
Pathogenic T-Cell Responses in Immune-Mediated Glomerulonephritis

Abstract
1. Introduction
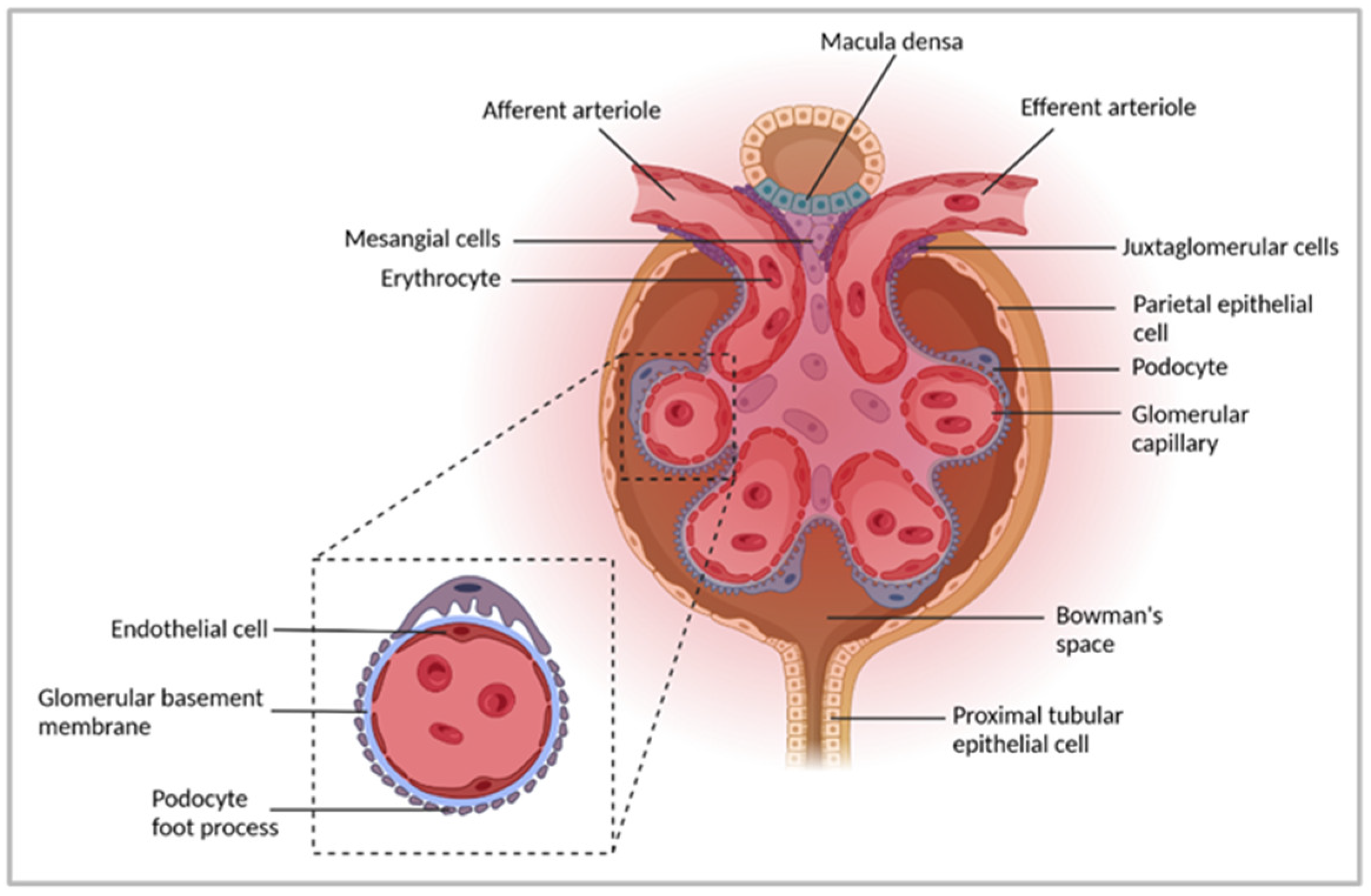
2. Immune-Mediated GN
3. Experimental Models to Study Pathogenic Mechanisms in Immune-Mediated GN
4. Pathogenic CD4+ T-Cell Responses in Immune-Mediated GN
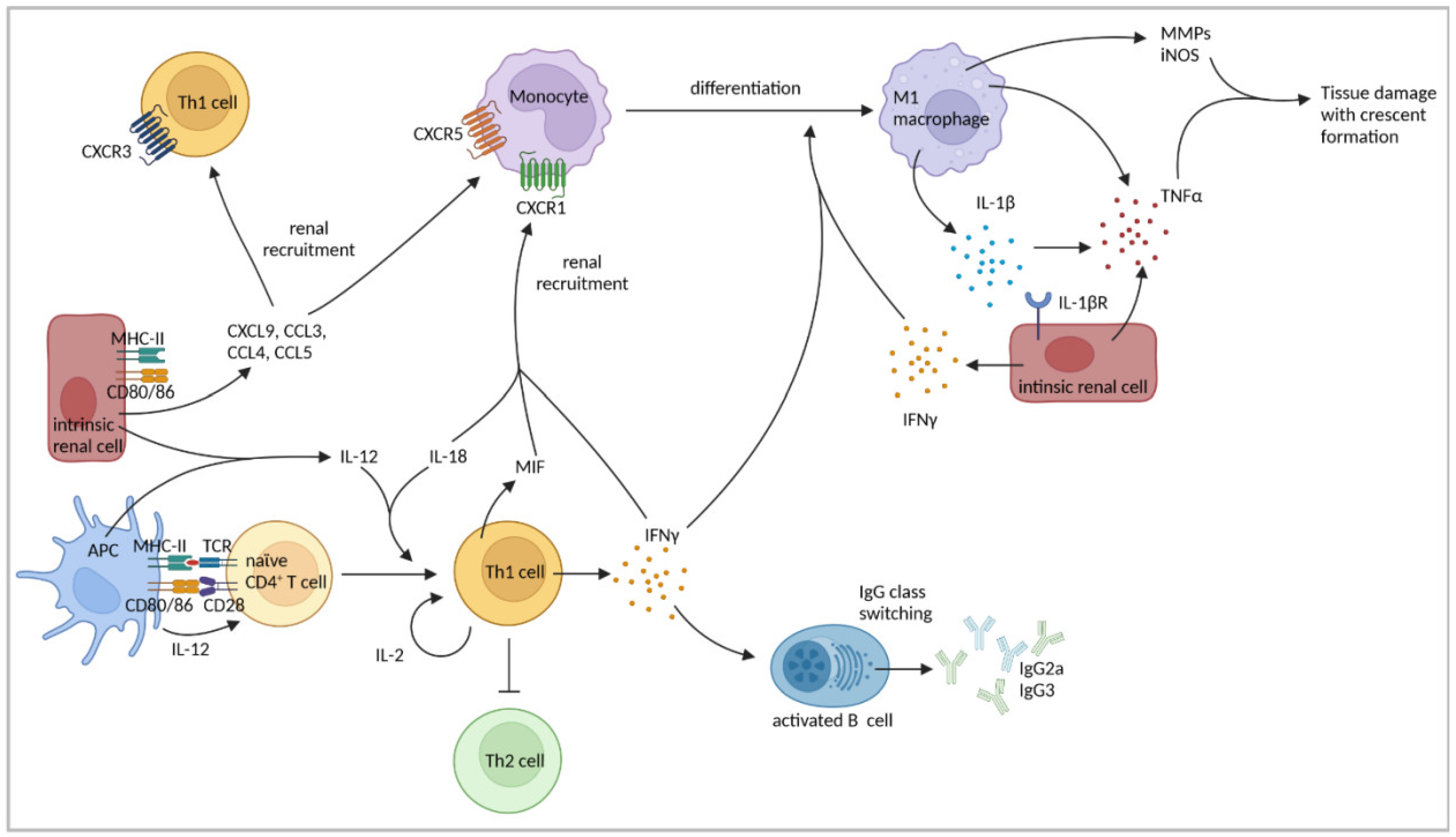
4.1. Th1-Cell Response
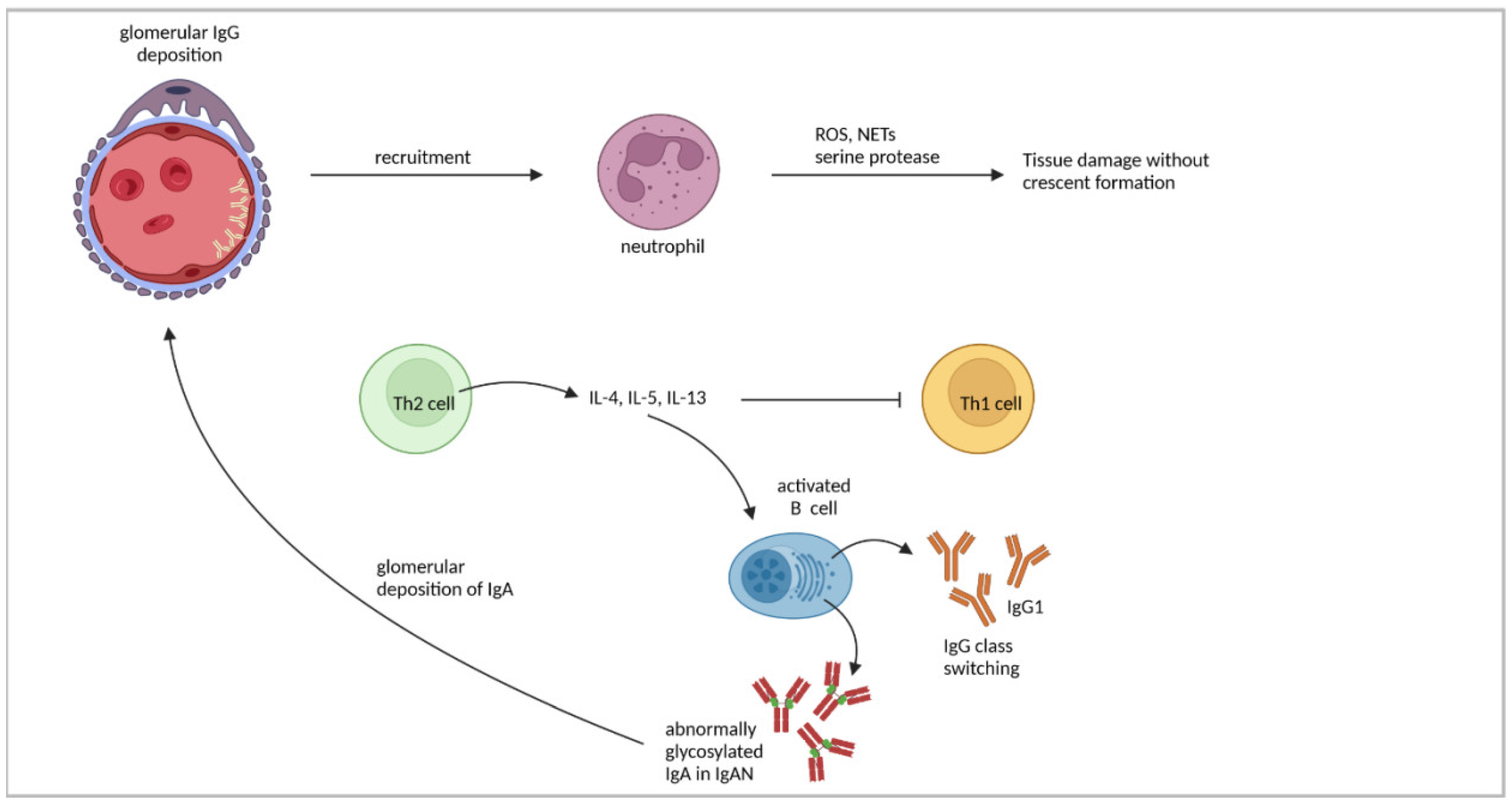
4.2. Th2-Cell Response
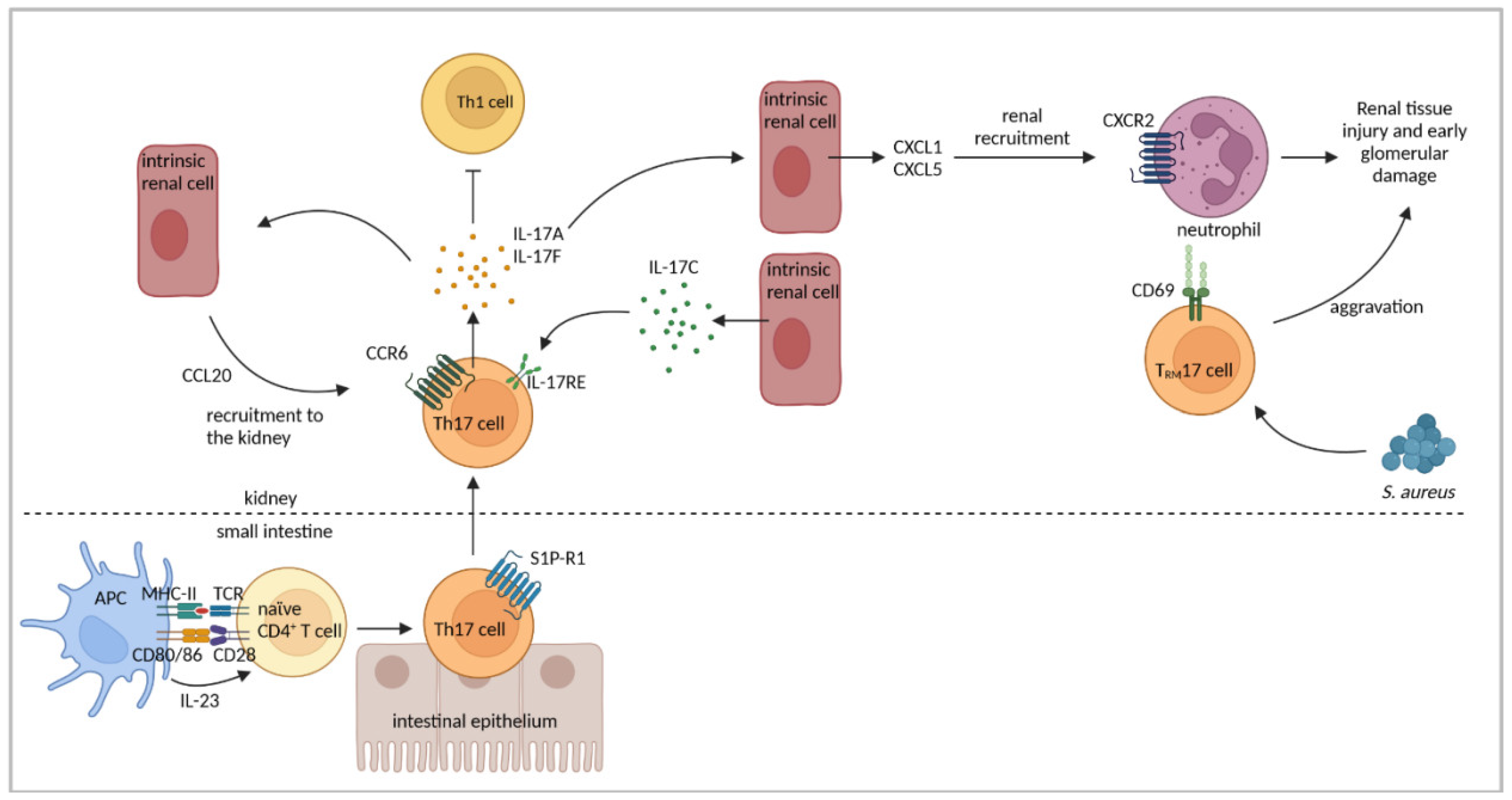
4.3. Th17-Cell Response
4.4. Tissue-Resident Memory CD4+ T Cells
4.5. CD4+ T-Cell Responses in GN Patients
5. Pathogenic CD8+ T-Cell Responses in cGN
5.1. Effector CD8+ T Cells
5.2. Tissue-Resident Memory CD8+ T Cells
6. Conclusions and Therapeutic Considerations
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Couser, W.G. Glomerulonephritis. Lancet 1999, 353, 1509–1515. [Google Scholar] [CrossRef]
- Smeets, B.; Uhlig, S.; Fuss, A.; Mooren, F.; Wetzels, J.F.M.; Floege, J.; Moeller, M.J. Tracing the Origin of Glomerular Extracapillary Lesions from Parietal Epithelial Cells. J. Am. Soc. Nephrol. 2009, 20, 2604–2615. [Google Scholar] [CrossRef]
- Kant, S.; Kronbichler, A.; Sharme, P.; Geetha, D. Advances in Understanding of Pathogenesis and Treatment of Immune-Mediated Kidney Disease: A Review. Am. J. Kidney Dis. 2021, 79, 582–600. [Google Scholar] [CrossRef]
- O’Sullivan, K.M.; Lo, C.Y.; Summers, S.A.; Elgass, K.D.; McMillan, P.J.; Longano, A.; Ford, S.L.; Gan, P.Y.; Kerr, P.G.; Kitching, A.R.; et al. Renal Participation of Myeloperoxidase in Antineutrophil Cytoplasmic Antibody (ANCA)-Associated Glomerulonephritis. Kidney Int. 2015, 88, 1030–1046. [Google Scholar] [CrossRef]
- Alexopoulos, E.; Seron, D.; Hartley, R.B.; Cameron, J.S. Lupus Nephritis: Correlation of Interstitial Cells with Glomerular Function. Kidney Int. 1990, 37, 100–109. [Google Scholar]
- Winchester, R.; Wiesendanger, M.; Zhang, H.Z.; Steshenko, V.; Peterson, K.; Geraldino-Pardilla, L.; Ruiz-Vazquez, E.; D’Agati, V. Immunologic Characteristics of Intrarenal T Cells: Trafficking of Expanded CD8+ T Cell β-Chain Clonotypes in Progressive Lupus Nephritis. Arthritis Rheum. 2012, 64, 1589–1600. [Google Scholar] [CrossRef]
- Couzi, L.; Merville, P.; Deminière, C.; Moreau, J.F.; Combe, C.; Pellegrin, J.L.; Viallard, J.F.; Blanco, P. Predominance of CD8+ T Lymphocytes among Periglomerular Infiltrating Cells and Link to the Prognosis of Class III and Class IV Lupus Nephritis. Arthritis Rheum. 2007, 56, 2362–2370. [Google Scholar] [CrossRef]
- Anguiano, L.; Kain, R.; Anders, H.-J. The Glomerular Crescent: Triggers, Evolution, Resolution, and Implications for Therapy. Curr. Opin. Nephrol. Hypertens. 2020, 29, 302–309. [Google Scholar]
- Kaissling, B.; Le Hir, M. Characterization and Distribution of Interstitial Cell Types in the Renal Cortex of Rats. Kidney Int. 1994, 45, 709–720. [Google Scholar] [CrossRef]
- Kurts, C.; Panzer, U.; Anders, H.J.; Rees, A.J. The Immune System and Kidney Disease: Basic Concepts and Clinical Implications. Nat. Rev. Immunol. 2013, 13, 738–753. [Google Scholar] [CrossRef]
- Al Mushafi, A.; Ooi, J.D.; Odobasic, D. Crescentic Glomerulonephritis: Pathogenesis and Therapeutic Potential of Human Amniotic Stem Cells. Front. Immunol. 2021, 12, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, T.; Luebbe, J.; Paust, H.J.; Panzer, U. Mechanisms and Functions of IL-17 Signaling in Renal Autoimmune Diseases. Mol. Immunol. 2018, 104, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Jennette, J.C.; Thomas, D.B. Crescentic Glomerulonephritis. Nephrol. Dial. Transplant. 2001, 16, 80–82. [Google Scholar] [CrossRef]
- Chen, A.; Lee, K.; Guan, T.; He, J.C.; Schlondorff, D. Role of CD8+ T Cells in Crescentic Glomerulonephritis. Nephrol. Dial. Transplant. 2020, 35, 564–572. [Google Scholar] [CrossRef]
- Chen, A.; Lee, K.; D’Agati, V.D.; Wei, C.; Fu, J.; Guan, T.J.; He, J.C.; Schlondorff, D.; Agudo, J. Bowman’s Capsule Provides a Protective Niche for Podocytes from Cytotoxic CD8+ T Cells. J. Clin. Investig. 2018, 128, 3413–3424. [Google Scholar] [CrossRef]
- Cattran, D.C.; Feehally, J.; Cook, H.T.; Liu, Z.H.; Fervenza, F.C.; Mezzano, S.A.; Floege, J.; Nachman, P.H.; Gipson, D.S.; Praga, M.; et al. Kidney Disease: Improving Global Outcomes (KDIGO) Glomerulonephritis Work Group. KDIGO Clinical Practice Guideline for Glomerulonephritis. Kidney Int. 2012, 2, 139–274. [Google Scholar]
- Orth, S.R.; Ritz, E. The Nephrotic Syndrome. N. Engl. J. Med. 1998, 338, 1202–1211. [Google Scholar] [CrossRef]
- Hull, R.P.; Goldsmith, D.J.A. Nephrotic Syndrome in Adults. BMJ 2008, 336, 1185–1189. [Google Scholar] [CrossRef]
- Llach, F. Thromboembolic Complications in Nephrotic Syndrome. Postgrad. Med. 1984, 76, 111–123. [Google Scholar] [CrossRef]
- Khanna, R. Clinical Presentation & Management of Glomerular Diseases: Hematuria, Nephritic & Nephrotic Syndrome. Mo. Med. 2011, 108, 33–36. [Google Scholar]
- Couser, W.G.; Salant, D.J. In Situ Immune Complex Formation and Glomerular Injury. Kidney Int. 1980, 17, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Jennette, J.C.; Falk, R.J. Pathogenesis of the Vascular and Glomerular Damage in ANCA-Positive Vasculitis. Nephrol. Dial. Transplant. 1998, 13, 16–20. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cornec, D.; Cornec Le-Gall, E.; Fervenza, F.C.; Specks, U. ANCA-Associated Vasculitis—Clinical Utility of Using ANCA Specificity to Classify Patients. Nat. Rev. Immunol. 2016, 12, 570–579. [Google Scholar] [CrossRef] [PubMed]
- Mcadoo, S.P.; Pusey, C.D. Anti-Glomerular Basement Membrane Disease. Clin. J. Am. Soc. Nephrol. 2017, 12, 1162–1172. [Google Scholar] [CrossRef]
- McGaha, T.L.; Madaio, M.P. Lupus Nephritis: Animal Modeling of a Complex Disease Syndrome Pathology. Drug Discov. Today Dis. Model. 2014, 11, 13–18. [Google Scholar] [CrossRef]
- Suzuki, H.; Suzuki, Y. Murine Models of Human IgA Nephropathy. Semin. Nephrol. 2018, 38, 513–520. [Google Scholar] [CrossRef]
- Imai, H.; Nakamoto, Y.; Asakura, K.; Miki, K.; Yasuda, T.; Miura, A.B. Spontaneous Glomerular IgA Deposition in DdY Mice: An Animal Model of IgA Nephritis. Kidney Int. 1985, 27, 756–761. [Google Scholar] [CrossRef]
- Ooi, J.D.; Phoon, R.K.S.; Holdsworth, S.R.; Kitching, A.R. IL-23, Not IL-12, Directs Autoimmunity to the Goodpasture Antigen. J. Am. Soc. Nephrol. 2009, 20, 980–989. [Google Scholar] [CrossRef]
- Reynolds, J.; Mavromatidis, K.; Cashman, S.J.; Evans, D.J.; Pusey, C.D. Experimental Autoimmune Glomerulonephritis (EAG) Induced by Homologous and Heterologous Glomerular Basement Membrane in Two Substrains of Wistar-Kyoto Rat. Nephrol. Dial. Transplant. 1998, 13, 44–52. [Google Scholar] [CrossRef]
