
Engineered Liposomes Protect Immortalized Immune Cells from Cytolysins Secreted by Group A and Group G Streptococci †


Abstract
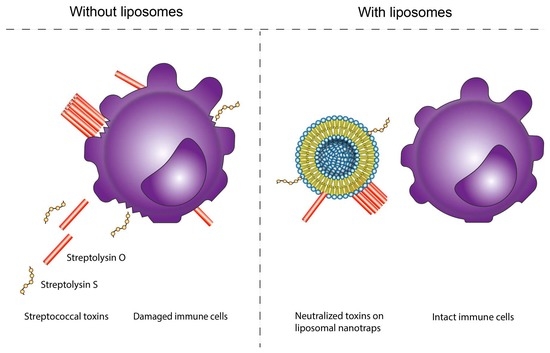
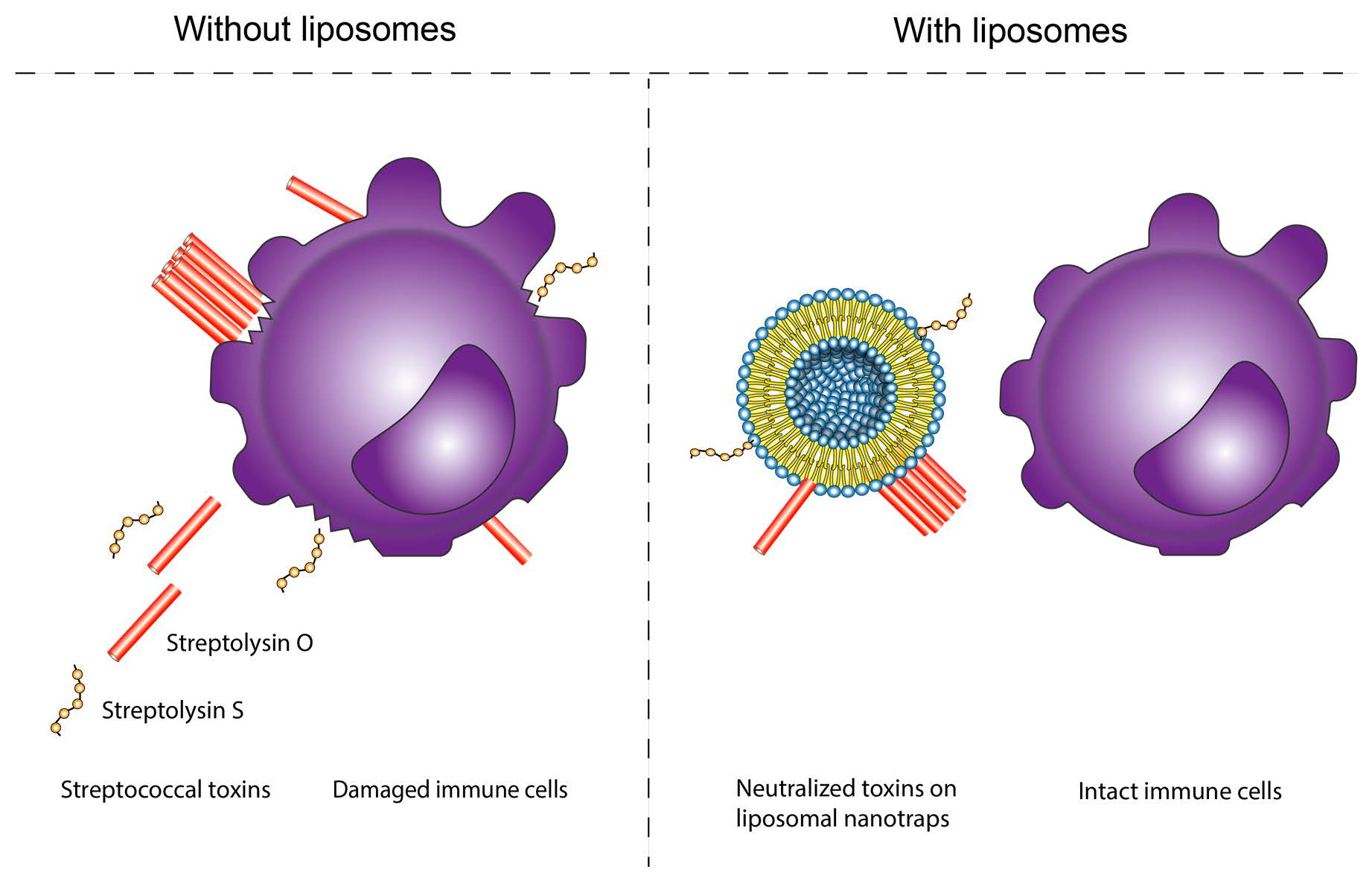
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Bacterial Cultures
2.2. Cell Cultures
2.3. Liposomal Nanotraps
2.4. Cells Survival
2.5. Mass Spectrometry
2.6. Statistical Analysis
3. Results
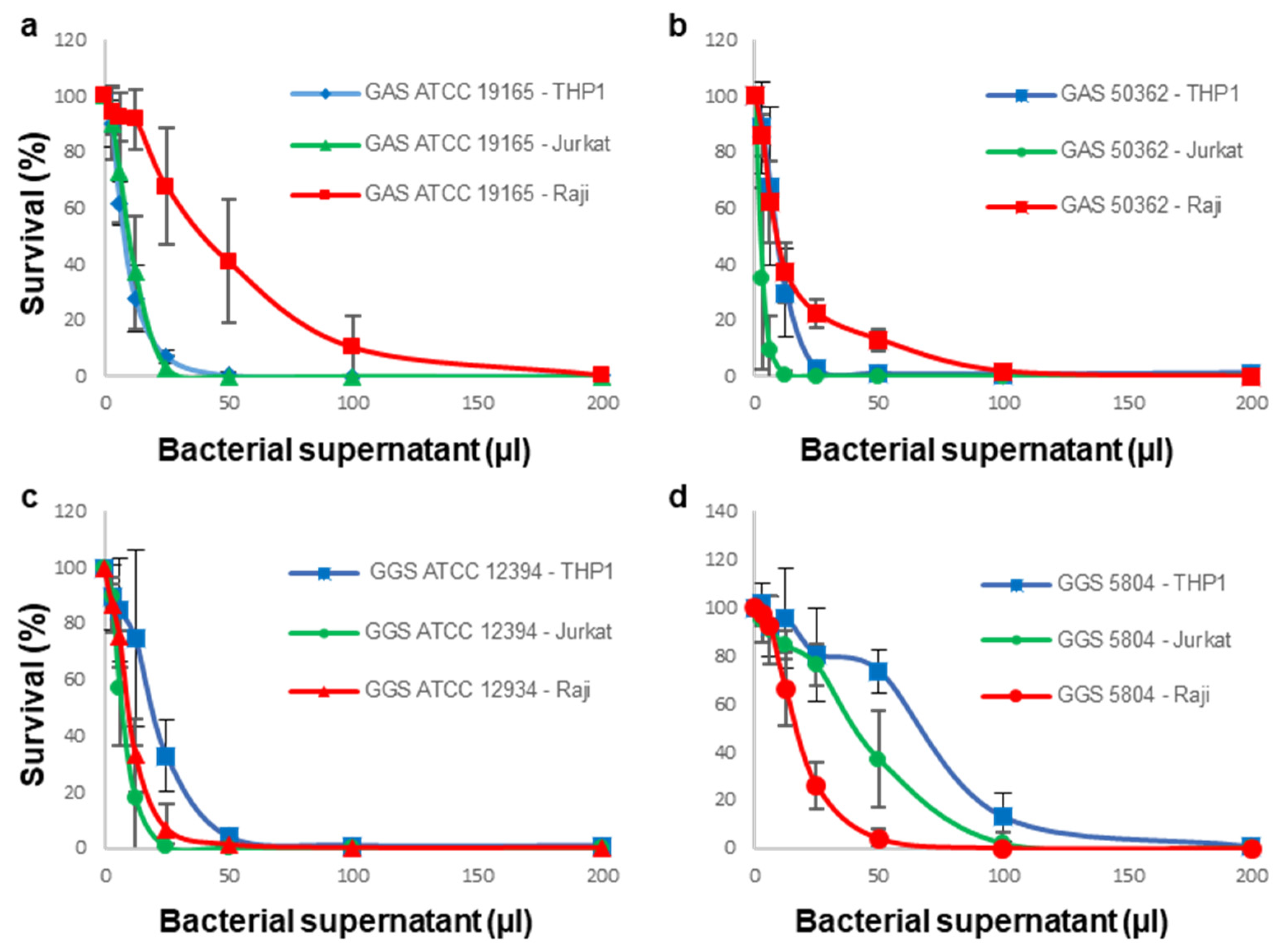
3.1. Sensitivity of Immune Cells to GAS or GGS Supernatants Is Strain and Cell Type Dependent
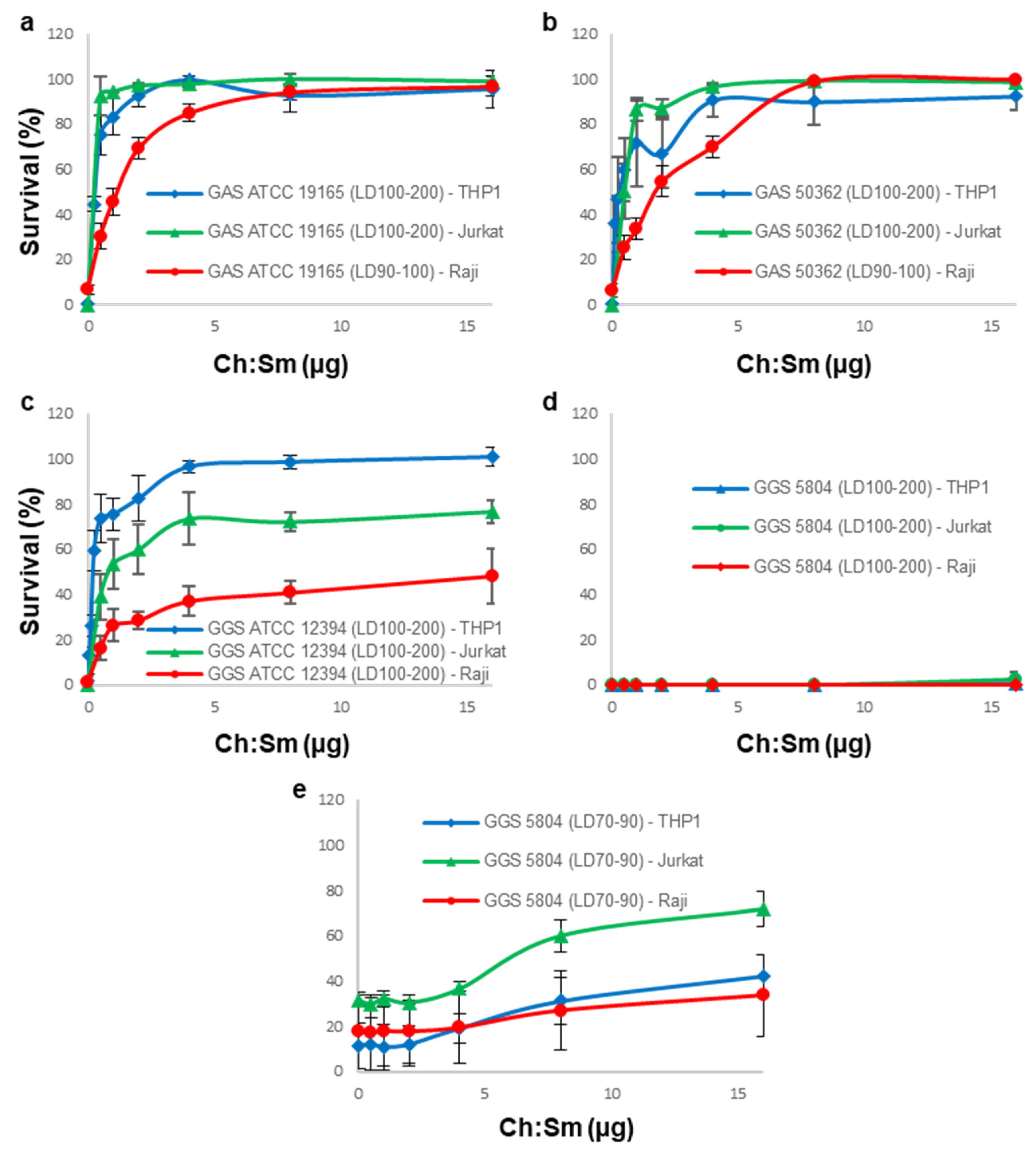
3.2. Cholesterol-Containing Liposomes Fully Neutralize Cytotoxic Activities of GAS Supernatants but Only Partially That of GGS Supernatants
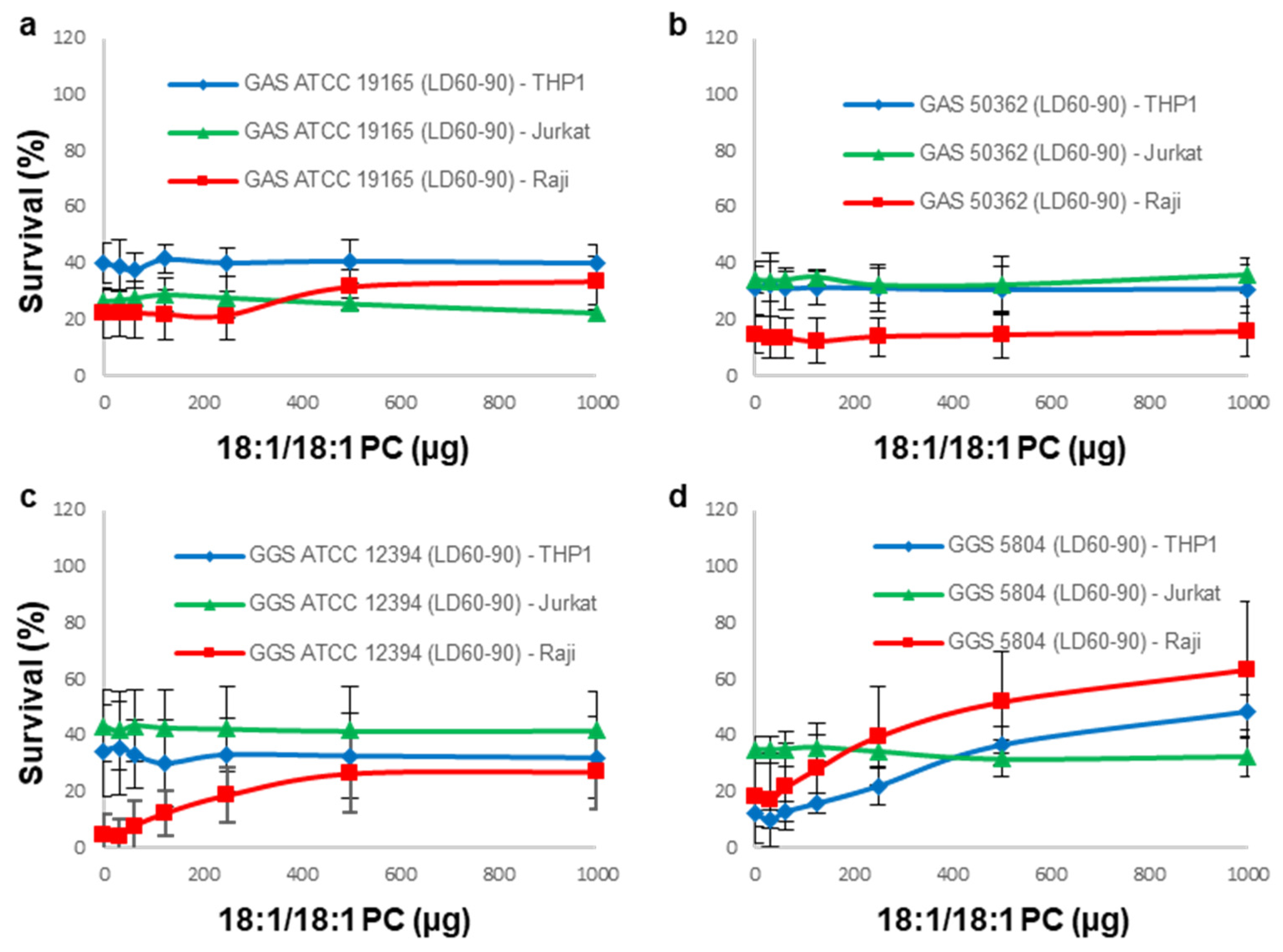
3.3. 18:1/18:1 PC-Liposomes Protect Raji and THP-1 but Not Jurkat Cells against SLS Secreted by GGS
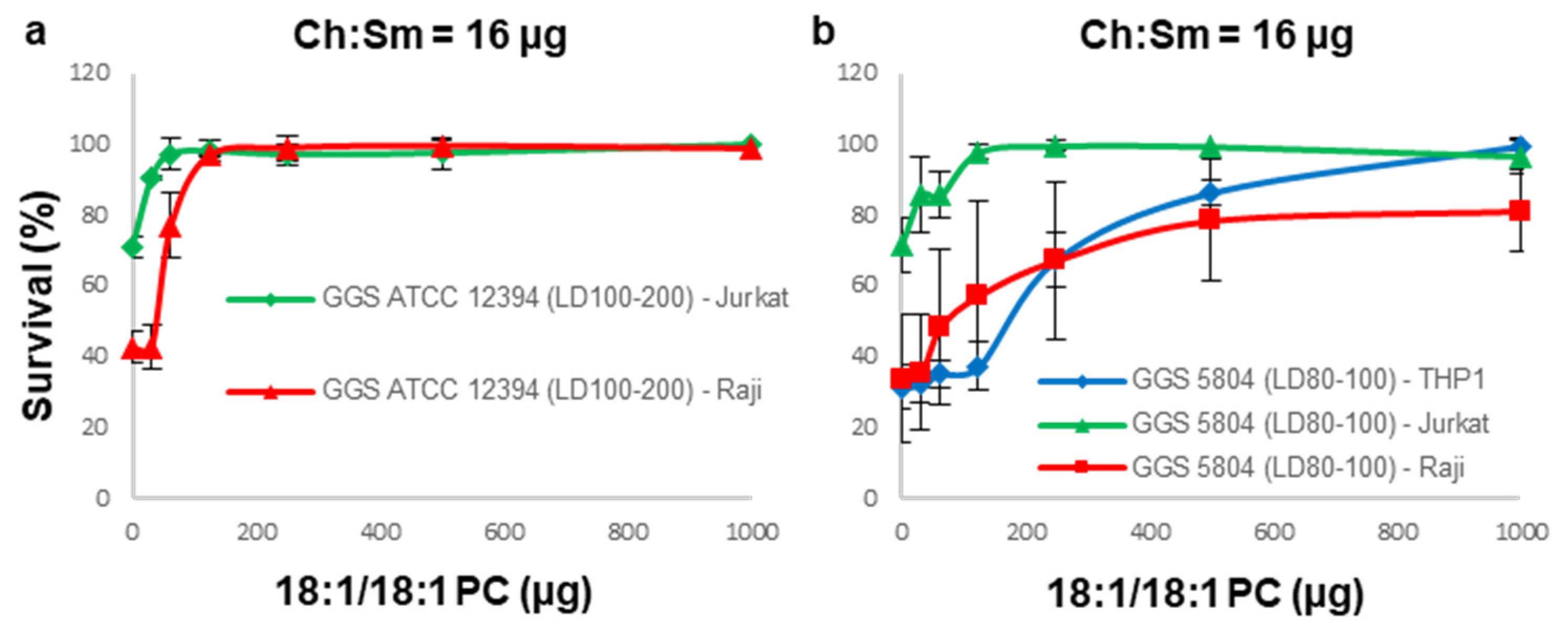
3.4. A Combination of Ch:Sm and 18:1/18:1 PC-Liposomes Provides Superior Protection against GGS Strains
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fernandes, P. Antibacterial discovery and development—the failure of success? Nat. Biotechnol. 2006, 24, 1497–1503. [Google Scholar] [CrossRef]
- Bush, K.; Courvalin, P.; Dantas, G.; Davies, J.; Eisenstein, B.; Huovinen, P.; Jacoby, G.A.; Kishony, R.; Kreiswirth, B.N.; Kutter, E.; et al. Tackling antibiotic resistance. Nat. Rev. Microbiol. 2011, 9, 894–896. [Google Scholar] [CrossRef] [PubMed]
- Clatworthy, A.; Pierson, E.; Hung, D.T. Targeting virulence: A new paradigm for antimicrobial therapy. Nat. Chem. Biol. 2007, 3, 541–548. [Google Scholar] [CrossRef]
