
Hepatocyte Thorns, A Novel Drug-Induced Stress Response in Human and Mouse Liver Spheroids

 ,
,  , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. ATP Assay
2.3. Thorn Isolation
2.4. Deglycosylation and Glycosylation Staining
2.5. Whole Spheroid Staining
2.6. RT-qPCR
2.7. Proteomics
2.7.1. Qualitative Proteomics
2.7.2. Quantitative Proteomics
2.7.3. Proteome Integral Solubility Alteration (PISA) Analysis
2.8. Scanning Electron Microscopy
2.9. Animal Experiments
2.10. Masson’s Trichrome Staining
3. Results
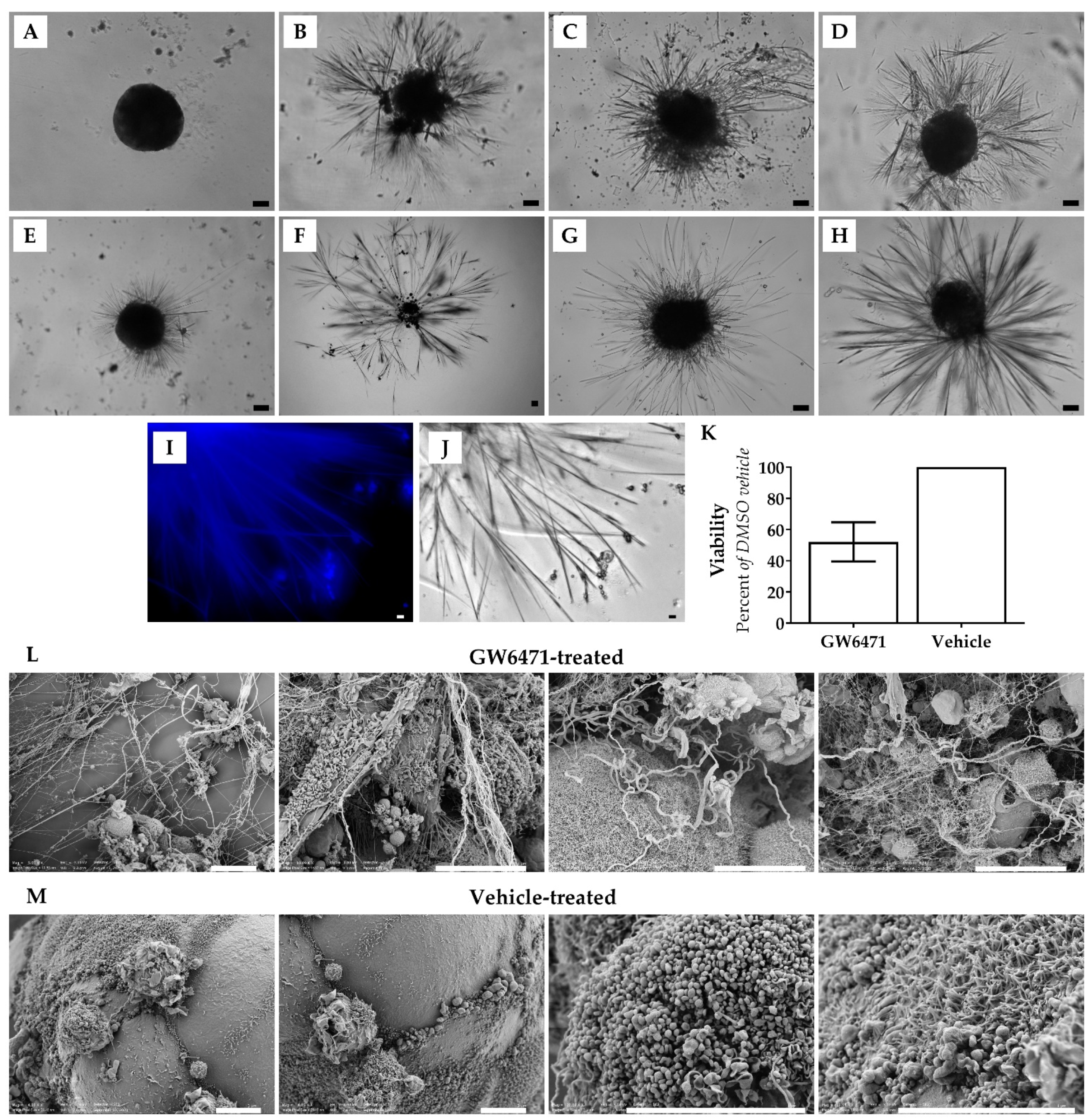
3.1. Chronic Drug Treatment Causes the Appearance of Thorn-Like Structures in Spheroid 3D Liver Model
3.2. Thorns Are Composed of Glycosylated Protein Fibres
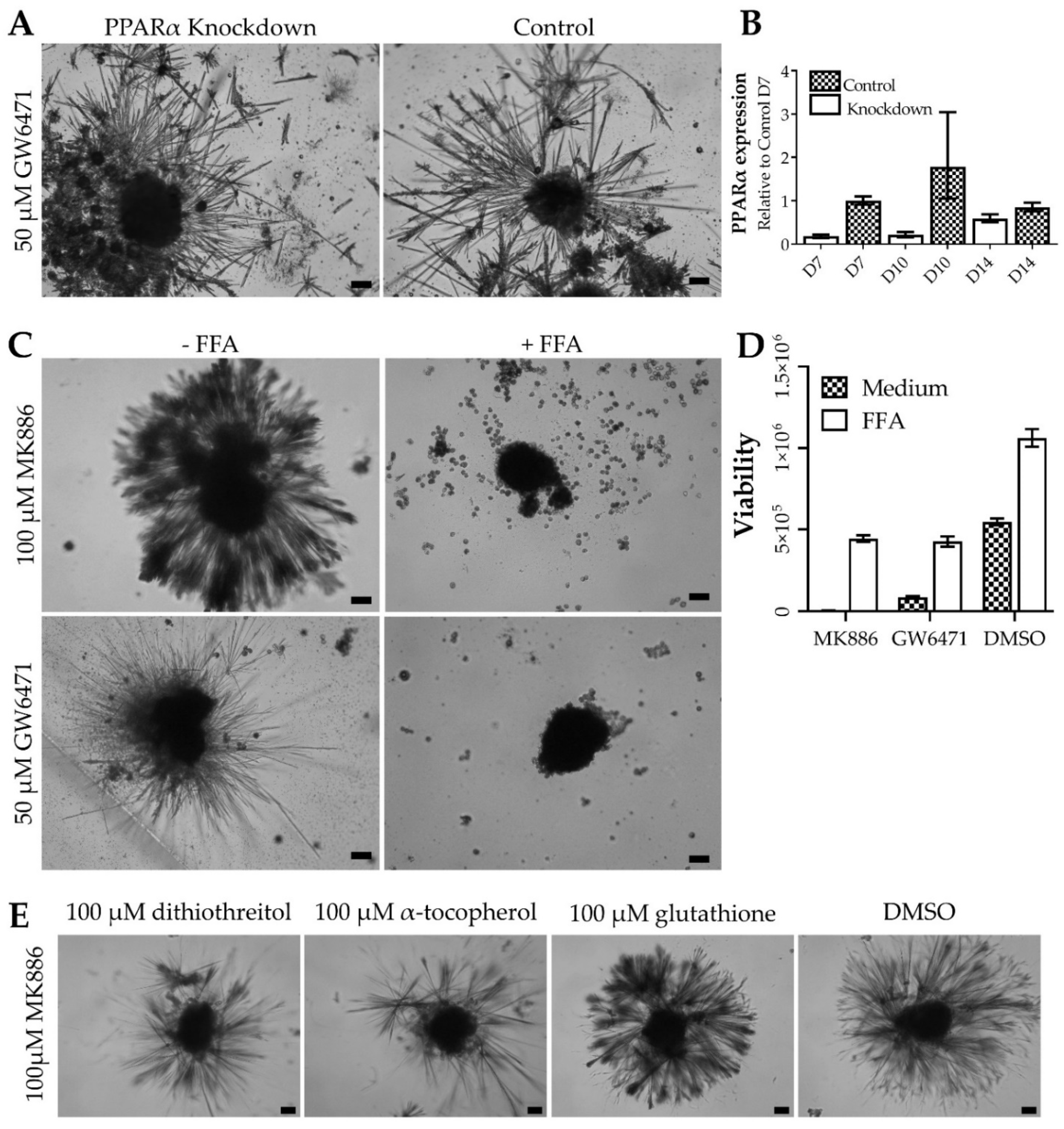
3.3. Thorns Form Due to Lipid Metabolism Disturbances and Their Creation Is Stimulated by Reactive Oxygen Species
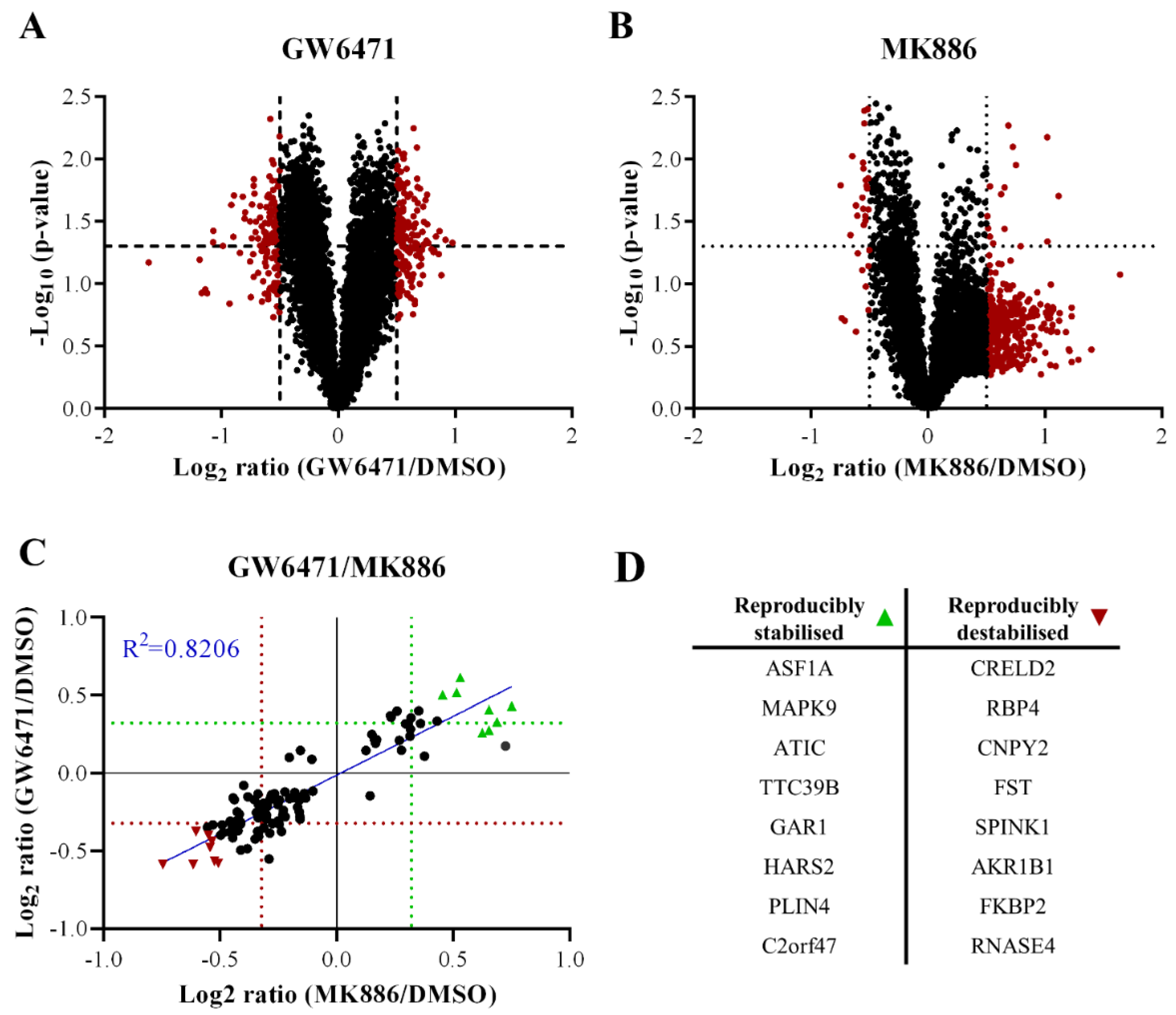
3.4. Thorns Represent Chronic Drug Treatment-Induced Hepatocyte Stress Response
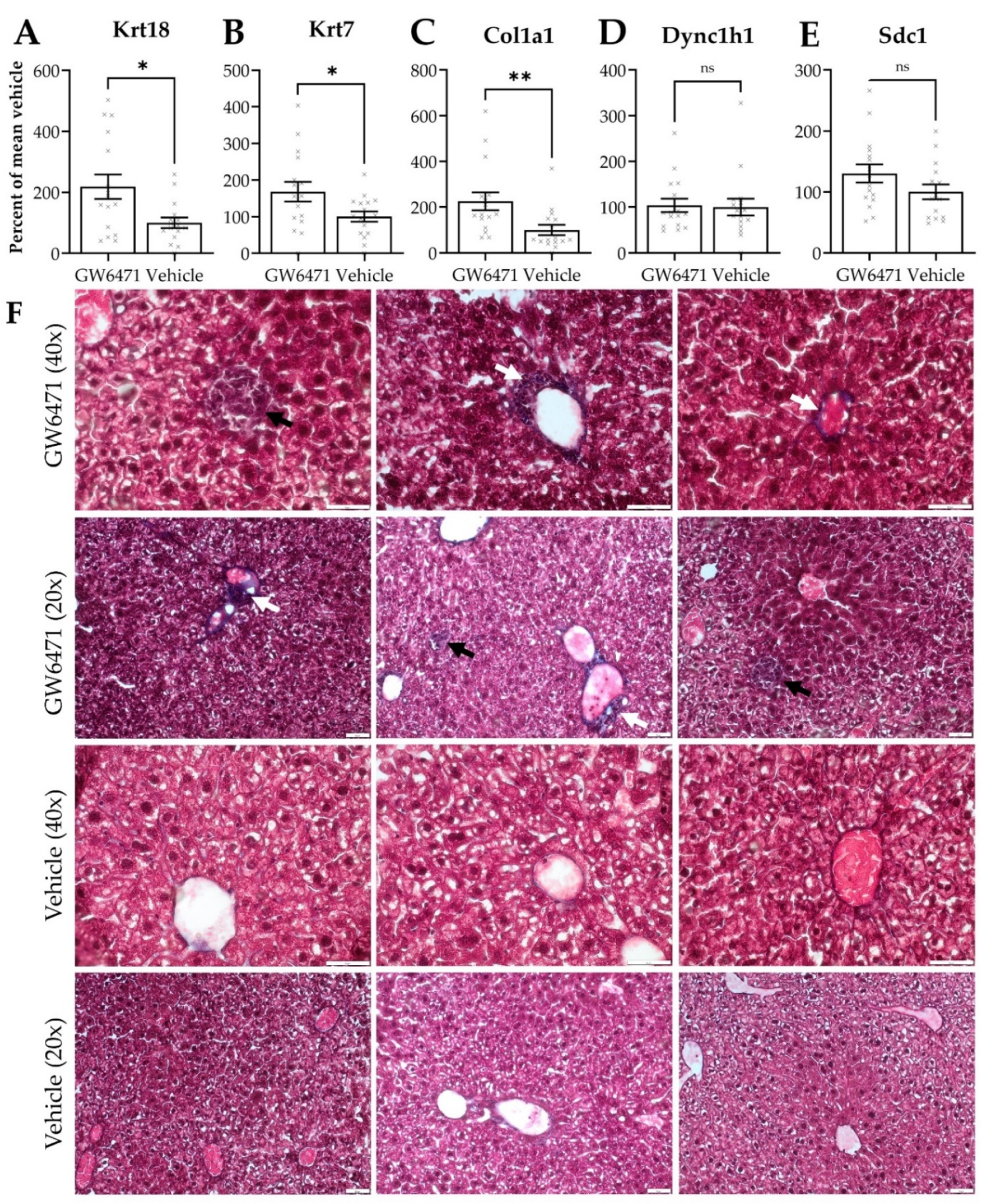
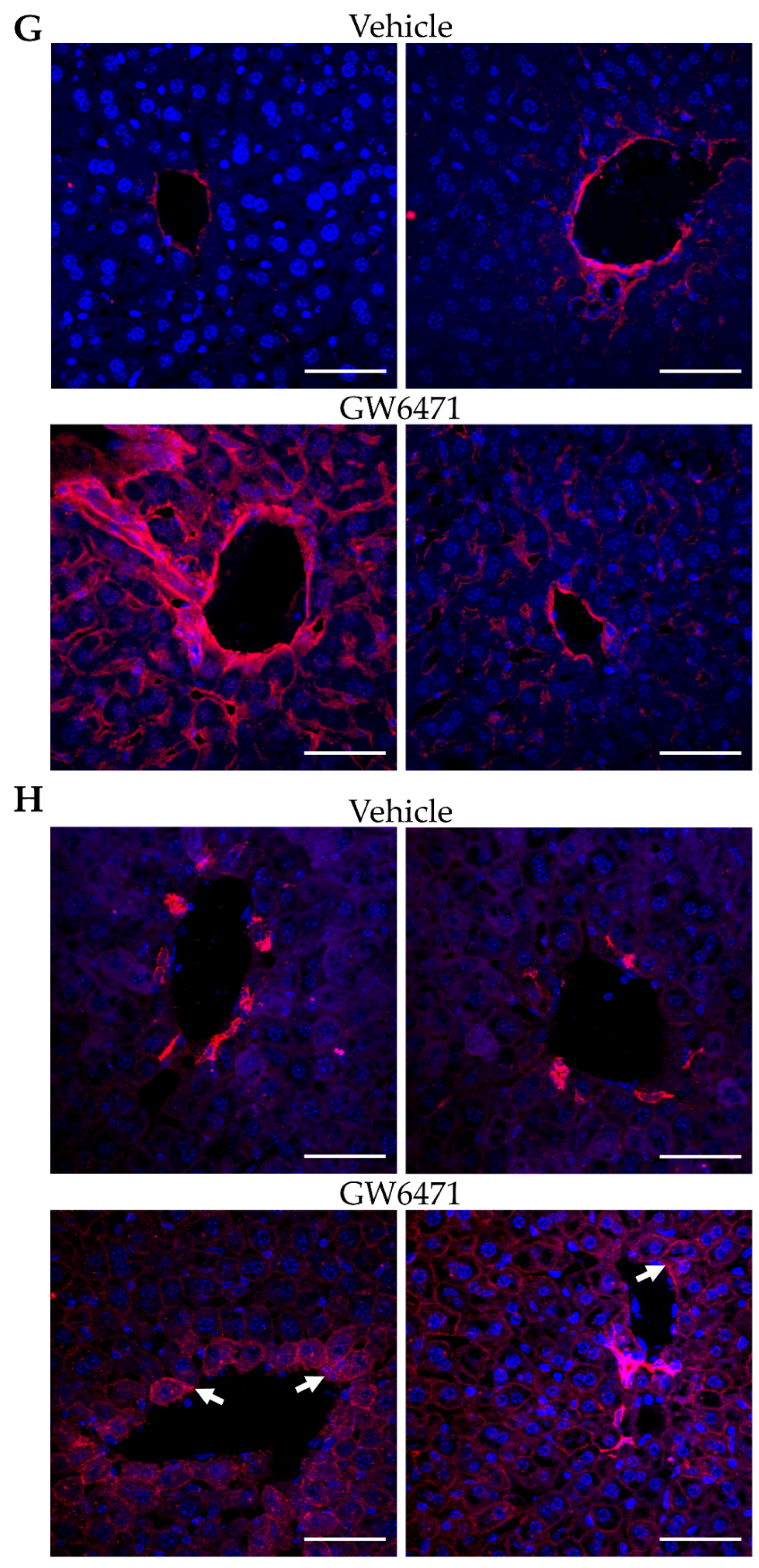
3.5. Chronically GW6471-Treated Mice Exhibit Fibre Accumulation in the Liver
4. Discussion
4.1. Thorn Characteristics
4.2. Proposed Mechanisms of Thorn Formation
4.3. Comparable Effects of GW6471 in Mice
4.4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bell, C.C.; Hendriks, D.F.G.; Moro, S.M.L.; Ellis, E.; Walsh, J.; Renblom, A.; Fredriksson Puigvert, L.; Dankers, A.C.A.; Jacobs, F.; Snoeys, J.; et al. Characterization of primary human hepatocyte spheroids as a model system for drug-induced liver injury, liver function and disease. Sci. Rep. 2016, 6, 25187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bell, C.C.; Dankers, A.C.A.; Lauschke, V.M.; Sison-Young, R.; Jenkins, R.; Rowe, C.; Goldring, C.E.; Park, K.; Regan, S.L.; Walker, T.; et