M.P.; Ikehata, M.; Armelloni, S.; Pignatari, C.; Giardino, L.A.; Li, M.; Alfieri, C.M.; Regalia, A.; Riccardi, D.; et al. FGF23-regulated production of Fetuin-A (AHSG) in osteocytes. Bone 2016, 83, 35–47. [Google Scholar] [CrossRef]
- Mattinzoli, D.; Ikehata, M.; Tsugawa, K.; Alfieri, C.M.; Dongiovanni, P.; Trombetta, E.; Valenti, L.; Puliti, A.; Lazzari, L.; Messa, P. FGF23 and Fetuin-A Interaction in the Liver and in the Circulation. Int. J. Biol. Sci. 2018, 14, 586–598. [Google Scholar] [CrossRef] [Green Version]
- Li, D.J.; Fu, H.; Zhao, T.; Ni, M.; Shen, F.M. Exercise-stimulated FGF23 promotes exercise performance via controlling the excess reactive oxygen species production and enhancing mitochondrial function in skeletal muscle. Metab. Clin. Exp. 2016, 65, 747–756. [Google Scholar] [CrossRef]
- Liu, S.; Guo, R.; Simpson, L.G.; Xiao, Z.S.; Burnham, C.E.; Quarles, L.D. Regulation of fibroblastic growth factor 23 expression but not degradation by PHEX. J. Biol. Chem. 2003, 278, 37419–37426. [Google Scholar] [CrossRef] [Green Version]
- Bacchetta, J.; Bardet, C.; Prié, D. Physiology of FGF23 and overview of genetic diseases associated with renal phosphate wasting. Metabolism 2020, 103, 153865. [Google Scholar] [CrossRef]
- Chong, W.H.; Molinolo, A.A.; Chen, C.C.; Collins, M.T. Tumor-induced osteomalacia. Endocr. Relat. Cancer 2011, 18, R53–R77. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Zhou, J.; Tang, W.; Jiang, X.; Rowe, D.W.; Quarles, L.D. Pathogenic role of Fgf23 in Hyp mice. Am. J. Physiol.-Endocrinol. Metab. 2006, 291, E38–E49. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, K.; Kimura, T.; Shiizaki, K. Biological and Clinical Effects of Calciprotein Particles on Chronic Kidney Disease-Mineral and Bone Disorder. Int. J. Endocrinol. 2018, 2018, 5282389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouksila, M.; Mrad, M.; Kaabachi, W.; Kalai, E.; Smaoui, W.; Rekik, S.; Krir, A.; Issaoui, N.; Hamzaoui, K.; Sahli, H.; et al. Correlation of Fgf23 and Balp with Bone Mineral Density in Hemodialysis Patients. J. Med. Biochem. 2019, 38, 418–426. [Google Scholar] [CrossRef]
- Wolf, M. Forging Forward with 10 Burning Questions on FGF23 in Kidney Disease. J. Am. Soc. Nephrol. 2010, 21, 1427–1435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benet-Pagès, A.; Orlik, P.; Strom, T.M.; Lorenz-Depiereux, B. An FGF23 missense mutation causes familial tumoral calcinosis with hyperphosphatemia. Hum. Mol. Genet. 2004, 14, 385–390. [Google Scholar] [CrossRef] [PubMed]
- Faul, C.; Amaral, A.P.; Oskouei, B.; Hu, M.-C.; Sloan, A.; Isakova, T.; Gutiérrez, O.M.; Aguillon-Prada, R.; Lincoln, J.; Hare, J.M.; et al. FGF23 induces left ventricular hypertrophy. J. Clin. Investig. 2011, 121, 4393–4408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leifheit-Nestler, M.; Große Siemer, R.; Flasbart, K.; Richter, B.; Kirchhoff, F.; Ziegler, W.H.; Klintschar, M.; Becker, J.U.; Erbersdobler, A.; Aufricht, C.; et al. Induction of cardiac FGF23/FGFR4 expression is associated with left ventricular hypertrophy in patients with chronic kidney disease. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transpl. Assoc. Eur. Ren. Assoc. 2016, 31, 1088–1099. [Google Scholar] [CrossRef] [Green Version]
- Mhatre, K.N.; Wakula, P.; Klein, O.; Bisping, E.; Völkl, J.; Pieske, B.; Heinzel, F.R. Crosstalk between FGF23- and angiotensin II-mediated Ca2+ signaling in pathological cardiac hypertrophy. Cell. Mol. Life Sci. 2018, 75, 4403–4416. [Google Scholar] [CrossRef]
- Böckmann, I.; Lischka, J.; Richter, B.; Deppe, J.; Rahn, A.; Fischer, D.-C.; Heineke, J.; Haffner, D.; Leifheit-Nestler, M. FGF23-Mediated Activation of Local RAAS Promotes Cardiac Hypertrophy and Fibrosis. Int. J. Mol. Sci. 2019, 20, 4634. [Google Scholar] [CrossRef] [Green Version]
- Itoh, N.; Nakayama, Y.; Konishi, M. Roles of FGFs As Paracrine or Endocrine Signals in Liver Development, Health, and Disease. Front. Cell Dev. Biol. 2016, 4, 30. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Li, Y. Understanding the structure-function relationship between FGF19 and its mitogenic and metabolic activities. Adv. Exp. Med. Biol. 2012, 728, 195–213. [Google Scholar] [CrossRef]