- Cegelski, L.; Marshall, G.R.; Eldridge, G.R.; Hultgren, S.J. The biology and future prospects of antivirulence therapies. Nat. Rev. Genet. 2008, 6, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Peraro, M.D.; van der Goot, F.G. Pore-forming toxins: Ancient, but never really out of fashion. Nat. Rev. Microbiol. 2016, 14, 77–92. [Google Scholar] [CrossRef] [PubMed]
- Rojko, N.; Anderluh, G. How Lipid Membranes Affect Pore Forming Toxin Activity. Acc. Chem. Res. 2015, 48, 3073–3079. [Google Scholar] [CrossRef]
- Rai, A.K.; Chattopadhyay, K. Revisiting the membrane interaction mechanism of a membrane-damaging β-barrel pore-forming toxin Vibrio cholerae cytolysin. Mol. Microbiol. 2015, 97, 1051–1062. [Google Scholar] [CrossRef] [Green Version]
- Kathuria, R.; Chattopadhyay, K. Vibrio cholerae cytolysin: Multiple facets of the membrane interaction mechanism of a β-barrel pore-forming toxin. IUBMB Life 2018, 70, 260–266. [Google Scholar] [CrossRef] [Green Version]
- González-Juarbe, N.; Bradley, K.M.; Shenoy, A.; Gilley, R.P.; Reyes, L.F.; Hinojosa, C.A.; Restrepo, M.I.; Dube, P.H.; Bergman, M.A.; Orihuela, C.J. Pore-forming toxin-mediated ion dysregulation leads to death receptor-independent necroptosis of lung epithelial cells during bacterial pneumonia. Cell Death Differ. 2017, 24, 917–928. [Google Scholar] [CrossRef] [Green Version]
- Bien, J.; Sokolova, O.; Bozko, P. Characterization of Virulence Factors of Staphylococcus aureus: Novel Function of Known Virulence Factors That Are Implicated in Activation of Airway Epithelial Proinflammatory Response. J. Pathog. 2011, 2011, 601905. [Google Scholar] [CrossRef] [Green Version]