al. Comparison of Hepatic 2D Sandwich Cultures and 3D Spheroids for Long-term Toxicity Applications: A Multicenter Study. Toxicol. Sci. 2018, 162, 655–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.H.; Batey, R.G.; George, J. Role of ethanol in the regulation of hepatic stellate cell function. World J. Gastroenterol. 2006, 12, 6926–6932. [Google Scholar] [CrossRef] [PubMed]
- Thor, H.; Mirabelli, F.; Salis, A.; Cohen, G.M.; Bellomo, G.; Orrenius, S. Alterations in hepatocyte cytoskeleton caused by redox cycling and alkylating quinones. Arch. Biochem. Biophys. 1988, 266, 397–407. [Google Scholar] [CrossRef]
- Puli, S.R.; Fraley, M.A.; Puli, V.; Kuperman, A.B.; Alpert, M.A. Hepatic cirrhosis caused by low-dose oral amiodarone therapy. Am. J. Med. Sci. 2005, 330, 257–261. [Google Scholar] [CrossRef] [PubMed]
- Harjumäki, R.; Pridgeon, C.S.; Ingelman-Sundberg, M. CYP2E1 in Alcoholic and Non-Alcoholic Liver Injury. Roles of ROS, Reactive Intermediates and Lipid Overload. Int. J. Mol. Sci. 2021, 22, 8221. [Google Scholar] [CrossRef]
- Denk, H.; Gschnait, F.; Wolff, K. Hepatocellular hyalin (Mallory bodies) in long term griseofulvin treated mice: A new experimental model for the study of hyalin formation. Lab. Investig. 1975, 32, 773–776. [Google Scholar]
- Hurrell, T.; Kastrinou-Lampou, V.; Fardellas, A.; Hendriks, D.F.G.; Nordling, Å.; Johansson, I.; Baze, A.; Parmentier, C.; Richert, L.; Ingelman-Sundberg, M. Human Liver Spheroids as a Model to Study Aetiology and Treatment of Hepatic Fibrosis. Cells 2020, 9, 964. [Google Scholar] [CrossRef] [Green Version]