- Shochet, L.; Holdsworth, S.; Kitching, A.R. Animal Models of ANCA Associated Vasculitis. Front. Immunol. 2020, 11, 1–20. [Google Scholar] [CrossRef]
- Falk, R.J.; Terrell, R.S.; Charles, L.A.; Jennette, J.C. Anti-Neutrophil Cytoplasmic Autoantibodies Induce Neutrophils to Degranulate and Produce Oxygen Radicals in Vitro. Proc. Natl. Acad. Sci. USA 1990, 87, 4115–4119. [Google Scholar] [CrossRef] [PubMed]
- Brouwer, E.; Huitema, M.G.; Mulder, A.H.; Heeringa, P.; van Goor, H.; Tervaert, J.W.; Weening, J.J.; Kallenberg, C.G. Neutrophil Activation in Vitro and in Vivo in Wegener’s Granulomatosis. Kidney Int. 1994, 45, 1120–1131. [Google Scholar] [CrossRef] [PubMed]
- Kessenbrock, K.; Krumbholz, M.; Schönermarck, U.; Back, W.; Gross, W.L.; Werb, Z.; Gröne, H.-J.; Brinkmann, V.; Jenne, D.E. Netting Neutrophils in Autoimmune Small-Vessel Vasculitis. Nat. Med. 2009, 15, 623–625. [Google Scholar] [CrossRef] [PubMed]
- Sheerin, N.S.; Springall, T.; Abe, K.; Sacks, S.H. Protection and Injury: The Differing Roles of Complement in the Development of Glomerular Injury. Eur. J. Immunol. 2001, 31, 1255–1260. [Google Scholar] [CrossRef]
- Cochrane, C.G.; Unanue, E.R.; Dixon, F.J. Role of Polymorphonuclear Leukocytes and Complement in Nephrotoxic Nephritis. J. Exp. Med. 1965, 122, 99–116. [Google Scholar] [CrossRef] [PubMed]
- Artinger, K.; Kirsch, A.H.; Aringer, I.; Moschovaki-filippidou, F.; Eller, P.; Rosenkranz, A.R. Innate and Adaptive Immunity in Experimental Glomerulonephritis: A Pathfinder Tale. Pediatr. Nephrol. 2017, 32, 943–947. [Google Scholar] [CrossRef] [PubMed]
- Turner, J.E.; Paust, H.J.; Steinmetz, O.M.; Peters, A.; Riedel, J.H.; Erhardt, A.; Wegscheid, C.; Velden, J.; Fehr, S.; Mittrücker, H.W.; et al. CCR6 Recruits Regulatory T Cells and Th17 Cells to the Kidney in Glomerulonephritis. J. Am. Soc. Nephrol. 2010, 21, 974–985. [Google Scholar] [CrossRef]
- Ougaard, M.K.E.; Kvist, P.H.; Jensen, H.E.; Hess, C.; Rune, I.; Søndergaard, H. Murine Nephrotoxic Nephritis as a Model of Chronic Kidney Disease. Int. J. Nephrol. 2018, 2018, 8–10. [Google Scholar] [CrossRef]
- Huang, X.R.; Holdsworth, S.R.; Tipping, P.G. Th2 Responses Induce Humorally Mediated Injury Experimental Basement Membrane Glomerulonephritis. J. Am. Soc. Nephrol. 1997, 8, 1101–1108. [Google Scholar] [CrossRef]
- Bolton, W.K.; Benton, F.R.; Lobo, P.I. Requirement of Functional T-Cells in the Production of Autoimmune Glomerulotubular Nephropathy in Mice. Clin. Exp. Immunol. 1978, 33, 474–477. [Google Scholar]
- Tipping, P.G.; Neale, T.J.; Holdsworth, S.R. T Lymphocyte Participation in Antibody-Induced Experimental Glomerulonephritis. Kidney Int. 1985, 27, 530–537. [Google Scholar] [CrossRef] [PubMed]
- Wofsy, D.; Ledbetter, J.A.; Hendler, P.L.; Seaman, W.E. Treatment of Murine Lupus with Monoclonal Anti-T Cell Antibody. J. Immunol. 1985, 134, 852–857. [Google Scholar] [PubMed]
- Huang, X.R.; Holdsworth, S.R.; Tipping, P.G. Evidence for Delayed-Type Hypersensitivity Mechanisms in Glomerular Crescent Formation. Kidney Int. 1994, 46, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Tipping, P.G.; Huang, X.R.; Qi, M.; Van, G.Y.; Tang, W.W. Crescentic Glomerulonephritis in CD4- and CD8-Deficient Mice: Requirement for CD4 but Not CD8 Cells. Am. J. Pathol. 1998, 152, 1541–1548. [Google Scholar] [PubMed]
- Ruth, A.-J.; Kitching, A.R.; Kwan, R.Y.Q.; Odobasic, D.; Ooi, J.D.K.; Timoshanko, J.R.; Hickey, M.J.; Holdsworth, S.R. Anti-Neutrophil Cytoplasmic Antibodies and Effector CD4+ Cells Play Nonredundant Roles in Anti-Myeloperoxidase Crescentic Glomerulonephritis. J. Am. Soc. Nephrol. 2006, 17, 1940–1949. [Google Scholar] [CrossRef] [PubMed]
- Gan, P.-Y.; Holdsworth, S.R.; Kitching, A.R.; Ooi, J.D. Myeloperoxidase (MPO)-Specific CD4+ T Cells Contribute to MPO-Anti-Neutrophil Cytoplasmic Antibody (ANCA) Associated Glomerulonephritis. Cell Immunol. 2013, 282, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.R.; Tipping, P.G.; Shuo, L.I.; Holdsworth, S.R. Th1 Responsiveness to Nephritogenic Antigens Determines Susceptibility to Crescentic Glomerulonephritis in Mice. Kidney Int. 1997, 51, 94–103. [Google Scholar] [CrossRef]
- Kitching, A.R.; Holdsworth, S.R.; Tipping, P.G. IFN-Gamma Mediates Crescent Formation and Cell-Mediated Immune Injury in Murine Glomerulonephritis. J. Am. Soc. Nephrol. 1999, 10, 752–759. [Google Scholar] [CrossRef]
- Timoshanko, J.R.; Holdsworth, S.R.; Kitching, A.R.; Tipping, P.G. IFN- γ Production by Intrinsic Renal Cells and Bone Marrow-Derived Cells Is Required for Full Expression of Crescentic Glomerulonephritis in Mice. J. Immunol. 2002, 168, 4135–4141. [Google Scholar] [CrossRef]
- Ozmen, L.; Roman, D.; Fountoulakis, M.; Schmid, G.; Ryffel, B.; Garotta, G. Experimental Therapy of Systemic Lupus Erythematosus: The Treatment of NZB/W Mice with Mouse Soluble Interferon-Gamma Receptor Inhibits the Onset of Glomerulonephritis. Eur. J. Immunol. 1995, 25, 6–12. [Google Scholar] [CrossRef]
- Manolios, N.; Schreiber, L.; Nelson, M.; Geczy, C.L. Enhanced Interferon-γ (IFN) Production by Lymph Node Cells from Autoimmune (MRL/l, MRL/n) Mice. Clin. Exp. Immunol. 1989, 76, 301–306. [Google Scholar] [PubMed]
- Haas, C.; Ryffel, B.; Michel, L. IFN-Gamma Is Essential for the Development of Autoimmune Glomerulonephritis in MRL/Ipr Mice. J. Immunol. 1997, 158, 5484–5491. [Google Scholar] [PubMed]
- Schwarting, A.; Wada, T.; Kinoshita, K.; Tesch, G.; Kelley, V.R. IFN-Gamma Receptor Signaling Is Essential for the Initiation, Acceleration, and Destruction of Autoimmune Kidney Disease in MRL-Fas(Lpr) Mice. J. Immunol. 1998, 161, 494–503. [Google Scholar] [PubMed]
- Richards, H.B.; Satoh, M.; Jennette, J.C.; Croker, B.P.; Yoshida, H.; Reeves, W.H. Interferon-Gamma Is Required for Lupus Nephritis in Mice Treated with the Hydrocarbon Oil Pristane. Kidney Int. 2001, 60, 2173–2180. [Google Scholar] [CrossRef] [PubMed]
- Summers, S.A.; Steinmetz, O.M.; Gan, P.-Y.; Ooi, J.D.; Odobasic, D.; Kitching, A.R.; Holdsworth, S.R. Toll-like Receptor 2 Induces Th17 Myeloperoxidase Autoimmunity While Toll-like Receptor 9 Drives Th1 Autoimmunity in Murine Vasculitis. Arthritis Rheum. 2011, 63, 1124–1135. [Google Scholar] [CrossRef]
- Suzuki, H.; Suzuki, Y.; Aizawa, M.; Yamanaka, T.; Kihara, M.; Pang, H.; Horikoshi, S.; Tomino, Y. Th1 Polarization in Murine IgA Nephropathy Directed by Bone Marrow-Derived Cells. Kidney Int. 2007, 72, 319–327. [Google Scholar] [CrossRef]