- Schmitz, M.; Roux, X.; Huttner, B.; Pugin, J. Streptococcal toxic shock syndrome in the intensive care unit. Ann. Intensiv. Care 2018, 8, 88. [Google Scholar] [CrossRef]
- Rolston, K.V. Group G streptococcal infections. Arch. Intern. Med. 1986, 146, 857–858. [Google Scholar] [CrossRef]
- Cook, L.; Chatterjee, N.; Li, Y.; Andrade, J.; Federle, M.J.; Eichenbaum, Z. Transcriptomic Analysis of Streptococcus pyogenes Colonizing the Vaginal Mucosa Identifies hupY, an MtsR-Regulated Adhesin Involved in Heme Utilization. mBio 2019, 10, e00848-19. [Google Scholar] [CrossRef] [Green Version]
- Ferreti, J.; Stevens, D.; Fischetti, V. Streptococcus pyogenes Basic Biology to Clinical Manifestations; University of Oklahoma Health Sciences Center: Oklahoma City, OK, USA, 2016. [Google Scholar] [PubMed]
- Cunningham, M.W. Pathogenesis of Group A Streptococcal Infections. Clin. Microbiol. Rev. 2000, 13, 470–511. [Google Scholar] [CrossRef]
- Carapetis, J.R.; Steer, A.C.; Mulholland, E.K.; Weber, M. The global burden of group A streptococcal diseases. Lancet Infect. Dis. 2005, 5, 685–694. [Google Scholar] [CrossRef]
- Sylvetsky, N.; Raveh, D.; Schlesinger, Y.; Rudensky, B.; Yinnon, A.M. Bacteremia due to beta-hemolytic streptococcus group g: Increasing incidence and clinical characteristics of patients. Am. J. Med. 2002, 112, 622–626. [Google Scholar] [CrossRef]
- Tweten, R.K. Cholesterol-Dependent Cytolysins, a Family of Versatile Pore-Forming Toxins. Infect. Immun. 2005, 73, 6199–6209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhakdi, S.; Tranum-Jensen, J.; Sziegoleit, A. Mechanism of membrane damage by streptolysin-O. Infect. Immun. 