- Gaetani, M.; Sabatier, P.; Saei, A.A.; Beusch, C.M.; Yang, Z.; Lundström, S.L.; Zubarev, R.A. Proteome Integral Solubility Alteration: A High-Throughput Proteomics Assay for Target Deconvolution. J. Proteome Res. 2019, 18, 4027–4037. [Google Scholar] [CrossRef]
- Li, J.; Van Vranken, J.G.; Pontano Vaites, L.; Schweppe, D.K.; Huttlin, E.L.; Etienne, C.; Nandhikonda, P.; Viner, R.; Robitaille, A.M.; Thompson, A.H.; et al. TMTpro reagents: A set of isobaric labeling mass tags enables simultaneous proteome-wide measurements across 16 samples. Nat. Methods 2020, 17, 399–404. [Google Scholar] [CrossRef]
- Hynes, R.O.; Naba, A. Overview of the matrisome-An inventory of extracellular matrix constituents and functions. Cold Spring Harb. Perspect. Biol. 2012, 4, a004903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, M.; Burk, O.; Klumpp, B.; Kandel, B.A.; Damm, G.; Weiss, T.S.; Klein, K.; Schwab, M.; Zanger, U.M. Direct transcriptional regulation of human hepatic cytochrome P450 3A4 (CYP3A4) by peroxisome proliferator-activated receptor alpha (PPARa)s. Mol. Pharmacol. 2013, 83, 709–718. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, L.; Kirk, R. High drug attrition rates—Where are we going wrong? Nat. Rev. Clin. Oncol. 2011, 8, 189–190. [Google Scholar] [CrossRef] [PubMed]
- Myasnikova, D.; Osaki, T.; Onishi, K.; Kageyama, T.; Zhang Molino, B.; Fukuda, J. Synergic effects of oxygen supply and antioxidants on pancreatic β-cell spheroids. Sci. Rep. 2019, 9, 1802. [Google Scholar] [CrossRef] [Green Version]
- Tsai, J.H.; Chen, H.W.; Chen, Y.W.; Liu, J.Y.; Lii, C.K. The protection of hepatocyte cells from the effects of oxidative stress by treatment with vitamin e in conjunction with DTT. J. Biomed. Biotechnol. 2010, 2010, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Yoon, M. The role of PPARalpha in lipid metabolism and obesity: Focusing on the effects of estrogen on PPARalpha actions. Pharmacol. Res. 2009, 60, 151–159. [Google Scholar] [CrossRef]
