
TNFα Mediates Inflammation-Induced Effects on PPARG Splicing in Adipose Tissue and Mesenchymal Precursor Cells

 ,
,  ,
,  , ,
, ,  ,
,  and
and 
Abstract
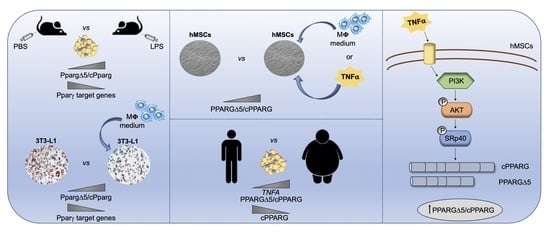
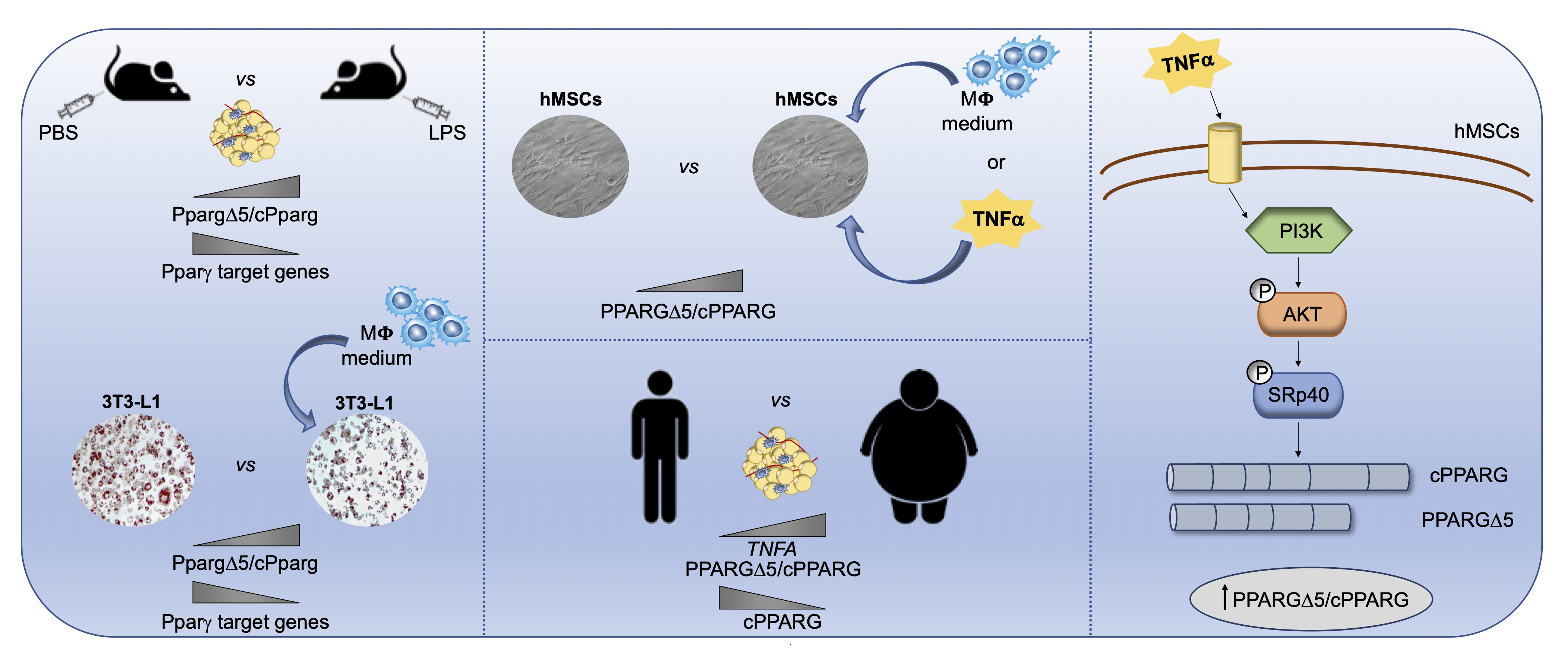
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Human Samples
2.2. Animals
2.3. Cell Cultures
2.4. Human Adipocyte Differentiation
2.5. Differentiation of Primary and InVitro Human Macrophages
2.6. Conditioned Medium Preparation
2.7. Murine Adipocyte Differentiation
2.8. Oil Red O Staining
2.9. Cell Treatments
2.10. Cell Transfection
2.11. RNA Isolation, RT-PCR and Quantitative PCR
2.12. Western Blot
2.13. ELISA
2.14. Statistics
3. Results
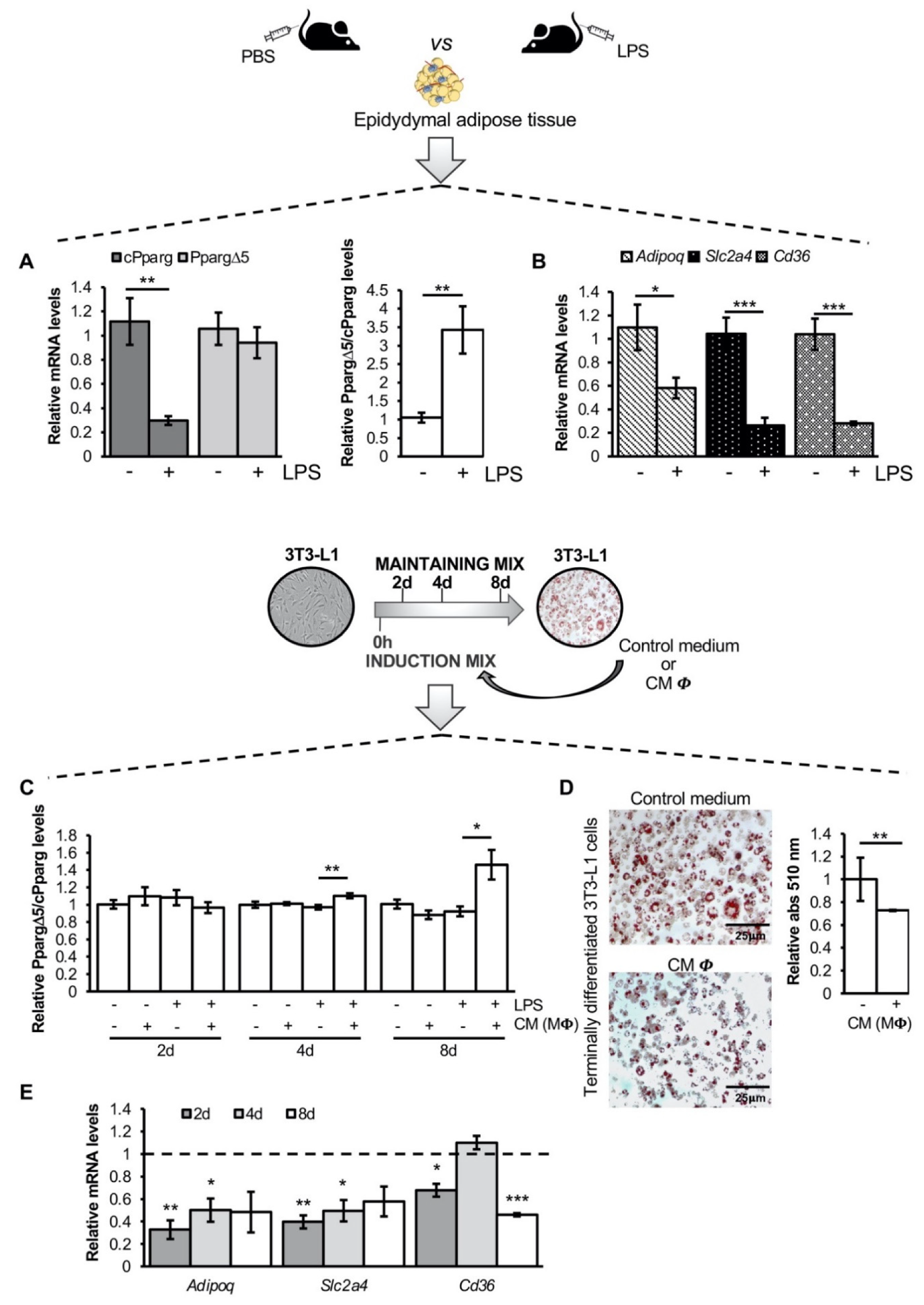
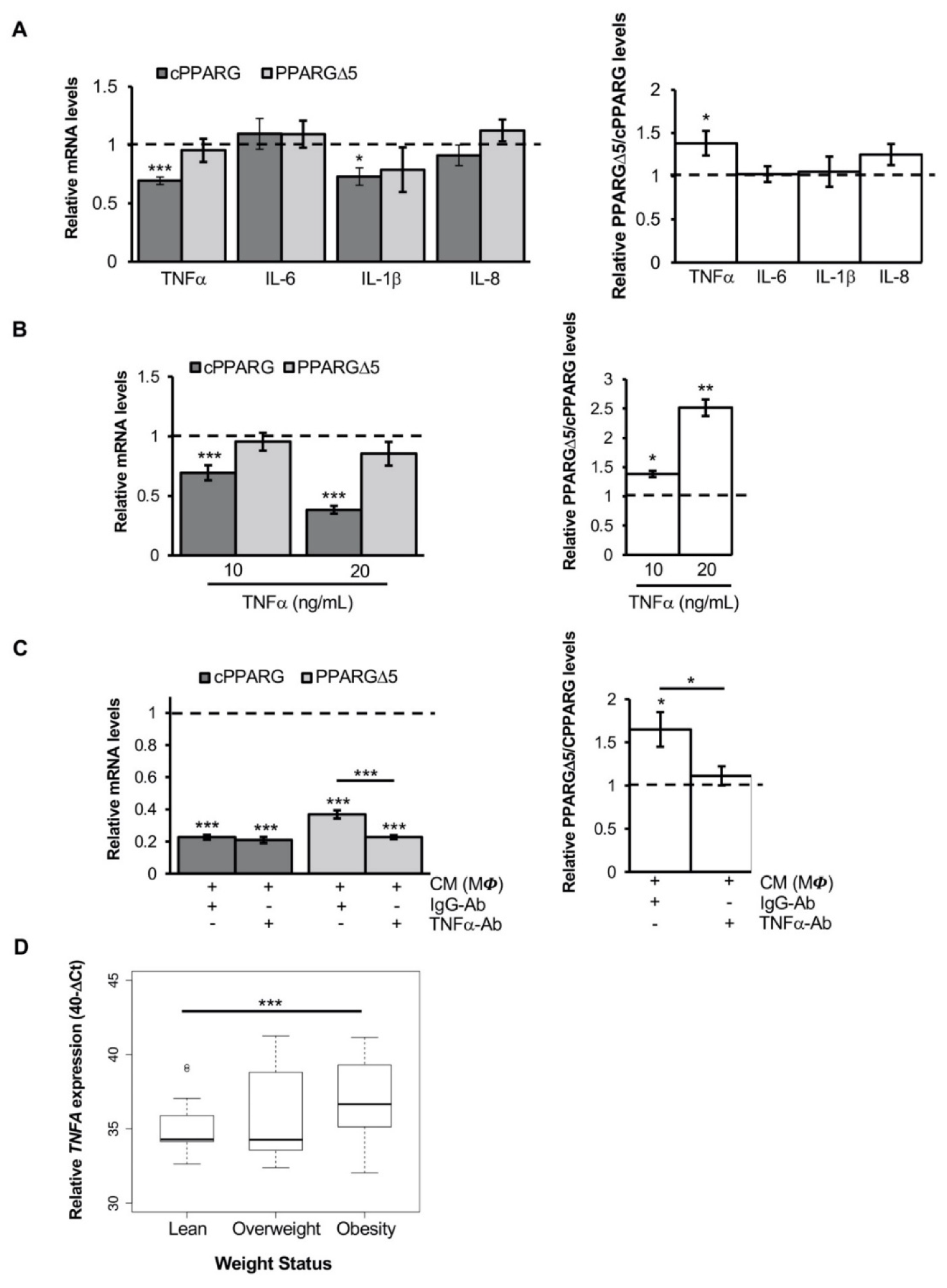
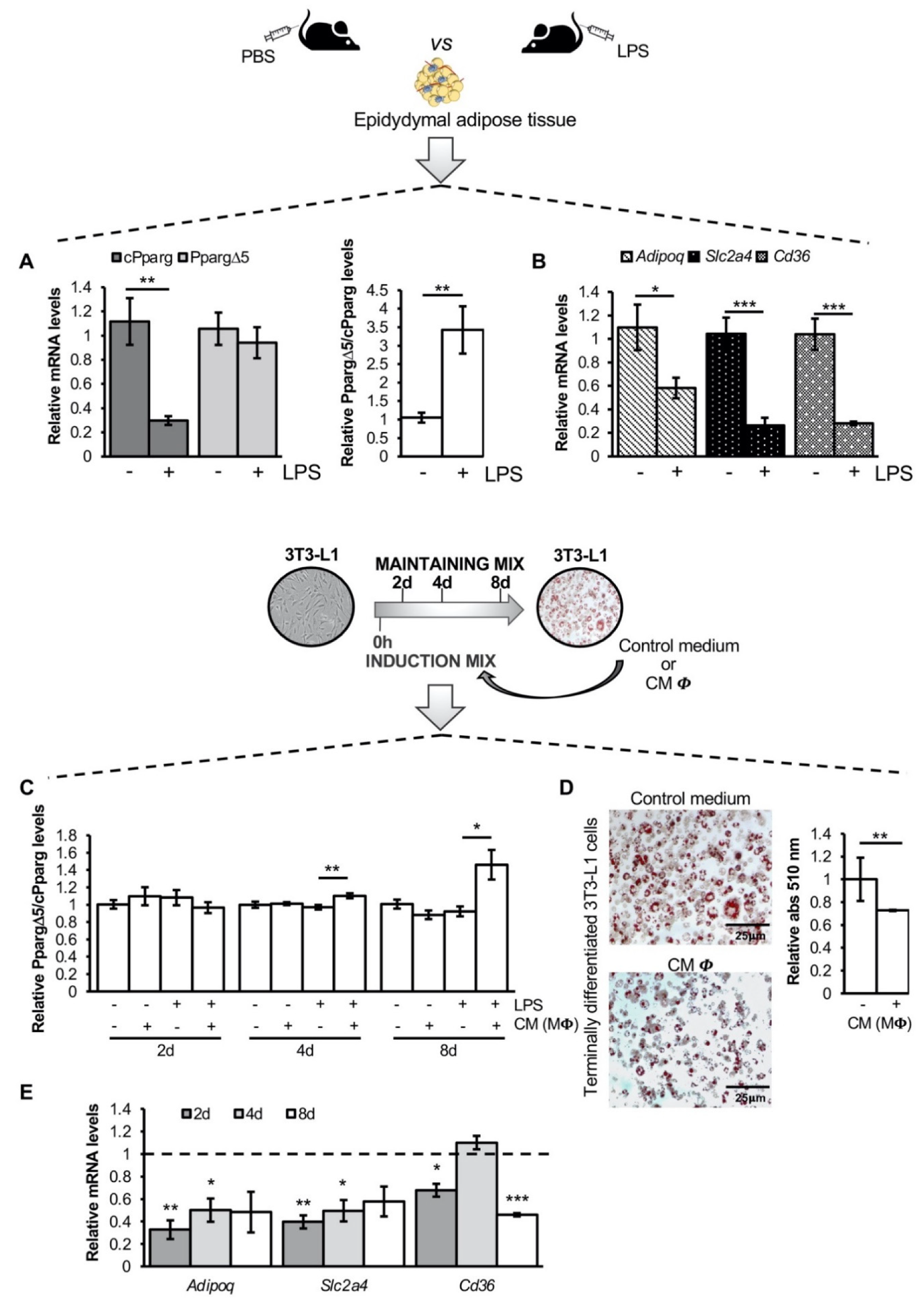
3.1. The Inflammatory Milieu Affects Pparg Expression and Splicing In Vitro and In Vivo
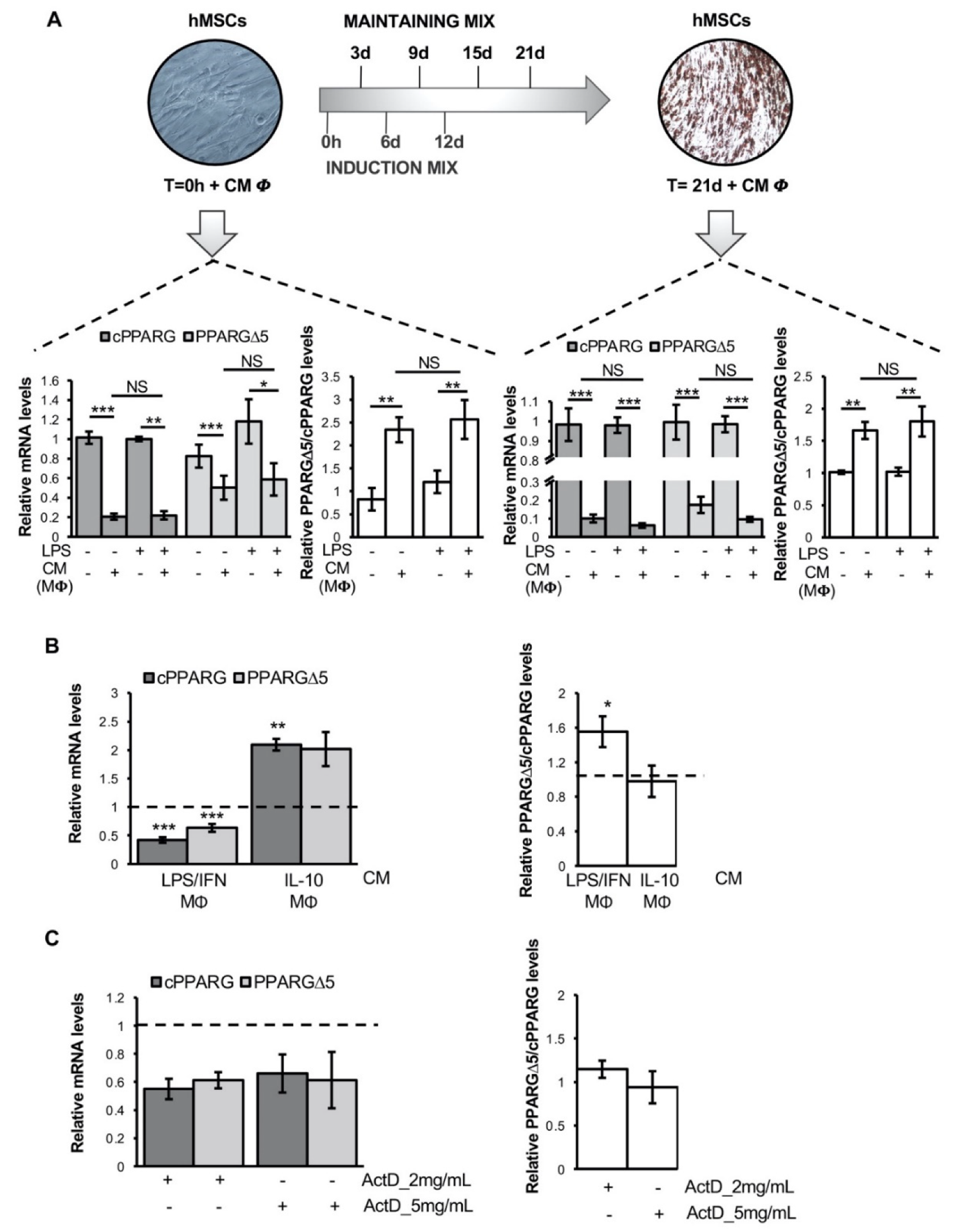
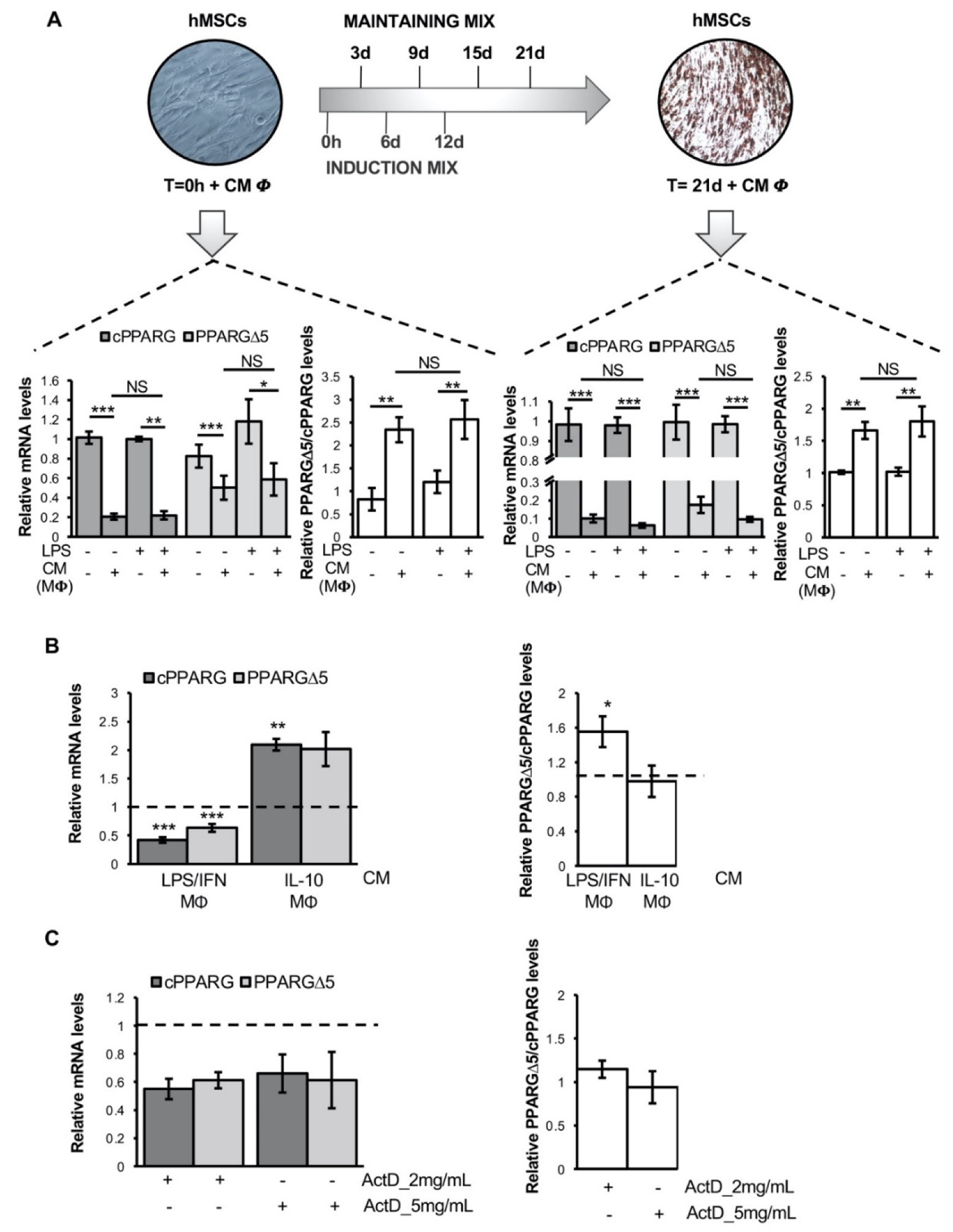
3.2. Pro-Inflammatory Macrophage Secretome Perturbs PPARG Splicing in Human Mesenchymal Stem Cells
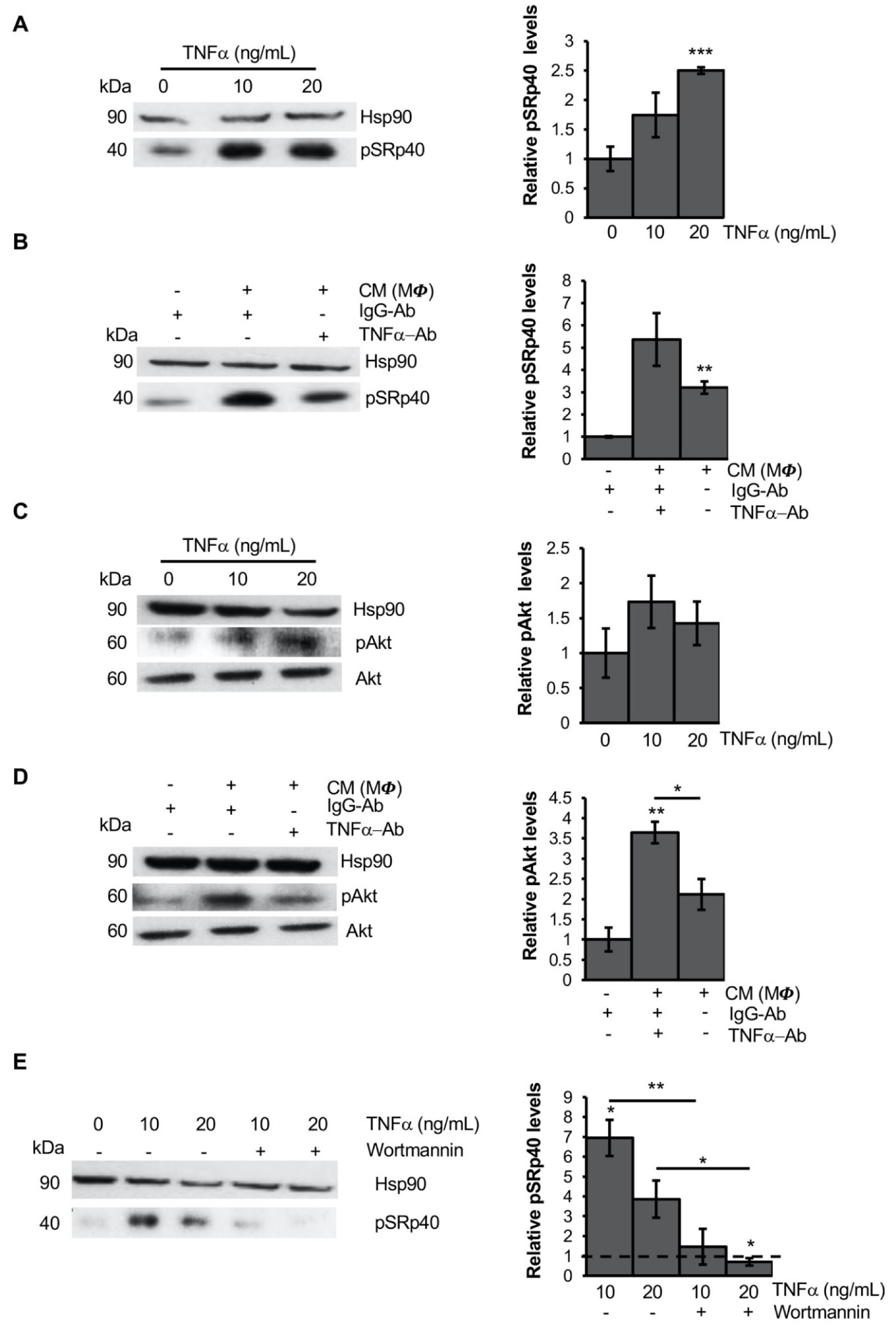
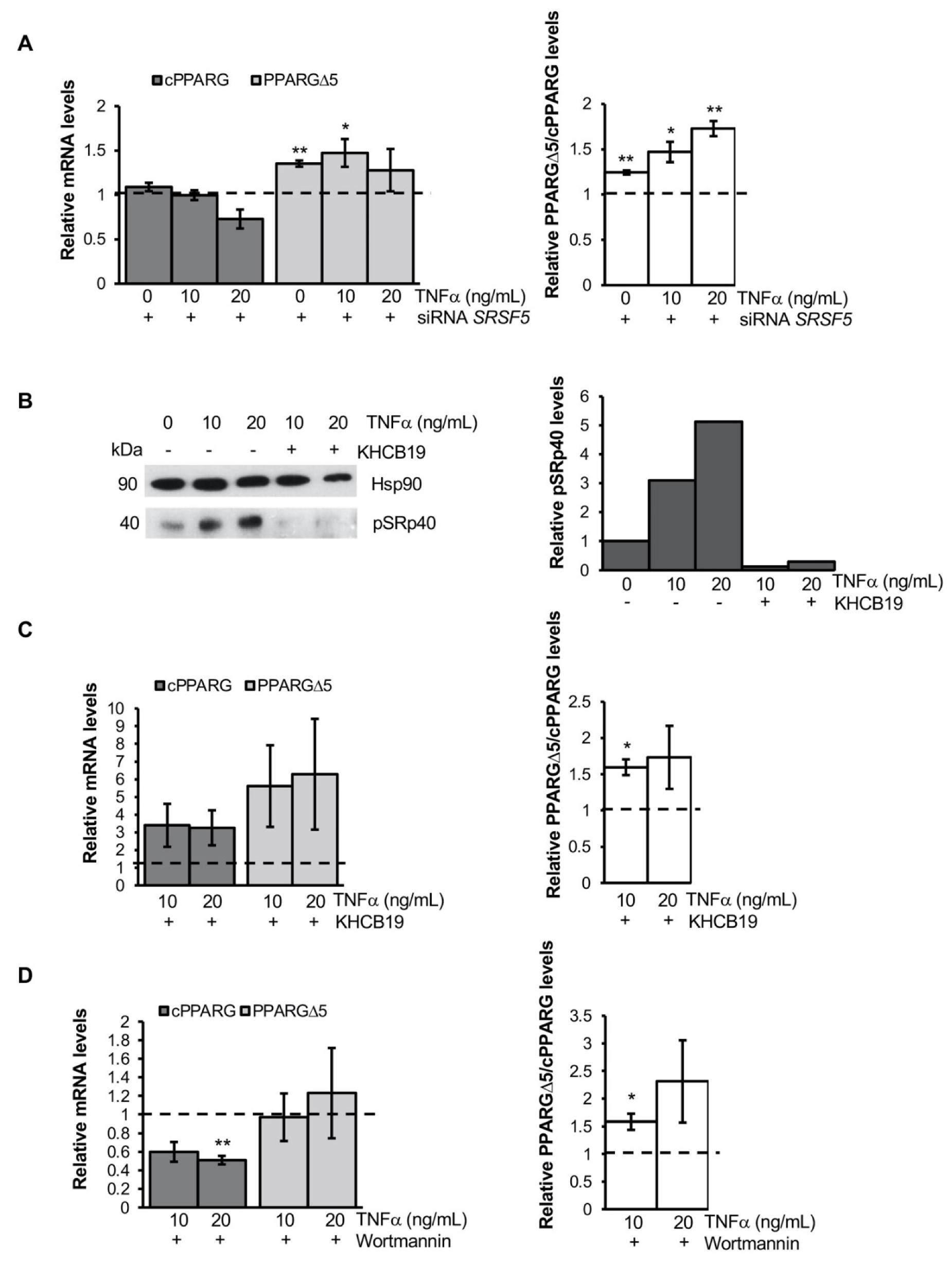
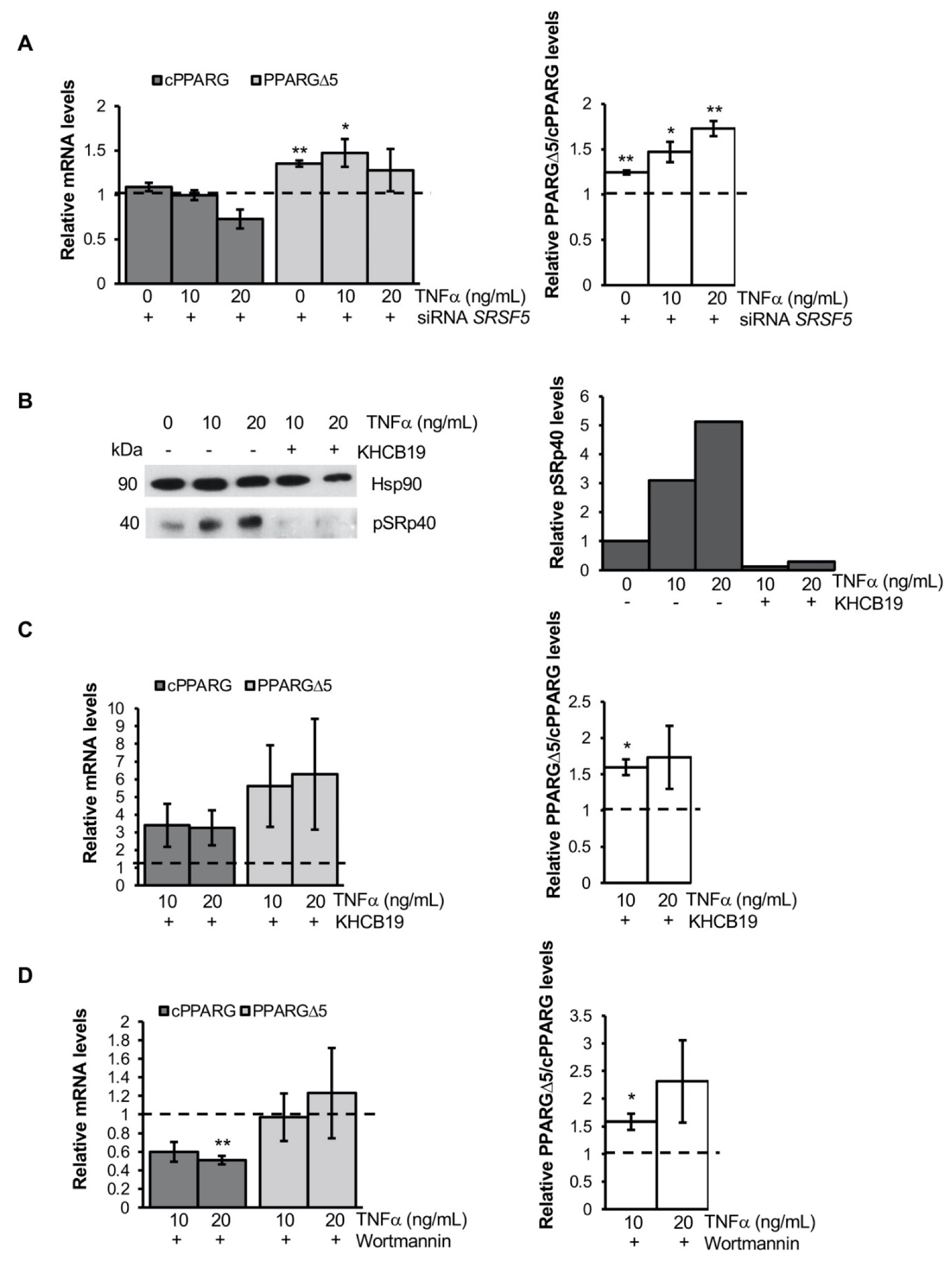
3.3. TNFα Alters PPARG Splicing in Human Mesenchymal Stem Cells
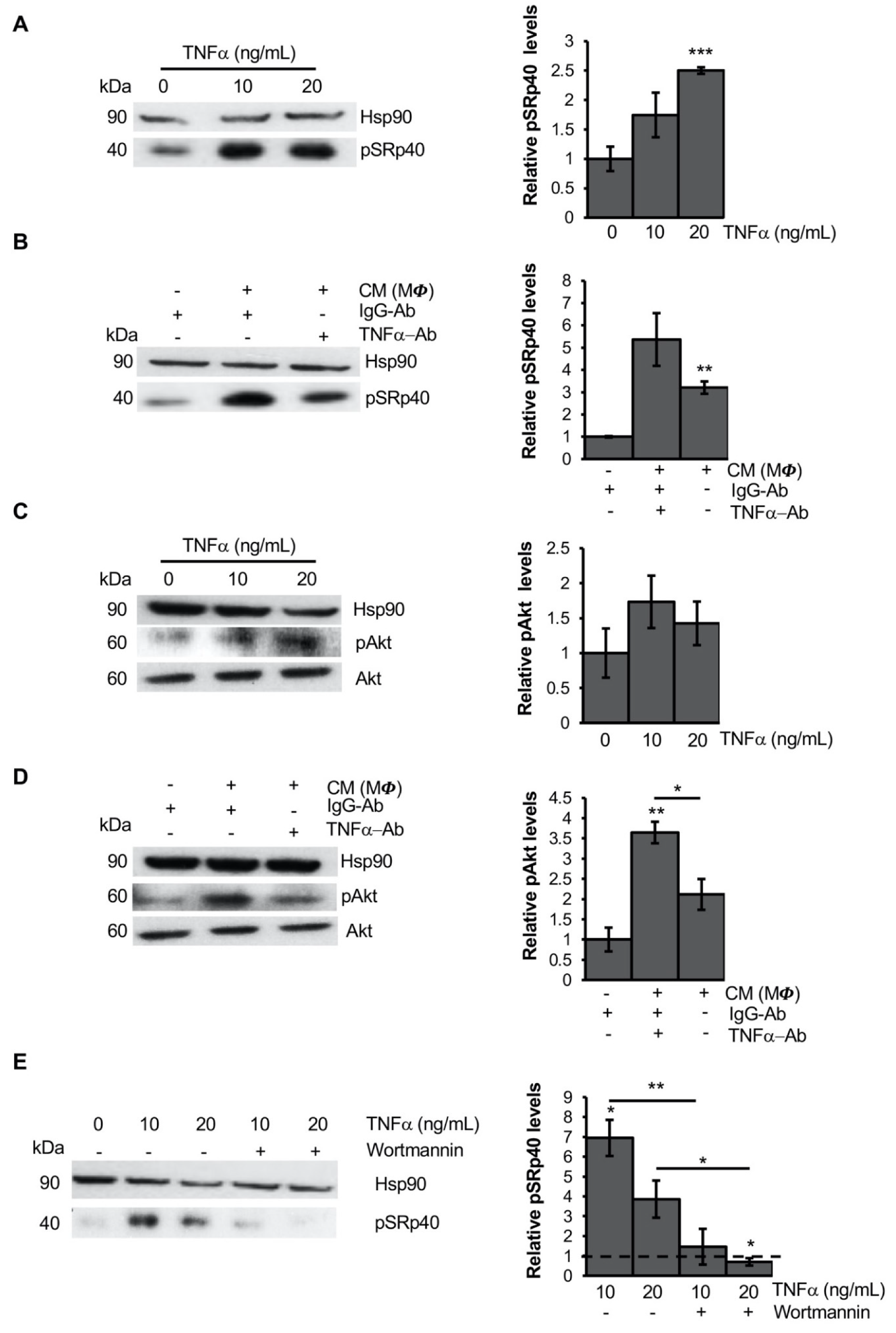
3.4. TNFα Modifies PPARG Splicing through PI3K/Akt Signaling
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Blüher, M. Metabolically Healthy Obesity. Endocr. Rev. 2020, 41, 405–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bray, G.A.; Kim, K.K.; Wilding, J.P.H.; World Obesity Federation. Obesity: A chronic relapsing progressive disease process. A position statement of the World Obesity Federation. Obes. Rev. 2017, 18, 715–723. [Google Scholar] [CrossRef] [Green Version]
- Lumeng, C.N.; Saltiel, A. Inflammatory links between obesity and metabolic disease. J. Clin. Investig. 2011, 121, 2111–2117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olefsky, J.M.; Glass, C.K. Macrophages, Inflammation, and Insulin Resistance. Annu. Rev. Physiol. 2010, 72, 219–246. