
Modelling α-Synuclein Aggregation and Neurodegeneration with Fibril Seeds in Primary Cultures of Mouse Dopaminergic Neurons



 ,
,
Abstract
: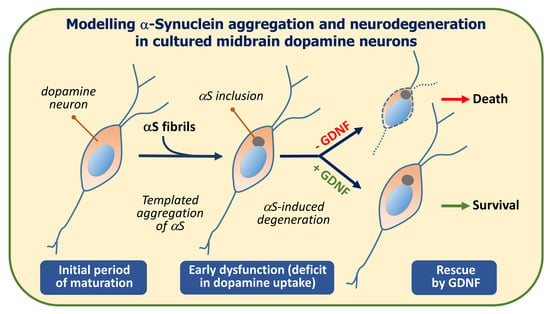
1. Introduction
2. Materials and Methods
2.1. Use of Animals
2.2. Midbrain Cell Culture Protocol
2.3. Maintenance and Treatment of Midbrain Cultures
2.4. Production of Astrocyte-Conditioned Medium
2.5. Immunodetection Protocols
2.6. Production of αS Fibrils
2.7. Manual Cell Counting Operations
2.8. Confocal Imaging and 3D Image Analysis
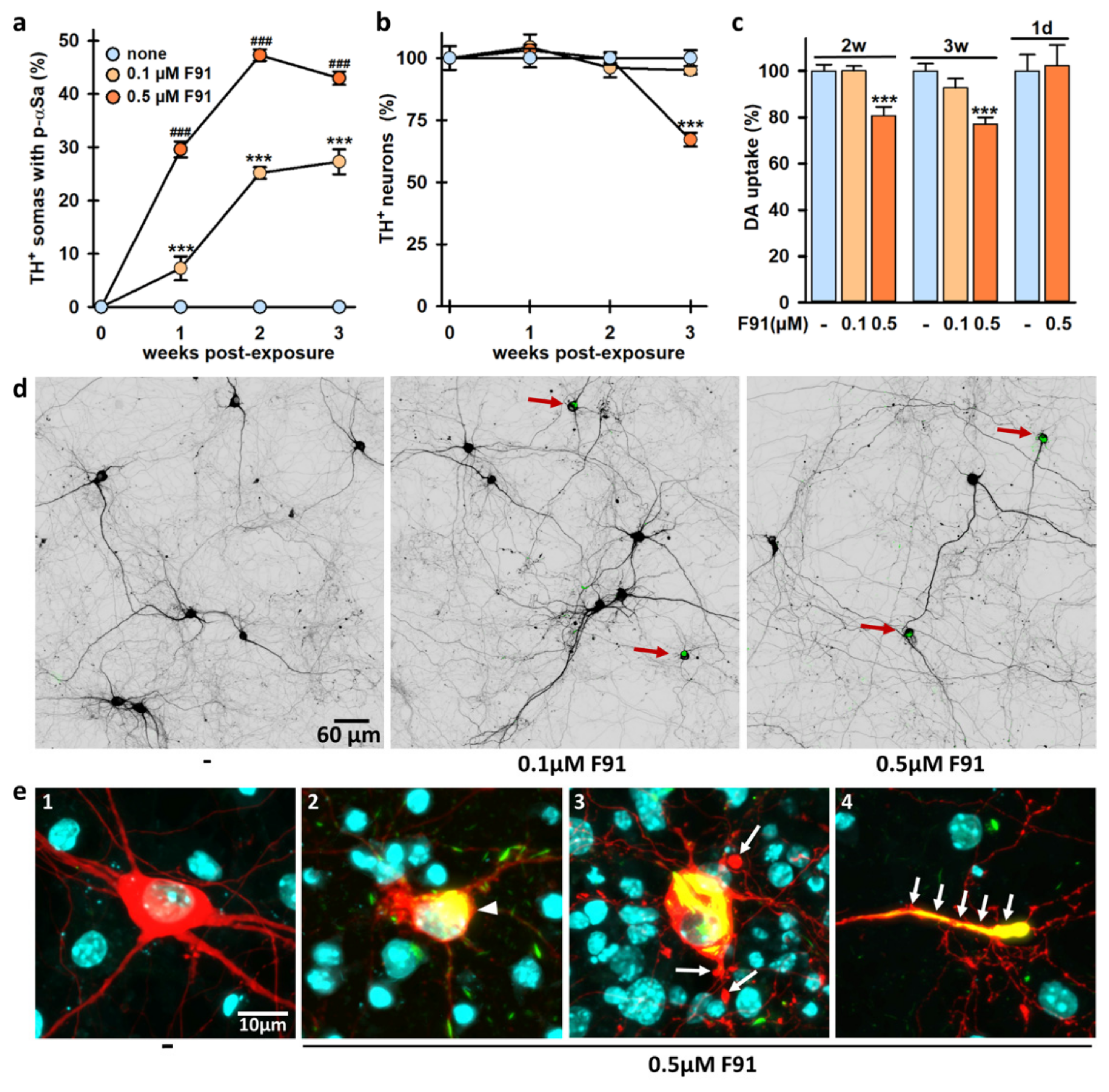
2.9. DA Uptake Measurements
2.10. Statistical Analysis
3. Results
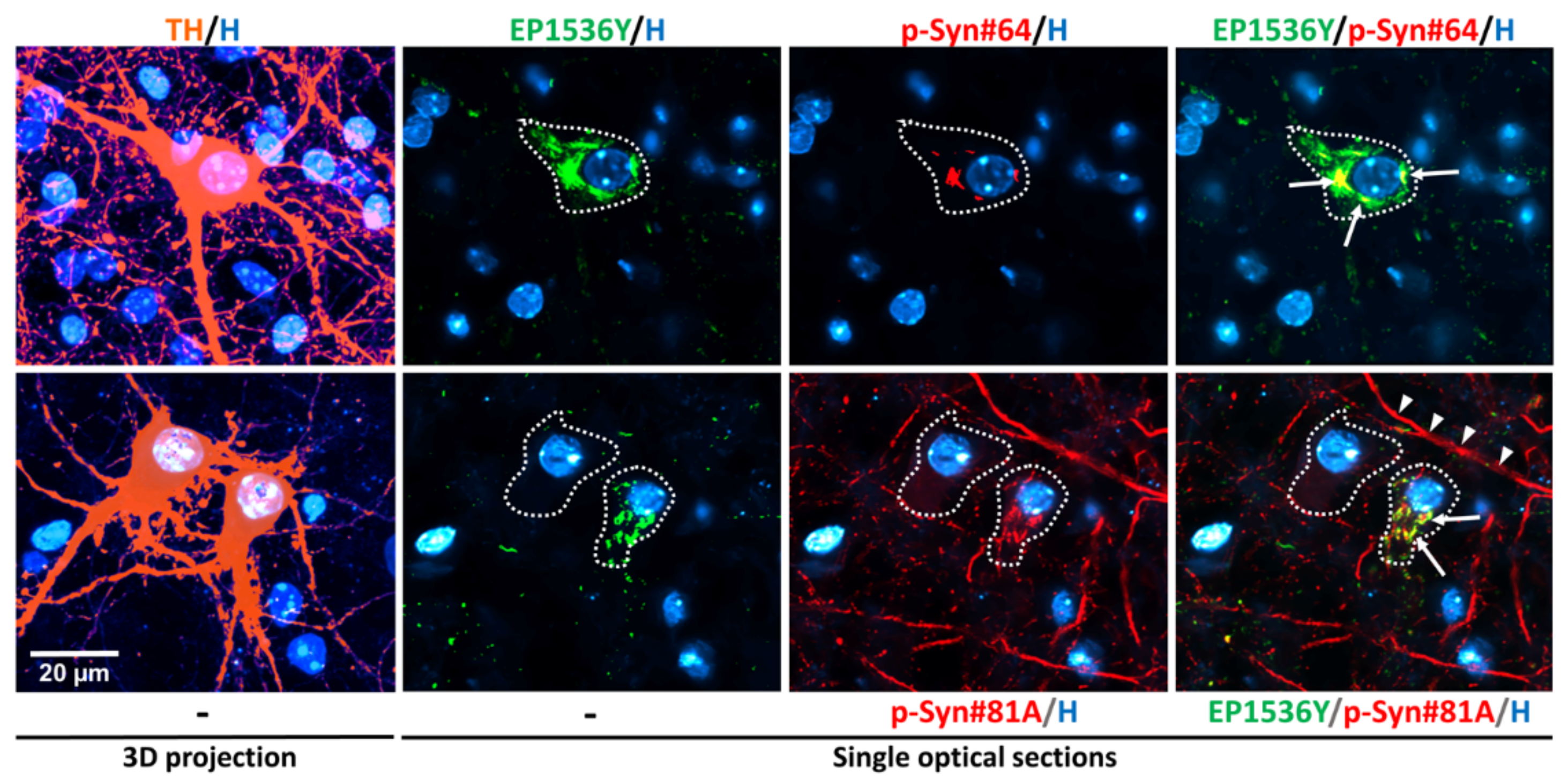
3.1. Detection of αS Aggregation in DA Neurons with Anti-αS (Phospho-Ser129) Antibodies
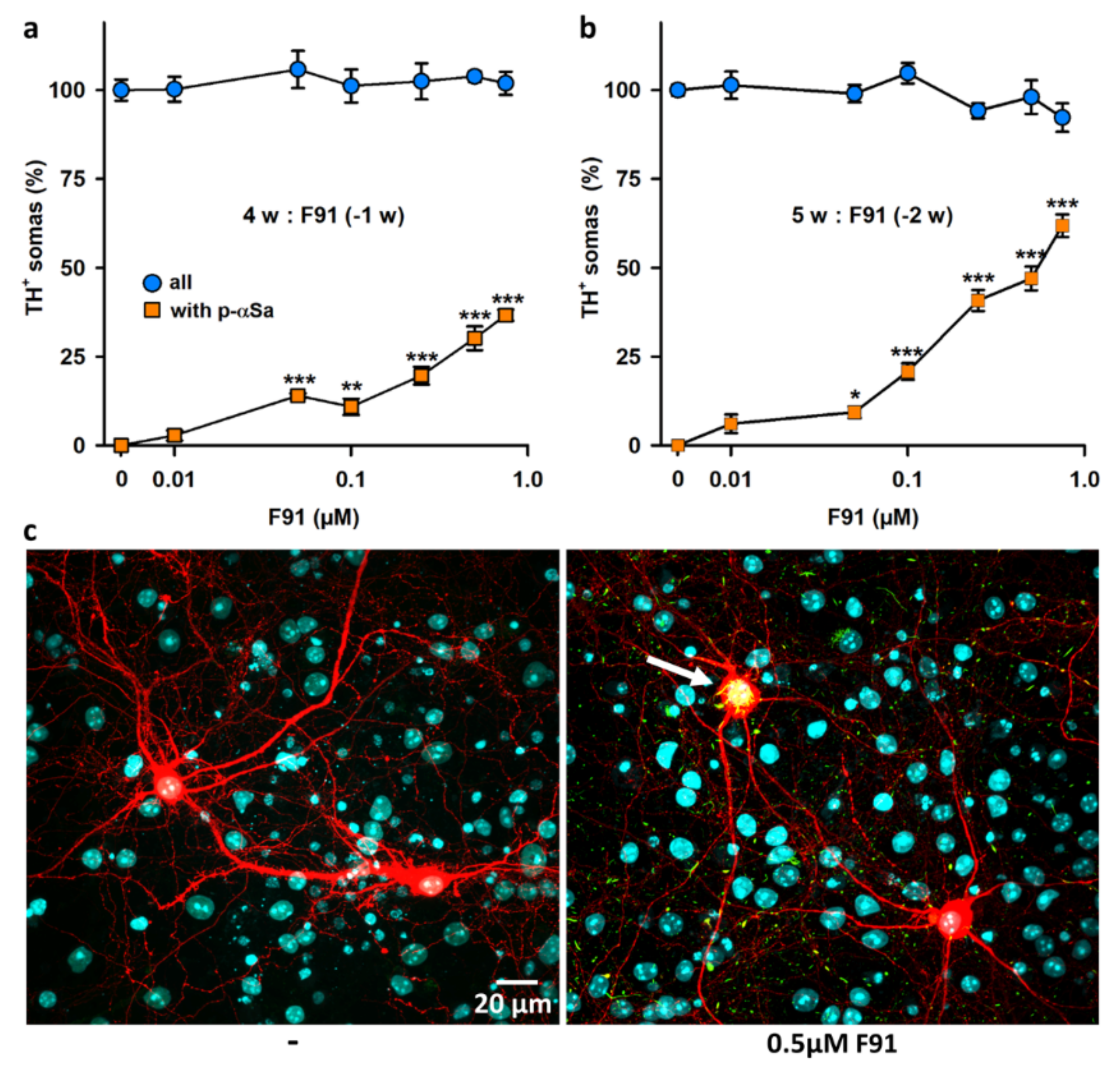
3.2. Seeded αS Aggregation in DA Neurons Increases Primarily with Fibril Concentrations and Post-Exposure Time to Fibrils
3.3. Seeded αS Aggregation in DA Neurons Is Not Influenced by the Degree of Maturation of the Midbrain Cultures
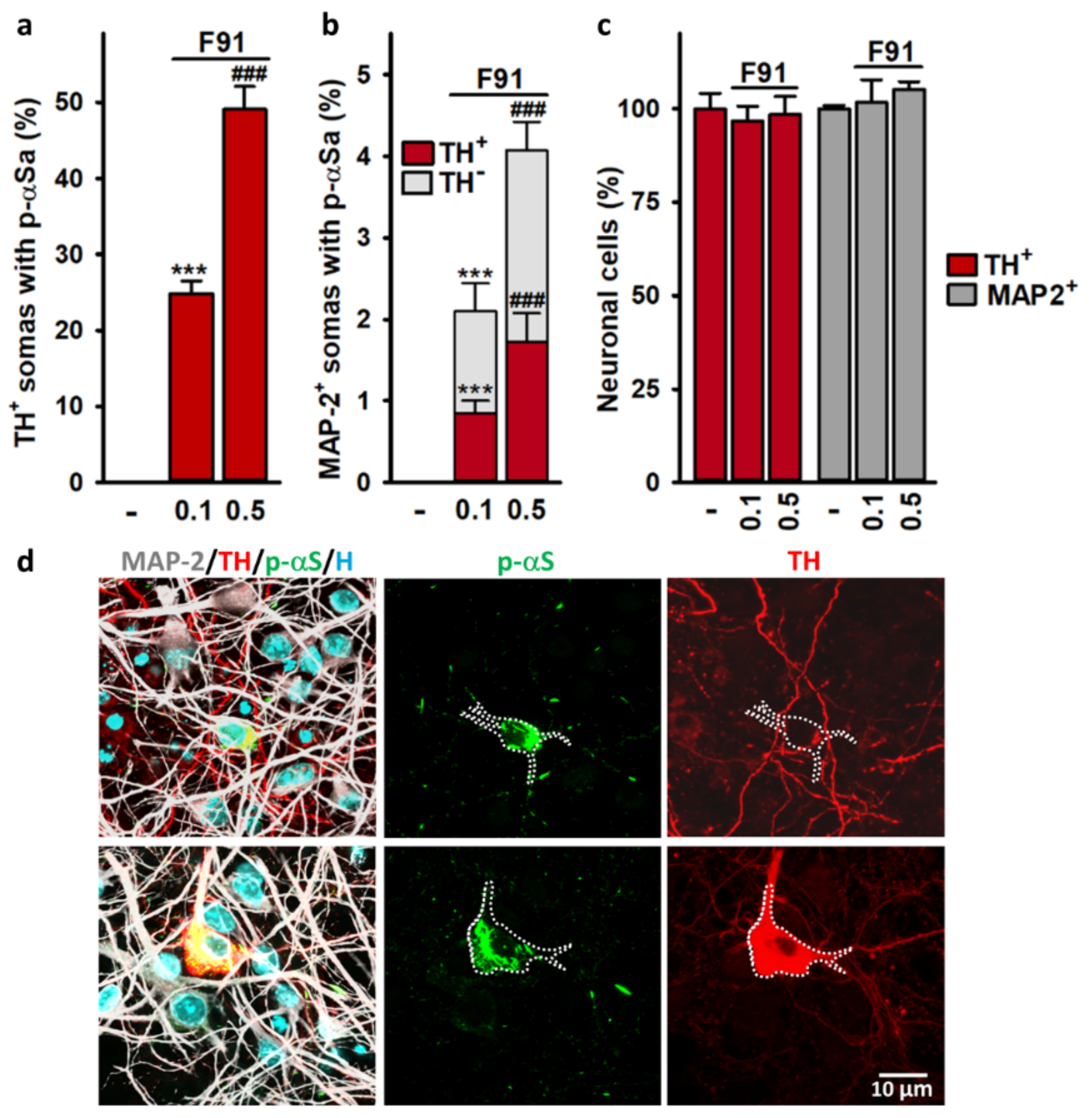
3.4. Somal αSa Are Preferentially Observed in DA Neurons
3.5. Impact of DA and DA Oxidative Metabolism on αS Seeded Aggregation
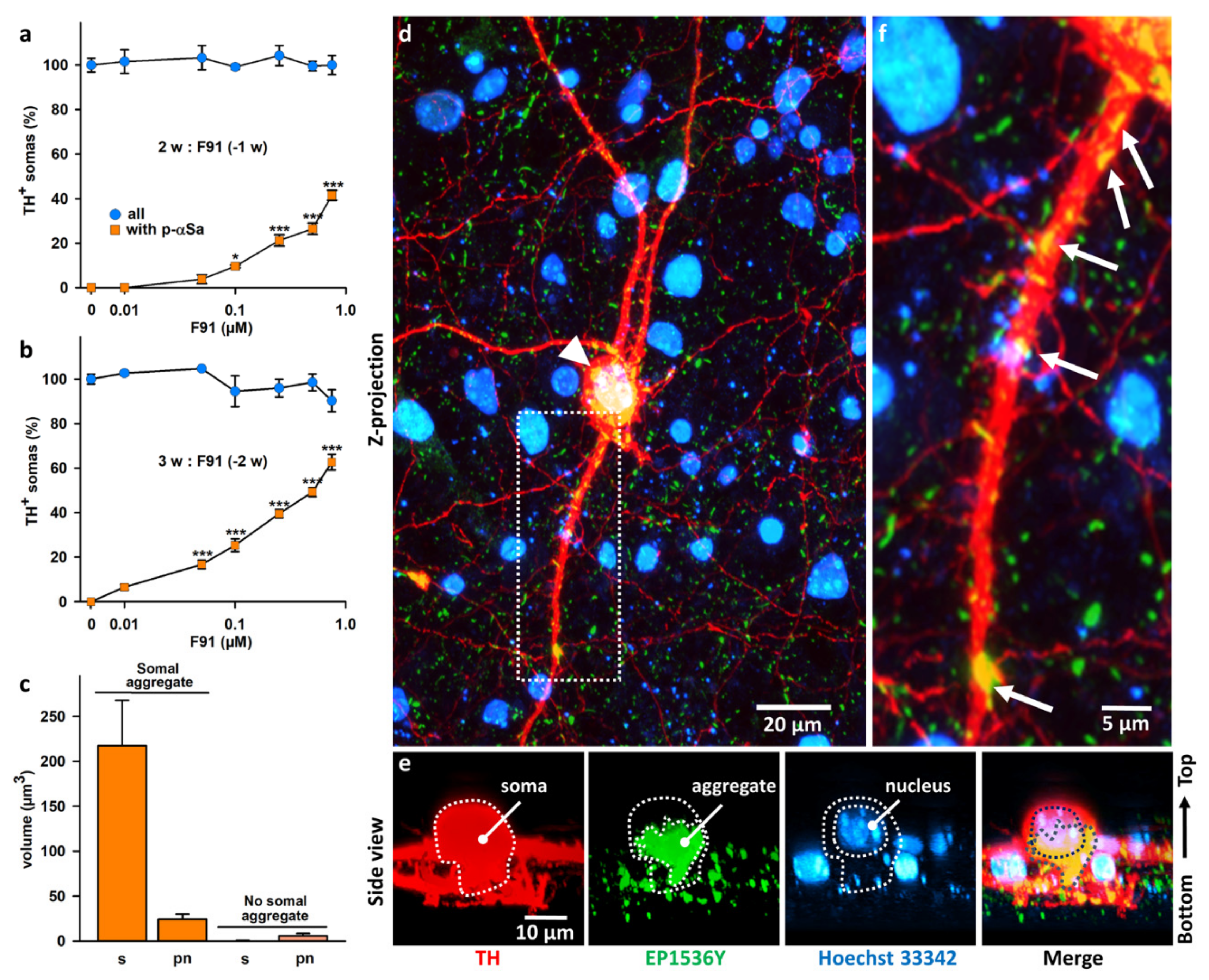
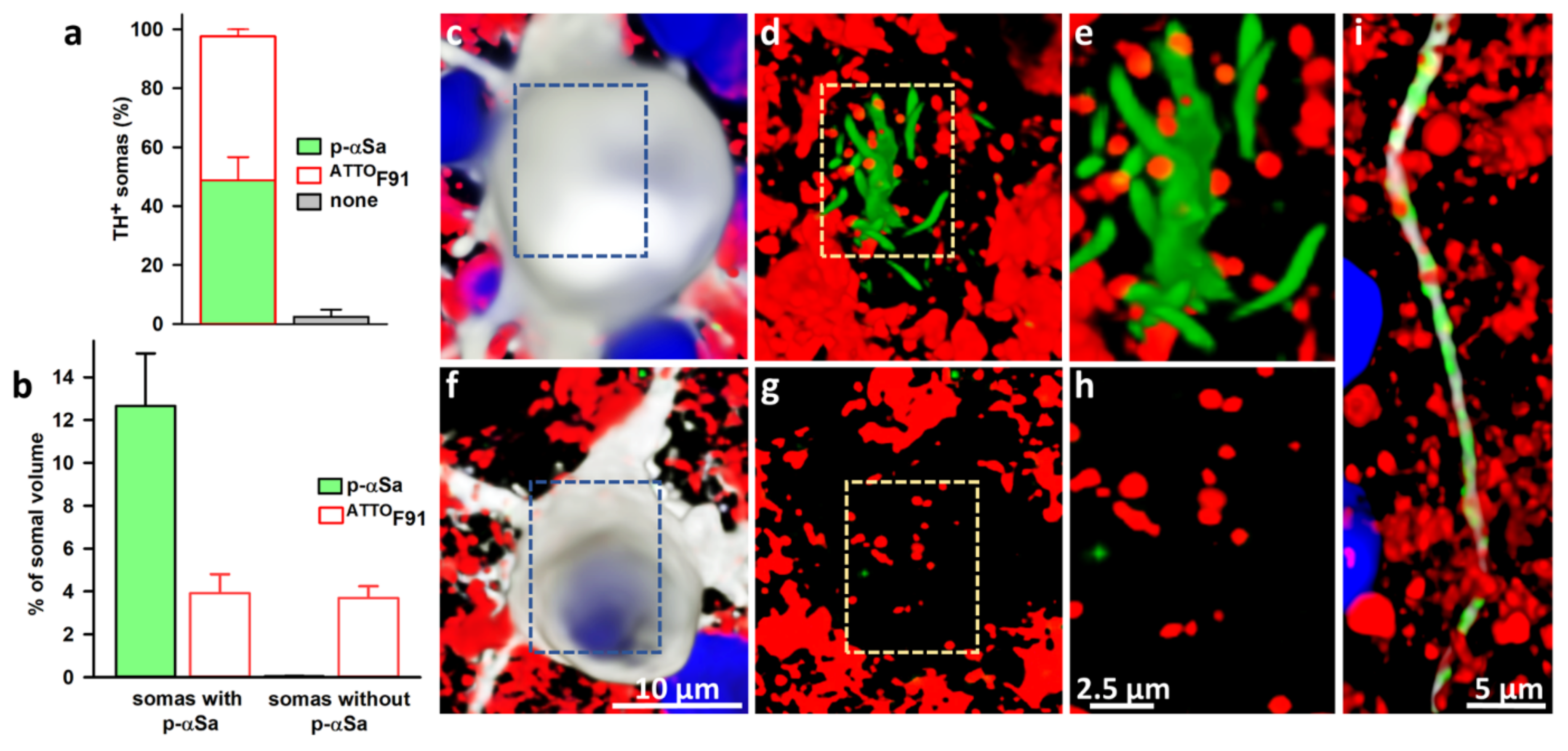
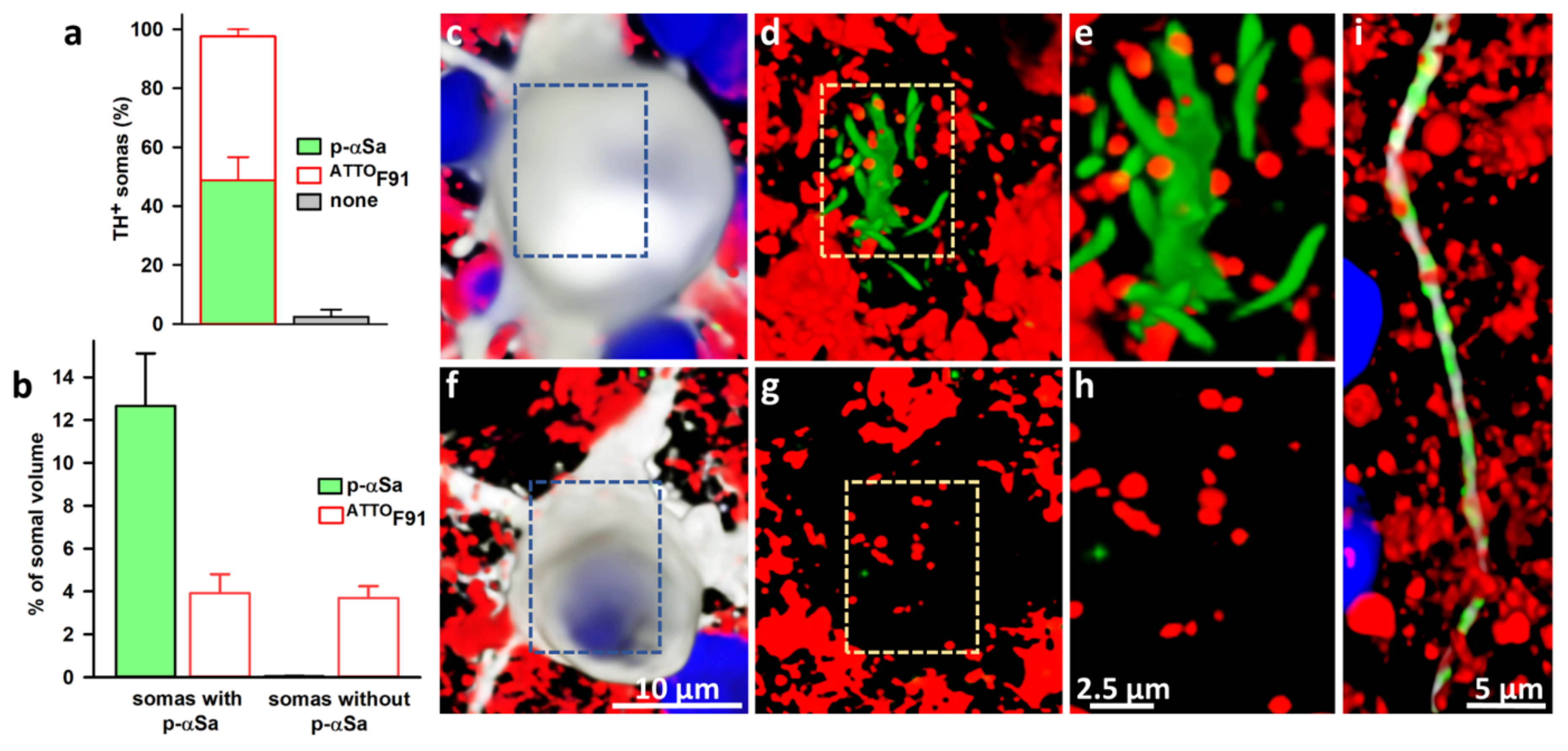
3.6. Subcellular Monitoring of αS Seeded Aggregation in DA Neurons with ATTOF91
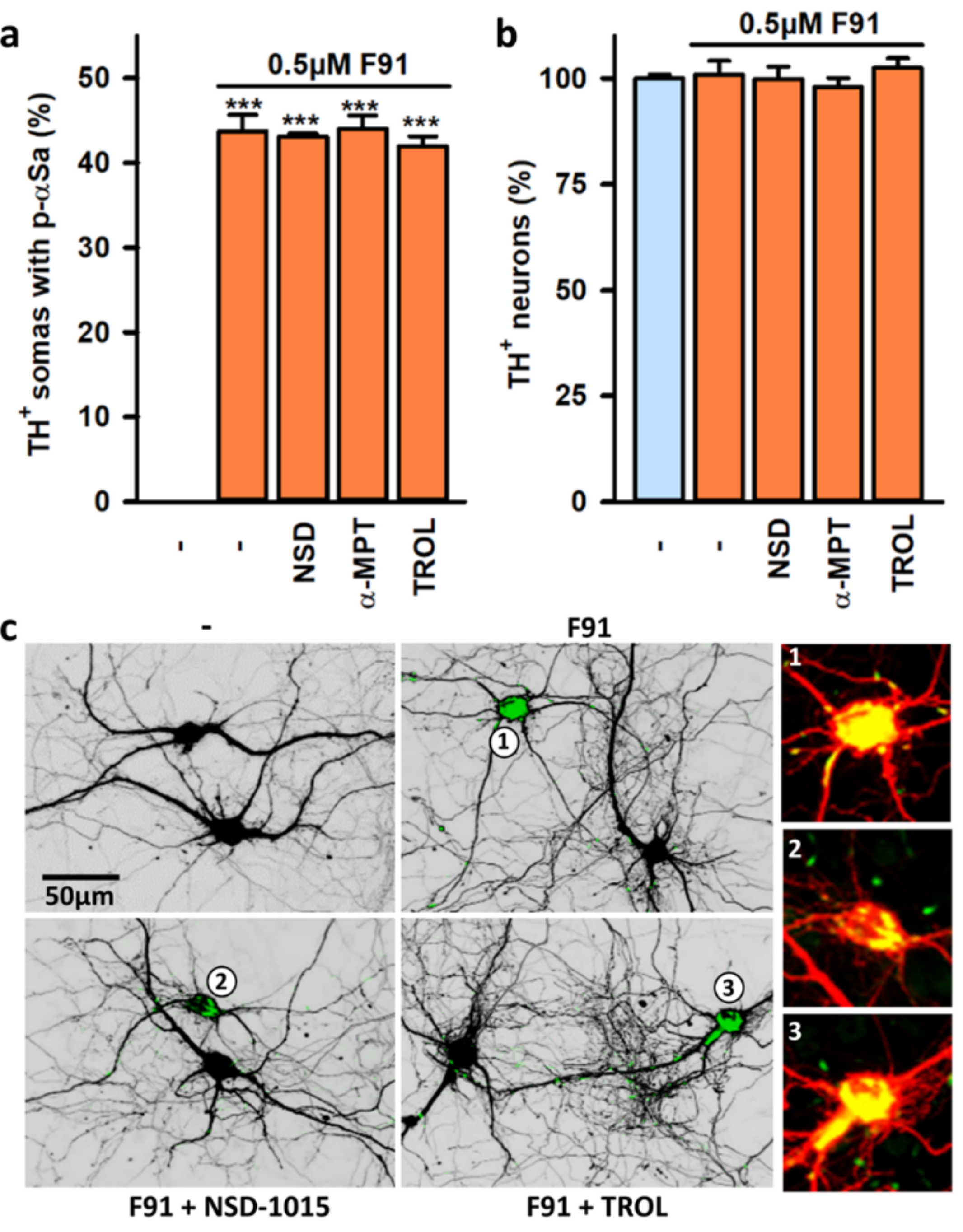
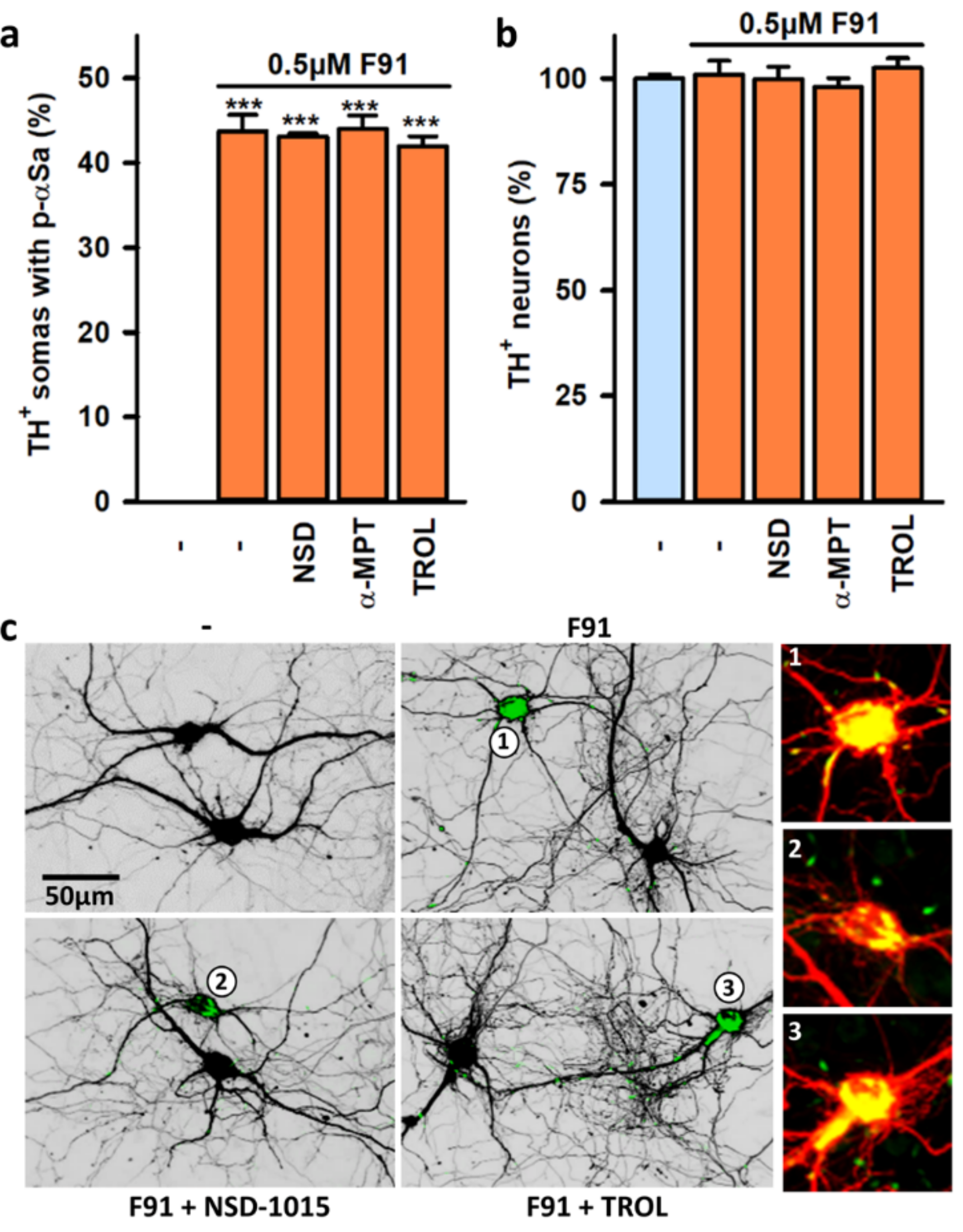
3.7. Impact of Mitochondrial Dysfunction on αS Seeded Aggregation
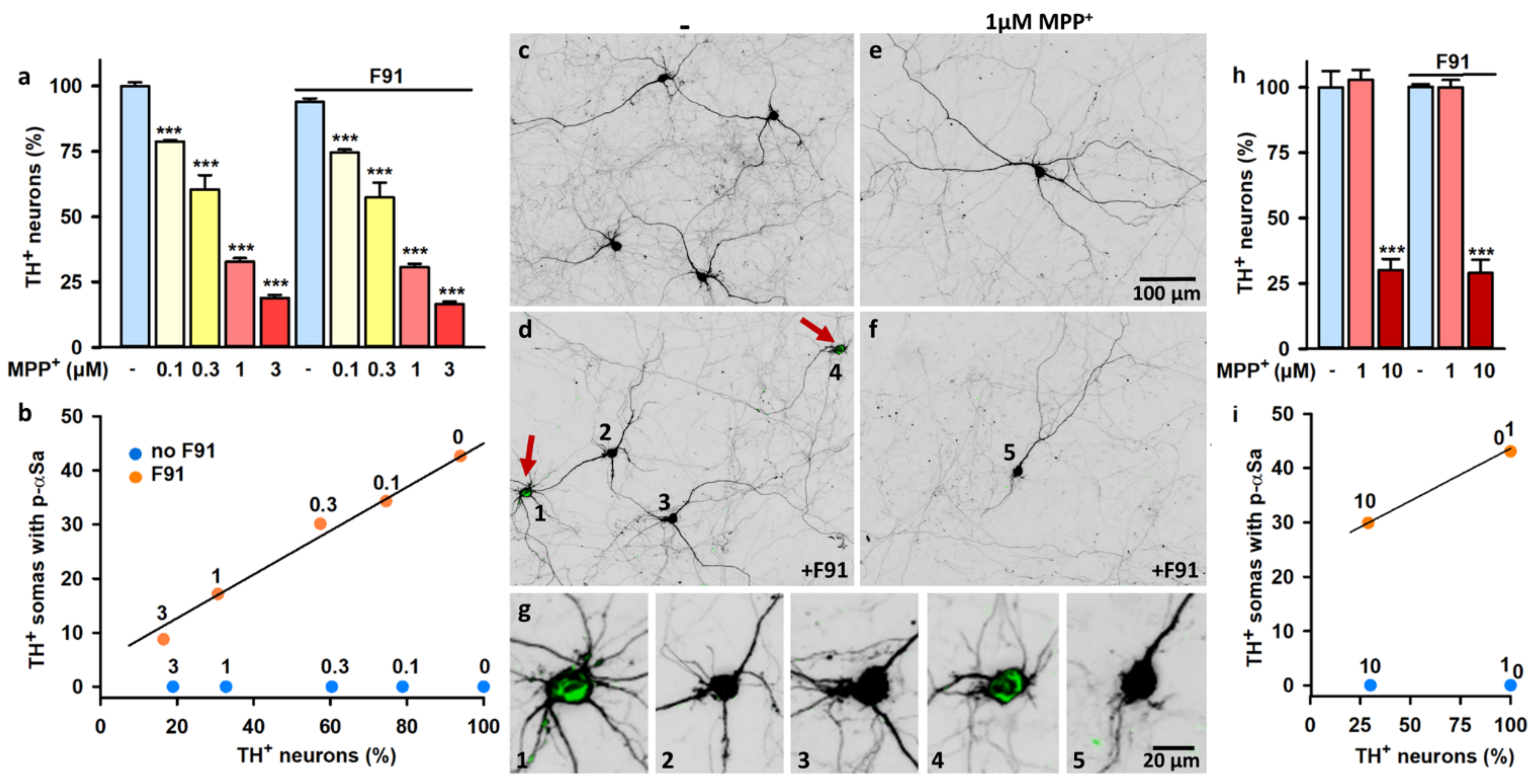
3.8. DA Cell Loss Occurs When Increasing the Lag Phase after Exposure to F91
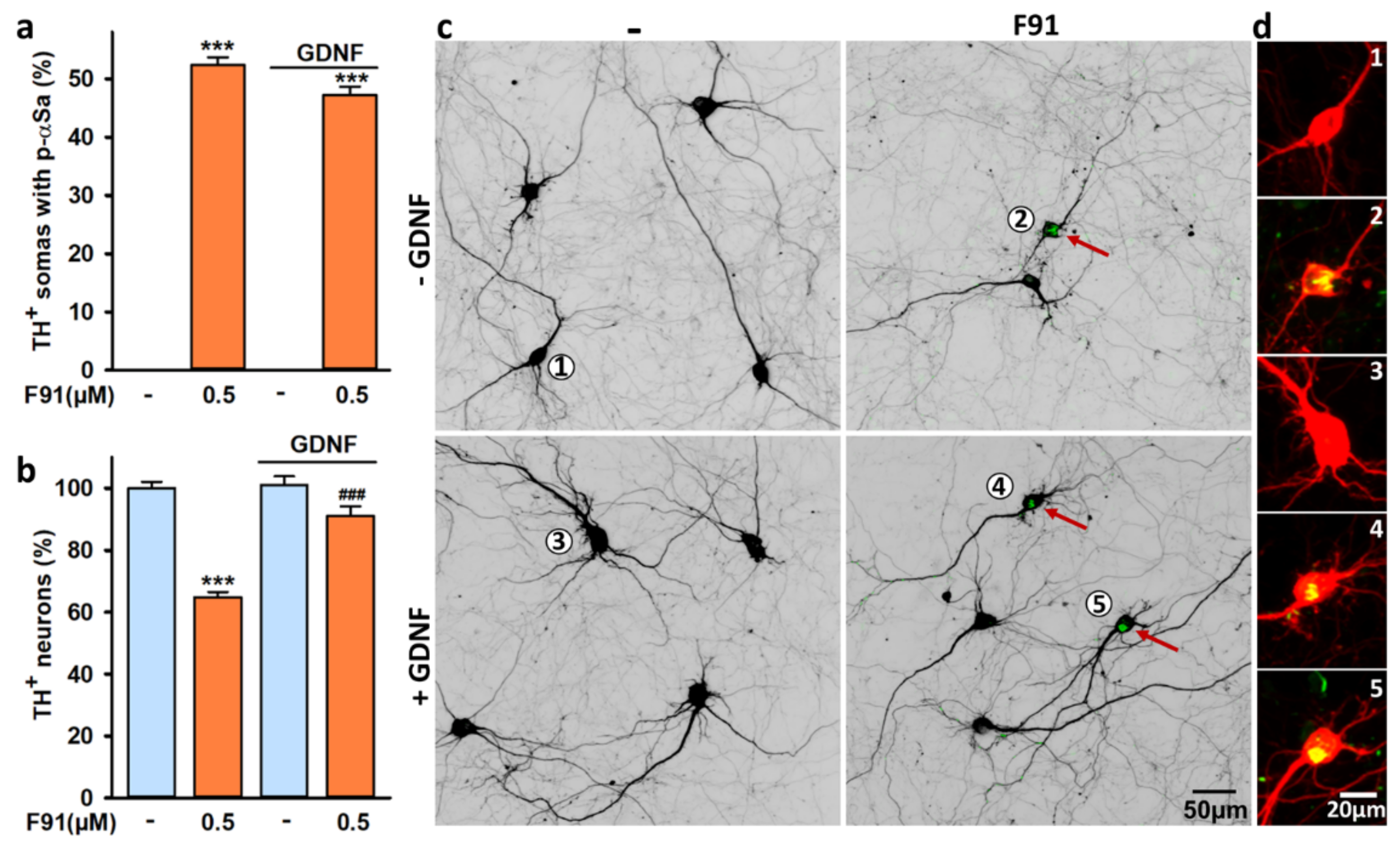
3.9. DA Cell Loss Induced by F91 Is Preventable by Trophic Stimulation with GDNF
4. Discussion
4.1. A Large Proportion of Midbrain DA Neurons Can Potentially Develop Somal αSa upon F91 Exposure
4.2. Impact of Culture Age on αS Seeded Aggregation
4.3. Subcellular Characterization of the Seeding Process in DA Neurons
4.4. αSa Are Preferentially Observed in the Soma of DA Neurons
4.5. Impact of Mitochondrial Dysfunction on Seeded αS Aggregation and Its Consequences
4.6. Factors Governing αS-Dependent Neurodegeneration in F91-Treated Midbrain Cultures
4.7. FαS Aggregation-Dependent DA Cell Loss Is Preventable by Trophic Stimulation with GDNF
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Michel, P.P.; Hirsch, E.C.; Hunot, S. Understanding Dopaminergic Cell Death Pathways in Parkinson Disease. Neuron 2016, 90, 675–691. [Google Scholar] [CrossRef] [Green Version]
- Przedborski, S. The Two-Century Journey of Parkinson Disease Research. Nat. Rev. Neurosci. 2017, 18, 251–259. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Goedert, M. Neurodegeneration and the Ordered Assembly of α-Synuclein. Cell Tissue Res. 2018, 373, 137–148. [Google Scholar] [CrossRef] [Green Version]
- Wakabayashi, K.; Tanji, K.; Mori, F.; Takahashi, H. The Lewy Body in Parkinson’s Disease: Molecules Implicated in the Formation and Degradation of α-Synuclein Aggregates: Molecular Components of Lewy Body. Neuropathology 2007, 27, 494–506. [Google Scholar] [CrossRef]
- Sulzer, D.; Edwards, R.H. The Physiological Role of a-synuclein and Its Relationship to Parkinson’s Disease. J. Neurochem. 2019, 150, 475–486. [Google Scholar] [CrossRef] [Green Version]