- Gervois, P.; Torra, I.P.; Fruchart, J.C.; Staels, B. Regulation of lipid and lipoprotein metabolism by PPAR activators. Clin. Chem. Lab. Med. 2000, 38, 3–11. [Google Scholar] [CrossRef]
- Vorrink, S.U.; Zhou, Y.; Ingelman-Sundberg, M.; Lauschke, V.M. Prediction of drug-induced hepatotoxicity using long-term stable primary hepatic 3D spheroid cultures in chemically defined conditions. Toxicol. Sci. 2018, 163, 655–665. [Google Scholar] [CrossRef]
- Aloe, L.; Alleva, E.; Bohm, A.; Levi-Montalcini, R. Aggressive behavior induces release of nerve growth factor from mouse salivary gland into the bloodstream. Proc. Natl. Acad. Sci. USA 1986, 83, 6184–6187. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.; Guo, Z.; Hopkins, C.D.; Wei, N.; Chu, E.; Wipf, P.; Schmitz, J.C. Bis-cyclopropane analog of disorazole C1 is a microtubuledestabilizing agent active in ABCB1-overexpressing human colon cancer cells. Oncotarget 2015, 6, 40866–40879. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Qu, J.Y. Autofluorescence spectroscopy of epithelial tissues. J. Biomed. Opt. 2006, 11, 054023. [Google Scholar] [CrossRef] [PubMed]
- Novikov, I.A.; Grusha, I.O.; Kiriushchenkova, N.P. Autofluorescent diagnostics of skin and mucosal tumors. Vestn. Oftalmol. 2013, 129, 147–153. [Google Scholar] [PubMed]
- Croce, A.C.; Ferrigno, A.; Bottiroli, G.; Vairetti, M. Autofluorescence-based optical biopsy: An effective diagnostic tool in hepatology. Liver Int. 2018, 38, 1160–1174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemoinne, S.; Cadoret, A.; El Mourabit, H.; Thabut, D.; Housset, C. Origins and functions of liver myofibroblasts. Biochim. Biophys. Acta Mol. Basis Dis. 2013, 1832, 948–954. [Google Scholar] [CrossRef] [Green Version]