- Wu, X.; Ge, H.; Lemon, B.; Vonderfecht, S.; Baribault, H.; Weiszmann, J.; Gupte, J.; Gardner, J.; Lindberg, R.; Wang, Z.; et al. Separating mitogenic and metabolic activities of fibroblast growth factor 19 (FGF19). Proc. Natl. Acad. Sci. USA 2010, 107, 14158–14163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mori, S.; Hatori, N.; Kawaguchi, N.; Hamada, Y.; Shih, T.C.; Wu, C.Y.; Lam, K.S.; Matsuura, N.; Yamamoto, H.; Takada, Y.K.; et al. The integrin-binding defective FGF2 mutants potently suppress FGF2 signalling and angiogenesis. Biosci. Rep. 2017, 37, BSR20170173. [Google Scholar] [CrossRef] [Green Version]
- Paluck, S.J.; Nguyen, T.H.; Lee, J.P.; Maynard, H.D. A Heparin-Mimicking Block Copolymer Both Stabilizes and Increases the Activity of Fibroblast Growth Factor 2 (FGF2). Biomacromolecules 2016, 17, 3386–3395. [Google Scholar] [CrossRef]
- Motomura, K.; Hagiwara, A.; Komi-Kuramochi, A.; Hanyu, Y.; Honda, E.; Suzuki, M.; Kimura, M.; Oki, J.; Asada, M.; Sakaguchi, N.; et al. An FGF1:FGF2 chimeric growth factor exhibits universal FGF receptor specificity, enhanced stability and augmented activity useful for epithelial proliferation and radioprotection. Biochim. Biophys. Acta 2008, 1780, 1432–1440. [Google Scholar] [CrossRef]
- Beenken, A.; Mohammadi, M. The structural biology of the FGF19 subfamily. Adv. Exp. Med. Biol. 2012, 728, 1–24. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Niu, J.; Lin, H.; Zhao, J.; Liu, Y.; Song, Z.; Xiang, C.; Wang, X.; Yang, Y.; Li, X.; et al. Paracrine-endocrine FGF chimeras as potent therapeutics for metabolic diseases. EBioMedicine 2019, 48, 462–477. [Google Scholar] [CrossRef] [Green Version]
- Plotnikov, A.N.; Hubbard, S.R.; Schlessinger, J.; Mohammadi, M. Crystal structures of two FGF-FGFR complexes reveal the determinants of ligand-receptor specificity. Cell 2000, 101, 413–424. [Google Scholar] [CrossRef] [Green Version]
- Niu, J.; Zhao, J.; Wu, J.; Qiao, G.; Gu, J.; Zhou, C.; Li, Q.; Ying, L.; Wang, D.; Lin, H.; et al. Curtailing FGF19′s mitogenicity by suppressing its receptor dimerization ability. Proc. Natl. Acad. Sci. USA 2020, 117, 29025–29034. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, A.; Parlee, S.; Perez-Tilve, D.; Li, P.; Pan, J.; Mroz, P.A.; Kruse Hansen, A.M.; Andersen, B.; Finan, B.; Kharitonenkov, A.; et al. Molecular elements in FGF19 and FGF21 defining KLB/FGFR activity and specificity. Mol. Metab. 2018, 13, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Krejci, P.; Prochazkova, J.; Bryja, V.; Kozubik, A.; Wilcox, W.R. Molecular pathology of the fibroblast growth factor family. Hum. Mutat. 2009, 30, 1245–1255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuro-o, M. Endocrine FGFs and Klothos: Emerging concepts. Trends Endocrinol. Metab. TEM 2008, 19, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Maity, S.; Al-Ameer, M.; Gundampati, R.K.; Agrawal, S.; Kumar, T.K.S. Heparin-Binding Affinity Tag: A Novel Affinity Tag for Simple and Efficient Purification of Recombinant Proteins. Methods Mol. Biol. 2021, 2178, 311–328. [Google Scholar] [CrossRef] [PubMed]
- Kerr, R.; Agrawal, S.; Maity, S.; Koppolu, B.; Jayanthi, S.; Suresh Kumar, G.; Gundampati, R.K.; McNabb, D.S.; Zaharoff, D.A.; Kumar, T.K.S. Design of a thrombin resistant human acidic fibroblast growth factor (hFGF1) variant that exhibits enhanced cell proliferation activity. Biochem. Biophys. Res. Commun. 2019, 518, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Thallapuranam, S.K.; Agarwal, S.; Gindampati, R.K.; Jayanthi, S.; Wang, T.; Jones, J.; Kolenc, O.; Lam, N.; Niyonshuti, I.; Balachandran, K.; et al. Engineered Fgf1 And Fgf2 Compositions and Methods of Use Thereof. U.S. Patent Application 16/356,872, 19 September 2019. [Google Scholar]
- Blaber, M.; DiSalvo, J.; Thomas, K.A. X-ray crystal structure of human acidic fibroblast growth factor. Biochemistry 1996, 35, 2086–2094. [Google Scholar] [CrossRef] [Green Version]