1985, 47, 52–60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molloy, E.; Cotter, P.D.; Hill, C.; Mitchell, D.; Ross, R. Streptolysin S-like virulence factors: The continuing sagA. Nat. Rev. Genet. 2011, 9, 670–681. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Besançon, H.; Babiychuk, V.; Larpin, Y.; Köffel, R.; Schittny, D.; Brockhus, L.; Hathaway, L.J.; Sendi, P.; Draeger, A.; Babiychuk, E. Tailored liposomal nanotraps for the treatment of Streptococcal infections. J. Nanobiotechnol. 2021, 19, 46. [Google Scholar] [CrossRef]
- Goldmann, O.; Rohde, M.; Chhatwal, G.S.; Medina, E. Role of Macrophages in Host Resistance to Group A Streptococci. Infect. Immun. 2004, 72, 2956–2963. [Google Scholar] [CrossRef] [Green Version]
- Larpin, Y.; Besançon, H.; Iacovache, M.; Babiychuk, V.S.; Babiychuk, E.B.; Zuber, B.; Draeger, A.; Köffel, R. Bacterial pore-forming toxin pneumolysin: Cell membrane structure and microvesicle shedding capacity determines differential survival of immune cell types. FASEB J. 2020, 34, 1665–1678. [Google Scholar] [CrossRef] [Green Version]
- Timmer, A.M.; Timmer, J.C.; Pence, M.A.; Hsu, L.-C.; Ghochani, M.; Frey, T.G.; Karin, M.; Salvesen, G.S.; Nizet, V. Streptolysin O Promotes Group A Streptococcus Immune Evasion by Accelerated Macrophage Apoptosis. J. Biol. Chem. 2009, 284, 862–871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyoshi-Akiyama, T.; Takamatsu, D.; Koyanagi, M.; Zhao, J.; Imanishi, K.; Uchiyama, T. Cytocidal Effect of Streptococcus pyogenes on Mouse Neutrophils In Vivo and the Critical Role of Streptolysin S. J. Infect. Dis. 2005, 192, 107–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geny, B.; Popoff, M.R. Bacterial protein toxins and lipids: Pore formation or toxin entry into cells. Biol. Cell 2006, 98, 667–678. [Google Scholar] [CrossRef] [PubMed]