- Gillard, J.; Ford-Hutchinson, A.W.; Chan, C.; Charleson, S.; Denis, D.; Foster, A.; Fortin, R.; Leger, S.; McFarlane, C.S.; Morton, H.; et al. L-663,536 (MK-886)(3-[1-(4-chlorobenzyl)-3-t-butyl-thio-5-isopropylindol-2-yl]-, 2-dimethylpropanoic acid), a novel, orally active leukotriene biosynthesis inhibitor. Can. J. Physiol. Pharmacol. 1989, 67, 456–464. [Google Scholar] [CrossRef]
- Bischoff, E.; Stasch, J.P. Effects of the sGC stimulator BAY 41-2272 are not mediated by phosphodiesterase 5 inhibition. Circulation 2004, 110, e320–e321. [Google Scholar] [CrossRef] [Green Version]
- De Jong, E.M.G.J.; Van Vlijmen, I.M.M.J.; Scholte, J.C.M.; Buntinx, A.; Friedman, B.; Tanaka, W.; Van De Kerkhof, P.C.M. Clinical and biochemical effects of an oral leukotriene biosynthesis inhibitor (MK886) in psoriasis. Skin Pharmacol. Physiol. 1991, 4, 278–285. [Google Scholar] [CrossRef]
- Friedman, B.S.; Bel, E.H.; Buntinx, A.; Tanaka, W.; Han, Y.H.R.; Shingo, S.; Spector, R.; Sterk, P. Oral leukotriene inhibitor (MK-886) blocks allergen-induced airway responses. Am. Rev. Respir. Dis. 1993, 147, 839–844. [Google Scholar] [CrossRef]
- Sakellariou, S.; Michaelides, C.; Voulgaris, T.; Vlachogiannakos, J.; Manesis, E.; Tiniakos, D.G.; Delladetsima, I. Keratin 7 expression in hepatic cholestatic diseases. Virchows Arch. 2021, 479, 815–824. [Google Scholar] [CrossRef]
- Ren, C.; Paronetto, F.; Mak, K.M.; Leo, M.A.; Lieber, C.S. Cytokeratin 7 staining of hepatocytes predicts progression to more severe fibrosis in alcohol-fed baboons. J. Hepatol. 2003, 38, 770–775. [Google Scholar] [CrossRef]