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S. Inflammation and metabolic disorders. Nature 2006, 444, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Reilly, S.M.; Saltiel, A.R. Adapting to obesity with adipose tissue inflammation. Nat. Rev. Endocrinol. 2017, 13, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Kanda, H.; Tateya, S.; Tamori, Y.; Kotani, K.; Hiasa, K.I.; Kitazawa, R.; Kitazawa, S.; Miyachi, H.; Maeda, S.; Egashira, K.; et al. MCP-1 contributes to macrophageinfiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J. Clin. Investig. 2006, 116, 1494–1505. [Google Scholar] [CrossRef] [PubMed]
- Cinti, S.; Mitchell, G.; Barbatelli, G.; Murano, I.; Ceresi, E.; Faloia, E.; Wang, S.; Fortier, M.; Greenberg, A.S.; Obin, M.S. Adipocyte death defines macrophage localization and function in adipose tissue of obese mice and humans. J. Lipid Res. 2005, 46, 2347–2355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wellen, K.E.; Hotamisligil, G.S. Obesity-induced inflammatory changes in adipose tissue. J. Clin. Investig. 2003, 112, 1785–1788. [Google Scholar] [CrossRef]
- Wensveen, F.; Valentić, S.; Šestan, M.; Wensveen, T.T.; Polić, B. The “Big Bang” in obese fat: Events initiating obesity-induced adipose tissue inflammation. Eur. J. Immunol. 2015, 45, 2446–2456. [Google Scholar] [CrossRef] [PubMed]
- Russo, L.; Muir, L.; Geletka, L.; Delproposto, J.; Baker, N.; Flesher, C.; O’Rourke, R.; Lumeng, C.N. Cholesterol 25-hydroxylase (CH25H) as a promoter of adipose tissue inflammation in obesity and diabetes. Mol. Metab. 2020, 39, 100983. [Google Scholar] [CrossRef]
- Gustafson, B.; Gogg, S.; Hedjazifar, S.; Jenndahl, L.; Hammarstedt, A.; Smith, U. Inflammation and impaired adipogenesis in hypertrophic obesity in man. Am. J. Physiol. Metab. 2009, 297, E999–E1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suganami, T.; Nishida, J.; Ogawa, Y. A paracrine loop between adipocytes and macrophages aggravates inflammatory changes: Role of free fatty acids and tumor necrosis factor alpha. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 2062–2068. [Google Scholar] [CrossRef] [Green Version]
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W., Jr. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Investig. 2003, 12, 1796–1808. [Google Scholar] [CrossRef]