- Parker, M.W.; Feil, S.C. Pore-forming protein toxins: From structure to function. Prog. Biophys. Mol. Biol. 2005, 88, 91–142. [Google Scholar] [CrossRef]
- Hu, C.-M.J.; Fang, R.H.; Copp, J.; Luk, B.T.; Zhang, L. A biomimetic nanosponge that absorbs pore-forming toxins. Nat. Nanotechnol. 2013, 8, 336–340. [Google Scholar] [CrossRef] [Green Version]
- Henry, B.D.; Neill, D.R.; Becker, K.A.; Gore, S.; Bricio-Moreno, L.; Ziobro, R.; Edwards, M.J.; Mühlemann, K.; Steinmann, J.; Kleuser, B.; et al. Engineered liposomes sequester bacterial exotoxins and protect from severe invasive infections in mice. Nat. Biotechnol. 2015, 33, 81–88. [Google Scholar] [CrossRef] [Green Version]
- Laterre, P.-F.; Colin, G.; Dequin, P.-F.; Dugernier, T.; Boulain, T.; da Silveira, S.A.; Lajaunias, F.; Perez, A.; François, B. CAL02, a novel antitoxin liposomal agent, in severe pneumococcal pneumonia: A first-in-human, double-blind, placebo-controlled, randomised trial. Lancet Infect. Dis. 2019, 19, 620–630. [Google Scholar] [CrossRef]
- Ruppen, C.; Rasmussen, M.; Casanova, C.; Sendi, P. A 10-year observational study of Streptococcus dysgalactiae bacteraemia in adults: Frequent occurrence among female intravenous drug users. Swiss Med. Wkly. 2017, 147, 4–7. [Google Scholar] [CrossRef]