- Tarlow, B.D.; Pelz, C.; Naugler, W.E.; Wakefield, L.; Wilson, E.M.; Finegold, M.J.; Grompe, M. Bipotential adult liver progenitors are derived from chronically injured mature hepatocytes. Cell Stem Cell 2014, 15, 605–618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perez-Riverol, Y.; Bai, J.; Bandla, C.; García-Seisdedos, D.; Hewapathirana, S.; Kamatchinathan, S.; Kundu, D.J.; Prakash, A.; Frericks-Zipper, A.; Eisenacher, M.; et al. The PRIDE database resources in 2022: A hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 2022, 50, D543–D552. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Identified Proteins | Sum of MK886 | Sum of DMSO |
|---|---|---|
| Spectrin alpha chain, non-erythrocytic 1 | 355 | 17 |
| Cytoplasmic dynein 1 heavy chain 1 | 331 | 5 |
| Filamin-A | 327 | 10 |
| Spectrin beta chain, non-erythrocytic 1 | 319 | 32 |
| CK, type I cytoskeletal 18 | 297 | 24 |
| Filamin-B | 276 | 7 |
| Clathrin heavy chain 1 | 266 | 18 |
| Plectin | 261 | 3 |
| Fatty acid synthase | 223 | 9 |
| Talin-1 | 213 | 8 |
| DNA-dependent protein kinase catalytic subunit | 207 | 1 |
| Elongation factor 1-alpha 1 | 202 | 19 |
| Tubulin beta-2B chain | 201 | 16 |
| Tubulin beta-4A | 191 | 17 |
| Ras GTPase-activating-like protein IQGAP2 | 186 | 7 |
| Neuroblast differentiation-associated protein AHNAK | 175 | 9 |
| Ras GTPase-activating-like protein IQGAP1 | 147 | 1 |
| Aldo-keto reductase family 1 member C2 | 144 | 7 |
| eIF-2-alpha kinase activator GCN1 | 128 | 2 |
| Complement C4-B | 121 | 3 |
| Elongation factor 2 | 118 | 3 |
| Fructose-bisphosphate aldolase B | 112 | 11 |
| Alpha-enolase | 108 | 8 |
| Ribosome-binding protein 1 | 104 | 5 |
| UTP--glucose-1-phosphate uridylyltransferase | 104 | 3 |
| NADPH--cytochrome P450 reductase | 103 | 10 |
| Cytochrome P450 2C8 | 101 | 10 |