- Shahmoradian, S.H.; Lewis, A.J.; Genoud, C.; Hench, J.; Moors, T.E.; Navarro, P.P.; Castaño-Díez, D.; Schweighauser, G.; Graff-Meyer, A.; Goldie, K.N.; et al. Lewy Pathology in Parkinson’s Disease Consists of Crowded Organelles and Lipid Membranes. Nat. Neurosci. 2019, 22, 1099–1109. [Google Scholar] [CrossRef] [Green Version]
- Logan, T.; Bendor, J.; Toupin, C.; Thorn, K.; Edwards, R.H. α-Synuclein Promotes Dilation of the Exocytotic Fusion Pore. Nat. Neurosci. 2017, 20, 681–689. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Crowther, R.A.; Jakes, R.; Hasegawa, M.; Goedert, M. α-Synuclein in Filamentous Inclusions of Lewy Bodies from Parkinson’s Disease and Dementia with Lewy Bodies. Proc. Natl. Acad. Sci. USA 1998, 95, 6469–6473. [Google Scholar] [CrossRef] [Green Version]
- Waxman, E.A.; Giasson, B.I. Molecular Mechanisms of α-Synuclein Neurodegeneration. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2009, 1792, 616–624. [Google Scholar] [CrossRef] [Green Version]
- Satake, W.; Nakabayashi, Y.; Mizuta, I.; Hirota, Y.; Ito, C.; Kubo, M.; Kawaguchi, T.; Tsunoda, T.; Watanabe, M.; Takeda, A.; et al. Genome-Wide Association Study Identifies Common Variants at Four Loci as Genetic Risk Factors for Parkinson’s Disease. Nat. Genet. 2009, 41, 1303–1307. [Google Scholar] [CrossRef]
- Theillet, F.-X.; Binolfi, A.; Bekei, B.; Martorana, A.; Rose, H.M.; Stuiver, M.; Verzini, S.; Lorenz, D.; van Rossum, M.; Goldfarb, D.; et al. Structural Disorder of Monomeric α-Synuclein Persists in Mammalian Cells. Nature 2016, 530, 45–50. [Google Scholar] [CrossRef] [Green Version]
- Pieri, L.; Madiona, K.; Melki, R. Structural and Functional Properties of Prefibrillar α-Synuclein Oligomers. Sci. Rep. 2016, 6, 24526. [Google Scholar] [CrossRef] [Green Version]
- Cascella, R.; Chen, S.W.; Bigi, A.; Camino, J.D.; Xu, C.K.; Dobson, C.M.; Chiti, F.; Cremades, N.; Cecchi, C. The Release of Toxic Oligomers from α-Synuclein Fibrils Induces Dysfunction in Neuronal Cells. Nat. Commun. 2021, 12, 1814. [Google Scholar] [CrossRef]
- Gracia, P.; Camino, J.D.; Volpicelli-Daley, L.; Cremades, N. Multiplicity of α-Synuclein Aggregated Species and Their Possible Roles in Disease. Int. J. Mol. Sci. 2020, 21, 8043. [Google Scholar] [CrossRef]
- Alam, P.; Bousset, L.; Melki, R.; Otzen, D.E. α-synuclein Oligomers and Fibrils: A Spectrum of Species, a Spectrum of Toxicities. J. Neurochem. 2019, 150, 522–534. [Google Scholar] [CrossRef] [Green Version]
- Marotta, N.P.; Ara, J.; Uemura, N.; Lougee, M.G.; Meymand, E.S.; Zhang, B.; Petersson, E.J.; Trojanowski, J.Q.; Lee, V.M.-Y. α-Synuclein from Patient Lewy Bodies Exhibits Distinct Pathological Activity That Can Be Propagated in Vitro. Acta Neuropathol. Commun. 2021, 9, 188. [Google Scholar] [CrossRef]
- Killinger, B.A.; Kordower, J.H. Spreading of Alpha-synuclein–Relevant or Epiphenomenon? J. Neurochem. 2019, 150, 605–611. [Google Scholar] [CrossRef]
- Henrich, M.T.; Geibl, F.F.; Lakshminarasimhan, H.; Stegmann, A.; Giasson, B.I.; Mao, X.; Dawson, V.L.; Dawson, T.M.; Oertel, W.H.; Surmeier, D.J. Determinants of Seeding and Spreading of α-Synuclein Pathology in the Brain. Sci. Adv. 2020, 6, eabc2487. [Google Scholar] [CrossRef]