- Xu, H.; Barnes, G.T.; Yang, Q.; Tan, G.; Yang, D.; Chou, C.J.; Sole, J.; Nichols, A.; Ross, J.S.; Tartaglia, L.A.; et al. Chronic inflammation in fat plays a crucial role in the de-velopment of obesity-related insulin resistance. J. Clin. Investig. 2003, 12, 1821–1830. [Google Scholar] [CrossRef]
- Lumeng, C.N.; Deyoung, S.M.; Saltiel, A.R. Macrophages block insulin action in adipocytes by altering expression of signal-ing and glucose transport proteins. Am. J. Physiol. Endocrinol. Metab. 2007, 292, E166–E174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armoni, M.; Harel, C.; Karnieli, E. Transcriptional regulation of the GLUT4 gene: From PPAR-gamma and FOXO1 to FFA and inflammation. Trends Endocrinol. Metab. 2007, 18, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.F.; Craig, C.; Tolentino, L.L.; Choi, O.; Morton, J.; Rivas, H.; Cushman, S.W.; Engleman, E.G.; McLaughlin, T. Adipose tissue macrophages impair preadipocyte differentiation in humans. PLoS ONE 2017, 12, e0170728. [Google Scholar] [CrossRef] [Green Version]
- Yin, R.; Fang, L.; Li, Y.; Xue, P.; Li, Y.; Guan, Y.; Chang, Y.; Chen, C.; Wang, N. Pro-inflammatory Macrophages suppress PPARγ activity in Adipocytes via S-nitrosylation. Free Radic. Biol. Med. 2015, 89, 895–905. [Google Scholar] [CrossRef]
- Isakson, P.; Hammarstedt, A.; Gustafson, B.; Smith, U. Impaired Preadipocyte Differentiation in Human Abdominal Obesity: Role of Wnt, Tumor Necrosis Factor-, and Inflammation. Diabetes 2009, 58, 1550–1557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruan, H.; Hacohen, N.; Golub, T.R.; Van Parijs, L.; Lodish, H.F. Tumor Necrosis Factor-α Suppresses Adipocyte-Specific Genes and Activates Expression of Preadipocyte Genes in 3T3-L1 Adipocytes: Nuclear Factor-κ B Activation by TNF-α Is Obligatory. Diabetes 2002, 51, 1319–1336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; Berger, J.; Hu, E.; Szalkowski, D.; White-Carrington, S.; Spiegelman, B.M.; Moller, D.E. Negative regulation of peroxisome proliferator-activated receptor-gamma gene expression contributes to the antiadipogenic effects of tumor necrosis factor-alpha. Mol. Endocrinol. 1996, 10, 1457–1466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lefterova, M.I.; Haakonsson, A.K.; Lazar, M.A.; Mandrup, S. PPARγ and the global map of adipogenesis and beyond. Trends Endocrinol. Metab. 2014, 25, 293–302. [Google Scholar] [CrossRef] [Green Version]