| Cytosolic 10-formyltetrahydrofolate dehydrogenase | 101 | 4 |
| Heat shock protein HSP 90-alpha | 96 | 9 |
| Tubulin beta chain | 241 | 41 |
| CK, type II cytoskeletal 7 | 114 | 16 |
| Identified Proteins | Sum of MK886 | Sum of DMSO |
|---|---|---|
| Filamin-A | 480 | 26 |
| CK, type I cytoskeletal 18 | 424 | 42 |
| Cytoplasmic dynein 1 heavy chain 1 | 420 | 6 |
| Plectin | 397 | 3 |
| Filamin-B | 393 | 15 |
| Talin-1 | 308 | 12 |
| Fatty acid synthase | 307 | 12 |
| Tubulin beta-2B chain | 266 | 16 |
| Ras GTPase-activating-like protein IQGAP2 | 261 | 25 |
| DNA-dependent protein kinase catalytic subunit | 252 | 1 |
| Elongation factor 1-alpha 1 | 239 | 24 |
| Tubulin beta-4A chain | 235 | 17 |
| CK, type II cytoskeletal 7 | 216 | 18 |
| Ras GTPase-activating-like protein IQGAP1 | 205 | 5 |
| Aldo-keto reductase family 1 member C2 | 182 | 12 |
| Complement C4-B | 167 | 16 |
| Elongation factor 2 | 163 | 6 |
| Ribosome-binding protein 1 | 159 | 5 |
| UTP--glucose-1-phosphate uridylyltransferase | 157 | 10 |
| Alpha-enolase | 156 | 10 |
| Coatomer subunit alpha | 153 | 1 |
| eIF-2-alpha kinase activator GCN1 | 153 | 5 |
| Formimidoyltransferase-cyclodeaminase | 141 | 13 |
| C-1-tetrahydrofolate synthase, cytoplasmic | 140 | 8 |
| Gelsolin | 134 | 9 |
| Staphylococcal nuclease domain-containing protein 1 | 133 | 1 |
| Cytosolic 10-formyltetrahydrofolate dehydrogenase | 133 | 4 |
| Aldo-keto reductase family 1 member C3 | 121 | 9 |
| Heat shock 70 kDa protein 1A | 113 | 6 |
| Long-chain-fatty-acid--CoA ligase 5 | 112 | 8 |
| Alpha-actinin-1 | 105 | 7 |
| T-complex protein 1 subunit beta | 100 | 2 |
| Glutamine--fructose-6-phosphate aminotransferase 1 | 99 | 3 |
| ATP-dependent RNA helicase A | 96 | 1 |
| Common Proteins | Cytoskeleton | Filament |