- Mezias, C.; Rey, N.; Brundin, P.; Raj, A. Neural Connectivity Predicts Spreading of Alpha-Synuclein Pathology in Fibril-Injected Mouse Models: Involvement of Retrograde and Anterograde Axonal Propagation. Neurobiol. Dis. 2020, 134, 104623. [Google Scholar] [CrossRef]
- Freundt, E.C.; Maynard, N.; Clancy, E.K.; Roy, S.; Bousset, L.; Sourigues, Y.; Covert, M.; Melki, R.; Kirkegaard, K.; Brahic, M. Neuron-to-Neuron Transmission of α-Synuclein Fibrils through Axonal Transport. Ann. Neurol. 2012, 72, 517–524. [Google Scholar] [CrossRef] [Green Version]
- Volpicelli-Daley, L.A.; Gamble, K.L.; Schultheiss, C.E.; Riddle, D.M.; West, A.B.; Lee, V.M.-Y. Formation of α-Synuclein Lewy Neurite–like Aggregates in Axons Impedes the Transport of Distinct Endosomes. MBoC 2014, 25, 4010–4023. [Google Scholar] [CrossRef]
- Desplats, P.; Lee, H.-J.; Bae, E.-J.; Patrick, C.; Rockenstein, E.; Crews, L.; Spencer, B.; Masliah, E.; Lee, S.-J. Inclusion Formation and Neuronal Cell Death through Neuron-to-Neuron Transmission of α-Synuclein. Proc. Natl. Acad. Sci. USA 2009, 106, 13010–13015. [Google Scholar] [CrossRef] [Green Version]
- Vekrellis, K.; Xilouri, M.; Emmanouilidou, E.; Stefanis, L. Inducible Over-Expression of Wild Type α-Synuclein in Human Neuronal Cells Leads to Caspase-Dependent Non-Apoptotic Death. J. Neurochem. 2009, 109, 1348–1362. [Google Scholar] [CrossRef]
- Xin, W.; Emadi, S.; Williams, S.; Liu, Q.; Schulz, P.; He, P.; Alam, N.; Wu, J.; Sierks, M. Toxic Oligomeric α-Synuclein Variants Present in Human Parkinson’s Disease Brains Are Differentially Generated in Mammalian Cell Models. Biomolecules 2015, 5, 1634–1651. [Google Scholar] [CrossRef] [Green Version]
- Scholz, D.; Pöltl, D.; Genewsky, A.; Weng, M.; Waldmann, T.; Schildknecht, S.; Leist, M. Rapid, Complete and Large-Scale Generation of Post-Mitotic Neurons from the Human LUHMES Cell Line: LUHMES as Widely Applicable Neuronal Model System. J. Neurochem. 2011, 119, 957–971. [Google Scholar] [CrossRef]
- Jiang, P.; Gan, M.; Yen, S.-H.; Moussaud, S.; McLean, P.J.; Dickson, D.W. Proaggregant Nuclear Factor(s) Trigger Rapid Formation of α-Synuclein Aggregates in Apoptotic Neurons. Acta Neuropathol. 2016, 132, 77–91. [Google Scholar] [CrossRef] [Green Version]
- Byers, B.; Lee, H.; Reijo Pera, R. Modeling Parkinson’s Disease Using Induced Pluripotent Stem Cells. Curr. Neurol. Neurosci. Rep. 2012, 12, 237–242. [Google Scholar] [CrossRef] [Green Version]
- Deas, E.; Cremades, N.; Angelova, P.R.; Ludtmann, M.H.R.; Yao, Z.; Chen, S.; Horrocks, M.H.; Banushi, B.; Little, D.; Devine, M.J.; et al. α-Synuclein Oligomers Interact with Metal Ions to Induce Oxidative Stress and Neuronal Death in Parkinson’s Disease. Antioxid. Redox Signal. 2016, 24, 376–391. [Google Scholar] [CrossRef] [Green Version]
- Tanudjojo, B.; Shaikh, S.S.; Fenyi, A.; Bousset, L.; Agarwal, D.; Marsh, J.; Zois, C.; Heman-Ackah, S.; Fischer, R.; Sims, D.; et al. Phenotypic Manifestation of α-Synuclein Strains Derived from Parkinson’s Disease and Multiple System Atrophy in Human Dopaminergic Neurons. Nat. Commun. 2021, 12, 3817. [Google Scholar] [CrossRef]
- Schapira, A.H.V.; Patel, S. Targeting Mitochondria for Neuroprotection in Parkinson Disease. JAMA Neurol. 2014, 71, 537–538. [Google Scholar] [CrossRef]