- Raman, R.; Venkataraman, G.; Ernst, S.; Sasisekharan, V.; Sasisekharan, R. Structural specificity of heparin binding in the fibroblast growth factor family of proteins. Proc. Natl. Acad. Sci. USA 2003, 100, 2357–2362. [Google Scholar] [CrossRef] [Green Version]
- Ago, H.; Kitagawa, Y.; Fujishima, A.; Matsuura, Y.; Katsube, Y. Crystal structure of basic fibroblast growth factor at 1.6 A resolution. J. Biochem. 1991, 110, 360–363. [Google Scholar] [CrossRef] [PubMed]
- Schlessinger, J.; Plotnikov, A.N.; Ibrahimi, O.A.; Eliseenkova, A.V.; Yeh, B.K.; Yayon, A.; Linhardt, R.J.; Mohammadi, M. Crystal structure of a ternary FGF-FGFR-heparin complex reveals a dual role for heparin in FGFR binding and dimerization. Mol. Cell 2000, 6, 743–750. [Google Scholar] [CrossRef]
- Yeh, B.K.; Igarashi, M.; Eliseenkova, A.V.; Plotnikov, A.N.; Sher, I.; Ron, D.; Aaronson, S.A.; Mohammadi, M. Structural basis by which alternative splicing confers specificity in fibroblast growth factor receptors. Proc. Natl. Acad. Sci. USA 2003, 100, 2266–2271. [Google Scholar] [CrossRef] [Green Version]
- Pellegrini, L.; Burke, D.F.; von Delft, F.; Mulloy, B.; Blundell, T.L. Crystal structure of fibroblast growth factor receptor ectodomain bound to ligand and heparin. Nature 2000, 407, 1029–1034. [Google Scholar] [CrossRef]
- Ornitz, D.M.; Yayon, A.; Flanagan, J.G.; Svahn, C.M.; Levi, E.; Leder, P. Heparin is required for cell-free binding of basic fibroblast growth factor to a soluble receptor and for mitogenesis in whole cells. Mol. Cell. Biol. 1992, 12, 240–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Ge, H.; Gupte, J.; Weiszmann, J.; Shimamoto, G.; Stevens, J.; Hawkins, N.; Lemon, B.; Shen, W.; Xu, J.; et al. Co-receptor requirements for fibroblast growth factor-19 signaling. J. Biol. Chem. 2007, 282, 29069–29072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Choi, J.; Mohanty, J.; Sousa, L.P.; Tome, F.; Pardon, E.; Steyaert, J.; Lemmon, M.A.; Lax, I.; Schlessinger, J. Structures of β-klotho reveal a ‘zip code’-like mechanism for endocrine FGF signalling. Nature 2018, 553, 501–505. [Google Scholar] [CrossRef]
- Goetz, R.; Beenken, A.; Ibrahimi, O.A.; Kalinina, J.; Olsen, S.K.; Eliseenkova, A.V.; Xu, C.; Neubert, T.A.; Zhang, F.; Linhardt, R.J.; et al. Molecular insights into the klotho-dependent, endocrine mode of action of fibroblast growth factor 19 subfamily members. Mol. Cell. Biol. 2007, 27, 3417–3428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunshee, D.R.; Bainbridge, T.W.; Kljavin, N.M.; Zavala-Solorio, J.; Schroeder, A.C.; Chan, R.; Corpuz, R.; Wong, M.; Zhou, W.; Deshmukh, G.; et al. Fibroblast Activation Protein Cleaves and Inactivates Fibroblast Growth Factor 21. J. Biol. Chem. 2016, 291, 5986–5996. [Google Scholar] [CrossRef] [Green Version]
- Kuro-o, M.; Matsumura, Y.; Aizawa, H.; Kawaguchi, H.; Suga, T.; Utsugi, T.; Ohyama, Y.; Kurabayashi, M.; Kaname, T.; Kume, E.; et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature 1997, 390, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Li, S.A.; Watanabe, M.; Yamada, H.; Nagai, A.; Kinuta, M.; Takei, K. Immunohistochemical localization of Klotho protein in brain, kidney, and reproductive organs of mice. Cell Struct. Funct. 2004, 29, 91–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urakawa, I.; Yamazaki, Y.; Shimada, T.; Iijima, K.; Hasegawa, H.; Okawa, K.; Fujita, T.; Fukumoto, S.; Yamashita, T. Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature 2006, 444, 770–774. [Google Scholar] [CrossRef] [PubMed]
- Kurosu, H.; Ogawa, Y.; Miyoshi, M.; Yamamoto, M.; Nandi, A.; Rosenblatt, K.P.; Baum, M.G.; Schiavi, S.; Hu, M.C.; Moe, O.W.; et al. Regulation of fibroblast growth factor-23 signaling by klotho. J. Biol. Chem. 2006, 281, 6120–6123. [Google Scholar] [CrossRef] [Green Version]