|---|---|---|
| Cytoplasmic dynein 1 heavy chain 1 | ✓ | ✓ |
| Filamin-A | ✓ | ✓ |
| CK, type I cytoskeletal 18 | ✓ | ✓ |
| Filamin-B | ✓ | ✓ |
| Plectin | ✓ | ✓ |
| Fatty acid synthase | ||
| Talin-1 | ✓ | |
| DNA-dependent protein kinase catalytic subunit | ||
| Elongation factor 1-alpha 1 | ||
| Tubulin beta-2B chain | ✓ | ✓ |
| Tubulin beta-4A | ✓ | ✓ |
| Ras GTPase-activating-like protein IQGAP2 | ||
| Neuroblast differentiation-associated protein AHNAK | ||
| Ras GTPase-activating-like protein IQGAP1 | ✓ | |
| Aldo-keto reductase family 1 member C2 | ||
| eIF-2-alpha kinase activator GCN1 | ||
| Complement C4-B | ||
| Elongation factor 2 | ||
| CK, type II cytoskeletal 7 | ✓ | ✓ |
| Alpha-enolase | ||
| UTP--glucose-1-phosphate uridylyltransferase | ||
| Cytosolic 10-formyltetrahydrofolate dehydrogenase |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pridgeon, C.S.; Bolhuis, D.P.; Milosavljević, F.; Manojlović, M.; Végvári, Á.; Gaetani, M.; Jukić, M.M.; Ingelman-Sundberg, M. Hepatocyte Thorns, A Novel Drug-Induced Stress Response in Human and Mouse Liver Spheroids. Cells 2022, 11, 1597. https://doi.org/10.3390/cells11101597
Pridgeon CS, Bolhuis DP, Milosavljević F, Manojlović M, Végvári Á, Gaetani M, Jukić MM, Ingelman-Sundberg M. Hepatocyte Thorns, A Novel Drug-Induced Stress Response in Human and Mouse Liver Spheroids. Cells. 2022; 11(10):1597. https://doi.org/10.3390/cells11101597
Chicago/Turabian StylePridgeon, Chris S., Dian P. Bolhuis, Filip Milosavljević, Marina Manojlović, Ákos Végvári, Massimiliano Gaetani, Marin M. Jukić, and Magnus Ingelman-Sundberg. 2022. "Hepatocyte Thorns, A Novel Drug-Induced Stress Response in Human and Mouse Liver Spheroids" Cells 11, no. 10: 1597. https://doi.org/10.3390/cells11101597