- González-Rodríguez, P.; Zampese, E.; Stout, K.A.; Guzman, J.N.; Ilijic, E.; Yang, B.; Tkatch, T.; Stavarache, M.A.; Wokosin, D.L.; Gao, L.; et al. Disruption of Mitochondrial Complex I Induces Progressive Parkinsonism. Nature 2021, 599, 650–656. [Google Scholar] [CrossRef]
- Mouton-Liger, F.; Rosazza, T.; Sepulveda-Diaz, J.; Ieang, A.; Hassoun, S.-M.; Claire, E.; Mangone, G.; Brice, A.; Michel, P.P.; Corvol, J.-C.; et al. Parkin Deficiency Modulates NLRP3 Inflammasome Activation by Attenuating an A20-Dependent Negative Feedback Loop. Glia 2018, 66, 1736–1751. [Google Scholar] [CrossRef] [Green Version]
- Ferreira Junior, N.C.; dos Santos Pereira, M.; Francis, N.; Ramirez, P.; Martorell, P.; González-Lizarraga, F.; Figadère, B.; Chehin, R.; Del Bel, E.; Raisman-Vozari, R.; et al. The Chemically-Modified Tetracycline COL-3 and Its Parent Compound Doxycycline Prevent Microglial Inflammatory Responses by Reducing Glucose-Mediated Oxidative Stress. Cells 2021, 10, 2163. [Google Scholar] [CrossRef]
- Michel, P.P.; Ruberg, M.; Agid, Y. Rescue of Mesencephalic Dopamine Neurons by Anticancer Drug Cytosine Arabinoside. J. Neurochem. 1997, 69, 1499–1507. [Google Scholar] [CrossRef]
- Vancha, A.R.; Govindaraju, S.; Parsa, K.V.; Jasti, M.; González-García, M.; Ballestero, R.P. Use of polyethyleneimine polymer in cell culture as attachment factor and lipofection enhancer. BMC Biotechnol. 2004, 4, 23. [Google Scholar] [CrossRef] [Green Version]
- Pozzi, D.; Ban, J.; Iseppon, F.; Torre, V. An improved method for growing neurons: Comparison with standard protocols. J. Neurosci. Methods 2017, 280, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos Pereira, M.; Abreu, G.H.D.; Rocca, J.; Hamadat, S.; Raisman-Vozari, R.; Michel, P.P.; Del Bel, E. Contributive Role of TNF-α to L-DOPA-Induced Dyskinesia in a Unilateral 6-OHDA Lesion Model of Parkinson’s Disease. Front. Pharmacol. 2021, 2204, 617085. [Google Scholar] [CrossRef]
- Sepulveda-Diaz, J.E.; Ouidja, M.O.; Socias, S.B.; Hamadat, S.; Guerreiro, S.; Raisman-Vozari, R.; Michel, P.P. A Simplified Approach for Efficient Isolation of Functional Microglial Cells: Application for Modeling Neuroinflammatory Responses in vitro: Simplified Approach for Microglia Isolation. Glia 2016, 64, 1912–1924. [Google Scholar] [CrossRef]
- Makky, A.; Bousset, L.; Polesel-Maris, J.; Melki, R. Nanomechanical Properties of Distinct Fibrillar Polymorphs of the Protein α-Synuclein. Sci. Rep. 2016, 6, 37970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghee, M.; Melki, R.; Michot, N.; Mallet, J. PA700, the regulatory complex of the 26S proteasome, interferes with a-synuclein assembly. FEBS J. 2005, 272, 4023–4033. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An Open-Source Platform for Biological-Image Analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, S.; Liu, M.; Mozgova, O.I.; Yu, W.; Baas, P.W. Mitotic Motors Coregulate Microtubule Patterns in Axons and Dendrites. J. Neurosci. 2012, 32, 14033–14049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rousseau, E.; Michel, P.P.; Hirsch, E.C. The Iron-Binding Protein Lactoferrin Protects Vulnerable Dopamine Neurons from Degeneration by Preserving Mitochondrial Calcium Homeostasis. Mol. Pharmacol. 2013, 84, 888–898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Deng, Y.; Qing, H. The Phosphorylation of α-Synuclein: Development and Implication for the Mechanism and Therapy of the Parkinson’s Disease. J. Neurochem. 2015, 135, 4–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujiwara, H.; Hasegawa, M.; Dohmae, N.; Kawashima, A.; Masliah, E.; Goldberg, M.S.; Shen, J.; Takio, K.; Iwatsubo, T. α-Synuclein Is Phosphorylated in Synucleinopathy Lesions. Nat. Cell Biol. 2002, 4, 160–164. [Google Scholar] [CrossRef]