- Fernandes-Freitas, I.; Owen, B.M. Metabolic roles of endocrine fibroblast growth factors. Curr. Opin. Pharmacol. 2015, 25, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Imel, E.A.; Biggin, A.; Schindeler, A.; Munns, C.F. FGF23, Hypophosphatemia, and Emerging Treatments. JBMR Plus 2019, 3, e10190. [Google Scholar] [CrossRef] [Green Version]
- Araya, K.; Fukumoto, S.; Backenroth, R.; Takeuchi, Y.; Nakayama, K.; Ito, N.; Yoshii, N.; Yamazaki, Y.; Yamashita, T.; Silver, J.; et al. A novel mutation in fibroblast growth factor 23 gene as a cause of tumoral calcinosis. J. Clin. Endocrinol. Metab. 2005, 90, 5523–5527. [Google Scholar] [CrossRef] [Green Version]
- Yamazaki, Y.; Okazaki, R.; Shibata, M.; Hasegawa, Y.; Satoh, K.; Tajima, T.; Takeuchi, Y.; Fujita, T.; Nakahara, K.; Yamashita, T.; et al. Increased circulatory level of biologically active full-length FGF-23 in patients with hypophosphatemic rickets/osteomalacia. J. Clin. Endocrinol. Metab. 2002, 87, 4957–4960. [Google Scholar] [CrossRef] [PubMed]
- Shimada, T.; Muto, T.; Urakawa, I.; Yoneya, T.; Yamazaki, Y.; Okawa, K.; Takeuchi, Y.; Fujita, T.; Fukumoto, S.; Yamashita, T. Mutant FGF-23 responsible for autosomal dominant hypophosphatemic rickets is resistant to proteolytic cleavage and causes hypophosphatemia in vivo. Endocrinology 2002, 143, 3179–3182. [Google Scholar] [CrossRef] [PubMed]
- White, K.E.; Evans, W.E.; O’Riordan, J.L.H.; Speer, M.C.; Econs, M.J.; Lorenz-Depiereux, B.; Grabowski, M.; Meitinger, T.; Strom, T.M. Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. Nat. Genet. 2000, 26, 345–348. [Google Scholar] [CrossRef] [PubMed]
- Berndt, T.J.; Craig, T.A.; McCormick, D.J.; Lanske, B.; Sitara, D.; Razzaque, M.S.; Pragnell, M.; Bowe, A.E.; O’Brien, S.P.; Schiavi, S.C.; et al. Biological activity of FGF-23 fragments. Pflug. Arch. Eur. J. Physiol. 2007, 454, 615–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, K.; Jeanneau, C.; Tarp, M.A.; Benet-Pagès, A.; Lorenz-Depiereux, B.; Bennett, E.P.; Mandel, U.; Strom, T.M.; Clausen, H. Polypeptide GalNAc-transferase T3 and familial tumoral calcinosis. Secretion of fibroblast growth factor 23 requires O-glycosylation. J. Biol. Chem. 2006, 281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, E.R.; McMahon, L.P.; Holt, S.G. Fibroblast growth factor 23. Ann. Clin. Biochem. 2014, 51, 308–309. [Google Scholar] [CrossRef]
- Tagliabracci, V.S.; Engel, J.L.; Wiley, S.E.; Xiao, J.; Gonzalez, D.J.; Nidumanda Appaiah, H.; Koller, A.; Nizet, V.; White, K.E.; Dixon, J.E. Dynamic regulation of FGF23 by Fam20C phosphorylation, GalNAc-T3 glycosylation, and furin proteolysis. Proc. Natl. Acad. Sci. USA 2014, 111, 5520–5525. [Google Scholar] [CrossRef] [Green Version]
- Tomiyama, K.; Maeda, R.; Urakawa, I.; Yamazaki, Y.; Tanaka, T.; Ito, S.; Nabeshima, Y.; Tomita, T.; Odori, S.; Hosoda, K.; et al. Relevant use of Klotho in FGF19 subfamily signaling system in vivo. Proc. Natl. Acad. Sci. USA 2010, 107, 1666–1671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, B.C.; Wang, M.; Blackmore, C.; Desnoyers, L.R. Liver-specific activities of FGF19 require Klotho beta. J. Biol. Chem. 2007, 282, 27277–27284. [Google Scholar] [CrossRef] [Green Version]
- Kuro-o, M. The Klotho proteins in health and disease. Nat. Rev. Nephrol. 2019, 15, 27–44. [Google Scholar] [CrossRef] [PubMed]
- Kurosu, H.; Choi, M.; Ogawa, Y.; Dickson, A.S.; Goetz, R.; Eliseenkova, A.V.; Mohammadi, M.; Rosenblatt, K.P.; Kliewer, S.A.; Kuro-o, M. Tissue-specific expression of betaKlotho and fibroblast growth factor (FGF) receptor isoforms determines metabolic activity of FGF19 and FGF21. J. Biol. Chem. 2007, 282, 26687–26695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, S.Y.; Lu, Y.W.; Richardson, J.; Min, X.; Weiszmann, J.; Richards, W.G.; Wang, Z.; Zhang, Z.; Zhang, J.; Li, Y. A systematic dissection of sequence elements determining β-Klotho and FGF interaction and signaling. Sci. Rep. 2018, 8, 11045. [Google Scholar] [CrossRef]