- Ahmadian, M.; Suh, J.M.; Hah, N.; Liddle, C.; Atkins, A.R.; Downes, M.; Evans, R.M. PPARγ signaling and metabolism: The good, the bad and the future. Nat. Med. 2013, 19, 557–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosen, E.D.; MacDougald, O.A. Adipocyte differentiation from the inside out. Nat. Rev. Mol. Cell Biol. 2006, 7, 885–896. [Google Scholar] [CrossRef]
- Lehrke, M.; Lazar, M.A. The many faces of PPARgamma. Cell. Rev. 2005, 123, 993–999. [Google Scholar] [CrossRef] [Green Version]
- Imai, T.; Takakuwa, R.; Marchand, S.; Dentz, E.; Bornert, J.-M.; Messaddeq, N.; Wendling, O.; Mark, M.; Desvergne, B.; Wahli, W.; et al. Peroxisome proliferator-activated receptor is required in mature white and brown adipocytes for their survival in the mouse. Proc. Natl. Acad. Sci. USA 2004, 101, 4543–4547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tontonoz, P.; Hu, E.; Spiegelman, B.M. Stimulation of adipogenesis in fibroblasts by PPAR gamma 2, a lipid-activated transcription factor. Cell 1995, 79, 1147–1156. [Google Scholar] [CrossRef]
- Aprile, M.; Cataldi, S.; Ambrosio, M.R.; D’Esposito, V.; Lim, K.; Dietrich, A.; Blüher, M.; Savage, D.B.; Formisano, P.; Ciccodicola, A.; et al. PPARγΔ5, a Naturally Occurring Dominant-Negative Splice Isoform, Impairs PPARγ Function and Adipocyte Differentiation. Cell Rep. 2018, 25, 1577–1592.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aprile, M.; Cataldi, S.; Perfetto, C.; Ambrosio, M.R.; Italiani, P.; Tatè, R.; Blüher, M.; Ciccodicola, A.; Costa, V. In Vitro-Generated Hypertrophic-Like Adipocytes Displaying PPARG Isoforms Unbalance Recapitulate Adipocyte Dysfunctions In Vivo. Cells 2020, 9, 1284. [Google Scholar] [CrossRef]
- Keller, M.; Hopp, L.; Liu, X.; Wohland, T.; Rohde, K.; Cancello, R.; Klös, M.; Bacos, K.; Kern, M.; Eichelmann, F.; et al. Genome-wide DNA promoter methylation and transcriptome analysis in human adipose tissue unravels novel candidate genes for obesity. Mol. Metab. 2016, 6, 86–100. [Google Scholar] [CrossRef] [PubMed]