- Shrivastava, A.N.; Bousset, L.; Renner, M.; Redeker, V.; Savistchenko, J.; Triller, A.; Melki, R. Differential Membrane Binding and Seeding of Distinct α-Synuclein Fibrillar Polymorphs. Biophys. J. 2020, 118, 1301–1320. [Google Scholar] [CrossRef] [Green Version]
- Larsen, K.E.; Fon, E.A.; Hastings, T.G.; Edwards, R.H.; Sulzer, D. Methamphetamine-Induced Degeneration of Dopaminergic Neurons Involves Autophagy and Upregulation of Dopamine Synthesis. J. Neurosci. 2002, 22, 8951–8960. [Google Scholar] [CrossRef]
- Lavaur, J.; Le Nogue, D.; Lemaire, M.; Pype, J.; Farjot, G.; Hirsch, E.C.; Michel, P.P. The Noble Gas Xenon Provides Protection and Trophic Stimulation to Midbrain Dopamine Neurons. J. Neurochem. 2017, 142, 14–28. [Google Scholar] [CrossRef]
- Michel, P.P.; Dandapani, B.K.; Knusel, B.; Sanchez-Ramos, J.; Hefti, F. Toxicity of 1-Methyl-4-Phenylpyridinium for Rat Dopaminergic Neurons in Culture: Selectivity and Irreversibility. J. Neurochem. 1990, 54, 1102–1109. [Google Scholar] [CrossRef]
- Danias, P.; Nicklas, W.J.; Ofori, S.; Shen, J.; Mytilineou, C. Mesencephalic Dopamine Neurons Become Less Sensitive to 1-Methyl-4-Phenyl-l,2,3,6-Tetrahydropyridine Toxicity during Development In Vitro. J. Neurochem. 1989, 53, 1149–1155. [Google Scholar] [CrossRef]
- Corti, O.; Lesage, S.; Brice, A. What Genetics Tells Us About the Causes and Mechanisms of Parkinson’s Disease. Physiol. Rev. 2011, 91, 1161–1218. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.-F.H.; Doherty, D.H.; Lile, J.D.; Bektesh, S.; Collins, F. GDNF: A Glial Cell Line-Derived Neurotrophic Factor for Midbrain Dopaminergic Neurons. Science 1993, 260, 1130–1132. [Google Scholar] [CrossRef] [PubMed]
- Kramer, E.R.; Liss, B. GDNF-Ret Signaling in Midbrain Dopaminergic Neurons and Its Implication for Parkinson Disease. FEBS Lett. 2015, 589, 3760–3772. [Google Scholar] [CrossRef] [PubMed]
- Guerreiro, S.; Toulorge, D.; Hirsch, E.; Marien, M.; Sokoloff, P.; Michel, P.P. Paraxanthine, the primary metabolite of caffeine, provides protection against dopaminergic cell death via stimulation of ryanodine receptor channels. Mol. Pharm. 2008, 74, 980–989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delic, V.; Chandra, S.; Abdelmotilib, H.; Maltbie, T.; Wang, S.; Kem, D.; Scott, H.J.; Underwood, R.N.; Liu, Z.; Volpicelli-Daley, L.A.; et al. Sensitivity and Specificity of Phospho-Ser129 α-Synuclein Monoclonal Antibodies. J. Comp. Neurol. 2018, 526, 1978–1990. [Google Scholar] [CrossRef] [PubMed]
- Koller, W.; O’Hara, R.; Weiner, W.; Lang, A.; Nutt, J.; Agid, Y.; Bonnet, A.M.; Jankovic, J. Relationship of Aging to Parkinson’s Disease. Adv. Neurol. 1987, 45, 317–321. [Google Scholar]
- Collier, T.J.; Kanaan, N.M.; Kordower, J.H. Aging and Parkinson’s Disease: Different Sides of the Same Coin? Mov. Disord. 2017, 32, 983–990. [Google Scholar] [CrossRef] [Green Version]