- Kuzina, E.S.; Ung, P.M.; Mohanty, J.; Tome, F.; Choi, J.; Pardon, E.; Steyaert, J.; Lax, I.; Schlessinger, A.; Schlessinger, J.; et al. Structures of ligand-occupied β-Klotho complexes reveal a molecular mechanism underlying endocrine FGF specificity and activity. Proc. Natl. Acad. Sci. USA 2019, 116, 7819–7824. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Lemon, B.; Li, X.; Gupte, J.; Weiszmann, J.; Stevens, J.; Hawkins, N.; Shen, W.; Lindberg, R.; Chen, J.L.; et al. C-terminal tail of FGF19 determines its specificity toward Klotho co-receptors. J. Biol. Chem. 2008, 283, 33304–33309. [Google Scholar] [CrossRef] [Green Version]
- Matei, A.; Bilha, S.C.; Constantinescu, D.; Pavel-Tanasa, M.; Cianga, P.; Covic, A.; Branisteanu, D.D. Body composition, adipokines, FGF23-Klotho and bone in kidney transplantation: Is there a link? J. Nephrol. 2021, 1–12. [Google Scholar] [CrossRef]
- Ho, B.B.; Bergwitz, C. FGF23 signalling and physiology. J. Mol. Endocrinol. 2021, 66, R23–R32. [Google Scholar] [CrossRef]
- Feger, M.; Ewendt, F.; Strotmann, J.; Schäffler, H.; Kempe-Teufel, D.; Glosse, P.; Stangl, G.I.; Föller, M. Glucocorticoids dexamethasone and prednisolone suppress fibroblast growth factor 23 (FGF23). J. Mol. Med. 2021, 99, 699–711. [Google Scholar] [CrossRef]
- Hensel, N.; Schön, A.; Konen, T.; Lübben, V.; Förthmann, B.; Baron, O.; Grothe, C.; Leifheit-Nestler, M.; Claus, P.; Haffner, D. Fibroblast growth factor 23 signaling in hippocampal cells: Impact on neuronal morphology and synaptic density. J. Neurochem. 2016, 137, 756–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, G.; Liu, Y.; Goetz, R.; Fu, L.; Jayaraman, S.; Hu, M.C.; Moe, O.W.; Liang, G.; Li, X.; Mohammadi, M. α-Klotho is a non-enzymatic molecular scaffold for FGF23 hormone signalling. Nature 2018, 553, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Min, X.; Weiszmann, J.; Johnstone, S.; Wang, W.; Yu, X.; Romanow, W.; Thibault, S.; Li, Y.; Wang, Z. Agonistic β-Klotho antibody mimics fibroblast growth factor 21 (FGF21) functions. J. Biol. Chem. 2018, 293, 14678–14688. [Google Scholar] [CrossRef] [Green Version]
- Imamura, T. Physiological functions and underlying mechanisms of fibroblast growth factor (FGF) family members: Recent findings and implications for their pharmacological application. Biol. Pharm. Bull. 2014, 37, 1081–1089. [Google Scholar] [CrossRef] [Green Version]
- Yie, J.; Wang, W.; Deng, L.; Tam, L.T.; Stevens, J.; Chen, M.M.; Li, Y.; Xu, J.; Lindberg, R.; Hecht, R.; et al. Understanding the physical interactions in the FGF21/FGFR/β-Klotho complex: Structural requirements and implications in FGF21 signaling. Chem. Biol. Drug Des. 2012, 79, 398–410. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Tan, Y.; Gu, J.; Liu, Y.; Song, L.; Niu, J.; Zhao, L.; Srinivasan, L.; Lin, Q.; Deng, J.; et al. Uncoupling the Mitogenic and Metabolic Functions of FGF1 by Tuning FGF1-FGF Receptor Dimer Stability. Cell Rep. 2017, 20, 1717–1728. [Google Scholar] [CrossRef] [Green Version]
- Wei, W.; Dutchak, P.A.; Wang, X.; Ding, X.; Wang, X.; Bookout, A.L.; Goetz, R.; Mohammadi, M.; Gerard, R.D.; Dechow, P.C.; et al. Fibroblast growth factor 21 promotes bone loss by potentiating the effects of peroxisome proliferator-activated receptor γ. Proc. Natl. Acad. Sci. USA 2012, 109, 3143–3148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, C.; Jin, C.; Li, X.; Wang, F.; McKeehan, W.L.; Luo, Y. Differential specificity of endocrine FGF19 and FGF21 to FGFR1 and FGFR4 in complex with KLB. PLoS ONE 2012, 7, e33870. [Google Scholar] [CrossRef] [Green Version]
- Goetz, R.; Nakada, Y.; Hu, M.C.; Kurosu, H.; Wang, L.; Nakatani, T.; Shi, M.; Eliseenkova, A.V.; Razzaque, M.S.; Moe, O.W.; et al. Isolated C-terminal tail of FGF23 alleviates hypophosphatemia by inhibiting FGF23-FGFR-Klotho complex formation. Proc. Natl. Acad. Sci. USA 2010, 107, 407–412. [Google Scholar] [CrossRef] [Green Version]