- Gunasekera, K.; Wüthrich, D.; Braga-Lagache, S.; Heller, M.; Ochsenreiter, T. Proteome remodelling during development from blood to insect-form Trypanosoma brucei quantified by SILAC and mass spectrometry. BMC Genom. 2012, 13, 556. [Google Scholar] [CrossRef] [Green Version]
- Braga-Lagache, S.; Buchs, N.; Iacovache, M.-I.; Zuber, B.; Jackson, C.B.; Heller, M. Robust Label-free, Quantitative Profiling of Circulating Plasma Microparticle (MP) Associated Proteins. Mol. Cell. Proteom. 2016, 15, 3640–3652. [Google Scholar] [CrossRef] [Green Version]
- Colinge, J.; Chiappe, D.; Lagache, S.; Moniatte, M.; Bougueleret, L. Differential Proteomics via Probabilistic Peptide Identification Scores. Anal. Chem. 2005, 77, 596–606. [Google Scholar] [CrossRef]
- Liu, H.; Sadygov, R.G.; Yates, J.R. A Model for Random Sampling and Estimation of Relative Protein Abundance in Shotgun Proteomics. Anal. Chem. 2004, 76, 4193–4201. [Google Scholar] [CrossRef]
- Rittirsch, D.; Flierl, M.A.; Ward, P.A. Harmful molecular mechanisms in sepsis. Nat. Rev. Immunol. 2008, 8, 776–787. [Google Scholar] [CrossRef] [Green Version]
- Sohlenkamp, C.; Geiger, O. Bacterial membrane lipids: Diversity in structures and pathways. FEMS Microbiol. Rev. 2016, 40, 133–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krueger, E.; Brown, A.C. Inhibition of bacterial toxin recognition of membrane components as an anti-virulence strategy. J. Biol. Eng. 2019, 13, 4. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, M.R.; Bischofberger, M.; Pernot, L.; Van Der Goot, F.G.; Frêche, B. Bacterial pore-forming toxins: The whole story? Cell. Mol. Life Sci. 2007, 65, 493–507. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Wang, J.; Jia, L.; Huang, J.; He, C.; Hu, F.; Yuan, L.; Wang, G.; Yu, M.; Li, Z. Transmembrane TNF-α promotes activation-induced cell death by forward and reverse signaling. Oncotarget 2017, 8, 63799–63812. [Google Scholar] [CrossRef] [Green Version]