- Guiu-Jurado, E.; Unthan, M.; Böhler, N.; Kern, M.; Landgraf, K.; Dietrich, A.; Schleinitz, D.; Ruschke, K.; Klöting, N.; Faßhauer, M.; et al. Bone morphogenetic protein 2 (BMP2) may contribute to partition of energy storage into visceral and subcutaneous fat depots. Obesity 2016, 24, 2092–2100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastor, F.; Dumas, K.; Barthélémy, M.-A.; Regazzetti, C.; Druelle, N.; Peraldi, P.; Cormont, M.; Tanti, J.-F.; Giorgetti-Peraldi, S. Implication of REDD1 in the activation of inflammatory pathways. Sci. Rep. 2017, 7, 7023. [Google Scholar] [CrossRef] [PubMed]
- Appari, M.; Channon, K.M.; McNeill, E. Metabolic Regulation of Adipose Tissue Macrophage Function in Obesity and Diabetes. Antioxid. Redox Signal. 2018, 29, 297–312. [Google Scholar] [CrossRef] [PubMed]
- Nawaz, A.; Tobe, K. M2-like macrophages serve as a niche for adipocyte progenitors in adipose tissue. J. Diabetes Investig. 2019, 10, 1394–1400. [Google Scholar] [CrossRef] [PubMed]
- Arango Duque, G.; Descoteaux, A. Macrophage Cytokines: Involvement in Immunity and Infectious Diseases. Front. Immunol. 2014, 5, 491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batson, J.; Toop, H.D.; Redondo, C.; Babaei-Jadidi, R.; Chaikuad, A.; Wearmouth, S.; Gibbons, B.; Allen, C.; Tallant, C.; Zhang, J.; et al. Development of Potent, Selective SRPK1 Inhibitors as Potential Topical Therapeutics for Neovascular Eye Disease. ACS Chem. Biol. 2017, 12, 825–832. [Google Scholar] [CrossRef]
- Nowak, D.G.; Amin, E.M.; Rennel, E.S.; Hoareau-Aveilla, C.; Gammons, M.; Damodoran, G.; Hagiwara, M.; Harper, S.J.; Woolard, J.; Ladomery, M.R.; et al. Regulation of Vascular Endothelial Growth Factor (VEGF) Splicing from Pro-angiogenic to Anti-angiogenic Isoforms: A novel therapeutic strategy for angiogenesis. J. Biol. Chem. 2010, 285, 5532–5540. [Google Scholar] [CrossRef] [Green Version]