- Hopfer, H.; Holzer, J.; Hünemörder, S.; Paust, H.J.; Sachs, M.; Meyer-Schwesinger, C.; Turner, J.E.; Panzer, U.; Mittrücker, H.W. Characterization of the Renal CD4+T-Cell Response in Experimental Autoimmune Glomerulonephritis. Kidney Int. 2012, 82, 60–71. [Google Scholar] [CrossRef]
- Hünemörder, S.; Treder, J.; Ahrens, S.; Schumacher, V.; Paust, H.J.; Menter, T.; Matthys, P.; Kamradt, T.; Meyer-Schwesinger, C.; Panzer, U.; et al. TH1 and TH17 Cells Promote Crescent Formation in Experimental Autoimmune Glomerulonephritis. J. Pathol. 2015, 237, 62–71. [Google Scholar] [CrossRef]
- Kitching, A.R.; Turner, A.L.; Semple, T.; Li, M.; Edgtton, K.L.; Wilson, G.R.; Timoshanko, J.R.; Hudson, B.G.; Holdsworth, S.R. Experimental Autoimmune Anti-Glomerular Basement Membrane Glomerulonephritis: A Protective Role for IFN-Gamma. J. Am. Soc. Nephrol. 2004, 15, 1764–1774. [Google Scholar] [CrossRef]
- Kitching, A.R.; Tipping, P.G.; Holdsworth, S.R. IL-12 Directs Severe Renal Injury, Crescent Formation and Th1 Responses in Murine Glomerulonephritis. Eur. J. Immunol. 1999, 29, 1–10. [Google Scholar] [CrossRef]
- Kitching, A.R.; Tipping, P.G.; Kurimoto, M.; Holdsworth, S.R. IL-18 Has IL-12-Independent Effects in Delayed-Type Hypersensitivity: Studies in Cell-Mediated Crescentic Glomerulonephritis. J. Immunol. 2000, 165, 4649–4657. [Google Scholar] [CrossRef] [PubMed]
- Timoshanko, J.R.; Kitching, A.R.; Holdsworth, S.R.; Tipping, P.G. Interleukin-12 from Intrinsic Cells Is an Effector of Renal Injury in Crescentic Glomerulonephritis. J. Am. Soc. Nephrol. 2001, 12, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Alleva, D.G.; Kaser, S.B.; Beller, D.I. Intrinsic Defects in Macrophage IL-12 Production Associated with Immune Dysfunction in the MRL/++ and New Zealand Black/White F1 Lupus-Prone Mice and the Leishmania Major-Susceptible BALB/c Strain. J. Immunol. 1998, 161, 6878–6884. [Google Scholar] [PubMed]
- Min, J.D.; Cho, M.L.; Cho, C.S.; Min, S.Y.; Kim, W.U.; Yang, S.Y.; Min, J.K.; Hong, Y.S.; Lee, S.H.; Park, S.H.; et al. Decreased Production of Interleukin-12 and Interferon-Gamma Is Associated with Renal Involvement in Systemic Lupus Erythematosus. Scand. J. Rheumatol. 2001, 30, 159–163. [Google Scholar] [PubMed]
- Calvani, N.; Satoh, M.; Croker, B.P.; Reeves, W.H.; Richards, H.B. Nephritogenic Autoantibodies but Absence of Nephritis in Il-12p35-Deficient Mice with Pristane-Induced Lupus. Kidney Int. 2003, 64, 897–905. [Google Scholar] [CrossRef] [PubMed]
- Annunziato, F.; Romagnani, C.; Romagnani, S. The 3 Major Types of Innate and Adaptive Cell-Mediated Effector Immunity. J. Allergy Clin. Immunol. 2015, 135, 626–635. [Google Scholar] [CrossRef] [PubMed]
- Esfandiari, E.; McInnes, I.B.; Lindop, G.; Huang, F.P.; Field, M.; Komai-Koma, M.; Wei, X.; Liew, F.Y. A Proinflammatory Role of IL-18 in the Development of Spontaneous Autoimmune Disease. J. Immunol. 2001, 167, 5338–5347. [Google Scholar] [CrossRef]
- Kinoshita, K.; Yamagata, T.; Nozaki, Y.; Sugiyama, M.; Ikoma, S.; Funauchi, M.; Kanamaru, A. Blockade of IL-18 Receptor Signaling Delays the Onset of Autoimmune Disease in MRL-Faslpr Mice. J. Immunol. 2004, 173, 5312–5318. [Google Scholar] [CrossRef]
- Schitmer, B.; Wedekind, D.; Glage, S.; Neumann, D. Deletion of IL-18 Expression Ameliorates Spontaneous Kidney Failure in MRLlpr Mice. PLoS ONE 2015, 10, e0140173. [Google Scholar]
- Panzer, U.; Steinmetz, O.M.; Paust, H.-J.; Meyer-Schwesinger, C.; Peters, A.; Turner, J.-E.; Zahner, G.; Heymann, F.; Kurts, C.; Hopfer, H.; et al. Chemokine Receptor CXCR3 Mediates T Cell Recruitment and Tissue Injury in Nephrotoxic Nephritis in Mice. J. Am. Soc. Nephrol. 2007, 18, 2071–2084. [Google Scholar] [CrossRef]
- Menke, J.; Zeller, G.C.; Kikawada, E.; Means, T.K.; Huang, X.R.; Lan, H.Y.; Lu, B.; Farber, J.; Luster, A.D.; Kelley, V.R. CXCL9, but Not CXCL10, Promotes CXCR3-Dependent Immune-Mediated Kidney Disease. J. Am. Soc. Nephrol. 2008, 19, 1177–1189. [Google Scholar] [CrossRef]
- Schadde, E.; Kretzler, M.; Banas, B.; Luckow, B.; Assmann, K. Expression of Chemokines and Their Receptors in Nephrotoxic Serum Nephritis. Nephrol. Dial. Transplant. 2000, 15, 1046–1053. [Google Scholar] [CrossRef] [PubMed]
- Chung, A.C.K.; Lan, H.Y. Chemokines in Renal Injury. J. Am. Soc. Nephrol. 2011, 22, 802–809. [Google Scholar] [CrossRef] [PubMed]
- Cochrane, C.G.; Unanue, E.R.; Dixon, F.J. A Mononuclear Cell Component in Experimental Immunological Glomerulonephritis. J. Exp. Med. 1965, 147, 369–384. [Google Scholar]
- Holdsworth, S.R.; Neale, T.J.; Wilson, C.B. Abrogation of Macrophage-Dependent Injury in Experimental Glomerulonephritis in the Rabbit. Use of an Antimacrophage Serum. J. Clin. Investig. 1981, 68, 686–698. [Google Scholar] [CrossRef]
- Holdsworth, S.R. Fc Dependence of Macrophage Induced Experimental Anti- Glomerular Basement Membrane Antibody Nephritis. J. Immunol. 1983, 130, 35–39. [Google Scholar]
- Boyce, N.W.; Tipping, P.G.; Holdsworth, S.R. Lymphokine (MIF) Production by Glomerular T-Lymphocytes in Experimental Glomerulonephritis. Kidney Int. 1986, 30, 673–677. [Google Scholar] [CrossRef]
- Meng, X.; Tang, P.M.; Yao, H. Macrophage Phenotype in Kidney Injury and Repair. Kidney Dis. 2015, 1, 138–146. [Google Scholar] [CrossRef]
- Tipping, P.G.; Holdsworth, S.R. T Cells in Crescentic Glomerulonephritis. J. Am. Soc. Nephrol. 2006, 17, 1253–1263. [Google Scholar] [CrossRef]
- Timoshanko, J.R.; Kitching, A.R.; Iwakura, Y.; Holdsworth, S.R.; Tipping, P.G. Leukocyte-Derived Interleukin-1 Beta Interacts with Renal Interleukin-1 Receptor I to Promote Renal Tumor Necrosis Factor and Glomerular Injury in Murine Crescentic Glomerulonephritis. Am. J. Pathol. 2004, 164, 1967–1977. [Google Scholar] [CrossRef]
- Li, S.; Kurts, C.; Köntgen, F.; Holdsworth, S.R.; Tipping, P.G. Major Histocompatibility Complex Class II Expression by Intrinsic Renal Cells Is Required for Crescentic Glomerulonephritis. J. Exp. Med. 1998, 188, 597–602. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, Y.; Sakatsume, M.; Xie, Y.; Kuroda, T.; Igashima, M.; Narita, I.; Gejyo, F. Macrophage Metalloelastase as a Major Factor for Glomerular Injury in Anti-Glomerular Basement Membrane Nephritis. J. Immunol. 2003, 170, 3377–3385. [Google Scholar] [CrossRef] [PubMed]
- Odobasic, D.; Kitching, A.R.; Semple, T.J.; Timoshanko, J.R.; Tipping, P.G.; Holdsworth, S.R. Glomerular Expression of CD80 and CD86 Is Required for Leukocyte Accumulation and Injury in Crescentic Glomerulonephritis. J. Am. Soc. Nephrol. 2012, 16, 2012–2022. [Google Scholar] [CrossRef]