- Zheng, L.; Fisher, G.H.; Miller, R.E.; Peschon, J.J.; Lynch, D.H.; Lenardo, M.J. Induction of apoptosis in mature T cells by tumour necrosis factor. Nature 1995, 377, 348–351. [Google Scholar] [CrossRef]
- Ashkenazi, A.; Dixit, V.M. Death Receptors: Signaling and Modulation. Science 1998, 281, 1305–1308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flusberg, D.A.; Sorger, P.K. Surviving apoptosis: Life–death signaling in single cells. Trends Cell Biol. 2015, 25, 446–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siemens, N.; INFECT Study Group; Kittang, B.R.; Chakrakodi, B.; Oppegaard, O.; Johansson, L.; Bruun, T.; Mylvaganam, H.; Svensson, M.; Skrede, S.; et al. Increased cytotoxicity and streptolysin O activity in group G streptococcal strains causing invasive tissue infections. Sci. Rep. 2015, 5, 16945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramanian, K.; Iovino, F.; Tsikourkitoudi, V.; Merkl, P.; Ahmed, S.; Berry, S.B.; Aschtgen, M.; Svensson, M.; Bergman, P.; Sotiriou, G.; et al. Mannose receptor-derived peptides neutralize pore-forming toxins and reduce inflammation and development of pneumococcal disease. EMBO Mol. Med. 2020, 12, e12695. [Google Scholar] [CrossRef] [PubMed]
- Larpin, Y.; Besançon, H.; Babiychuk, V.; Babiychuk, E.; Köffel, R. Small Pore-Forming Toxins Different Membrane Area Binding and Ca2+ Permeability of Pores Determine Cellular Resistance of Monocytic Cells. Toxins 2021, 13, 126. [Google Scholar] [CrossRef] [PubMed]