- Nowak, D.G.; Woolard, J.; Amin, E.M.; Konopatskaya, O.; Saleem, M.A.; Churchill, A.J.; Ladomery, M.R.; Harper, S.J.; Bates, D.O. Expression of pro- and anti-angiogenic isoforms of VEGF is differentially regulated by splicing and growth factors. J. Cell Sci. 2008, 121, 3487–3495. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Z.; Gengaro, P.; Wang, W.; Wang, X.-Q.; Li, C.; Faubel, S.; Rivard, C.; Schrier, R.W. Role of NF-κB and PI 3-kinase/Akt in TNF-α-induced cytotoxicity in microvascular endothelial cells. Am. J. Physiol. Physiol. 2008, 295, F932–F941. [Google Scholar] [CrossRef]
- Faurschou, A.; Gniadecki, R. TNF-α stimulates Akt by a distinct aPKC-dependent pathway in premalignant keratinocytes. Exp. Dermatol. 2008, 17, 992–997. [Google Scholar] [CrossRef]
- Patel, N.A.; Kaneko, S.; Apostolatos, H.S.; Bae, S.S.; Watson, J.E.; Davidowitz, K.; Chappell, D.S.; Birnbaum, M.; Cheng, J.Q.; Cooper, D.R. Molecular and Genetic Studies Imply Akt-mediated Signaling Promotes Protein Kinase CβII Alternative Splicing via Phosphorylation of Serine/Arginine-rich Splicing Factor SRp40. J. Biol. Chem. 2005, 280, 14302–14309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahebi, M.; Hanafi, M.M.; van Wijnen, A.J.; Azizi, P.; Abiri, R.; Ashkani, S.; Taheri, S. Towards understanding pre-mRNA splicing mechanisms and the role of SR proteins. Gene 2016, 587, 107–119. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Fu, X.-D. Regulation of splicing by SR proteins and SR protein-specific kinases. Chromosoma 2013, 122, 191–207. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, V.; Matos, P.; Jordan, P. Antagonistic SR proteins regulate alternative splicing of tumor-related Rac1b downstream of the PI3-kinase and Wnt pathways. Hum. Mol. Genet. 2009, 18, 3696–3707. [Google Scholar] [CrossRef] [PubMed]
- Erkelenz, S.; Mueller, W.F.; Evans, M.S.; Busch, A.; Schöneweis, K.; Hertel, K.J.; Schaal, H. Position-dependent splicing activation and repression by SR and hnRNP proteins rely on common mechanisms. RNA 2012, 19, 96–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cáceres, J.F.; Stamm, S.; Helfman, D.M.; Krainer, A.R. Regulation of Alternative Splicing in Vivo by Overexpression of Antagonistic Splicing Factors. Science 1994, 265, 1706–1709. [Google Scholar] [CrossRef] [Green Version]
- Mayeda, A.; Krainer, A.R. Regulation of alternative pre-mRNA splicing by hnRNP A1 and splicing factor SF2. Cell 1992, 68, 365–375. [Google Scholar] [CrossRef]