- Goetz, R.; Ohnishi, M.; Kir, S.; Kurosu, H.; Wang, L.; Pastor, J.; Ma, J.; Gai, W.; Kuro-o, M.; Razzaque, M.S.; et al. Conversion of a paracrine fibroblast growth factor into an endocrine fibroblast growth factor. J. Biol. Chem. 2012, 287, 29134–29146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurosu, H.; Yamamoto, M.; Clark, J.D.; Pastor, J.V.; Nandi, A.; Gurnani, P.; McGuinness, O.P.; Chikuda, H.; Yamaguchi, M.; Kawaguchi, H.; et al. Suppression of aging in mice by the hormone Klotho. Science 2005, 309, 1829–1833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powers, C.J.; McLeskey, S.W.; Wellstein, A. Fibroblast growth factors, their receptors and signaling. Endocr. Relat. Cancer 2000, 7, 165–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kilkenny, D.M.; Rocheleau, J.V. The FGF21 Receptor Signaling Complex: Klothoβ, FGFR1c, and Other Regulatory Interactions. Vitam. Horm. 2016, 101, 17–58. [Google Scholar] [CrossRef] [PubMed]
- Camilleri, M.; Nord, S.L.; Burton, D.; Oduyebo, I.; Zhang, Y.; Chen, J.; Im, K.; Bhad, P.; Badman, M.K.; Sanders, D.S.; et al. Randomised clinical trial: Significant biochemical and colonic transit effects of the farnesoid X receptor agonist tropifexor in patients with primary bile acid diarrhoea. Aliment. Pharmacol. Ther. 2020, 52, 808–820. [Google Scholar] [CrossRef] [PubMed]
- Neuschwander-Tetri, B.A.; Loomba, R.; Sanyal, A.J.; Lavine, J.E.; Van Natta, M.L.; Abdelmalek, M.F.; Chalasani, N.; Dasarathy, S.; Diehl, A.M.; Hameed, B.; et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): A multicentre, randomised, placebo-controlled trial. Lancet 2015, 385, 956–965. [Google Scholar] [CrossRef] [Green Version]
- Walters, J.R.F.; Johnston, I.M.; Nolan, J.D.; Vassie, C.; Pruzanski, M.E.; Shapiro, D.A. The response of patients with bile acid diarrhoea to the farnesoid X receptor agonist obeticholic acid. Aliment. Pharmacol. Ther. 2014, 41, 54–64. [Google Scholar] [CrossRef]
- Harrison, S.A.; Rossi, S.J.; Paredes, A.H.; Trotter, J.F.; Bashir, M.R.; Guy, C.D.; Banerjee, R.; Jaros, M.J.; Owers, S.; Baxter, B.A.; et al. NGM282 Improves Liver Fibrosis and Histology in 12 Weeks in Patients With Nonalcoholic Steatohepatitis. Hepatology 2020, 71, 1198–1212. [Google Scholar] [CrossRef] [Green Version]
- Zhou, M.; Learned, R.M.; Rossi, S.J.; DePaoli, A.M.; Tian, H.; Ling, L. Engineered FGF19 eliminates bile acid toxicity and lipotoxicity leading to resolution of steatohepatitis and fibrosis in mice. Hepatol. Commun. 2017, 1, 1024–1042. [Google Scholar] [CrossRef]
- Harrison, S.A.; Rinella, M.E.; Abdelmalek, M.F.; Trotter, J.F.; Paredes, A.H.; Arnold, H.L.; Kugelmas, M.; Bashir, M.R.; Jaros, M.J.; Ling, L.; et al. NGM282 for treatment of non-alcoholic steatohepatitis: A multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet 2018, 391, 1174–1185. [Google Scholar] [CrossRef]
- DePaoli, A.M.; Zhou, M.; Kaplan, D.D.; Hunt, S.C.; Adams, T.D.; Learned, R.M.; Tian, H.; Ling, L. FGF19 Analog as a Surgical Factor Mimetic That Contributes to Metabolic Effects Beyond Glucose Homeostasis. Diabetes 2019, 68, 1315–1328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Stanislaus, S.; Chinookoswong, N.; Lau, Y.Y.; Hager, T.; Patel, J.; Ge, H.; Weiszmann, J.; Lu, S.C.; Graham, M.; et al. Acute glucose-lowering and insulin-sensitizing action of FGF21 in insulin-resistant mouse models-association with liver and adipose tissue effects. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E1105–E1114. [Google Scholar] [CrossRef] [Green Version]
- Keuper, M.; Häring, H.-U.; Staiger, H. Circulating FGF21 Levels in Human Health and Metabolic Disease. Exp. Clin. Endocrinol. Diabetes Off. J. Ger. Soc. Endocrinol. Ger. Diabetes Assoc. 2020, 128, 752–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coskun, T.; Bina, H.A.; Schneider, M.A.; Dunbar, J.D.; Hu, C.C.; Chen, Y.; Moller, D.E.; Kharitonenkov, A. Fibroblast growth factor 21 corrects obesity in mice. Endocrinology 2008, 149, 6018–6027. [Google Scholar] [CrossRef]