- Banker, G.A. Trophic Interactions Between Astroglial Cells and Hippocampal Neurons in Culture. Science 1980, 209, 809–810. [Google Scholar] [CrossRef] [Green Version]
- Courte, J.; Bousset, L.; Boxberg, Y.V.; Villard, C.; Melki, R.; Peyrin, J.-M. The Expression Level of Alpha-Synuclein in Different Neuronal Populations Is the Primary Determinant of Its Prion-like Seeding. Sci. Rep. 2020, 10, 4895. [Google Scholar] [CrossRef]
- Vasili, E.; Dominguez-Meijide, A.; Flores-León, M.; Al-Azzani, M.; Kanellidi, A.; Melki, R.; Stefanis, L.; Outeiro, T.F. Endogenous Levels of Alpha-Synuclein Modulate Seeding and Aggregation in Cultured Cells. Mol. Neurobiol. 2022, 59, 1273–1284. [Google Scholar] [CrossRef]
- Flavin, W.P.; Bousset, L.; Green, Z.C.; Chu, Y.; Skarpathiotis, S.; Chaney, M.J.; Kordower, J.H.; Melki, R.; Campbell, E.M. Endocytic Vesicle Rupture Is a Conserved Mechanism of Cellular Invasion by Amyloid Proteins. Acta Neuropathol. 2017, 134, 629–653. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, E.C.; Jenner, P.; Przedborski, S. Pathogenesis of Parkinson’s Disease: Pathogenesis of Parkinson’s Disease. Mov. Disord. 2013, 28, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Brichta, L.; Greengard, P. Molecular Determinants of Selective Dopaminergic Vulnerability in Parkinson’s Disease: An Update. Front. Neuroanat. 2014, 8, 152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gribaudo, S.; Tixador, P.; Bousset, L.; Fenyi, A.; Lino, P.; Melki, R.; Peyrin, J.-M.; Perrier, A.L. Propagation of α-Synuclein Strains within Human Reconstructed Neuronal Network. Stem Cell Rep. 2019, 12, 230–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braak, H.; Tredici, K.D.; Rüb, U.; de Vos, R.A.I.; Jansen Steur, E.N.H.; Braak, E. Staging of Brain Pathology Related to Sporadic Parkinson’s Disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Giguère, N.; Burke Nanni, S.; Trudeau, L.-E. On Cell Loss and Selective Vulnerability of Neuronal Populations in Parkinson’s Disease. Front. Neurol. 2018, 9, 455. [Google Scholar] [CrossRef]
- Ordonez, D.G.; Lee, M.K.; Feany, M.B. α-Synuclein Induces Mitochondrial Dysfunction through Spectrin and the Actin Cytoskeleton. Neuron 2018, 97, 108–124.e6. [Google Scholar] [CrossRef]
- Burtscher, J.; Copin, J.-C.; Sandi, C.; Lashuel, H.A. Pronounced α-Synuclein Pathology in a Seeding-Based Mouse Model Is Not Sufficient to Induce Mitochondrial Respiration Deficits in the Striatum and Amygdala. ENeuro 2020, 7, ENEURO.0110-20.2020. [Google Scholar] [CrossRef]
- Park, J.-H.; Burgess, J.D.; Faroqi, A.H.; DeMeo, N.N.; Fiesel, F.C.; Springer, W.; Delenclos, M.; McLean, P.J. α-Synuclein-Induced Mitochondrial Dysfunction Is Mediated via a Sirtuin 3-Dependent Pathway. Mol. Neurodegener. 2020, 15, 5. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.-J.; Shin, S.Y.; Choi, C.; Lee, Y.H.; Lee, S.-J. Formation and Removal of α-Synuclein Aggregates in Cells Exposed to Mitochondrial Inhibitors. J. Biol. Chem. 2002, 277, 5411–5417. [Google Scholar] [CrossRef] [Green Version]
- Kalivendi, S.V.; Cunningham, S.; Kotamraju, S.; Joseph, J.; Hillard, C.J.; Kalyanaraman, B. α-Synuclein Up-Regulation and Aggregation during MPP+-Induced Apoptosis in Neuroblastoma Cells. J. Biol. Chem. 2004, 279, 15240–15247. [Google Scholar] [CrossRef] [Green Version]