- Wolfmeier, H.; Schoenauer, R.; Atanassoff, A.P.; Neill, D.; Kadioglu, A.; Draeger, A.; Babiychuk, E.B. Ca2+-dependent repair of pneumolysin pores: A new paradigm for host cellular defense against bacterial pore-forming toxins. Biochim. Biophys. Acta (BBA)—Bioenerg. 2015, 1853, 2045–2054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buckingham, L.; Duncan, J.L. Approximate dimensions of membrane lesions produced by streptolysin S and streptolysin O. Biochim. Biophys. Acta (BBA)—Biomembr. 1983, 729, 115–122. [Google Scholar] [CrossRef]
- Andreeva-Kovalevskaya, Z.I.; Solonin, A.S.; Sineva, E.V.; Ternovsky, V.I. Pore-forming proteins and adaptation of living organisms to environmental conditions. Biochemistry 2008, 73, 1473–1492. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Besançon, H.; Larpin, Y.; Babiychuk, V.S.; Köffel, R.; Babiychuk, E.B. Engineered Liposomes Protect Immortalized Immune Cells from Cytolysins Secreted by Group A and Group G Streptococci. Cells 2022, 11, 166. https://doi.org/10.3390/cells11010166
Besançon H, Larpin Y, Babiychuk VS, Köffel R, Babiychuk EB. Engineered Liposomes Protect Immortalized Immune Cells from Cytolysins Secreted by Group A and Group G Streptococci. Cells. 2022; 11(1):166. https://doi.org/10.3390/cells11010166
Chicago/Turabian StyleBesançon, Hervé, Yu Larpin, Viktoria S. Babiychuk, René Köffel, and Eduard B. Babiychuk. 2022. "Engineered Liposomes Protect Immortalized Immune Cells from Cytolysins Secreted by Group A and Group G Streptococci" Cells 11, no. 1: 166. https://doi.org/10.3390/cells11010166