- Breda, P.C.; Wiech, T.; Meyer-Schwesinger, C.; Grahammer, F.; Huber, T.; Panzer, U.; Tiegs, G.; Neumann, K. Renal Proximal Tubular Epithelial Cells Exert Immunomodulatory Function by Driving Inflammatory CD4+ T Cell Responses. Am. J. Physiol. Physiol. 2019, 317, F77–F89. [Google Scholar] [CrossRef] [PubMed]
- Timoshanko, J.R.; Sedgwick, J.D.; Holdsworth, S.R.; Tipping, P.G. Intrinsic Renal Cells Are the Major Source of Tumor Necrosis Factor Contributing to Renal Injury in Murine Crescentic Glomerulonephritis. J. Am. Soc. Nephrol. 2003, 14, 1785–1793. [Google Scholar] [CrossRef] [PubMed]
- Odobasic, D.; Gan, P.; Summers, S.A.; Semple, T.J.; Muljadi, R.C.M.; Iwakura, Y.; Kitching, A.R.; Holdsworth, S.R. Interleukin-17A Promotes Early but Attenuates Established Disease in Crescentic Glomerulonephritis in Mice. Am. J. Pathol. 2011, 179, 1188–1198. [Google Scholar] [CrossRef]
- Summers, S.A.; Steinmetz, O.M.; Li, M.; Kausman, J.Y.; Semple, T.; Edgtton, K.L.; Borza, D.B.; Braley, H.; Holdsworth, S.R.; Kitching, A.R. Th1 and Th17 Cells Induce Proliferative Glomerulonephritis. J. Am. Soc. Nephrol. 2009, 20, 2518–2524. [Google Scholar] [CrossRef]
- Wong, C.K.; Ho, C.Y.; Li, E.K.; Lam, C.W. Elevation of Proinflammatory Cytokine (IL-18, IL-17, IL-12) and Th2 Cytokine (IL-4) Concentrations in Patients with Systemic Lupus Erythematosus. Lupus 2000, 9, 589–593. [Google Scholar] [CrossRef]
- Wong, C.K.; Lit, L.C.W.; Tam, L.S.; Li, E.K.M.; Wong, P.T.Y.; Lam, C.W.K. Hyperproduction of IL-23 and IL-17 in Patients with Systemic Lupus Erythematosus: Implications for Th17-Mediated Inflammation in Auto-Immunity. Clin. Immunol. 2008, 127, 385–393. [Google Scholar] [CrossRef]
- Sigdel, K.R.; Duan, L.; Wang, Y.; Hu, W.; Wang, N.; Sun, Q.; Liu, Q.; Liu, X.; Hou, X.; Cheng, A.; et al. Serum Cytokines Th1, Th2, and Th17 Expression Profiling in Active Lupus Nephritis-IV: From a Southern Chinese Han Population. Mediat. Inflamm. 2016, 2016, 4927530. [Google Scholar] [CrossRef]
- Gan, P.-Y.; Chan, A.; Ooi, J.D.; Dick, J.; Nagai, K.; O’Sullivan, K.M.; Oudin, V.; Shim, R.; Kitching, A.R.; Holdsworth, S.R. Biologicals Targeting T Helper Cell Subset Differentiating Cytokines Are Effective in the Treatment of Murine Anti-Myeloperoxidase Glomerulonephritis. Kidney Int. 2019, 96, 1121–1133. [Google Scholar] [CrossRef] [PubMed]
- Krebs, C.F.; Reimers, D.; Zhao, Y.; Paust, H.; Bartsch, P.; Nuñez, S.; Rosemblatt, M.V.; Hellmig, M.; Kilian, C.; Borchers, A.; et al. Pathogen-Induced Tissue-Resident Memory TH 17 (TRM 17) Cells Amplify Autoimmune Kidney Disease. Sci. Immunol. 2020, 17, 1–16. [Google Scholar]
- Yoshida, M.; Iwahori, T.; Nakabayashi, I.; Akashi, M.; Watanabe, T.; Yoshikawa, N. In Vitro Production of Myeloperoxidase Anti-Neutrophil Cytoplasmic Antibody and Establishment of Th1-Type T Cell Lines from Peripheral Blood Lymphocytes of Patients. Clin. Exp. Rheumatol. 2005, 23, 227–230. [Google Scholar] [PubMed]
- Lin, F.-J.; Jiang, G.-R.; Shan, J.-P.; Zhu, C.; Zou, J.; Wu, X.-R. Imbalance of Regulatory T Cells to Th17 Cells in IgA Nephropathy. Scand. J. Clin. Lab. Investig. 2012, 72, 221–229. [Google Scholar] [CrossRef]
- Peng, Z.; Tian, J.; Cui, X.; Xian, W.; Sun, H.; Li, E.; Geng, L.; Zhang, L.; Zhao, P. Increased Number of Th22 Cells and Correlation with Th17 Cells in Peripheral Blood of Patients with IgA Nephropathy. Hum. Immunol. 2013, 74, 1586–1591. [Google Scholar] [CrossRef]
- Yang, L.; Zhang, Y.; Peng, W.; Wei, M.; Qin, W. MicroRNA-155-Induced T Lymphocyte Subgroup Drifting in IgA Nephropathy. Int. Urol. Nephrol. 2017, 49, 353–361. [Google Scholar] [CrossRef]
- Schena, F.P.; Cerullo, G.; Torres, D.D.; Scolari, F.; Foramitti, M.; Amoroso, A.; Pitulli, D.; Floege, J.; Mertens, P.R.; Zerres, K.; et al. Role of Interferon-Gamma Gene Polymorphisms in Susceptibility to IgA Nephropathy: A Family-Based Association Study. Eur. J. Hum. Genet. 2006, 14, 488–496. [Google Scholar] [CrossRef][Green Version]
- Antonelou, M.; Evans, R.D.R.; Henderson, S.R.; Salama, A.D. Neutrophils Are Key Mediators in Crescentic Glomerulonephritis and Targets for New Therapeutic Approaches. Nephrol. Dial. Transplant. 2020, 37, 230–238. [Google Scholar] [CrossRef]
- Morris, S.C.; Lees, A.; Inman, J.; Finkelman, F.D. Role of Antigen-Specific T Cell Help in the Generation of in Vivo Antibody Responses: I. Antigen-Specific T Cell Help Is Required to Generate a Polyclonal IgG1 Response in Anti-IgD Antibody-Injected Mice. J. Immunol. 1992, 149, 3836–3844. [Google Scholar]
- Racke, B.M.K.; Bonomo, A.; Scott, D.E.; Cannella, B.; Levine, S.A.; Raine, I.I.C.S.; Shevach, E.M.; Röcken, M. Cytokine-Induced Immune Deviation as a Therapy for Inflammatory Autoimmune Disease. J. Exp. Med. 1994, 180, 1961–1966. [Google Scholar] [CrossRef]
- Tipping, P.G.; Kitching, A.R.; Huang, X.R.; Mulch, D.A.; Holdsworth, S.R. Immune Modulation with Interleukin-4 and Interleukin-10 Prevents Crescent Formation and Glomerular Injury in Experimental Glomerulonephritis. Eur. J. Immunol. 1997, 27, 530–537. [Google Scholar] [CrossRef]
- Ebihara, I.; Hirayama, K.; Yamamoto, S.; Muro, K.; Yamagata, K.; Koyama, A. Th2 Predominance at the Single-Cell Level in Patients with IgA Nephropathy. Nephrol. Dial. Transplant. 2001, 16, 1783–1789. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Inoshita, H.; Kim, B.-G.; Yamashita, M.; Choi, S.H.; Tomino, Y.; Lettiro, J.J. Disruption of Smad4 Expression in T Cells Leads to IgA Nephropathy-like Manifestations. PLoS ONE 2013, 8, e78736. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chintalacharuvu, S.R.; Emancipator, S.N. Differential Glycosylation of Two Glycoproteins Synthesized by Murine B Cells in Response to IL-4 plus IL-5. Cytokine 2000, 12, 1182–1188. [Google Scholar] [CrossRef]
- Mestecky, J.; Tomana, M.; Crowley-Nowick, P.A.; Moldoveanu, Z.; Julian, B.A.; Jackson, S. Defective Galactosylation and Clearance of IgA1 Molecules as a Possible Etiopathogenic Factor in IgA Nephropathy. Contrib. Nephrol. 1993, 104, 172–182. [Google Scholar] [PubMed]
- Hiki, Y.; Odani, H.; Takahashi, M.; Yasuda, Y.; Nishimoto, A.; Iwase, H.; Shinzato, T.; Kobayashi, Y.; Maeda, K. Mass Spectrometry Proves Under-O-Glycosylation of Glomerular IgA1 in IgA Nephropathy. Kidney Int. 2001, 59, 1077–1085. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; Kobayashi, N.; Ikeda, T.; Suzuki, Y.; Tsuge, T.; Hirokoshi, S.; Emancipator, S.N.; Tomino, Y. Down-Regulation of Core 1 Beta1,3-Galactosyltransferase and Cosmc by Th2 Cytokine Alters O-Glycosylation of IgA1. Nephrol. Dial. Transplant. 2010, 25, 3890–3897. [Google Scholar] [CrossRef]
- Sun, Q.; Zhang, J.; Zhou, N.; Liu, X.; Shen, Y. DNA Methylation in Cosmc Promoter Region and Aberrantly Glycosylated IgA1 Associated with Pediatric IgA Nephropathy. PLoS ONE 2015, 10, e0112305. [Google Scholar] [CrossRef]