- Reeve, A.K.; Ludtmann, M.H.; Angelova, P.R.; Simcox, E.M.; Horrocks, M.H.; Klenerman, D.; Gandhi, S.; Turnbull, D.M.; Abramov, A.Y. Aggregated α-Synuclein and Complex I Deficiency: Exploration of Their Relationship in Differentiated Neurons. Cell Death Dis. 2015, 6, e1820. [Google Scholar] [CrossRef] [PubMed]
- Ludtmann, M.H.R.; Angelova, P.R.; Horrocks, M.H.; Choi, M.L.; Rodrigues, M.; Baev, A.Y.; Berezhnov, A.V.; Yao, Z.; Little, D.; Banushi, B.; et al. α-Synuclein Oligomers Interact with ATP Synthase and Open the Permeability Transition Pore in Parkinson’s Disease. Nat. Commun. 2018, 9, 2293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neystat, M.; Lynch, T.; Przedborski, S.; Kholodilov, N.; Rzhetskaya, M.; Burke, R.E. α-Synuclein expression in substantia nigra and cortex in Parkinson’s disease. Mov. Disord. 1999, 14, 417–422. [Google Scholar] [CrossRef]
- Kingsbury, A.E.; Daniel, S.E.; Sangha, H.; Eisen, S.; Lees, A.J.; Foster, O.J. Alteration in a-synuclein mRNA expression in Parkinson’s disease. Mov. Disord. 2004, 19, 162–170. [Google Scholar] [CrossRef]
- Su, X.; Fischer, D.L.; Li, X.; Bankiewicz, K.; Sortwell, C.E.; Federoff, H.J. α-Synuclein mRNA Is Not Increased in Sporadic PD and α-Synuclein Accumulation Does Not Block GDNF Signaling in Parkinson’s Disease and Disease Models. Mol. Ther. 2017, 25, 2231–2235. [Google Scholar] [CrossRef] [Green Version]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the Parkin Gene Cause Autosomal Recessive Juvenile Parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [CrossRef] [PubMed]
- Van Rompuy, A.-S.; Oliveras-Salvá, M.; Van der Perren, A.; Corti, O.; Van den Haute, C.; Baekelandt, V. Nigral Overexpression of α-Synuclein in the Absence of Parkin Enhances α-Synuclein Phosphorylation but Does Not Modulate Dopaminergic Neurodegeneration. Mol. Neurodegener. 2015, 10, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halliday, G.; Herrero, M.T.; Murphy, K.; McCann, H.; Ros-Bernal, F.; Barcia, C.; Mori, H.; Blesa, F.J.; Obeso, J.A. No Lewy Pathology in Monkeys with over 10 Years of Severe MPTP Parkinsonism: No α-Synuclein Deposition in MPTP Monkeys. Mov. Disord. 2009, 24, 1519–1523. [Google Scholar] [CrossRef]
- Vermilyea, S.C.; Emborg, M.E. α-Synuclein and Nonhuman Primate Models of Parkinson’s Disease. J. Neurosci. Methods 2015, 255, 38–51. [Google Scholar] [CrossRef] [Green Version]
- Patterson, J.R.; Duffy, M.F.; Kemp, C.J.; Howe, J.W.; Collier, T.J.; Stoll, A.C.; Miller, K.M.; Patel, P.; Levine, N.; Moore, D.J.; et al. Time Course and Magnitude of α-Synuclein Inclusion Formation and Nigrostriatal Degeneration in the Rat Model of Synucleinopathy Triggered by Intrastriatal α-Synuclein Preformed Fibrils. Neurobiol. Dis. 2019, 130, 104525. [Google Scholar] [CrossRef] [PubMed]
- Luna, E.; Decker, S.C.; Riddle, D.M.; Caputo, A.; Zhang, B.; Cole, T.; Caswell, C.; Xie, S.X.; Lee, V.M.Y.; Luk, K.C. Differential α-Synuclein Expression Contributes to Selective Vulnerability of Hippocampal Neuron Subpopulations to Fibril-Induced Toxicity. Acta Neuropathol. 2018, 135, 855–875. [Google Scholar] [CrossRef] [PubMed]