- Pascual, G.; Fong, A.L.; Ogawa, S.; Gamliel, A.; Li, A.C.; Perissi, V.; Rose, D.W.; Willson, T.M.; Rosenfeld, M.G.; Glass, C.K. A SUMOylation-dependent pathway mediates transrepression of inflammatory response genes by PPAR-γ. Nature 2005, 437, 759–763. [Google Scholar] [CrossRef] [PubMed]
- Bassaganya-Riera, J.; Misyak, S.; Guri, A.J.; Hontecillas, R. PPAR γ is highly expressed in F4/80hi adipose tissue macrophages and dampens adipose-tissue inflammation. Cell. Immunol. 2009, 258, 138–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charo, I.F. Macrophage Polarization and Insulin Resistance: PPARγ in Control. Cell Metab. 2007, 6, 96–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, N.H.; Kwang Tan, A.Y.; Chen, Y.; Sekar, K.; Tan, P.Y.; Chan, D.; Chiang, K.; Wong, N.C. Thermal evaluation of vertical greenery systems for building walls. Build. Environ. 2010, 45, 663–672. [Google Scholar] [CrossRef]
- Viglietti, A.I.P.; Giambartolomei, G.H.; Quarleri, J.; Delpino, M.V. Brucella abortus Infection Modulates 3T3-L1 Adipocyte Inflammatory Response and Inhibits Adipogenesis. Front. Endocrinol. 2020, 11, 755. [Google Scholar] [CrossRef] [PubMed]
- Lacasa, D.; Taleb, S.; Keophiphath, M.; Miranville, A.; Clement, K. Macrophage-Secreted Factors Impair Human Adipogenesis: Involvement of Proinflammatory State in Preadipocytes. Endocrinology 2007, 148, 868–877. [Google Scholar] [CrossRef] [Green Version]
- Batista, T.M.; Jayavelu, A.K.; Albrechtsen, N.J.W.; Iovino, S.; Lebastchi, J.; Pan, H.; Dreyfuss, J.M.; Krook, A.; Zierath, J.R.; Mann, M.; et al. A Cell-Autonomous Signature of Dysregulated Protein Phosphorylation Underlies Muscle Insulin Resistance in Type 2 Diabetes. Cell Metab. 2020, 32, 844–859.e5. [Google Scholar] [CrossRef]
- Vernia, S.; Edwards, Y.J.; Han, M.S.; Cavanagh-Kyros, J.; Barrett, T.; Kim, J.K.; Davis, R.J. An alternative splicing program promotes adipose tissue thermogenesis. eLife 2016, 5, e17672. [Google Scholar] [CrossRef] [PubMed]