- Steinmetz, O.M.; Summers, S.A.; Gan, P.-Y.; Semple, T.; Holdsworth, S.R.; Kitching, A.R. The Th17-Defining Transcription Factor RORγt Promotes Glomerulonephritis. J. Am. Soc. Nephrol. 2011, 22, 472–483. [Google Scholar] [CrossRef]
- Paust, H.-J.; Turner, J.-E.; Steinmetz, O.M.; Peters, A.; Heymann, F.; Holscher, C.; Wolf, G.; Kurts, C.; Mittrucker, H.-W.; Stahl, R.A.K.; et al. The IL-23/Th17 Axis Contributes to Renal Injury in Experimental Glomerulonephritis. J. Am. Soc. Nephrol. 2009, 20, 969–979. [Google Scholar] [CrossRef]
- Krebs, C.F.; Turner, J.-E.; Paust, H.-J.; Kapffer, S.; Koyro, T.; Krohn, S.; Ufer, F.; Friese, M.A.; Flavell, R.A.; Stockinger, B.; et al. Plasticity of Th17 Cells in Autoimmune Kidney Diseases. J. Immunol. 2016, 197, 449–457. [Google Scholar] [CrossRef] [PubMed]
- Gan, P.-Y.; Steinmetz, O.M.; Tan, D.S.Y.; O’Sullivan, K.M.; Ooi, J.D.; Iwakura, Y.; Kitching, A.R.; Holdsworth, S.R. Th17 Cells Promote Autoimmune Anti-Myeloperoxidase Glomerulonephritis. J. Am. Soc. Nephrol. 2010, 21, 925–931. [Google Scholar] [CrossRef] [PubMed]
- Meng, T.; Li, X.; Ao, X.; Zhong, Y.; Tang, R.; Peng, W.; Yang, J.; Zou, M.; Zhou, Q. Hemolytic Streptococcus May Exacerbate Kidney Damage in IgA Nephropathy through CCL20 Response to the Effect of Th17 Cells. PLoS ONE 2014, 9, e108723. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Kyttaris, V.C.; Tsokos, G.C. The Role of IL-23/IL-17 Axis in Lupus Nephritis. J. Immunol. 2009, 183, 3160–3169. [Google Scholar] [CrossRef]
- Kyttaris, V.C.; Kampagianni, O.; Tsokos, G.C. Treatment with Anti-Interleukin 23 Antibody Ameliorates Disease in Lupus-Prone Mice. Biomed. Res. Int. 2013, 2013, 861028. [Google Scholar] [CrossRef]
- Jacob, N.; Yang, H.; Pricop, L.; Liu, Y.; Gao, X.; Zheng, S.G.; Wang, J.; Gao, H.-X.; Putterman, C.; Koss, M.N.; et al. Accelerated Pathological and Clinical Nephritis in Systemic Lupus Erythematosus-Prone New Zealand Mixed 2328 Mice Doubly Deficient in TNF Receptor 1 and TNF Receptor 2 via a Th17-Associated Pathway. J. Immunol. 2009, 182, 2532–2541. [Google Scholar] [CrossRef]
- Martin-Orozco, N.; Chung, Y.; Chang, S.H.; Wang, Y.-H.; Dong, C. Th17 Cells Promote Pancreatic Inflammation but Only Induce Diabetes Efficiently in Lymphopenic Hosts after Conversion into Th1 Cells. Eur. J. Immunol. 2009, 29, 216–224. [Google Scholar] [CrossRef]
- Hirota, K.; Yoshimoto, H.; Hashimoto, M.; Maeda, S.; Teradaira, S.; Sugimoto, N.; Yamaguchi, T.; Nomura, T.; Ito, H.; Nakamura, T.; et al. Preferential Recruitment of CCR6-Expressing Th17 Cells to Inflamed Joints via CCL20 in Rheumatoid Arthritis and Its Animal Model. J. Exp. Med. 2007, 204, 2803–2812. [Google Scholar] [CrossRef]
- Stockinger, B.; Omenetti, S. The Dichotomous Nature of T Helper 17 Cells. Nat. Rev. Immunol. 2017, 17, 535–544. [Google Scholar] [CrossRef]
- Krebs, C.F.; Paust, H.J.; Krohn, S.; Koyro, T.; Brix, S.R.; Riedel, J.H.; Bartsch, P.; Wiech, T.; Meyer-Schwesinger, C.; Huang, J.; et al. Autoimmune Renal Disease Is Exacerbated by S1P-Receptor-1-Dependent Intestinal Th17 Cell Migration to the Kidney. Immunity 2016, 45, 1078–1092. [Google Scholar] [CrossRef]
- Riedel, J.-H.; Paust, H.-J.; Krohn, S.; Turner, J.-E.; Kluger, M.A.; Steinmetz, O.M.; Krebs, C.F.; Stahl, R.A.K.; Panzer, U. IL-17F Promotes Tissue Injury in Autoimmune Kidney Diseases. J. Am. Soc. Nephrol. 2016, 27, 3666–3677. [Google Scholar] [CrossRef] [PubMed]
- Krohn, S.; Nies, J.F.; Kapffer, S.; Schmidt, T.; Riedel, J.H.; Kaffke, A.; Peters, A.; Borchers, A.; Steinmetz, O.M.; Krebs, C.F.; et al. IL-17C/IL-17 Receptor E Signaling in CD4 + T Cells Promotes T H 17 Cell-Driven Glomerular Inflammation. J. Am. Soc. Nephrol. 2018, 29, 1210–1222. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, T.; Luebbe, J.; Kilian, C.; Riedel, J.-H.; Hiekmann, S.; Asada, N.; Ginsberg, P.; Robben, L.; Song, N.; Kaffke, A.; et al. IL-17 Receptor C Signaling Controls CD4+ TH17 Immune Responses and Tissue Injury in Immune-Mediated Kidney Diseases. J. Am. Soc. Nephrol. 2021, 32, 3081–3098. [Google Scholar] [CrossRef] [PubMed]
- Hirota, K.; Duarte, J.H.; Veldhoen, M.; Hornsby, E.; Li, Y.; Cua, D.J.; Ahlfors, H.; Wilhelm, C. Fate Mapping of Interleukin 17-Producing T Cells in Inflammatory Responses. Nat. Immunol. 2011, 12, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Reboldi, A.; Coisne, C.; Baumjohann, D.; Benvenuto, F.; Bottinelli, D.; Lira, S.; Uccelli, A.; Lanzavecchia, A.; Engelhardt, B.; Sallusto, F. C-C Chemokine Receptor 6-Regulated Entry of TH-17 Cells into the CNS through the Choroid Plexus Is Required for the Initiation of EAE. Nat. Immunol. 2009, 10, 514–523. [Google Scholar] [CrossRef] [PubMed]
- Bending, D.; De La Peña, H.; Veldhoen, M.; Phillips, J.M.; Uyttenhove, C.; Stockinger, B.; Cooke, A. Highly Purified Th17 Cells from BDC2.5NOD Mice Convert into Th1-like Cells in NOD/SCID Recipient Mice. J. Clin. Investig. 2009, 119, 565–572. [Google Scholar] [CrossRef] [PubMed]
- Murphy, K.M.; Stockinger, B. Effector T Cell Plasticity: Flexibility in the Face of Changing Circumstances. Nat. Immunol. 2010, 11, 674–680. [Google Scholar] [CrossRef]
- Kamanaka, M.; Kim, S.T.; Wan, Y.Y.; Sutterwala, F.S.; Lara-Tejero, M.; Galán, J.E.; Harhaj, E.; Flavell, R.A. Expression of Interleukin-10 in Intestinal Lymphocytes Detected by an Interleukin-10 Reporter Knockin Tiger Mouse. Immunity 2006, 25, 941–952. [Google Scholar] [CrossRef]
- Soukou, S.; Huber, S.; Krebs, C.F. T Cell Plasticity in Renal Autoimmune Disease. Cell Tissue Res. 2021, 385, 323–333. [Google Scholar] [CrossRef]
- Talaat, R.M.; Mohamed, S.F.; Bassyouni, I.H.; Raouf, A.A. Th1/Th2/Th17/Treg Cytokine Imbalance in Systemic Lupus Erythematosus (SLE) Patients: Correlation with Disease Activity. Cytokine 2015, 72, 146–153. [Google Scholar] [CrossRef]
- Vukelic, M.; Laloo, A.; Kyttaris, V.C. Interleukin 23 Is Elevated in the Serum of Patients with SLE. Lupus 2020, 29, 1943–1947. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ito, S.; Chino, Y.; Goto, D.; Matsumoto, I.; Murata, H.; Tsutsumi, A.; Hayashi, T.; Uchida, K.; Usui, J.; et al. Laser Microdissection-Based Analysis of Cytokine Balance in the Kidneys of Patients with Lupus Nephritis. Clin. Exp. Immunol. 2010, 159, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Crispín, J.C.; Oukka, M.; Bayliss, G.; Cohen, R.A.; Van Beek, C.A.; Stillman, I.E.; Kyttaris, V.C.; Juang, Y.-T.; Tsokos, G.C. Expanded Double Negative T Cells in Patients with Systemic Lupus Erythematosus Produce IL-17 and Infiltrate the Kidneys. J. Immunol. 2008, 181, 8761–8766. [Google Scholar] [CrossRef] [PubMed]