- Olanow, C.W.; Perl, D.P.; DeMartino, G.N.; McNaught, K.S.P. Lewy-Body Formation Is an Aggresome-Related Process: A Hypothesis. Lancet Neurol. 2004, 3, 496–503. [Google Scholar] [CrossRef]
- Carmo-Gonçalves, P.; Romão, L.; Follmer, C. In vitro Protective Action of Monomeric and Fibrillar α-Synuclein on Neuronal Cells Exposed to the Dopaminergic Toxins Salsolinol and DOPAL. ACS Chem. Neurosci. 2020, 11, 3541–3548. [Google Scholar] [CrossRef]
- Gómez-Benito, M.; Granado, N.; García-Sanz, P.; Michel, A.; Dumoulin, M.; Moratalla, R. Modeling Parkinson’s Disease with the Alpha-Synuclein Protein. Front. Pharmacol. 2020, 11, 356. [Google Scholar] [CrossRef] [PubMed]
- Janezic, S.; Threlfell, S.; Dodson, P.D.; Dowie, M.J.; Taylor, T.N.; Potgieter, D.; Parkkinen, L.; Senior, S.L.; Anwar, S.; Ryan, B.; et al. Deficits in Dopaminergic Transmission Precede Neuron Loss and Dysfunction in a New Parkinson Model. Proc. Natl. Acad. Sci. USA 2013, 110, E4016–E4025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahul-Mellier, A.-L.; Burtscher, J.; Maharjan, N.; Weerens, L.; Croisier, M.; Kuttler, F.; Leleu, M.; Knott, G.W.; Lashuel, H.A. The Process of Lewy Body Formation, Rather than Simply α-Synuclein Fibrillization, Is One of the Major Drivers of Neurodegeneration. Proc. Natl. Acad. Sci. USA 2020, 117, 4971–4982. [Google Scholar] [CrossRef] [Green Version]
- Chmielarz, P.; Er, Ş.; Konovalova, J.; Bandres, L.; Hlushchuk, I.; Albert, K.; Panhelainen, A.; Luk, K.; Airavaara, M.; Domanskyi, A. GDNF/RET Signaling Pathway Activation Eliminates Lewy Body Pathology in midbrain dopamine neurons. Mov. Disord. 2020, 35, 2279–2289. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primary Antibodies | Host | Working Dilution | Source | Identifier |
|---|---|---|---|---|
| anti-p-S129 αS [EP1536Y] | Rabbit | 1:2500 | Abcam | ab51253 |
| anti-p-S129 αS [P-Syn#64] | Mouse | 1:50,000 | Wako | W1W015-25191 |
| anti-p-S129 αS [P-syn/81A] | Mouse | 1:1000 | Abcam | ab184674 |
| anti-TH [LNC1] | Mouse | 1:2500 | Immunostar | 22941 |
| anti-TH | Chicken | 1:1000 | Abcam | ab76442 |
| anti-MAP-2 | Chicken | 1:500 | Abcam | ab5392 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tourville, A.; Akbar, D.; Corti, O.; Prehn, J.H.M.; Melki, R.; Hunot, S.; Michel, P.P. Modelling α-Synuclein Aggregation and Neurodegeneration with Fibril Seeds in Primary Cultures of Mouse Dopaminergic Neurons. Cells 2022, 11, 1640. https://doi.org/10.3390/cells11101640
Tourville A, Akbar D, Corti O, Prehn JHM, Melki R, Hunot S, Michel PP. Modelling α-Synuclein Aggregation and Neurodegeneration with Fibril Seeds in Primary Cultures of Mouse Dopaminergic Neurons. Cells. 2022; 11(10):1640. https://doi.org/10.3390/cells11101640
Chicago/Turabian StyleTourville, Aurore, David Akbar, Olga Corti, Jochen H. M. Prehn, Ronald Melki, Stéphane Hunot, and Patrick P. Michel. 2022. "Modelling α-Synuclein Aggregation and Neurodegeneration with Fibril Seeds in Primary Cultures of Mouse Dopaminergic Neurons" Cells 11, no. 10: 1640. https://doi.org/10.3390/cells11101640
APA StyleTourville, A., Akbar, D., Corti, O., Prehn, J. H. M., Melki, R., Hunot, S., & Michel, P. P. (2022). Modelling α-Synuclein Aggregation and Neurodegeneration with Fibril Seeds in Primary Cultures of Mouse Dopaminergic Neurons. Cells, 11(10), 1640. https://doi.org/10.3390/cells11101640