- Dong, J.Q.; Rossulek, M.; Somayaji, V.R.; Baltrukonis, D.; Liang, Y.; Hudson, K.; Hernandez-Illas, M.; Calle, R.A. Pharmacokinetics and pharmacodynamics of PF-05231023, a novel long-acting FGF21 mimetic, in a first-in-human study. Br. J. Clin. Pharmacol. 2015, 80, 1051–1063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Ishino, T.; Chen, G.; Rolzin, P.; Osothprarop, T.F.; Retting, K.; Li, L.; Jin, P.; Matin, M.J.; Huyghe, B.; et al. Development of a novel long-acting antidiabetic FGF21 mimetic by targeted conjugation to a scaffold antibody. J. Pharmacol. Exp. Ther. 2013, 346, 270–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, A.M.; Somayaji, V.R.; Dong, J.Q.; Rolph, T.P.; Weng, Y.; Chabot, J.R.; Gropp, K.E.; Talukdar, S.; Calle, R.A. Once-weekly administration of a long-acting fibroblast growth factor 21 analogue modulates lipids, bone turnover markers, blood pressure and body weight differently in obese people with hypertriglyceridaemia and in non-human primates. Diabetes Obes. Metab. 2017, 19, 1762–1772. [Google Scholar] [CrossRef]
- Carpenter, T.O.; Imel, E.A.; Ruppe, M.D.; Weber, T.J.; Klausner, M.A.; Wooddell, M.M.; Kawakami, T.; Ito, T.; Zhang, X.; Humphrey, J.; et al. Randomized trial of the anti-FGF23 antibody KRN23 in X-linked hypophosphatemia. J. Clin. Investig. 2014, 124, 1587–1597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haffner, D.; Emma, F.; Eastwood, D.M.; Duplan, M.B.; Bacchetta, J.; Schnabel, D.; Wicart, P.; Bockenhauer, D.; Santos, F.; Levtchenko, E.; et al. Clinical practice recommendations for the diagnosis and management of X-linked hypophosphataemia. Nat. Rev. Nephrol. 2019, 15, 435–455. [Google Scholar] [CrossRef] [Green Version]
- Imel, E.A.; Zhang, X.; Ruppe, M.D.; Weber, T.J.; Klausner, M.A.; Ito, T.; Vergeire, M.; Humphrey, J.S.; Glorieux, F.H.; Portale, A.A.; et al. Prolonged Correction of Serum Phosphorus in Adults With X-Linked Hypophosphatemia Using Monthly Doses of KRN23. J. Clin. Endocrinol. Metab. 2015, 100, 2565–2573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Imel, E.A.; Ruppe, M.D.; Weber, T.J.; Klausner, M.A.; Ito, T.; Vergeire, M.; Humphrey, J.; Glorieux, F.H.; Portale, A.A.; et al. Pharmacokinetics and pharmacodynamics of a human monoclonal anti-FGF23 antibody (KRN23) in the first multiple ascending-dose trial treating adults with X-linked hypophosphatemia. J. Clin. Pharmacol. 2016, 56, 176–185. [Google Scholar] [CrossRef] [Green Version]
- Carpenter, T.O.; Whyte, M.P.; Imel, E.A.; Boot, A.M.; Högler, W.; Linglart, A.; Padidela, R.; van’t Hoff, W.; Mao, M.; Chen, C.-Y.; et al. Burosumab Therapy in Children with X-Linked Hypophosphatemia. N. Engl. J. Med. 2018, 378, 1987–1998. [Google Scholar] [CrossRef] [Green Version]
- Whyte, M.P.; Carpenter, T.O.; Gottesman, G.S.; Mao, M.; Skrinar, A.; San Martin, J.; Imel, E.A. Efficacy and safety of burosumab in children aged 1–4 years with X-linked hypophosphataemia: A multicentre, open-label, phase 2 trial. Lancet Diabetes Endocrinol. 2019, 7, 189–199. [Google Scholar] [CrossRef]
- Imel, E.A.; Glorieux, F.H.; Whyte, M.P.; Munns, C.F.; Ward, L.M.; Nilsson, O.; Simmons, J.H.; Padidela, R.; Namba, N.; Cheong, H.I.; et al. Burosumab versus conventional therapy in children with X-linked hypophosphataemia: A randomised, active-controlled, open-label, phase 3 trial. Lancet 2019, 393, 2416–2427. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef] [Green Version]
- Fu, T.; Choi, S.E.; Kim, D.H.; Seok, S.; Suino-Powell, K.M.; Xu, H.E.; Kemper, J.K. Aberrantly elevated microRNA-34a in obesity attenuates hepatic responses to FGF19 by targeting a membrane coreceptor β-Klotho. Proc. Natl. Acad. Sci. USA 2012, 109, 16137–16142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bárcena, C.; Mayoral, P.; Quirós, P.M. Mitohormesis, an Antiaging Paradigm. Int. Rev. Cell Mol. Biol. 2018, 340, 35–77. [Google Scholar] [CrossRef]
- Tyynismaa, H.; Carroll, C.J.; Raimundo, N.; Ahola-Erkkilä, S.; Wenz, T.; Ruhanen, H.; Guse, K.; Hemminki, A.; Peltola-Mjøsund, K.E.; Tulkki, V.; et al. Mitochondrial myopathy induces a starvation-like response. Hum. Mol. Genet. 2010, 19, 3948–3958. [Google Scholar] [CrossRef] [Green Version]