- Abdulahad, W.H.; Stegeman, C.A.; Limburg, P.C.; Kallenberg, C.G.M. Skewed Distribution of Th17 Lymphocytes in Patients with Wegener’s Granulomatosis in Remission. Arthritis Rheum. 2008, 58, 2196–2205. [Google Scholar] [CrossRef] [PubMed]
- Nogueira, E.; Hamour, S.; Sawant, D.; Henderson, S.; Mansfield, N.; Chavele, K.-M.; Pusey, C.D.; Salama, A.D. Serum IL-17 and IL-23 Levels and Autoantigen-Specific Th17 Cells Are Elevated in Patients with ANCA-Associated Vasculitis. Nephrol. Dial. Transpl. 2010, 25, 2209–2217. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.-R.; Wen, J.; Zhang, H.; Wang, L.; Gou, F.-F.; Yang, M.; Fan, J.-M. Interleukin-17 Promotes the Production of Underglycosylated IgA1 in DAKIKI Cells. Ren. Fail. 2018, 40, 60–67. [Google Scholar] [CrossRef]
- Masopust, D.; Soerens, A.G. Tissue-Resident T Cells and Other Resident Leukocytes. Annu. Rev. Immunol. 2019, 37, 521–546. [Google Scholar] [CrossRef]
- Kumar, B.V.; Ma, W.; Miron, M.; Granot, T.; Guyer, R.S.; Carpenter, D.J.; Senda, T.; Sun, X.; Ho, S.-H.; Lerner, H.; et al. Human Tissue-Resident Memory T Cells Are Defined by Core Transcriptional and Functional Signatures in Lymphoid and Mucosal Sites. Cell Rep. 2017, 20, 2921–2934. [Google Scholar] [CrossRef]
- Wucherpfennig, K.W. Mechanisms for the Induction of Autoimmunity by Infectious Agents. J. Clin. Investig. 2001, 108, 1097–1104. [Google Scholar] [CrossRef]
- Abdulahad, W.H.; van der Geld, Y.M.; Stegeman, C.A.; Kallenberg, C.G.M. Persistent Expansion of CD4+ Effector Memory T Cells in Wegener’s Granulomatosis. Kidney Int. 2006, 70, 938–947. [Google Scholar] [CrossRef]
- Abdulahad, W.H.; Stegeman, C.A.; Kallenberg, C.G.M. The Role of CD4(+) T Cells in ANCA-Associated Systemic Vasculitis. Nephrology 2009, 14, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Weidner, S.; Carl, M.; Riess, R.; Rupprecht, H.D. Histologic Analysis of Renal Leukocyte Infiltration in Antineutrophil Cytoplasmic Antibody-Associated Vasculitis: Importance of Monocyte and Neutrophil Infiltration in Tissue Damage. Arthritis Rheum. 2004, 50, 3651–3657. [Google Scholar] [CrossRef] [PubMed]
- Arazi, A.; Rao, D.A.; Berthier, C.C.; Davidson, A.; Liu, Y.; Hoover, P.J.; Chicoine, A.; Eisenhaure, T.M.; Jonsson, A.H.; Li, S.; et al. The Immune Cell Landscape in Kidneys of Patients with Lupus Nephritis. Nat. Immunol. 2019, 20, 902–914. [Google Scholar] [CrossRef] [PubMed]
- McKinney, E.F.; Lyons, P.A.; Carr, E.J.; Hollis, J.L.; Jayne, D.R.W.; Willcocks, L.C.; Koukoulaki, M.; Brazma, A.; Jovanovic, V.; Kemeny, D.M.; et al. A CD8+ T Cell Transcription Signature Predicts Prognosis in Autoimmune Disease. Nat. Med. 2010, 16, 586–591. [Google Scholar] [CrossRef]
- McKinney, E.F.; Lee, J.C.; Jayne, D.R.W.; Lyons, P.A.; Smith, K.G.C. T-Cell Exhaustion, Co-Stimulation and Clinical Outcome in Autoimmunity and Infection. Nature 2015, 523, 612–616. [Google Scholar] [CrossRef]
- Kawasaki, K.; Yaoita, E.; Yamamoto, T.; Kihara, I. Depletion of CD8 Positive Cells in Nephrotoxic Serum Nephritis of WKY Rats. Kidney Int. 1992, 41, 1517–1526. [Google Scholar] [CrossRef][Green Version]
- Reynolds, J.; Norgan, V.A.; Bhambra, U.; Smith, J.; Cook, H.T.; Pusey, C.D. Anti-CD8 Monoclonal Antibody Therapy Is Effective in the Prevention and Treatment of Experimental Autoimmune Glomerulonephritis. J. Am. Soc. Nephrol. 2002, 13, 359–369. [Google Scholar] [CrossRef]
- Li, S.; Holdsworth, S.R.; Tipping, P.G. MHC Class I Pathway Is Not Required for the Development of Crescentic Glomerulonephritis in Mice. Clin. Exp. Immunol. 2000, 122, 453–458. [Google Scholar] [CrossRef]
- Chang, J.; Eggenhuizen, P.; O’Sullivan, K.M.; Alikhan, M.A.; Holdsworth, S.R.; Ooi, J.D.; Kitching, A.R. CD8+ T Cells Effect Glomerular Injury in Experimental Anti-Myeloperoxidase GN. J. Am. Soc. Nephrol. 2017, 28, 47–55. [Google Scholar] [CrossRef]
- Goldwich, A.; Burkard, M.; Ölke, M.; Daniel, C.; Amann, K.; Hugo, C.; Kurts, C.; Steinkasserer, A.; Gessner, A. Podocytes Are Nonhematopoietic Professional Antigen-Presenting Cells. J. Am. Soc. Nephrol. 2013, 24, 906–916. [Google Scholar] [CrossRef]
- Macconi, D.; Chiabrando, C.; Schiarea, S.; Aiello, S.; Cassis, L.; Gagliardini, E.; Noris, M.; Buelli, S.; Zoja, C.; Corna, D.; et al. Proteasomal Processing of Albumin by Renal Dendritic Cells Generates Antigenic Peptides. J. Am. Soc. Nephrol. 2009, 20, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Zoja, C.; Benigni, A.; Remuzzi, G. Cellular Responses to Protein Overload: Key Event in Renal Disease Progression. Curr. Opin. Nephrol. Hypertens. 2004, 13, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Dornieden, T.; Sattler, A.; Pascual-Reguant, A.; Ruhm, A.H.; Thiel, L.G.; Bergmann, Y.S.; Thole, L.M.L.; Köhler, R.; Kühl, A.A.; Hauser, A.E.; et al. Signatures and Specificity of Tissue-Resident Lymphocytes Identified in Human Renal Peritumor and Tumor Tissue. J. Am. Soc. Nephrol. 2021, 32, 2223–2241. [Google Scholar] [CrossRef] [PubMed]
- Parga-Vidal, L.; van Alderen, M.C.; Stark, R.; van Gisbergen, K.P.J.M. Tissue-Resident Memory T Cells in the Urogenital Tract. Nat. Rev. Nephrol. 2022, 18, 209–223. [Google Scholar] [CrossRef] [PubMed]
- Tilstra, J.S.; Avery, L.; Menk, A.V.; Gordon, R.A.; Smita, S.; Kane, L.P.; Chikina, M.; Delgoffe, G.M.; Shlomchik, M.J. Kidney-Infiltrating T Cells in Murine Lupus Nephritis Are Metabolically and Functionally Exhausted. J. Clin. Investig. 2018, 128, 4884–4897. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Guo, C.; Li, X.; Huang, Y.; Li, M.; Zhang, T.; Zhao, S.; Wang, S.; Zhang, H.; Yang, N. JAK/STAT Signaling Controls the Fate of CD8(+)CD103(+) Tissue-Resident Memory T Cell in Lupus Nephritis. J. Autoimmun. 2020, 109, 102424. [Google Scholar] [CrossRef] [PubMed]
- Dudek, M.; Pfister, D.; Donakonda, S.; Filpe, P.; Schneider, A.; Laschinger, M.; Hartmann, D.; Hüser, N.; Meiser, P.; Bayerl, F.; et al. Auto-Aggressive CXCR6(+) CD8 T Cells Cause Liver Immune Pathology in NASH. Nature 2021, 592, 444–449. [Google Scholar] [CrossRef]
- Benfaremo, D.; Paci, V.; Luchetti, M.M.; Gabrielli, A. Novel Therapeutic Approaches and Treatment Targets for Psoriatic Arthritis. Curr. Pharm. Biotechnol. 2021, 22, 85–98. [Google Scholar] [CrossRef]
- Novartis. Study of Safety, Efficacy and Tolerability of Secukinumab Versus Placebo, in Combination with SoC Therapy, in Patients with Active Lupus Nephritis (SELUNE; NCT04181762). 2020. Available online: https://clinicaltrials.gov/ct2/show/NCT04181762 (accessed on 6 April 2022).