- Pihlajamäki, J.; Lerin, C.; Itkonen, P.; Boes, T.; Floss, T.; Schroeder, J.; Dearie, F.; Crunkhorn, S.; Burak, F.; Jimenez-Chillaron, J.C.; et al. Expression of the Splicing Factor Gene SFRS10 Is Reduced in Human Obesity and Contributes to Enhanced Lipogenesis. Cell Metab. 2011, 14, 208–218. [Google Scholar] [CrossRef] [Green Version]
- Prabhu, S.; Deng, H.; Cross, T.-W.L.; Shahoei, S.H.; Konopka, C.J.; Medina, N.G.; Applegate, C.C.; Wallig, M.A.; Dobrucki, L.W.; Nelson, E.R.; et al. Nanocarriers targeting adipose macrophages increase glucocorticoid anti-inflammatory potency to ameliorate metabolic dysfunction. Biomater. Sci. 2020, 9, 506–518. [Google Scholar] [CrossRef]
- Aprile, M.; Ambrosio, M.R.; D’Esposito, V.; Beguinot, F.; Formisano, P.; Costa, V.; Ciccodicola, A. PPARGin Human Adipogenesis: Differential Contribution of Canonical Transcripts and Dominant Negative Isoforms. PPAR Res. 2014, 2014, 537865. [Google Scholar] [CrossRef] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cataldi, S.; Aprile, M.; Melillo, D.; Mucel, I.; Giorgetti-Peraldi, S.; Cormont, M.; Italiani, P.; Blüher, M.; Tanti, J.-F.; Ciccodicola, A.; et al. TNFα Mediates Inflammation-Induced Effects on PPARG Splicing in Adipose Tissue and Mesenchymal Precursor Cells. Cells 2022, 11, 42. https://doi.org/10.3390/cells11010042
Cataldi S, Aprile M, Melillo D, Mucel I, Giorgetti-Peraldi S, Cormont M, Italiani P, Blüher M, Tanti J-F, Ciccodicola A, et al. TNFα Mediates Inflammation-Induced Effects on PPARG Splicing in Adipose Tissue and Mesenchymal Precursor Cells. Cells. 2022; 11(1):42. https://doi.org/10.3390/cells11010042
Chicago/Turabian StyleCataldi, Simona, Marianna Aprile, Daniela Melillo, Inès Mucel, Sophie Giorgetti-Peraldi, Mireille Cormont, Paola Italiani, Matthias Blüher, Jean-François Tanti, Alfredo Ciccodicola, and et al. 2022. "TNFα Mediates Inflammation-Induced Effects on PPARG Splicing in Adipose Tissue and Mesenchymal Precursor Cells" Cells 11, no. 1: 42. https://doi.org/10.3390/cells11010042
APA StyleCataldi, S., Aprile, M., Melillo, D., Mucel, I., Giorgetti-Peraldi, S., Cormont, M., Italiani, P., Blüher, M., Tanti, J.-F., Ciccodicola, A., & Costa, V. (2022). TNFα Mediates Inflammation-Induced Effects on PPARG Splicing in Adipose Tissue and Mesenchymal Precursor Cells. Cells, 11(1), 42. https://doi.org/10.3390/cells11010042