- Conte, M.; Ostan, R.; Fabbri, C.; Santoro, A.; Guidarelli, G.; Vitale, G.; Mari, D.; Sevini, F.; Capri, M.; Sandri, M.; et al. Human Aging and Longevity Are Characterized by High Levels of Mitokines. J. Gerontol. Ser. A Biol. Sci. Med Sci. 2019, 74, 600–607. [Google Scholar] [CrossRef]
- Ajaz, S.; McPhail, M.J.; Singh, K.K.; Mujib, S.; Trovato, F.M.; Napoli, S.; Agarwal, K. Mitochondrial metabolic manipulation by SARS-CoV-2 in peripheral blood mononuclear cells of patients with COVID-19. Am. J. Physiol. Cell Physiol. 2021, 320, C57–C65. [Google Scholar] [CrossRef] [PubMed]
- El-Hodhod, M.A.; Hamdy, A.M.; Abbas, A.A.; Moftah, S.G.; Ramadan, A.A. Fibroblast growth factor 23 contributes to diminished bone mineral density in childhood inflammatory bowel disease. BMC Gastroenterol. 2012, 12, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fitzpatrick, E.A.; Han, X.; Xiao, Z.; Quarles, L.D. Role of Fibroblast Growth Factor-23 in Innate Immune Responses. Front. Endocrinol. 2018, 9, 320. [Google Scholar] [CrossRef]
- Munoz Mendoza, J.; Isakova, T.; Ricardo, A.C.; Xie, H.; Navaneethan, S.D.; Anderson, A.H.; Bazzano, L.A.; Xie, D.; Kretzler, M.; Nessel, L.; et al. Fibroblast growth factor 23 and Inflammation in CKD. Clin. J. Am. Soc. Nephrol. CJASN 2012, 7, 1155–1162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanks, L.J.; Casazza, K.; Judd, S.E.; Jenny, N.S.; Gutiérrez, O.M. Associations of fibroblast growth factor-23 with markers of inflammation, insulin resistance and obesity in adults. PLoS ONE 2015, 10, e0122885. [Google Scholar] [CrossRef]
- Gutiérrez, O.M.; Mannstadt, M.; Isakova, T.; Rauh-Hain, J.A.; Tamez, H.; Shah, A.; Smith, K.; Lee, H.; Thadhani, R.; Jüppner, H.; et al. Fibroblast growth factor 23 and mortality among patients undergoing hemodialysis. N. Engl. J. Med. 2008, 359, 584–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richter, M.; Lautze, H.J.; Walther, T.; Braun, T.; Kostin, S.; Kubin, T. The failing heart is a major source of circulating FGF23 via oncostatin M receptor activation. J. Heart Lung Transplant. Off. Publ. Int. Soc. Heart Transplant. 2015, 34, 1211–1214. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Li, L.; Yang, J.; King, G.; Xiao, Z.; Quarles, L.D. Counter-regulatory paracrine actions of FGF-23 and 1,25(OH)2D in macrophages. FEBS Lett. 2016, 590, 53–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossaint, J.; Oehmichen, J.; Van Aken, H.; Reuter, S.; Pavenstädt, H.J.; Meersch, M.; Unruh, M.; Zarbock, A. FGF23 signaling impairs neutrophil recruitment and host defense during CKD. J. Clin. Investig. 2016, 126, 962–974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, Z.; Riccardi, D.; Velazquez, H.A.; Chin, A.L.; Yates, C.R.; Carrick, J.D.; Smith, J.C.; Baudry, J.; Quarles, L.D. A computationally identified compound antagonizes excess FGF-23 signaling in renal tubules and a mouse model of hypophosphatemia. Sci. Signal. 2016, 9, ra113. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D.; Knauf, C. How gut microbes talk to organs: The role of endocrine and nervous routes. Mol. Metab. 2016, 5, 743–752. [Google Scholar] [CrossRef] [PubMed]
- Boulangé, C.L.; Neves, A.L.; Chilloux, J.; Nicholson, J.K.; Dumas, M.E. Impact of the gut microbiota on inflammation, obesity, and metabolic disease. Genome Med. 2016, 8, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Phan, P.; Saikia, B.B.; Sonnaila, S.; Agrawal, S.; Alraawi, Z.; Kumar, T.K.S.; Iyer, S. The Saga of Endocrine FGFs. Cells 2021, 10, 2418. https://doi.org/10.3390/cells10092418
Phan P, Saikia BB, Sonnaila S, Agrawal S, Alraawi Z, Kumar TKS, Iyer S. The Saga of Endocrine FGFs. Cells. 2021; 10(9):2418. https://doi.org/10.3390/cells10092418
Chicago/Turabian StylePhan, Phuc, Bibhuti Ballav Saikia, Shivakumar Sonnaila, Shilpi Agrawal, Zeina Alraawi, Thallapuranam Krishnaswamy Suresh Kumar, and Shilpa Iyer. 2021. "The Saga of Endocrine FGFs" Cells 10, no. 9: 2418. https://doi.org/10.3390/cells10092418
APA StylePhan, P., Saikia, B. B., Sonnaila, S., Agrawal, S., Alraawi, Z., Kumar, T. K. S., & Iyer, S. (2021). The Saga of Endocrine FGFs. Cells, 10(9), 2418. https://doi.org/10.3390/cells10092418