- Xing, G.Q.; Chen, M.; Liu, G.; Heeringa, P.; Zhang, J.J.; Zheng, X.; Jie, E.; Kallenberg, C.G.M.; Zhao, M.H. Complement Activation Is Involved in Renal Damage in Human Antineutrophil Cytoplasmic Autoantibody Associated Pauci-Immune Vasculitis. J. Clin. Immunol. 2009, 29, 282–291. [Google Scholar] [CrossRef]
- Reich, K.; Iversen, L.; Puig, L.; Lambert, J.; Mrowietz, U.; Kaplan Saday, K.; Warren, R.B. Long-Term Efficacy and Safety of Brodalumab in Moderate-to-severe Plaque Psoriasis: A Post Hoc Pooled Analysis of AMAGINE-2 and -3. J. Eur. Acad. Dermatol. Venereol. 2022, 2022, 1–9. [Google Scholar] [CrossRef]
- van Vollenhoven, R.F.; Hahn, B.H.; Tsokos, G.C.; Wagner, C.L.; Lipsky, P.; Touma, Z.; Werth, V.P.; Gordon, R.M.; Zhou, B.; Hsu, B.; et al. Efficacy and Safety of Ustekinumab, an IL-12 and IL-23 Inhibitor, in Patients with Active Systemic Lupus Erythematosus: Results of a Multicentre, Double-Blind, Phase 2, Randomised, Controlled Study. Lancet 2018, 392, 1330–1339. [Google Scholar] [CrossRef]
- Santacruz, J.C.; Mantilla, M.J.; Rueda, I.; Pulido, S.; Rodriguez-Salas, G.; Londono, J. A Practical Perspective of the Hematologic Manifestations of Systemic Lupus Erythematosus. Cureus 2022, 14, e22938. [Google Scholar] [CrossRef] [PubMed]
- Mejia-Vilet, J.M.; Malvar, A.; Arazi, A.; Rovin, B.H. The Lupus Nephritis Management Renaissance. Kidney Int. 2022, 101, 242–255. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.S.W.; Rojas, O.L.; Gommerman, J.L. B Cell Depletion Therapies in Autoimmune Disease: Advances and Mechanistic Insights. Nat. Rev. Drug Discov. 2021, 20, 179–199. [Google Scholar] [CrossRef] [PubMed]
- Kraaij, T.; Arends, E.J.; Van Dam, L.S.; Kamerling, S.W.A.; Van Daele, P.L.A.; Bredewold, O.W.; Ray, A.; Bakker, J.A.; Scherer, H.U.; Huizinga, T.J.W.; et al. Long-Term Effects of Combined B-Cell Immunomodulation with Rituximab and Belimumab in Severe, Refractory Systemic Lupus Erythematosus: 2-Year Results. Nephrol. Dial. Transplant. 2021, 36, 1474–1483. [Google Scholar] [CrossRef]
- Brilland, B.; Garnier, A.S.; Chevailler, A.; Jeannin, P.; Subra, J.F.; Augusto, J.F. Complement Alternative Pathway in ANCA-Associated Vasculitis: Two Decades from Bench to Bedside. Autoimmun. Rev. 2020, 19, 102424. [Google Scholar] [CrossRef]
- Hilhorst, M.; Van Paassen, P.; Van Rie, H.; Bijnens, N.; Heerings-Rewinkel, P.; Van Breda Vriesman, P.; Cohen Tervaert, J.W. Complement in ANCA-Associated Glomerulonephritis. Nephrol. Dial. Transplant. 2017, 32, 1302–1313. [Google Scholar] [CrossRef]
- Sakaguchi, S.; Yamaguchi, T.; Nomura, T.; Ono, M. Regulatory T Cells and Immune Tolerance. Cell 2008, 133, 775–787. [Google Scholar] [CrossRef]
- Humrich, J.Y.; Morbach, H.; Undeutsch, R.; Enghard, P.; Rosenberger, S.; Weigert, O.; Kloke, L.; Heimann, J.; Gaber, T.; Brandenburg, S.; et al. Homeostatic Imbalance of Regulatory and Effector T Cells Due to IL-2 Deprivation Amplifies Murine Lupus. Proc. Natl. Acad. Sci. USA 2010, 107, 204–209. [Google Scholar] [CrossRef]
- Alcocer-Varela, J.; Alarcon-Segovia, D. Decreased Production of and Response to Interleukin-2 by Cultured Lymphocytes from Patients with Systemic Lupus Erythematosus. J. Clin. Investig. 1982, 69, 1388–1392. [Google Scholar] [CrossRef]
- Von Spee-Mayer, C.; Siegert, E.; Abdirama, D.; Rose, A.; Klaus, A.; Alexander, T.; Enghard, P.; Sawitzki, B.; Hiepe, F.; Radbruch, A.; et al. Low-Dose Interleukin-2 Selectively Corrects Regulatory T Cell Defects in Patients with Systemic Lupus Erythematosus. Ann. Rheum. Dis. 2016, 75, 1407–1415. [Google Scholar] [CrossRef] [PubMed]
- Neumann, K.; Tiegs, G. Immune Regulation in Renal Inflammation. Cell Tissue Res. 2021, 385, 305–322. [Google Scholar] [CrossRef] [PubMed]
- Venkatadri, R.; Sabapathy, V.; Dogan, M.; Sharma, R. Targeting Regulatory T Cells for Therapy of Lupus Nephritis. Front. Pharmacol. 2022, 12, 806612. [Google Scholar] [CrossRef] [PubMed]
- Fontenot, J.D.; Rasmussen, J.P.; Gavin, M.A.; Rudensky, A.Y. A Function for Interleukin 2 in Foxp3-Expressing Regulatory T Cells. Nat. Immunol. 2005, 6, 1142–1151. [Google Scholar] [CrossRef] [PubMed]
- D’Cruz, L.M.; Klein, L. Development and Function of Agonist-Induced CD25+Foxp3+ Regulatory T Cells in the Absence of Interleukin 2 Signaling. Nat. Immunol. 2005, 6, 1152–1159. [Google Scholar] [CrossRef]
- Setoguchi, R.; Hori, S.; Takahashi, T.; Sakaguchi, S. Homeostatic Maintenance of Natural Foxp3+ CD25+ CD4+ Regulatory T Cells by Interleukin (IL)-2 and Induction of Autoimmune Disease by IL-2 Neutralization. J. Exp. Med. 2005, 201, 723–735. [Google Scholar] [CrossRef]
- Yang, X.P.; Ghoreschi, K.; Steward-Tharp, S.M.; Rodriguez-Canales, J.; Zhu, J.; Grainger, J.R.; Hirahara, K.; Sun, H.W.; Wei, L.; Vahedi, G.; et al. Opposing Regulation of the Locus Encoding IL-17 through Direct, Reciprocal Actions of STAT3 and STAT5. Nat. Immunol. 2011, 12, 247–254. [Google Scholar] [CrossRef]
- He, J.; Zhang, X.; Wei, Y.; Sun, X.; Chen, Y.; Deng, J.; Jin, Y.; Gan, Y.; Hu, X.; Jia, R.; et al. Low-Dose Interleukin-2 Treatment Selectively Modulates CD4+ T Cell Subsets in Patients with Systemic Lupus Erythematosus. Nat. Med. 2016, 22, 991–993. [Google Scholar] [CrossRef]
- Zhao, C.; Chu, Y.; Liang, Z.; Zhang, B.; Wang, X.; Jing, X.; Hao, M.; Wang, Y.; An, J.; Zhang, X.; et al. Low Dose of IL-2 Combined with Rapamycin Restores and Maintains the Long-Term Balance of Th17/Treg Cells in Refractory SLE Patients. BMC Immunol. 2019, 20, 32. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of GN | Key Findings | Refs |
|---|---|---|
| NTN | IFNγ, secreted by Th1 cells, but also renal intrinsic cells, promoted crescent formation. | [47,48,49] |
| Neutralization of the p40 subunit of IL-12 attenuated crescent formation and glomerular CD4+ T-cell infiltration. | [60] | |
| IL-12p40−/− mice showed reduced crescent formation and proteinuria. | [61] | |
| Application of IL-12 in non-crescentic mice worsened disease towards cGN. | [60] | |
| Tubular epithelial cells and mesangial cells produced IL-12. | [62] | |
| IL-18 enhanced immune response induced by IL-12. | [66] | |
| Treatment of IL-12p40−/− mice with IL-18 restored crescent formation. | [61] | |
| CXCR3−/− mice developed less severe NTN with reduced IFNγ production and renal T-cell infiltration. | [70] | |
| CXCL9 induced CXCR3-mediated macrophage and T-cell recruitment. | [71] | |
| CCL3, CCL4, and CCL5 were upregulated, leading to renal monocyte and T-cell recruitment via CCR5 and CCR1. | [72,73] | |
| Macrophages induced glomerular injury. | [74,75,76] | |
| Glomerular macrophage accumulation was preceded by glomerular T-cell infiltration. | [41] | |
| T cell-derived MIF induced macrophage accumulation within glomeruli. | [77] | |
| Lack of CD4+ T cells resulted in reduced macrophage accumulation within glomeruli. | [42,43] | |
| Blocking of IFNγ, IL-12 or IL-18 diminished glomerular macrophage accumulation. | [47,48,60,61] | |
| IFNγ production by Th1 and renal intrinsic cells induced pro-inflammatory M1-macrophage polarization and production of IL-1β and TNFα by macrophages. | [78,79,80] | |
| Renal intrinsic cells were identified as major source of TNFα. | [85] | |
| MHC-II expression on renal intrinsic cells was important for T-cell and macrophage recruitment. | [81] | |
| Blockage of CD80/CD86 reduced intraglomerular accumulation of CD4+ T cells and macrophages. | [83] | |
| IL-12p35−/− mice showed less severe NTN on day 21 of disease, but increased IL-17A expression. | [86] | |
| OVA as GBM-fixed antigen in mice | Transfer of OVA-specific Th1 cells induced cGN, macrophage recruitment, and tissue injury at a later time point than after Th17-cell transfer. Renal CCL2 and CCL5 expression were elevated after Th1 cell transfer. | [87] |
| NZB/NZW mice | Treatment of mice with IFNγ promoted disease progression. | [50] |
| An IL-12 defect resulted in more severe LN. | [63] | |
| MRL-lpr mice | IFNγ induced apoptosis in tubular epithelial cells. | [53] |
| Defect in IL-12 production by macrophages led to high levels of type 2 cytokines and may drive LN. | [63] | |
| IL-18 levels were elevated and IL-18 accelerated GN. | [67] | |
| Lack of IL-18Rα or IL-18 improved proteinuria and survival. | [68,69] | |
| CXCR3-mediated macrophage and T-cell recruitment were dependent on CXCL9. | [71] | |
| Pristane-induced murine LN | IFNγ−/− mice did not develop LN. | [54] |
| IL-12p35−/− mice did not develop glomerular damage and proteinuria. | [65] | |
| Human LN | Reduced production of IL-12 and IFNγ resulted in higher levels of type 2 cytokines and may drive LN. | [64] |
| IL-12 serum levels were elevated in SLE patients. | [88,89,90] | |
| Murine anti-MPO GN | Neutralization of IFNγ led to less severe cGN. | [55] |
| The Th1 response developed following an early Th17 response. | [91] | |
| Treatment of mice with anti-IL-12p35 antibody blocked late GN. | [91] | |
| Human ANCA-GN | Identification of CD4+ TRM cells that showed a Th1 and Th17 signature and correlated with renal failure. | [92] |
| Patient-derived blood lymphocytes expressed IFNγ. | [93] | |
| Human IgAN | Th1 polarization was observed in IgAN patients. | [56] |
| Higher frequencies of blood Th2 and Th17 cells, but lower proportion of Th1 cells were shown. | [94,95,96] | |
| IFNγ polymorphism led to decreased NF-κB binding affinity and less IFNγ production associated with higher susceptibility to IgAN development. | [97] | |
| ddY mice | Strong Th1 response developed in early disease. | [56] |
| A Th1 polarization correlated with early renal injury. | [56] | |
| Murine EAG | Autoreactive Th1 cells led to the progression from mild to severe cGN. | [57,58] |
| IFNγ−/− mice developed more severe cGN. | [59] |
| Type of GN | Key Findings | Refs |
|---|---|---|
| NTN | Th2-prone BALB/c mice developed a proliferative, non-crescentic GN with renal neutrophil accumulation. Linear IgG deposition was observed in glomerular capillary loops. | [40] |
| IL-4 was produced by splenocytes. BALB/c mice developed glomerular injury characterized by thickened capillary walls, mesangial expansion, and glomerular hypercellularity. | [47] | |
| In C57BL/6 mice, IL-4 application attenuated glomerular immune cell accumulation and crescent formation and reduced serum levels of IgG2a and IgG3. | [101] | |
| Lack of IL-4 in BALB/c mice did not exacerbate GN towards cGN. | [60] | |
| Human IgAN | Imbalance of Th1/Th2 cytokines in favor of Th2 cytokines might lead to disease progression in IgAN. | [102] |
| Elevated levels of abnormally glycosylated IgA were present in patients. | [105,106] | |
| IL-4 promoted IgA1 production and higher secretion of aberrantly glycosylated IgA1. | [107,108] | |
| Murine IgAN | DdY mice with quiescent disease showed strong Th2 response. | [56] |
| Overproduction of type 2 cytokines correlated with increased serum levels of IgA, glomerular IgA deposition, aberrantly glycosylated IgA, and proteinuria in Smad4co/co;Lck-cre mice. | [103] |
| Type of GN | Key Findings | Refs |
|---|---|---|
| NTN | Th17 cells were detected in the kidney. IL-23p19−/− and IL17A−/− mice showed less crescent formation, reduced proteinuria, tubulointerstitial injury and glomerular immune cell infiltration. | [110] |
| Transfer of in vitro polarized Th17 cells to Rag1−/− mice led to crescent formation. Th17 cells were stable and did not start to produce IFNγ. | [87,111] | |
| Transfer of RORγt−/− CD4+ T cells to Rag1−/− mice resulted in reduced crescent formation. | [109] | |
| IL-17A−/− mice were protected from NTN on day six of disease but not on day 14, and cGN was more severe on day 21 because of an enhanced Th1 response. IL-23p19−/− showed more severe cGN on day 21 than WT mice. | [86] | |
| Renal CCL20 was upregulated and renal Th17-cell infiltration was abrogated in CCR6−/− mice. | [37] | |
| Th17 cells showed inflammation-induced migration from the intestine to the kidney in a sphingosine-1 phosphate receptor 1 and CCL20/CCR6-mediated fashion. Renal Th17 number was influenced by the intestinal microbiome. | [120] | |
| Il-17F−/− mice developed less severe GN associated with reduced renal neutrophil infiltration. | [121] | |
| Renal intrinsic cells expressed IL-17C and renal Th17 cells expressed IL-17RE. | [122] | |
| Lack of IL-17RC on Th17 cells aggravated GN. | [123] | |
| Anti-CD3 treatment induced IL-10 production in Th17 cells. | [111] | |
| Th17 cells converted to IL-10+ Tr1 cells under anti-CD3 treatment but did not seem to play a regulatory role. | [129] | |
| Infection with S. aureus induced TRM17 cells that persisted in the kidney after cleared infection and aggravated NTN. TRM17 cells were activated by Th17-polarizing cytokines. | [92] | |
| OVA as GBM-fixed antigen in mice | Transfer of OVA-specific Th17 cells induced non-crescentic GN with renal neutrophil recruitment and enhanced renal expression of CXCL1. Th17 cells induced early tissue injury and proteinuria. Transferred Th17 cells were phenotypically stable. | [87] |
| NZB/NZW mice | Elevated numbers of Th17 cells correlated with accelerated LN. | [116] |
| MRL-lpr mice | Progression of LN was associated with higher expression of IL-17A and IL-23R by T cells. | [114] |
| Transfer of IL-23-pre-treated lymphocytes from MRL-lpr mice to Rag1−/− mice induced nephritis. | [114] | |
| Blockage of IL-23 diminished IL-17A expression and proteinuria. | [115] | |
| Pristane-induced murine LN | Th17-cell phenotype was stable and not plastic in LN. | [111] |
| Human LN | Elevated serum levels of IL-17A were detected in SLE patients. | [88,90,130] |
| Elevated serum levels of IL-23 were detected in SLE patients | [89,131] | |
| Th17 cells were determined in glomeruli and tubulointerstitium in human LN and correlated with disease activity. | [132] | |
| IL-17A+ DN T cells were detected in kidneys of LN patients. | [133] | |
| Murine anti-MPO GN | Immunization of WT mice with MPO led to systemic IL-17A production. IL17A−/− mice were protected from anti-MPO GN and showed less renal neutrophil accumulation | [112] |
| Mice showed an early Th17 and a late Th1 response. | [91] | |
| Treatment of mice with anti-IL-23p19 Ab blocked early GN. | [91] | |
| Human ANCA-GN | Elevated numbers of blood Th17 cells and levels of Th17 cell-associated cytokines were determined and correlated with disease activity. | [134,135] |
| Human IgAN | IL-17A expression was present at renal tubular sites, correlating with renal damage and impaired renal function. | [95,96] |
| Enhanced numbers of circulating Th17 cells and increased serum levels of IL-17A were detected. | [94] | |
| Secretion of aberrantly glycosylated IgA1 was induced by IL-17A. | [136] | |
| ddY mice | Mice showed elevated numbers of renal Th17 cells and enhanced IL-17A expression. CCL20 was upregulated and neutralization of CCL20 decreased renal Th17 cell infiltration. | [113] |
| Murine EAG | IL-23p19−/− mice developed less severe EAG. | [28] |
| Renal Th17 cell infiltration led to disease exacerbation. | [57] | |
| Autoreactive Th17 cells were found in EAG. IL-17A−/− and IL-23p19−/− mice showed less crescent formation and reduced tubulointerstitial damage. | [58] |
| Type of GN | Key Findings | Refs |
|---|---|---|
| NTN | CD8+ T-cell depletion led to reduced crescent formation, proteinuria and glomerular macrophage infiltration in rats. | [146] |
| CD8−/− mice developed accelerated disease. | [44] | |
| Transporter associated with antigen processing-deficient mice with reduced CD8+ T-cell numbers were not protected from NTN. CD8+ T-cell depletion led to reduced proteinuria and recruitment of CD4+ T cells and macrophages to the kidney. | [148] | |
| Transfer of EGFP-specific CD8+ T cells to mice with EGFP-expression by podocytes together with NTN induction induced severe cGN and CD8+ T cells infiltrated glomeruli with a ruptured BC. | [15] | |
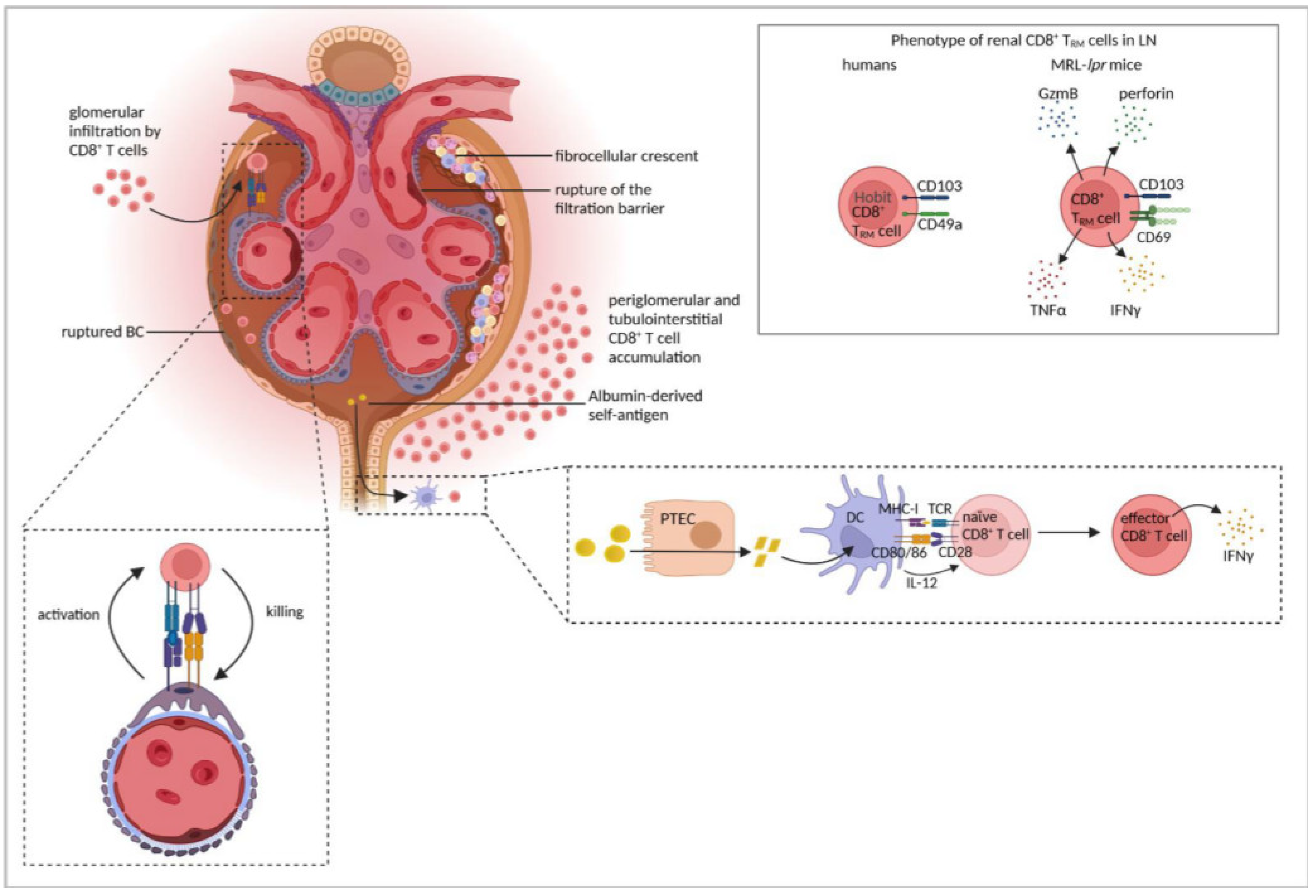
| Five-sixth nephrectomy | DCs, isolated from renal lymph nodes, presented albumin-derived peptides, processed through PTECs, to stimulate inflammatory CD8+ T-cell activation. | [151] |
| MRL-lpr mice | Renal CD8+ T cells were exhausted. | [155] |
| Renal CD8+ TRM cells expressed CD69 and CD103, were not exhausted and expressed GzmB, perforin, IFNγ, and TNFα and correlated with disease severity. | [156] | |
| Human LN | Periglomerular CD8+ T-cell accumulation correlated with disease activity, BC rupture, and crescent formation. | [7] |
| CD8+ T cells clonally expanded and persisted for years in the kidney. | [6] | |
| Cytotoxic CD8+ T cells were present in the kidney, either expressing GzmB and perforin or GzmK. CD8+ TRM cells expressed Hobit, CD103, and CD49a | [143] | |
| CD8+ TRM cells expressed CD103. | [156] | |
| Human anti-MPO GN | CD8+ T cells infiltrated the nephritic kidney. | [4,5,6,7,142] |
| Murine anti-MPO GN | CD8+ T-cell depletion after MPO immunization attenuated segmental necrosis, proteinuria, and CD4+ T cell- and macrophage recruitment to glomeruli. | [149] |
| Transfer of MPO-specific CD8+ T cells to Rag1−/− mice prior to disease induction aggravated GN. | [149] | |
| Human ANCA-GN | AAV patients with activated CD8+ T cells in blood had a poor prognosis. | [144,145] |
| Murine EAG | CD8+ T-cell depletion prevented EAG and ameliorated existing disease. | [147] |
| Biological | Disease | Approved/Clinical Development | Refs |
|---|---|---|---|
| Secukinumab (anti-IL-17A) | LN | Phase III clinical trial | [159] |
| Ustekinumab (anti-IL-12/IL-23-p40) | Psoriasis, psoriatic arthritis, Crohn’s disease SLE | Approved Phase II clinical trial | [162] |
| Belimumab (anti-BAFF) | SLE | Approved | [164] |
| Belimumab + rituximab (anti-CD20) | LN | Approved | [165,166] |
| Avacopan (C5a receptor antagonist) | ANCA-associated GN AAV, in combination with rituximab or cyclophosphamide | Pre-clinical development Approved | [167] |
| Low-dose IL-2 | SLE, LN | Clinical development | [172,173,174,175,176,177,178,179,180] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Linke, A.; Tiegs, G.; Neumann, K. Pathogenic T-Cell Responses in Immune-Mediated Glomerulonephritis. Cells 2022, 11, 1625. https://doi.org/10.3390/cells11101625
Linke A, Tiegs G, Neumann K. Pathogenic T-Cell Responses in Immune-Mediated Glomerulonephritis. Cells. 2022; 11(10):1625. https://doi.org/10.3390/cells11101625
Chicago/Turabian StyleLinke, Alexandra, Gisa Tiegs, and Katrin Neumann. 2022. "Pathogenic T-Cell Responses in Immune-Mediated Glomerulonephritis" Cells 11, no. 10: 1625. https://doi.org/10.3390/cells11101625
APA StyleLinke, A., Tiegs, G., & Neumann, K. (2022). Pathogenic T-Cell Responses in Immune-Mediated Glomerulonephritis. Cells, 11(10), 1625. https://doi.org/10.3390/cells11101625